

Abstract
Epigenetic modifications, in particular DNA methylation, are gaining increasing interest as complementary information to DNA mutations for cancer diagnostics and prognostics. We introduce a method to simultaneously profile DNA mutation and methylation events for an array of sites with single site specificity. Genomic (mutation) or bisulphite-treated (methylation) DNA is amplified using nondiscriminatory primers, and the amplicons are then hybridized to a giant magnetoresistive (GMR) biosensor array followed by melting curve measurements. The GMR biosensor platform offers scalable multiplexed detection of DNA hybridization, which is insensitive to temperature variation. The melting curve approach further enhances the assay specificity and tolerance to variations in probe length. We demonstrate the utility of this method by simultaneously profiling five mutation and four methylation sites in human melanoma cell lines. The method correctly identified all mutation and methylation events and further provided quantitative assessment of methylation density validated by bisulphite pyrosequencing.
Keywords: methylation, mutation, DNA array, GMR biosensor, melanoma, melting curve
Graphical abstract
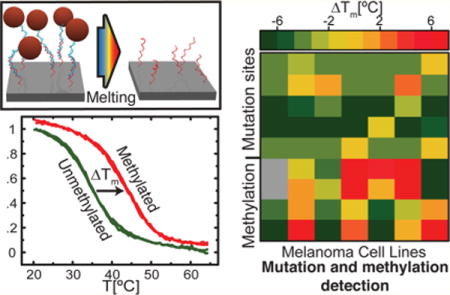
Cancer is a cellular disease caused by the stepwise accumulation of genetic and epigenetic alterations.1,2 Extensive sequencing efforts have identified recurrent genetic mutations that are useful as genetic biomarkers for assessing risk of developing cancer, classifying disease subtypes, predicting response to treatment, and monitoring efficacy of treatment. DNA methylation causes epigenetic silencing of tumor suppressor genes and is studied both for its direct implication in oncogenesis and for its utility as cancer biomarker.3–5 In bladder and colon cancer, the combination of genetic and epigenetic analyses has been proven to have a higher diagnostic value than either of the two approaches applied separately.6,7 However, compared to mutation genotyping, methylation profiling is not a yes-no result. Many promoter regions contain CpG islands, i.e., clusters of multiple CpG dinucleotides (the most common methylation site), and gene silencing mechanisms driven by promoter methylation are generally sensitive to the overall density of methylated sites. Finally, the methylation density may vary between alleles and cells within a single tumor, resulting in a heterogeneous pattern.4,8,9
A variety of techniques have been developed to detect single point mutations in DNA based on amplification,10,11 probe hybridization,12 enzymatic digestion,11 gel electrophoresis,13 or sequencing.14 DNA methylation information is lost during polymerase chain reaction (PCR) amplification, and DNA hybridization is insensitive to the methylation status of the target region. Therefore, a methylation sensitive pretreatment of the DNA has to be employed. The main DNA methylation analysis techniques are based on methylation sensitive enzymatic digestion, affinity enrichment using antibodies specific for methylated cytosine or bisulphite conversion of unmethylated cytosine into uracil (reviewed by Laird9 and Plongthongkum et al.8). Bisulphite conversion is most widely used since a methylation event is converted into a single base alteration (C/T) that can be detected with techniques derived from mutation detection including sequencing array hybridization, methylation sensitive PCR,9 and methylation sensitive melting curve analysis.15,16 Sequencing of bisulphite-converted DNA quantifies the methylation status and allows for comparison of data from different sequencing runs and batches, but it is costly and time-consuming.8 Amplification-based techniques are limited in multiplexing by the number of available fluorophores and the number of channels in the thermocycler. Similarly, melting-based techniques as high resolution melting (HRM) have limited multiplexing capabilities and are only weakly sensitive to A–T and C–G single base substitutions. Array-based methods, such as the Illumina BeadChip (Illumina Inc., San Diego, CA), offer a highly multiplexed site-specific assay. However, after bisulphite conversion and amplification of the template, DNA products comprise mostly three bases (guanine, adenine, and thymine plus residues of methylated cytosine). This reduced sequence complexity makes design of probes for end-point detection complicated and the decreased sequence variation reduces specificity.9
Our work aims to perform methylation and mutation profiling simultaneously in a scalable chip platform that offers highly specific and quantitative DNA methylation and mutation data on a compact, easy-to-use, and potentially low-cost platform. Our approach is based on hybridization of magnetically labeled target DNA to DNA probes tethered to the surface of a GMR biosensor array (Figure 1). The magnetic detection approach has been previously demonstrated for DNA detection17–19 as well as immunoassays.20 To increase the specificity of the DNA hybridization assay, we employed melting curve measurements of the surface-tethered DNA hybrids.21 This avoids conventional assay condition optimization since the target-probe hybrids are exposed to continuously increasing stringency during melting curve measurement.22 Melting curves for surface-tethered DNA probes have also been measured using fluorescence23 and surface plasmon resonance.24,25 Compared to these methods, the GMR biosensors offer high sensitivity, no dependence on temperature and are insensitive to the sample matrix as virtually all biological material is nonmagnetic.20,26
Figure 1.
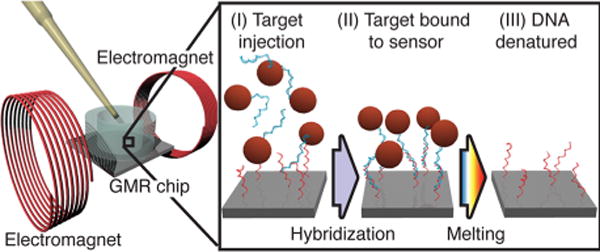
Schematic protocol for the detection of magnetically labeled DNA using GMR biosensors: (I) After denaturation of the reverse strand and labeling, PCR products are injected into the reaction well over the chip. The DNA labeled with MNPs hybridizes to complementary surface-tethered probes for 1 h at 37 °C. (II) The DNA is hybridized to the sensor surface and unbound sample is removed by washing. (III) The temperature is swept from 20 to 65 °C to measure the melting temperature, Tm.
Using our technique, we analyzed melanoma cell lines with known genetic and epigenetic alterations.27,28 More specifically, we investigated mutations in NRAS and BRAF, which are found in 15–25% and 50–70% of melanomas, respectively.29 In addition, we quantitatively measured the methylation status of the promoter regions of the genes encoding retinoic acid receptor β (RARB) and the receptor tyrosine kinase KIT (KIT), which are targeted by hypermethylation in 20–70% and 25–40% of melanomas, respectively.28,30,31
RESULTS AND DISCUSSION
DNA Mutation Analysis
To detect DNA mutations, we PCR amplified the genomic regions of interest using nondiscriminatory primers. The PCR products were then magnetically labeled using biotinylated primers and streptavidin-coated magnetic nanoparticles (MNPs). After magnetic column separation and denaturation of the double-stranded PCR products, ssDNA conjugated to MNPs (MNP-ssDNA) was introduced to the GMR biosensor array where multiple DNA probes were separately tethered to the surface of each sensor.19 Upon hybridization of the injected MNP-ssDNA to surface-tethered complementary probes, GMR biosensors produced changes in sensor magnetoresistive ratio (ΔMR) in proportion to the bound MNPs.32 To genotype a mutation, we employed a set of two probes complementary to the wild type (WT) and mutant type (MT) sequences of the sample (Supporting Information Table S1). During hybridization at low stringency, amplicons hybridized to both WT and MT probes with similar affinity. To obtain single base specificity, stringent washing is typically used after hybridization in DNA microarray. To achieve a more flexible system for detection of single-base mutations, we challenged the hybrids by increasing the temperature and continuously measuring DNA melting simultaneously for all probes on the GMR biosensor array (Figure 1).
Figure 2a shows real-time monitoring of ΔMR during hybridization (1 h at 37 °C) of a known WT sample to probes with a perfect match (WT) or a single-base mismatch (MT) for the BRAF c.1391 G > A mutation. In addition, a biotinylated DNA probe was used as positive reference and a DNA probe with an unspecific sequence was used as negative reference. After 1 h of hybridization, the MT probe gave a slightly higher ΔMR signal than the WT probe, indicating that low-stringency hybridization was insufficient to genotype the WT sample. The positive reference signal reached a higher level than the hybridization probes due to the faster kinetics of the biotin–streptavidin interaction compared to hybridization. After hybridization, the unbound sample was removed by a low-temperature wash at low stringency. Then, the temperature of the sensor was ramped at constant rate from 20 °C until all DNA hybrids melted at 65 °C (Figure 2a). The signal (ΔMR) from the GMR biosensors was corrected for its temperature dependence during ramping using the sensor resistance (R), which is linearly related to the sensor temperature.
Figure 2.
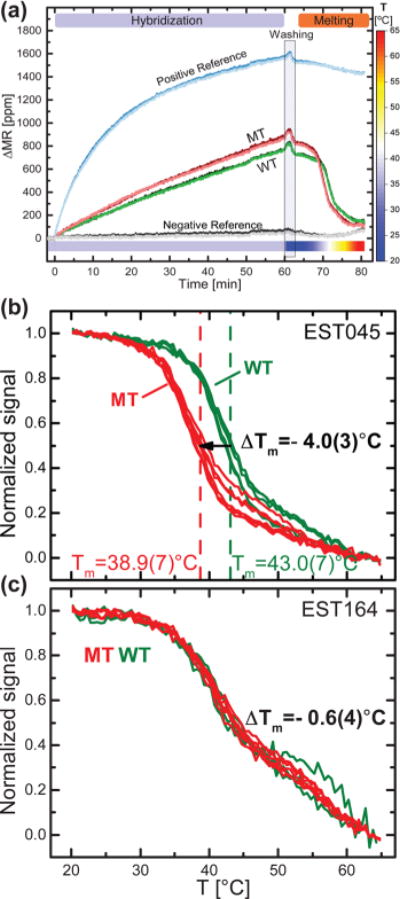
(a) Time series of the signal (ΔMR) from GMR biosensors functionalized with positive and negative references, wild type (WT) and mutant type (MT) probes for the BRAF c.1391 G > A mutation. Each line corresponds to up to three sensors functionalized with the same probe. The measurement was performed with PCR products from EST045 cell line that is WT for the investigated mutation. The sample was injected at t = 2 min. After 60 min hybridization at 37 °C, the chip was washed and the temperature was ramped from 20 to 65 °C while measuring the melting curves. (b) and (c) show melting curves from WT (green) and mutant type MT (red) probes targeting BRAF c.1391 G > A mutation obtained for the indicated cell lines, where the EST045 and EST164 cell lines were wild type and heterozygous mutant, respectively. Signals were normalized by the initial signal at T = 20 °C. The melting temperature Tm is defined as the temperatures at which the normalized curves cross 0.5. ΔTm is the difference in melting temperature between the MT and WT probes. The numbers in parentheses are standard deviations on the last significant digit (n = 4–6). Graphs of the first derivative of the melting curves are available in Supporting Information Figure S3.
Figure 2b shows the melting curve of WT BRAF amplicons hybridized to WT and MT probes for the c.1391G > A mutation. Here, the ΔMR signal was normalized by the initial signal at T = 20 °C. We defined the melting temperature Tm as the temperature at which the signal (ΔMR) dropped to the half of its initial signal (at 20 °C). Each melting experiment was repeated with two identical GMR biosensor chips. Three sensors were functionalized with each probe, thus generating up to six identical melting curves for each probe. The obtained melting curves were found to be highly reproducible—both from sensor to sensor and from chip to chip. The hybrids of the target DNA with WT and MT probes in Figure 2b showed melting temperatures of Tm(WT) = 43.0(7) °C and Tm(MT) = 38.9(7) °C, respectively, where the numbers in parentheses are standard deviations of Tm on the last digit (n ≥ 4). We defined the melting temperature difference, ΔTm, as the difference between the melting temperature from the MT probe and that from the WT probe, ΔTm = Tm(MT) − Tm(WT). Thus, ΔTm < 0 indicates a higher complementarity of the target to the WT probe than the MT probe, and hence that the target is WT. The obtained value ΔTm = −4.0(3) °C is in agreement with the expectation for a single base mismatch between the WT target and MT probe using a nearest neighbor calculation.33 We also note that the lower standard deviation of ΔTm compared to Tm indicates that differences in melting temperatures were more reproducible than their absolute values.
Figure 2c shows melting curves measured for a cell line heterozygous for the BRAF c.1391G > A mutation.27 The melting curves from WT and MT probes were found to overlap, resulting in ΔTm = −0.6(4) °C because the heterozygous sample contains both MT and WT targets, which hybridize to both WT and MT probes. The resulting melting curves from WT and MT probes were both given by the contribution of low-Tm and high-Tm DNA hybrids. Therefore, the melting curves overlapped and presented a lower slope.
DNA Methylation Analysis
We applied a similar detection scheme to analyze the methylation state of specific regions of the target. We employed bisulphite treatment of the genomic DNA (Figure 3a) to convert a methylation event into a single base substitution (C > T). After bisulphite conversion, we amplified the gene promoter region of interest by nondiscriminatory PCR.
Figure 3.
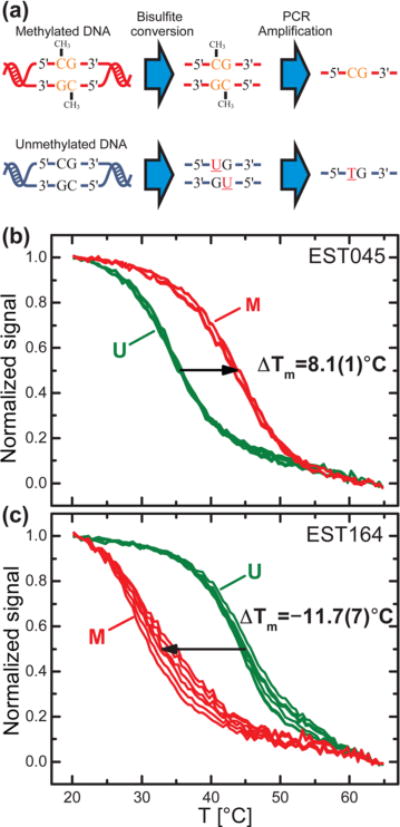
(a) Schematic of the bisulphite conversion process: Upon bisulphite treatment, unmethylated cytosines are converted to uracil whereas 5-methylcytosines are retained. In the subsequent PCR, uracil is substituted by thymine. Thus, the methylated cytosines are mapped to single base alterations (C > T) of the amplicons. (b) and (c) show melting curves from methylated (M, red) and unmethylated (U, green) probes targeting KIT methylation (site p1). The melting curves were measured for (b) the hypermethylated cell line EST045 and (c) the unmethylated cell line EST164. Graphs of the first derivative of the melting curves are available in Supporting Information Figure S3.
Figures 3b and c show melting curves to estimate methylation status of the KIT promoter (site p1) of hypermethylated (EST045) and wild-type (EST164) cell lines.28 The amplicons were hybridized to probes complementary to unmethylated (U) or methylated (M) target DNA. Melting curves were measured as described previously. Here, ΔTm was defined as the melting temperature of the M probe minus that of the U probe, ΔTm = Tm(M) − Tm(U). Thus, a negative ΔTm indicates a higher complementarity of the target to the U probe and a lower degree of methylation. The ∼20 bp region of the KIT promoter investigated includes three CpG sites that can be methylated (sequences in Supporting Information Table S1), and thus we expect higher ΔTm than for single base substitution. For the hyper-methylated cell line in Figure 3b, we found ΔTm = 8.1(1) °C, confirming the hypermethylation status of the KIT promoter, whereas we found ΔTm = −11.7(7) °C for the WT cell line in Figure 3c, indicating the unmethylated status.
Multiplex DNA Profiling of Melanoma Cell Lines
The GMR biosensor array comprises of 64 individual sensors that can be individually functionalized with amino-modified DNA probes. Using the mutation and methylation detection techniques described above, we simultaneously probed three mutation sites in BRAF, two mutation sites in NRAS, two methylation sites in the KIT promoter, and two methylation sites in the RARB promoter in triplicate. We performed mutation and methylation profiling of seven melanoma cell lines. For each cell line, the targeted regions of BRAF and of NRAS were amplified by non- discriminatory PCR. Also, the promoter regions of KIT and RARB were amplified by nondiscriminatory PCR after bisulphite conversion. After magnetic labeling, a mixture of all amplicons from a cell line was injected over the sensor surface. For each cell line, melting curve profiling was repeated with two nominally identical GMR biosensor arrays. The melting curves were analyzed in terms of melting temperatures, and we determined ΔTm for all investigated mutation and methylation sites.
Figure 4a shows the ΔTm values measured for the BRAF c.1391 G > A mutation for all cell lines (other mutation sites are shown in Supplementary Figure S1). Six cell lines showed ΔTm values around ΔTm = −4 °C, indicating a homozygous WT sequence. EST164 is known to be the only cell line with a heterozygous mutation in this site,27 showing ΔTm = −0.5(4) °C, which is significantly different from the other cell lines.
Figure 4.
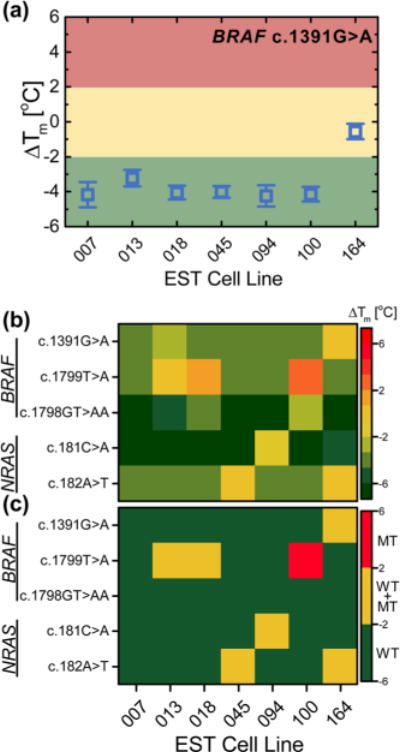
(a) Mutation profiling of melanoma cell lines. ΔTm measured for BRAF c.1391G > A mutation for the seven investigated EST cell lines. Error bars are one standard deviation (n = 4–6). The horizontal lines are threshold values used for genotyping: ΔTm < −2 °C (green) WT, −2 °C < ΔTm < 2 °C (yellow) heterozygous MT, ΔTm > 2 °C (red) homozygous MT. (b) Heat map of ΔTm measured for the mutation and for the investigated EST cell lines. (c) Heat map of measured ΔTm with applied threshold to genotype mutations: WT in green, heterozygous MT in yellow, homozygous MT in red.
The ΔTm values measured for all investigated mutations for each cell line are displayed in the heat map of Figure 4b. Classifying WT (ΔTm < −2 °C), heterozygous MT (−2 °C < ΔTm < 2 °C), and homozygous MT (ΔTm > 2 °C) resulted in the mutation map presented in Figure 4c. All mutations identified in the cell lines were consistent with previous genotyping data.27 For the NRAS c.182 A > T mutation in the cell line EST045, we measured ΔTm = −0.2(4) °C, genotyping the cell line as heterozygous for this mutation; however, the cell line is known to be heterozygous for an A > G substitution in that location (see Supplementary Figure S2). As an MT probe targeting an A > T mutation was employed, both the WT and MT probes were similarly mismatched to the target, resulting in ΔTm close to zero. The absolute values of Tm in Supplementary Figure S2 were comparable to the other investigated mismatched probes confirming the mismatch of the target to both the WT and MT probes. Therefore, an unknown mutation can be detected by a lower Tm from the WT probe, but probes targeting all possible mutations should be included in the assay to perform accurate genotyping.
DNA Methylation Density
Methylation profiling differs substantially from genotyping since the methylation status of each CpG site in the promoter region varies between alleles and within a cell population. Therefore, it requires a different data analysis in terms of methylated fraction of the sample DNA. We measured melting curves using surface-tethered probes targeting two locations of the KIT promoter and two locations of the RARB promoter. The targeted sequences contain one to four CpG sites. Combining multiple investigated sites with the intrinsic variation of the methylation pattern, we obtained a continuous variation of ΔTm for the analyzed cell lines. Figure 5a shows the measured ΔTm values for all cell lines. Here, higher ΔTm in yellow or red indicates higher affinity of the sample to the M probe, i.e., a hypermethylation event. The complex ΔTm pattern is a direct consequence of the intrinsic methylation variation.
Figure 5.
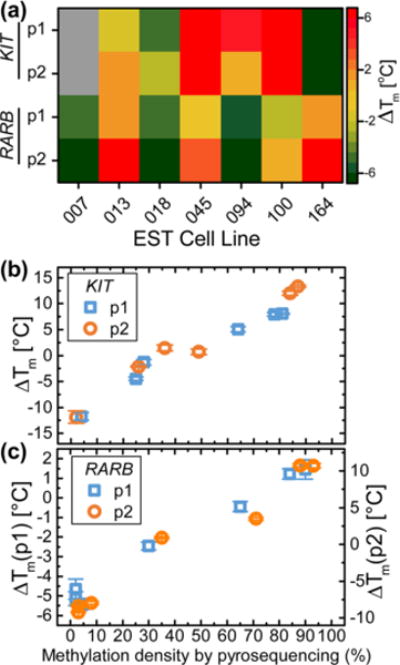
Mutation and methylation profiling of melanoma cell lines. (a) Heat map of ΔTm measured for KIT and RARB methylation probes for the seven investigated EST cell lines. Calculation of ΔTm for EST007 KIT was not possible due to low binding signal. (b) ΔTm values measured for KIT p1 (blue squares) and p2 (orange circles) methylation probe locations vs methylation density measured by pyrosequencing. (b) ΔTm measured for RARB p1 (blue squares) and p2 (orange circles) methylation probe locations vs methylation level measured by pyrosequencing. Error bars are one standard deviation (n = 4–6).
The methylation density was assessed independently by pyrosequencing of the bisulphite-converted DNA. To each target sequence corresponding to the probes, we calculated methylation density depending on both the fraction of methylated sample and the number (1–4) of methylated CpG sites in the region targeted by the probe. Figures 5b and 5c show ΔTm measured using the GMR biosensor versus the methylation density obtained by pyrosequencing for the KIT p1 and p2 probe locations and the RARB p1 and p2 probe locations, respectively. In these plots, each point corresponds to one of the measured cell lines. There is an evident linear correlation between ΔTm and the methylation density (R2 > 0.94 for all probe locations, results of linear regression are given in Supplementary Table S4). For the KIT p1 and p2 probes, the slopes are comparable (∼0.22 °C/%, Figure 5b), whereas for the RARB probes, the slopes for the p1 and p2 probes differ significantly (p1:0.076(5) °C/%, p2:0.22(2) °C/%). The slope for the p2 probe was three times that for the p1 probe because the p2 probe covers three CpG sites whereas the p1 probe only covers one CpG site. Nevertheless, three methylation sites allowed for a more complex pattern of methylation sites and thus the RARB p2 probe showed a broader spread of data around the best linear fit. The probes can be tailored to sacrifice linearity to favor higher values of ΔTm.
These results demonstrate the application of temperature melting on a GMR biosensor as a semiquantitative method for profiling methylation density. High-throughput profiling of genome wide methylation can be performed with single-base resolution using array-based methods like Illumina BeadChips but the specificity of such arrays is limited by lower sequence variability of bisulphite converted DNA.8,9 A quantification of the overall methylation density of a gene promoter can be obtained with methylation-specific melting curve analysis.15,16 Here, we combined the throughput and scalability of arrays with the specificity and flexibility of melting curve analysis. The obtained quantitative profiling was equivalent to the results of pyrosequencing.
CONCLUSION
We have presented an approach for simultaneous DNA mutation and methylation profiling. Our method combines the widespread DNA microarray techniques for both mutation and methylation analysis in a single platform. Melting curves measurements are used to increase the specificity of mutation detection. For methylation detection, the melting curve quantifies the methylation state at a level equivalent to pyrosequencing. The same technique could potentially be employed on a variety of other platforms capable of masuring DNA hybridization vs temperature.
The GMR biosensor platform has a low cross-sensitivity to temperature and provides a sensitive readout. Although it does not offer the extreme throughput as advanced bead microarray systems (e.g., Illumina), in its present format, the GMR biosensor platform can be used for the simultaneous triplicate investigation of about 20 mutation and methylation sites. Nevertheless, the GMR biosensor array has a modular design that can be scaled to include up to thousands of biosensors.32
METHODS
Cells and Reagents
Melanoma cell lines for this study were obtained from The European Searchable Tumour Line Database (ESTDAB: http://www.ebi.ac.uk/ipd/estdab) and were maintained in RPMI-1640 medium containing 10% FBS and antibiotics at 37 °C and 5% CO2. The PCR primers for this study have been modified from Dahl et al.27,28 and were obtained from DNA Technology A/S, Denmark. The sequences can be found in the Supporting Information Table S2. The amine modified DNA probes (sequences in Supporting Information Table S1) were matched for melting temperature calculated with nearest-neighbor model.33 The probes were obtained from DNA Technology A/S. The other reagents: poly(ethylene-alt-maleic anhydride) (Sigma-Aldrich), poly(allylamine hydrochloride) (Polyscience), distilled water (Invitrogen), 1-Ethyl-3-(3-(dimethylamino)propyl)-carbodiimide EDC (Sigma-Aldrich), N-hy-droxysuccinimide NHS (Sigma-Aldrich) 1% bovine serum albumin BSA (Sigma-Aldrich), phosphate buffered saline PBS (Gibco), distilled water (Invitrogen), Tween 20 (Sigma-Aldrich), Urea (Fisher Scientific), 20× saline sodium citrate SSC (Invitrogen), mineral oil (Sigma-Aldrich), MNPs Streptavidin MicroBeads (Miltenyi), magnetic separation columns μ Columns (Miltenyi).
DNA Extraction and Bisulphite Treatment
Genomic DNA was isolated using the Qiagen AllPrep DNA/RNA/Protein Mini kit (Qiagen GmbH, Hilden, Germany) and quantified using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). Bisulphite conversion of DNA (500 ng) was carried out using the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA) according to the manufacturer’s protocol.
PCR Amplification
Prior to the GMR biosensor assay, PCR was performed using a Veriti 96-Well Thermal Cycler (Applied Biosystems) and TEMPase Hot Start Polymerase (VWR). All amplifications were initiated with enzyme activation and DNA denaturation at 95 °C for 15 min, 40 cycles of 95 °C for 30 s, 56 °C for 30 s and 72 °C for 30 s, followed by a final incubation at 72 °C for 10 min. All primer sequences used are listed in the Supporting Information Table S2.
Magnetic Labeling
Products from PCR amplification of each cell line were processed as described by Rizzi et al.19 to obtain ss-DNA target conjugated with MNPs. Briefly, for each amplified region, 10 μL of PCR products were mixed with 10 μL of stock solution of streptavidin-coated MNPs (MACS Streptavidin Microbeads, cat: 130–048–102, Miltenyi Biotec Norden AB, Lund, Sweden) and incubated for 30 min at 37 °C. A magnetic separation column (μ Column, Miltenyi Biotec Norden AB, Lund, Sweden) was prepared by washing with 1 mL of 1% Tween 20 and 1 mL of 0.1% BSA containing 0.05% Tween 20, sequentially. After the conjugation with magnetic particles, the five PCR products were mixed and added to the column under an applied magnetic field for separation. While the target DNA-bead complexes were trapped in the column, the reverse strands were denatured and removed by adding 2 mL of 6 M Urea solution at 75 °C. Then, the applied magnetic field was removed, and the conjugated complexes were eluted with 100 μL of 2× SSC buffer.
Sensor Preparation
The GMR biosensor chip with an array of 8 × 8 sensors was fabricated as previously described.34 The chip surface was chemically activated following Kim et al.35 Briefly, the chip was sequentially washed with acetone, methanol, and isopropanol. After cleaned with oxygen plasma, the chip was treated with poly(ethylene-alt-maleic anhydride) for 5 min. Then, the chip was washed with distilled water, and baked at 110 °C for 1 h using a hot plate. After treatement with poly(allylamine hydrochloride) for 5 min, the chip was washed with the distilled water, and activated with a mixture of NHS and EDC for 1 h. After the chip was washed again with distilled water, a robotic arrayer (sciFlexarrayer, Scienion) was used to print the amino-modified DNA probes on different sensors (Supplementary Table S2). Each DNA probe was dissolved in 3× SSC buffer at 20 μM, and was used for printing in triplicate. Four sensors on the same chip were functionalized with biotinylated DNAs as positive references, and another set of four sensors was functionalized with DNA non-complementary to any of the PCR amplified regions as negative references. The chip was stored at room temperature until use.
Data Acquisition for Temperature Calibration
Prior to the assay, the thermal resistivity of each GMR sensor was characterized for temperature calibration. The temperature coefficient for each chip was obtained by linear fitting to resistance measurements at 20, 30, 40, 50, and 60 °C. The temperature coefficient was then used to trace the instantaneous temperature of each sensor.
GMR Biosensor Assay
The measurement setup described previously26,36 was employed to measure the response of GMR sensor to MNPs. The temperature of the GMR chips was controlled by means of a Peltier element coupled to the chip. The Peltier element was driven by a LFI3751 control unit (Wavelength Electronics, USA) with a Pt1000 thermoresistor (Supporting Information Figure S4). First, the chip was washed with 3 mL of 0.1% bovine serum albumin (BSA, Sigma-Aldrich) and 0.05% Tween20 (Sigma-Aldrich). The chip was then blocked with 100 μL of 1% BSA for 1 h in a shaker. After blocking, the chip was washed with 3 mL of 0.1% BSA and 0.05% Tween20, followed by washing with 3 mL of distilled water. Prior to sample injection, a baseline signal measurement was performed for 2 min at 37 °C. After sample injection, the DNA hybridization signals from different sensors were measured for 1 h at 37 °C. Then, the temperature was lowered to 20 °C to inhibit further binding of the sample and stabilize DNA hybrids. The chip was washed five times with 100 μL of 0.05× SSC to remove unbound sample. 150 μL of 0.05× SSC were left in the reaction well and covered with 50 μL of mineral oil to prevent evaporation. Melting curves for all the probes were measured while temperature was ramped from 20 to 65 °C at 0.05 °C/s. The temperature was then swept back to 20 °C to obtain temperature reference signals.
Data Processing
The MNPs are detected as a variation of the magnetoresistivity (ΔMR) of the GMR sensors. The temperature coefficients of ΔMR were calculated for each sensor as in Hall et al.26 using the temperature reference signals. It was necessary to use a fifth order polynomial to account for nonlinear temperature dependency at high temperature (Supporting Information Figure S5). After temperature correction, the melting curves measured between 20 to 65 °C were normalized by their initial value at 20 °C. The melting temperature Tm was defined as the temperature at which the normalized signal is 0.5. To calculate Tm, a first order polynomial was fitted to the melting curve in the region of interest. Each mutation (methylation) site was genotyped using two probes. ΔTm was defined as the difference between Tm measured for the probe complementary to the mutated (methylated) sequence minus Tm measured for the probe complementary to the Wild Type (unmethylated) sequence.
Pyrosequencing
The methylation status of the RARB and KIT promoter regions was analyzed by pyrosequencing using the PyroMark Q24 platform (Qiagen) and subsequent data analysis using the PyroMark Q24 software. Primer sequences are listed in Supplementary Table S3. DNA enzymatically methylated in vitro (CpGenome Universal Methylated DNA; Millipore) and unmethylated DNA prepared by whole genome amplification (WGA; GenomePlex, Sigma-Aldrich) was used as methylation-positive and -negative controls, respectively.
Supplementary Material
Acknowledgments
This work was supported by the Danish Council for Independent Research (Postdoc project, DFF-4005–00116), the Autoimmunity Center of Excellence (U19AI110491), and the Center for Cancer Nanotechnology Excellence (U54CA151459 and U54CA199075).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.7b03053.
Table S1, DNA probe sequences; Table S2, PCR primer sequences; Table S3, pyrosequencing primers sequences; Figure S1, ΔTm measured for all probes and for all cell-lines; Figure S2, absolute Tm values measured for NRAS c.182 A > T and denaturing gradient gel electrophoresis of NRAS exon2 PCR products; Figure S3, first temperature derivative of melting curves shown in Figures 2 and 3; Figure S4, pictures of temperature control unit with mounted chip and its mounting in Helmholtz coils for measurements; Figure S5, temperature dependence of center and side tones of GMR sensor (PDF)
ORCID
Giovanni Rizzi: 0000-0003-3512-7389
Notes
The authors declare the following competing financial interest(s): S.X.W. has related patents or patent applications assigned to the Stanford University and out-licensed for potential commercialization. S.X.W. has stock or stock options in MagArray, Inc., which has licensed relevant patents from the Stanford University for commercialization of GMR biosensor chips.
References
- 1.Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Esteller M, Corn PG, Baylin SB, Herman JG. A Gene Hypermethylation Profile of Human Cancer 1. Cancer. 2001;3:3225–3229. [PubMed] [Google Scholar]
- 4.Baylin SB. DNA Methylation and Gene Silencing in Cancer. Nat Clin Pract Oncol. 2005;2:S4–S11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- 5.Herman JG, Baylin SB. Gene Silencing in Cancer in Association with Promoter Hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 6.Serizawa RR, Ralfkiær U, Steven K, Lam GW, Schmiedel S, Schüz J, Hansen AB, Horn T, Guldberg P. Integrated Genetic Epigenetic and Analysis of Bladder Cancer Reveals an Additive Diagnostic Value of FGFR3Mutations and Hypermethylation Events. Int J Cancer. 2011;129:78–87. doi: 10.1002/ijc.25651. [DOI] [PubMed] [Google Scholar]
- 7.Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, Hamilton SR, Issa JJ. Integrated Genetic and Epigenetic Analysis Identifies Three Different Subclasses of Colon Cancer. Proc Natl Acad Sci USA. 2007;104:18654–18659. doi: 10.1073/pnas.0704652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plongthongkum N, Diep DH, Zhang K. Advances in the Profiling of DNA Modifications: Cytosine Methylation and beyond. Nat Rev Genet. 2014;15:647–661. doi: 10.1038/nrg3772. [DOI] [PubMed] [Google Scholar]
- 9.Laird PW. Principles and Challenges of Genome-Wide DNA Methylation Analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 10.Orita M, Suzuki Y, Sekiya T, Hayashi’ K. Rapid and Sensitive Detection of Point Mutations and DNA Polymorphisms Using the Polymerase Chain Reaction. Genomics. 1989;5:874–879. doi: 10.1016/0888-7543(89)90129-8. [DOI] [PubMed] [Google Scholar]
- 11.Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N. Enzymatic Amplification of Beta-Globin Genomic Sequences and Restriction Site Analysis for Diagnosis of Sickle Cell Anemia. Science. 1985;230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- 12.Wang DG, Fan J-B, Siao C-J, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, Kruglyak L, Stein L, Hsie L, Topaloglou T, Hubbell E, Robinson E, Mittmann M, Morris M, Shen N, Kilburn D, et al. Large-Scale Identification, Mapping and Genotyping of Single-Nucleotide Polymorphisms in the Human Genome. Science. 1998;280:1077–1082. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]
- 13.Orita M, Iwahana H, Kanazawat H, Hayashi K, Sekiya T. Detection of Polymorphisms of Human DNA by Gel Electrophoresis as Single-Strand Conformation Polymorphisms. Proc Natl Acad Sci USA. 1989;86:2766–2770. doi: 10.1073/pnas.86.8.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metzker ML. Sequencing Technologies — the next Generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 15.Worm J, Aggerholm A, Guldberg P. In-Tube DNA Methylation Profiling by Fluorescence Melting Curve Analysis. Clin Chem. 2001;47:1183–1189. [PubMed] [Google Scholar]
- 16.Wojdacz TK, Dobrovic A. Methylation-Sensitive High Resolution Melting (MS-HRM): A New Approach for Sensitive and High-Throughput Assessment of Methylation. Nucleic Acids Res. 2007;35:e41–e41. doi: 10.1093/nar/gkm013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu L, Yu H, Akhras MS, Han SJ, Osterfeld S, White RL, Pourmand N, Wang SX. Giant Magnetoresistive Biochip for DNA Detection and HPV Genotyping. Biosens Bioelectron. 2008;24:99–103. doi: 10.1016/j.bios.2008.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rizzi G, Østerberg FW, Dufva M, Fougt Hansen M. Magnetoresistive Sensor for Real-Time Single Nucleotide Polymorphism Genotyping. Biosens Bioelectron. 2014;52:445–451. doi: 10.1016/j.bios.2013.09.026. [DOI] [PubMed] [Google Scholar]
- 19.Rizzi G, Lee J-R, Guldberg P, Dufva M, Wang SX, Hansen MF. Denaturation Strategies for Detection of Double Stranded PCR Products on GMR Magnetic Biosensor Array. Biosens Bioelectron. 2017;93:155–160. doi: 10.1016/j.bios.2016.09.031. [DOI] [PubMed] [Google Scholar]
- 20.Gaster RS, Hall Da, Nielsen CH, Osterfeld SJ, Yu H, Mach KE, Wilson RJ, Murmann B, Liao JC, Gambhir SS, Wang SX. Matrix-Insensitive Protein Assays Push the Limits of Biosensors in Medicine. Nat Med. 2009;15:1327–1332. doi: 10.1038/nm.2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rizzi G, Østerberg FW, Henriksen AD, Dufva M, Hansen MF. On-Chip Magnetic Bead-Based DNA Melting Curve Analysis Using a Magnetoresistive Sensor. J Magn Magn Mater. 2015;380:215–220. [Google Scholar]
- 22.Dufva M, Petersen J, Poulsen L. Increasing the Specificity and Function of DNA Microarrays by Processing Arrays at Different Stringencies. Anal Bioanal Chem. 2009;395:669–677. doi: 10.1007/s00216-009-2848-z. [DOI] [PubMed] [Google Scholar]
- 23.Petersen J, Poulsen L, Petronis S, Birgens H, Dufva M. Use of a Multi-Thermal Washer for DNA Microarrays Simplifies Probe Design and Gives Robust Genotyping Assays. Nucleic Acids Res. 2008;36:e10. doi: 10.1093/nar/gkm1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiche JB, Buhot A, Calemczuk R, Livache T. Temperature Effects on DNA Chip Experiments from Surface Plasmon Resonance Imaging: Isotherms and Melting Curves. Biophys J. 2007;92:935–946. doi: 10.1529/biophysj.106.097790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagner CE, Macedo LJA, Opdahl A. Temperature Gradient Approach for Rapidly Assessing Sensor Binding Kinetics and Thermodynamics. Anal Chem. 2015;87:7825–7832. doi: 10.1021/acs.analchem.5b01518. [DOI] [PubMed] [Google Scholar]
- 26.Hall DA, Gaster RS, Osterfeld SJ, Murmann B, Wang SX. GMR Biosensor Arrays: Correction Techniques for Reproducibility and Enhanced Sensitivity. Biosens Bioelectron. 2010;25:2177–2181. doi: 10.1016/j.bios.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dahl C, Christensen C, Jönsson G, Lorentzen A, Skjødt ML, Borg A, Pawelec G, Guldberg P. Mutual Exclusivity Analysis of Genetic and Epigenetic Drivers in Melanoma Identifies a Link Between p14ARF and RARβ. Signaling Mol Cancer Res. 2013;11:1166–1178. doi: 10.1158/1541-7786.MCR-13-0006. [DOI] [PubMed] [Google Scholar]
- 28.Dahl C, Abildgaard C, Riber-Hansen R, Steiniche T, Lade-Keller J, Guldberg P. KIT Is a Frequent Target for Epigenetic Silencing in Cutaneous Melanoma. J Invest Dermatol. 2015;135:516–524. doi: 10.1038/jid.2014.372. [DOI] [PubMed] [Google Scholar]
- 29.Dahl C, Guldberg P. The Genome and Epigenome of Malignant Melanoma. APMIS. 2007;115:1161–1176. doi: 10.1111/j.1600-0463.2007.apm_855.xml.x. [DOI] [PubMed] [Google Scholar]
- 30.Hoon DSB, Spugnardi M, Kuo C, Huang SK, Morton DL, Taback B. Profiling Epigenetic Inactivation of Tumor Suppressor Genes in Tumors and Plasma from Cutaneous Melanoma Patients. Oncogene. 2004;23:4014–4022. doi: 10.1038/sj.onc.1207505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furuta J, Umebayashi Y, Miyamoto K, Kikuchi K, Otsuka F, Sugimura T, Ushijima T. Promoter Methylation Profiling of 30 Genes in Human Malignant Melanoma. Cancer Sci. 2004;95:962–968. doi: 10.1111/j.1349-7006.2004.tb03184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaster RS, Xu L, Han S-J, Wilson RJ, Hall DA, Osterfeld SJ, Yu H, Wang SX. Quantification of Protein Interactions and Solution Transport Using High-Density GMR Sensor Arrays. Nat Nanotechnol. 2011;6:314–320. doi: 10.1038/nnano.2011.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.SantaLucia J. A Unified View of Polymer, Dumbbell, and Oligonucleotide DNA Nearest-Neighbor Thermodynamics. Proc Natl Acad Sci U S A. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osterfeld SJ, Yu H, Gaster RS, Caramuta S, Xu L, Han S-J, Hall DA, Wilson RJ, Sun S, White RL, Davis RW, Pourmand N, Wang SX. Multiplex Protein Assays Based on Real-Time Magnetic Nanotag Sensing. Proc Natl Acad Sci U S A. 2008;105:20637–20640. doi: 10.1073/pnas.0810822105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim D, Marchetti F, Chen Z, Zaric S, Wilson RJ, Hall DA, Gaster RS, Lee JR, Wang J, Osterfeld SJ, Yu H, White RM, Blakely WF, Peterson LE, Bhatnagar S, Mannion B, Tseng S, Roth K, Coleman M, Snijders AM, et al. Nanosensor Dosimetry of Mouse Blood Proteins after Exposure to Ionizing Radiation. Sci Rep. 2013;3:1–8. doi: 10.1038/srep02234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hall DA, Gaster RS, Lin T, Osterfeld SJ, Han S, Murmann B, Wang SX. GMR Biosensor Arrays: A System Perspective. Biosens Bioelectron. 2010;25:2051–2057. doi: 10.1016/j.bios.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.