

Abstract
Cross-species transmission of viruses poses a sustained threat to public health. Due to increased contact between humans and other animal species the possibility exists for cross-species transmissions and ensuing disease outbreaks. By using conventional PCR amplification and next generation sequencing, we obtained 130 partial or full genome kobuvirus sequences from humans in a sentinel cohort in Vietnam and various mammalian hosts including bats, rodents, pigs, cats, and civets. The evolution of kobuviruses in different hosts was analysed using Bayesian phylogenetic methods. We estimated and compared time of origin of kobuviruses in different host orders; we also examined the cross-species transmission of kobuviruses within the same host order and between different host orders. Our data provide new knowledge of rodent and bat kobuviruses, which are most closely related to human kobuviruses. The novel bat kobuviruses isolated from bat roosts in Southern Vietnam were genetically distinct from previously described bat kobuviruses, but closely related to kobuviruses found in rodents. We additionally found evidence of frequent cross-species transmissions of kobuviruses within rodents. Overall, our phylogenetic analyses reveal multiple cross-species transmissions both within and among mammalian species, which increases our understanding of kobuviruses genetic diversity and the complexity of their evolutionary history.
Keywords: kobuvirus, next generation sequencing, Bayesian phylogenetics, cross-species transmission
1. Introduction
Many human pathogens that are of current public health concern are zoonotic or originate from domestic or wild animals before adapting to humans (Woolhouse and Gowtage-Sequeria 2005; Zhuo and Feschotte 2015). Pathogens that infect multiple host species are more likely to emerge in human populations than host-restricted pathogens (Taylor, Latham, and Woolhouse 2001). It is therefore important to understand the genetic diversity of viruses circulating in both human and animal populations and to identify possible cross-species transmissions.
Aichi Virus A-1 (hereafter ‘AiV-A1’) is one of several recently discovered enteric viruses associated with acute gastroenteritis in humans which belongs to the Kobuvirus genus (Yamashita et al. 1991). Kobuviruses have a single-stranded, positive-sense RNA genome 6.9–8.4 kb in length. The genome structure is composed of a leader protein (L), followed by structural capsid proteins (VP0, VP3, and VP1) and non-structural proteins (2 A–2 C and 3 A–3 D) (Yamashita et al. 1998, 2003; Ng et al. 2012). To date, kobuviruses have been found in multiple host taxa including humans, lagomorpha (rabbits), chiroptera (bats), rodentia, artiodactyla, carnivora, and aves, as well as in environmental samples (Yamashita et al. 2003; Reuter et al. 2008, 2010; Kapoor et al. 2011a; Wu et al. 2012; Lodder et al. 2013; Smits et al. 2013; Oem et al. 2014; Pankovics et al. 2015, 2016). Kobuviruses are currently grouped into six different species (AiV A to F) and unassigned kobuviruses according to the latest report of International Committee on Taxonomy of Viruses (https://talk.ictvonline.org/taxonomy/). However, it is still unclear whether the broad host range of kobuviruses is indicative of cross-species transmission events. Additionally, there may be novel and unknown kobuviruses present in other host species; their existence may increase the scope for further transmission events between animals and humans.
Vietnam has a large human population and high livestock density (Gerber et al. 2005). Approximately half of the Vietnamese population participate in small-scale animal production (GSO 2013; Rabaa et al. 2015). Many smallholdings have minimum biosecurity and house several different animal species within a small area. Housing different animal species within a single enclosure creates an environment where interspecies transmission of pathogens may be more likely. In addition, eating wild animals that have been hunted and commercial wildlife farming (such as bamboo rats, civets, porcupines, wild boars, and bats) are both popular in Vietnam as a result of population growth and urban prosperity (Cooper 1995; Drury 2011; Deutsch and Murakhver 2012). Such activities increase the likelihood of pathogen transfer between wildlife, humans and domestic animals (Krause 1994).
VIZIONS (The Vietnamese Initiative for Zoonotic Infections) is a multidisciplinary project aiming to understand the emergence of zoonotic viral pathogens by analysing virus populations in: 1, hospitalised patients, 2, people who maintain regular contact with animals, and 3, diverse animal populations (Rabaa et al. 2015). The aim of this study, conducted with VIZIONS, was to describe the genetic diversity of currently circulating kobuviruses in human and non-human hosts in Vietnam. In particular, we aimed to investigate the likelihood of putative cross-species transmissions of kobuviruses.
2. Materials and methods
2.1 Viral sequences
The VIZIONS project instigated the collection of faecal samples from diarrheal patients admitted to Dong Thap Provincial hospital in southern Vietnam (n = 671) along with rectal swabs and fecal samples from people who had occupational or residential exposure to animals (a ‘high risk’ cohort) from November 2012 to May 2016 (n = 551). Faecal samples and rectal swabs from domestic pigs (n = 278) and farmed wild boars (n = 7) were also collected. Rodent faecal samples (n = 315) were collected from rats that were purchased at the local wet market. Bat faecal samples (n = 179) were collected from roosting sites. Faecal samples from 45 dogs and 13 cats that belonged to members of the high-risk cohort were also collected (Carrique-Mas et al. 2015).
In total 2,059 samples were subjected to a viral diagnostic algorithm and put through a next generation sequencing (NGS) process (Rabaa et al. 2015). Samples were enriched for viruses, after which nucleic acid was extracted (de Vries et al. 2011, 2012). All nucleic acids were transcribed and subjected to second strand synthesis. Prepared dsDNA were sequenced on an Illumina HiSeq platform. The resulting dsDNA from each sample was sheared and fractionated to 400–500 bp in length, after which Illumina adapters with a unique barcode were ligated to the fragments. Resulting libraries were sequenced with the Illumina HiSeq platforms to generate 3–4 million 250 bp paired-end reads per sample (Munnink et al. 2017). The quality of the reads was assessed using FASTQC (Davis et al. 2013). Primers and lower quality bases were trimmed using Cutadapt (Marcel 2011). Read pairs likely to have derived from host organisms (limited to humans, pigs and rodents), bacteria or archaea were identified and removed with Kraken (Wood and Salzberg 2014). De novo assembly on remaining read pairs (presumed viral) was conducted using CLC Assembly Cell V 5.0.4 (https://www.qiagenbioinformatics.com/products/clc-assembly-cell/). The resultant contigs were compared with the NCBI GenBank nt database using BLASTn.
Additionally, we obtained kobuvirus sequences from a second VIZIONS study, which exploited PCR screening for multiple viruses in rodents and civets in Vietnam in April and May 2014. In this study, 375 rats were trapped from four different provinces: Dong Thap, Long An, An Giang, and Dak Lak. An additional 32 bamboo rats and 30 civets were purchased from farms in Dak Lak province. Faecal samples and tissue samples (e.g. liver, spleen, and lung) were aseptically collected during post-mortem and stored in sterile tubes at −20°C. RNA was extracted from 10% (w/v) of faecal sample suspension using a MagNA Pure 96 Viral NA small volume kit (Roche) and an automated extractor (Roche) as described previously (Van Dung et al. 2014). Reverse transcription was performed using SuperScript III Reverse Transcriptase (Invitrogen, UK) following the manufacturer recommendations. cDNA was screened for kobuviruses using a nested PCR protocol with generic primers targeting the VP1 region (Van Dung et al. 2016). Characterization of viral sequences in PCR amplification positive samples was performed by sequencing of the VP1 region using primers defined in Supplementary Table S1.
Newly identified sequences were aligned with kobuvirus reference sequences from NCBI database using ClustalW (Larkin et al. 2007). We used SSE V1.3 software to calculate uncorrected distance of new sequences against reference sequences (Simmonds 2012). All kobuvirus sequences determined in this study were deposited in GenBank under accession numbers KT944135 to KT944197, MF947381 to MF947447. GenBank accession numbers, hosts and geographic locations of all kobuvirus sequences used in this study are listed in Supplementary Table S2.
2.2 Phylogenetic analysis
We estimated the phylogenies of the dated samples using a Bayesian Markov chain Monte Carlo (MCMC) method which was implemented using the Bayesian evolutionary analysis in BEAST v.1.8.2 (Drummond et al. 2012). Different combinations of substitution models, clock models and population size models were evaluated by using the path sampling sampling to estimate marginal likelihoods (Baele et al. 2012). The best fitting model was a general time-reversible model with a gamma distribution (G) across sites as the substitution model, with an uncorrelated log-normal relaxed molecular clock model and with a constant size coalescent process prior over the phylogenies. The MCMC chains were run for 100 million iterations with sub-sampling every 10,000 iterations and 10% burn-in. MCMC convergence and effective sample size of parameter estimates were evaluated using Tracer 1.5 (http://beast.bio.ed.ac.uk). Our sequence data were obtained from samples collected over a short time period (within 5 years) so cannot be used to generate robust estimates of the substitution rate and time to the most recent common ancestor (TMRCA). Therefore, we applied an informative prior on the substitution rate parameter using a normal distribution (mean rate = 2 × 10−3 substitutions/site/year; SD =5 × 10−4) based on published evolution rates of the VP1 gene of related picornaviruses in both humans and non-humans, as is fully described in our previous study (Lu et al. 2016). An asymmetric model with Bayesian Stochastic Search Variable selection (BSSVS) was applied to identify statistically significant transition rates between host species and geographic locations. Maximum clade credibility (MCC) trees were summarized using Tree Annotator and visualized using FigTree v1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/).
3. Results
3.1 genetic diversity of Vietnamese kobuviruses
We obtained in total 130 partial genome sequences of kobuviruses from six different host populations in Vietnam: humans, pigs, rodents, bats, cats, and civets. PCR screening identified a high prevalence of kobuviruses in bamboo rats (81%; 26/32), rats (21%; 77/375), and civets (17%; 5/30). Sixty-three VP1 gene sequences were then amplified from bamboo rats (n = 23), rats (n = 39), and civets (n = 1) from samples which were PCR positive for kobuviruses. From the 2,097 extractions subjected to NGS, we detected 157 kobuvirus RNA sequences from total contigs with HSPs (high-scoring segment pairs) scores ≥500 by running BLASTn. The highest prevalence of kobuviruses was in bats (44%, 78/179), followed sequentially by rats (17%, 55/315), cats (15%, 2/13), and pigs (8%, 22/285). Kobuviruses had a much lower prevalence in humans (0.8%, 11/1, 222) than in the other mammalian species except in dogs (0, 0/45). From these contigs we obtained 19 kobuvirus genome sequences with length >5,000 bp and an additional 48 VP1 gene sequences with length 500–831 bp.
We next reconstructed a Bayesian MCMC tree for 416 kobuvirus VP1 sequences isolated from 7 different host orders and 15 countries, including newly identified Vietnamese sequences in this study (n = 130) and all available kobuvirus sequences with full length VP1 (n = 286) retrieved from GenBank (Supplementary Table S2). The kobuviruses VP1 sequences in our study clustered together with previously isolated kobuviruses from the same host taxon: human-derived kobuviruses fell into the AiV-A1 clade; pig-derived sequences fell into Clade AiV-C1; cat-derived kobuviruses grouped with the other feline kobuvirus sequences in Clade AiV-A4; the civet kobuvirus sequence fell between feline and canine kobuviruses clades; and rodent isolates grouped with previously described mouse kobuviruses. However, the kobuviruses found in Vietnamese bats were divergent from previously found bat kobuviruses, but were closely related to the clade of rodent kobuviruses (Fig. 1).
Figure 1.

Simplified MCC tree representing the time-scale phylogeny of kobuviruses from all hosts based on the VP1 gene. The nodes of Kobuvirus species that have multiple sequences are collapsed into polytomies. Branches are coloured according to their descendent nodes annotated by the different host orders, with the key for colours shown on the left. Bayesian posterior probabilities are given at the nodes of major clades. The names of kobuvirus species (AiV A to F and unassigned kobuvirus) are presented on the node if represented by multiple sequences or above the branch if represent by a single sequence. Red stars indicated Vietnamese sequences obtained in that host species in this study. Cross-species transmission with Bayes Factor ≥5 is indicated with arrows. The scale bars indicate the branch lengths in year. The tree being labelled with sequences accession numbers is in Supplementary Fig. S1.
To better determine the zoonotic origin and the cross-species transmission of kobuviruses, we applied a discrete trait model with BSSVS on MCMC time-scaled phylogenies. The estimated origin time of the entire Kobuvirus genus is over 1,000 years but with considerable uncertainty (mean = 1,091 years; 95% highest posterior density (HPD): 587–1,704 years) (Fig. 1). Three major clades can be seen in the simplified kobuvirus phylogeny (Fig. 1). One major clade (AiV-A), which is the largest species with a wider host range than other kobuvirus species was composed of kobuviruses found in humans (AiV-A1), canines (AiV-A2), sewage (AiV-A3), felines (AiV-A4), birds (AiV-A5), and rats (AiV-A6). The TMRCA of AiV-A was estimated as 200 years ago, but again with considerable uncertainty (mean = 198 years; 95% HPD: 112–750 years), with carnivora as the most likely ancestor host order (host posterior probability = 0.59). The second clade was comprised of AiV-B, C, and D, with the majority of kobuviruses from artiodactyla (swine, bovine, and ovine) and 1 sequence (KF006985) from ferrets (Smits et al. 2013). The recently discovered kobuviruses from rabbits (AiV-E) and bats (AiV-F and unassigned kobuviruses), were distinct from the two main clades.
We identified several statistically supported (Bayes Factor ≥ 5) transmission events between different host taxa during the evolution of kobuviruses (Fig. 1). There was a very strongly supported host jump from artiodactyla (sheep) to carnivora (ferret) with BF = 62. There was also a strongly supported jump from bats to rabbits with BF = 20. We additionally detected cross-species transmission between bats and rodents, but with only moderate support for either direction (BF = 8 from rodents to bats; BF = 5 from bats to rodents). We further observed that rodent kobuvirus clade and Vietnamese bat kobuvirus clade were the two clades that were the most closely related to human kobuvirus (AiV-A1) (Fig. 1). However, the genetic distance between rodent/bat kobuvirus clades and AiV-A1 clade was substantial providing no direct evidence that AiV-A1 originated directly from rodents or bats. In addition, there were possible historical host jumps from carnivora to humans, and from carnivora to birds, but with weak support only (both with BF < 5).
3.2 Kobuviruses in rodents
We isolated 77 rodent-derived kobuvirus sequences from Vietnam, including 41 sequences from six murine species (including Bandicota indica, Rattus argentiventer, Rattus norvegicus, Rattus tanezumi, Rattus exulans, and Rattus losea) and 23 sequences from bamboo rats (Rhizomys pruinosus), which belong to the family Spalacidae. Kobuvirus VP1 sequences recovered from the six murine species exhibited a varied nucleotide sequence identity of 78–100%; Bamboo rat-derived kobuvirus sequences Bamboo rat-derived kobuvirus sequences were highly similar to each other (nucleotide sequence distances in the range of 0–2.9%). Phylogenetic analysis showed that these sequences fell into a rodent kobuvirus clade together with five previously reported kobuvirus sequences (Phan et al. 2011; Ng et al. 2012; Firth et al. 2014) with a TMRCA of 73 years (with 95%HPD 55–236 years) (Figs 1 and 2). The entire rodent kobuvirus phylogeny could be grouped into four subclades. One subclade was composed of 41 murine Vietnamese sequences, the second subclade was composed of all bamboo rat sequences, 12 murine Vietnamese sequences, as well as three sequences isolated from brown rats (R.norvegicus) in USA (Firth et al., 2014). A sewage-derived kobuvirus KoV-SewKTM (Ng et al. 2012) and a Vietnamese rat kobuvirus (KT944172) grouped together and formed a third subclade. One kobuvirus (JF755427), isolated from a wild Canyon mouse (Peromyscus crinitus, Cricetidae) in the USA (Phan et al. 2011), was divergent from all the other sequences (Fig. 2). These results confirmed that rodent-derived kobuviruses are present in multiple rodent families across wide geographic distances.
Figure 2.

MCC tree representing the time-scale phylogeny of kobuviruses found in rodents based on the VP1 gene. Branches are coloured according to their descendent nodes annotated by the different rodent families, with the key for colours shown on the left. The accession number with the host species and isolation countries of sequences are showed at the tips. Sequences obtained in this study are highlighted in blue. Bayesian posterior probabilities are given at the nodes of major clades. The scale bars indicate the branch lengths in year.
Our ancestral reconstruction also revealed extremely frequent transmissions of kobuviruses between the six species within the Muridae family (Fig. 3 and Supplementary Table S3). Kobuviruses from B.indica may have been transmitted to four rodent species with mean transmission rate = 0.02–0.06 exchanges year−1 and BF = 9–85. Kobuviruses from R.argentiventer have been transmitted to two other rodent species with mean transmission rate = 0.01 and 0.05 exchanges year−1 and BF = 17 and 27, respectively. Kobuviruses from R.norvegicus have been transmitted to three other rodent species with mean transmission rate = 0.01–0.02 exchanges year−1 and BF = 5–6; transmissions in reverse direction were also supported (Supplementary Table S3). There was a notable host jump from Muridae to Spalacidae, with the specific species R.norvegicus acting as a donor for R.pruinosus, with mean transmission rate = 0.01 and BF = 5 (Figs 2 and 3). These results indicate that rodent-derived kobuviruses may be transmitted frequently within the same rodent family, and may also transmit between different rodent families.
Figure 3.
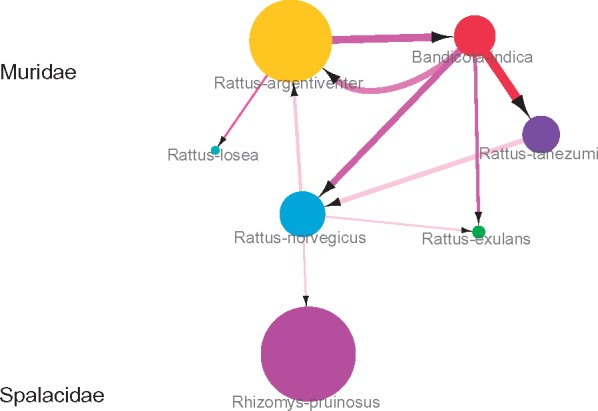
Transmission network of kobuviruses in 7 rodent species. Size of nodes describes the number of kobuviruses from each species. Arrows show the direction of transmission between species; the arrow width indicates per capita transmission rate (exchanges year−1). Transmission rates and BF support are given in Supplementary Table S3.
3.3 Kobuviruses in bats
We isolated 38 partial kobuvirus genome sequences (length = 523–6,859 nt) from bat faeces collected from bat roosts in Vietnam, which were closely related to rodent kobuviruses. Comparing the entire coding region (2, 164aa), a Vietnamese bat kobuvirus (MF947437) showed 87% aa identity to a Vietnamese rodent kobuvirus (MF947446) as well as to KoV-SewKTM. MF947437 also showed 74% aa identity to a mouse kobuvirus (JF755427), but only 40% aa identity to a known bat kobuvirus (KJ641686). Comparing the VP1 region only, the Vietnamese bat kobuvirus sequences shared 82–92% aa identity to rodent kobuviruses (Supplementary Fig. S2). The time-scaled phylogeny of VP1 regions showed that the clade of Vietnamese bat kobuviruses and the clade of rodent kobuviruses were close related with a TMRCA estimated at ∼100 years but with some uncertainty (mean 99 years, 95% HPD 55–164 years). Vietnamese bat sequences may have a more recent TMRCA with mean estimate of 43 years (95% HPD 10–78 years) (Fig. 1). Further metagenomic investigations indicated that the bat faecal samples belonged to two bat species: Scotophilus kuhlii and Murina ussuriensis. These two bat species are within the family Vespertilionidae and are commonly found in Asia (Parker, Rambaut, and Pybus 2008). Further ancestral reconstruction of Vietnamese bat kobuvirus VP1 sequences indicated possible cross-species transmission between the two species (with BF > 100), despite the limited sample size (Fig. 4). In addition, we observed that the Vietnamese bat kobuvirus VP1 sequences were separated into two subclades. It is unclear whether the clustering of the two subclades was associated with bat species differentiation as the uneven samples from two bat species (37 samples from S.kuhlii n = 37 and 1 sample from M.ussuriensis), and is also not related to geographic separation though the bat faecal samples, which were known to be collected from two districts (Cao Lanh and Chau Tang) in Dong Thap province (Supplementary Fig. S3).
Figure 4.

MCC tree representing the time-scale phylogeny of kobuviruses found in Vietnamese bats based on the VP1 gene. Branches are coloured according to their descendent nodes annotated by the different bat species, with the key for colours shown on the left. Sequences obtained in this study are highlighted in blue. Bayesian posterior probabilities are given at the nodes of major clades. The scale bars indicate the branch lengths in year.
3.4 Kobuviruses in humans
The human kobuviruses (AiV-A1) were closely related to rodent kobuviruses and Vietnamese bat kobuviruses (Fig. 1). Here, we obtained six human AiV-A1 sequences from Dong Thap province in Vietnam duriNg 2012–16. Four (MF947441, MF947407, MF947442, and MF947408) were identified in hospitalized young children (2 months to 2 years old) with diarrhoea, only one patients (where MF947442 were isolated from) had contact with animals (kept dogs and cats at home) in the record. Two (MF947409 and MF947410) were isolated from adult farm workers who had close contact with animals (including domestic pigs, chickens, ducks, dogs, and cats). None of them had contact with rodents and bats in the record. The estimated mean TMRCA of AiV-A1 overall (including three Genotypes A–C) was 73 years with 95% HPD= 41–218 years. Genotypes A and B have similar TMRCAs, originating in the early 1980 s (mean = 35 years and 95% HPD = 17–40 years for Genotype A; mean = 33 years and 95% HPD = 22–43 years for Genotype B). Phylogenetic analysis of VP1 gene sequences showed that the six AiV-A1 sequences found in our study all belonged to Genotype B with an estimated TMRCA around 2006. They were close related to other Genotype B sequences isolated from southeast China and South Korea in 2008 and 2010. Two AiV-A1 sequences (EU143279 and EU143278) have been previously detected in Dong Thap province in 2002 and 2003; they belong to Genotype A (Pham et al., 2008). This result may indicate the current dominant AiV-A1 in Dong Thap province in Vietnam is Genotype B, the ancestor of which existed and spread in this area over a 10-year time frame (Supplementary Fig. S4).
3.5 Kobuviruses in cats and civets
We identified one partial genome sequence of kobuvirus (MF947443) from a cat faecal sample in Dong Thap province. It had high nucleotide identity with other cat kobuviruses (90–93%). Asian palm civets (Paradoxurus hermaphrodites; Viverridae family) are farmed in Vietnam for the production of civet coffee (Deutsch and Murakhver 2012). We isolated a kobuvirus VP1 sequence (KT944174) from civet faecal samples taken at a civet farm in Dak Lak province. It represents the first reported kobuvirus in Asian palm civets. This civet kobuvirus sequence was comparable to canine and feline kobuviruses, with 70–77% nt identity. Phylogenetic analysis showed the civet sequence fell into the clade composed of kobuviruses isolated from carnivora, including cats (Felis catus), dogs (Canis lupus), and foxes (Vulpes vulpes), but it belonged to a separate sub-lineage from canine kobuviruses and feline kobuviruses (Figs 1 and 5). In addition, the phylogeny of kobuviruses found in carnivora (including Canidae, Felidae, and Viverridae families) matched the branching order of their host phylogeny (Cornelis et al. 2012), consistent with no cross-species transmission events.
Figure 5.

MCC tree representing the time-scale phylogeny of kobuviruses found in carnivora according to the VP1 gene. Branches are coloured according to their descendent nodes gannotated by the different carnivora families, with the key for colours shown on the left. The accession number with the host species and isolation countries of sequences are showed at the tips. Sequences obtained in this study are highlighted in blue. Bayesian posterior probabilities are given at the nodes of major clades. The scale bars indicate the branch lengths in year.
3.6 Kobuviruses in pigs
In a previous study, we described the genetic diversity and spatiotemporal transmission of porcine kobuviruses (AiV-C1) in Vietnam using the VP1 sequences isolated between February and April 2012. We identified three co-circulating subclades in domestic pigs in Dong Thap province (Lu et al. 2016). In the present study, we obtained seven VP1 gene sequences of AiV-C1 in Dong Thap province from March 2013 to October 2014: four sequences (MF947411, MF947412, MF947413, and MF947416) belong to Clade 1, two (MF947414 and MF947415) belong to Clades 2 and 1 sequence (MF947417) belongs to Clade 3, indicating the three sub-clades of AiV-C1 are continuously co-circulating in Vietnam (Supplementary Fig. S5).
4. Discussion
Kobuviruses have a broad host range and can be transmitted either by physical contact, or indirectly from an environment contaminated by faecal shedding (Reuter Reuter, Boros, and Pankovics 2011). Therefore, it is plausible that kobuviruses can be transmitted between different host taxa. Phylogenetic analysis supported the possibility of historical transmission events between different host orders, including jumps between artiodactyla and carnivora, between rabbits and bats, and between rodents and bats.
The rodentia is the largest order of mammals, comprising about 43% of all mammalian species (Huchon et al. 2002). Rodents are known to harbour pathogens that cause > 60 human diseases (Meerburg, Singleton, and Kijlstra 2009), as well as viruses with unknown zoonotic potential (e.g. kobuviruses, rosaviruses). Pathogens from rodents can be transmitted to humans or other animals either directly, by handling animals during trapping or farming, or indirectly, by occupying areas inhabited by rodents (Meerburg, Singleton, and Kijlstra 2009). In this study, although we found kobuviruses in both rodents and humans, they were phylogenetically distinct. We did not detect kobuviruses from people that had had close contact with rodents. We found that kobuviruses may transmit frequently between multiple wild rodent species in Vietnam. Notably, we found a high prevalence of kobuviruses (81%) in farmed bamboo rats, which likely originated from kobuviruses carried by wild rodents.
Similar to rodents, bats are one of the most diverse and widely distributed mammals. They are a natural reservoir for many important emerging infectious viruses such as severe acute respiratory syndrome-related coronaviruses, lyssaviruses, filoviruses, and mammalian paramyxovirus (Johara et al. 2001; Leroy et al., 2005; Calisher et al. 2006; Drexler et al. 2012a). Bat-derived viruses can be transmitted to other host species via contamination of food with bat excreta, or direct exposure to bat blood or excreta, or infection via intermediate hosts (Quan et al. 2013). Pathogens derived from other animals may also spill over into bats, as some bat species feed on small vertebrates such as rats and birds (Morrison 1983). Therefore, there is a possibility of a bat-derived virus transmitting to rodents living in the same environment or vice versa. In this study, we described a rodent-like kobuvirus from bat specimens in Dong Thap province in Vietnam. However, direct evidence of virus spill over between rodents and bats is still lacking. Ancestral reconstruction analysis indicated that kobuviruses are more likely to jump from rodents to bats, although this result had only weak support. Given that the common ancestor of the two lineages existed >100 years ago, there could also be an unknown host of both lineages and the host jump event may have occurred somewhere a long time before. In addition, the Vietnamese bat kobuviruses found in our study were distinct from the species AiV-F and unassigned kobuviruses previously found in bats, indicating that bat-derived kobuviruses are quite diverse. In comparison, high genetic diversity has also been observed among bat-derived hepaciviruses, which raises the possibility that other hepaciviruses, including HCV, may have originated from bats (Kapoor et al. 2011b; Drexler et al. 2012b; Quan et al. 2013). However, for kobuvirus, although the clade of Vietnamese bat kobuviruses is adjacent to the clade of human kobuviruses, there is no clear evidence to show that AiV-A1 originated from bats. Further investigation and discovery of kobuvirus sequences over larger geographical scales and in other host species may help to clarify the evolutionary connections.
In conclusion, we have identified kobuviruses in animal and human samples originating from Vietnam during between 2012 and 2016. This is the first large-scale study investigating kobuviruses in human and animals. The sequences obtained in this study provide new information on the occurrence of these viruses in a wider range of hosts than previously known. Cross-species transmission events are revealed at the host order, family and species levels. Further molecular and epidemiological studies are required to determine the relevance, distribution, and diversity of kobuviruses in larger geographic scales and over time.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
This work was funded by VIZIONS, a strategic award from the Wellcome Trust (ref. WT/093724). Stephen Baker is a Sir Henry Dale Fellow, jointly funded by the Wellcome Trust and the Royal Society (ref. 100087/Z/12/Z).
Data availability
GenBank accession numbers, hosts, and geographic locations of all kobuvirus sequences used in this study are listed in Supplementary Table S2. The tree file representing the time-scale phylogeny of kobuviruses from all hosts based on the VP1 gene (Fig. 1 and Supplementary Fig. S1) is available in Dryad (doi: 10.5061/dryad.98q55).
Conflict of interest: None declared.
Supplementary Material
References
- Baele G. et al. (2012) ‘Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty’, Molecular Biology and Evolution, 29: 2157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calisher C. H. et al. (2006) ‘Bats: Important Reservoir Hosts of Emerging Viruses’, Clinical Microbiology Reviews, 19: 531–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrique-Mas J. J. et al. (2015) ‘The Baseline Characteristics and Interim Analyses of the High-Risk Sentinel Cohort of the Vietnam Initiative on Zoonotic InfectiONS (VIZIONS)’, Scientific Reports, 5: 17965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper J. E. (1995) ‘Wildlife Species for Sustainable Food Production’, Biodiversity and Conservation, 4: 215–9. [Google Scholar]
- Cornelis G. et al. (2012) ‘Ancestral Capture of Syncytin-Car1, A Fusogenic Endogenous Retroviral Envelope Gene Involved in Placentation and Conserved in Carnivora’, Proceedings of the National Academy of Sciences of the United States of America, 109: E432–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. P. A. et al. (2013) ‘Kraken: A Set of Tools for Quality Control and Analysis of High-Throughput Sequence Data’, Methods, 63: 41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries M. et al. (2011) ‘A Sensitive Assay for Virus Discovery in Respiratory Clinical Samples’, PLoS One, 6: e16118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries M. et al. (2012) ‘Performance of VIDISCA-454 in Feces-Suspensions and Serum’, Viruses, 4: 1328–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch J., Murakhver N. (2012). They Eat That?: A Cultural Encyclopedia of Weird and Exotic Food from around the World. Santa Barbara, CA: ABC-CLIO. [Google Scholar]
- Drexler J. F. et al. (2012a) ‘Bats Host Major Mammalian Paramyxoviruses’, Nature Communications, 3: 796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler J. F. et al. (2012b) ‘Bats Worldwide Carry Hepatitis E Virus-Related Viruses That Form a Putative Novel Genus within the Family Hepeviridae’, Journal of Virology, 86: 9134–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J. et al. (2012) ‘Bayesian Phylogenetics with BEAUti and the BEAST 1.7’, Molecular Biology and Evolution, 29: 1969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury R. (2011) ‘Hungry for Success: Urban Consumer Demand for Wild Animal Products in Vietnam’, Conservation and Society, 9: 247–57. [Google Scholar]
- Firth C. et al. (2014) ‘Detection of Zoonotic Pathogens and Characterization of Novel Viruses Carried by Commensal Rattus norvegicus in New York City’, MBio, 5: e01933-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber P. et al. (2005) ‘Geographical Determinants and Environmental Implications of Livestock Production Intensification in Asia’, Bioresource Technology, 96: 263–76. [DOI] [PubMed] [Google Scholar]
- GSO. (2013). Statistical Yearbook of Vietnam 2013. Ha Noi, Vietnam: GSO. [Google Scholar]
- Huchon D. et al. (2002) ‘Rodent Phylogeny and a Timescale for the Evolution of Glires: Evidence from an Extensive Taxon Sampling Using Three Nuclear Genes’, Molecular Biology and Evolution, 19: 1053–65. [DOI] [PubMed] [Google Scholar]
- Johara M. Y. et al. (2001) ‘Nipah Virus Infection in Bats (Order Chiroptera) in Peninsular Malaysia’, Emerging Infect Dis, 7: 439–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A. et al. (2011a) ‘Characterization of a Canine Homolog of Human Aichivirus’, Journal of Virology, 85: 11520–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A. et al. (2011b) ‘Characterization of a Canine Homolog of Hepatitis C Virus’, Proceedings of the National Academy of Sciences of the United States of America, 108: 11608–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause R. M. (1994) ‘Dynamics of Emergence’, The Journal of Infectious Diseases, 170: 265–71. [DOI] [PubMed] [Google Scholar]
- Larkin M. A. et al. (2007) ‘Clustal W and Clustal X Version 2.0’, Bioinformatics (Oxford, England), 23: 2947–8. [DOI] [PubMed] [Google Scholar]
- Leroy E. M. et al. (2005) ‘Fruit Bats as Reservoirs of Ebola Virus’, Nature, 438: 575–6. [DOI] [PubMed] [Google Scholar]
- Lodder W. J. et al. (2013) ‘Aichi Virus in Sewage and Surface Water, The Netherlands’, Emerging Infectious Diseases, 19: 1222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L. et al. (2016) ‘Evolution and Phylogeographic Dissemination of Endemic Porcine Picornaviruses in Vietnam’, Virus Evolution, 2: vew001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcel M. (2011) ‘Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads’, EMBnetjournal, 17: 10–2. [Google Scholar]
- Meerburg B. G., Singleton G. R., Kijlstra A. (2009) ‘Rodent-Borne Diseases and Their Risks for Public Health’, Critical Review of Microbiology, 35: 221–70. [DOI] [PubMed] [Google Scholar]
- Morrison D. W. (1983) ‘Bat Ecology’, Science (New York, N.Y.), 219: 961–2. [DOI] [PubMed] [Google Scholar]
- Munnink B. B. O. et al. (2017) ‘Characterization of Posa and Posa-like Virus Genomes in Fecal Samples from Humans, Pigs, Rats, and Bats Collected from a Single Location in Vietnam’, Virus Evolution, 3: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T. F. et al. (2012) ‘High Variety of Known and New RNA and DNA Viruses of Diverse Origins in Untreated Sewage’, Journal of Virology, 86: 12161–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oem J. K. et al. (2014) ‘Novel Kobuvirus Species Identified from Black Goat with Diarrhea’, Veterinary Microbiology, 172: 563–7. [DOI] [PubMed] [Google Scholar]
- Pankovics P. et al. (2016) ‘Novel Picornavirus in Domestic Rabbits (Oryctolagus Cuniculus Var. domestica)’, Infection, Genetics and Evolution, 37: 117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankovics P. et al. (2015) ‘Identification and Complete Genome Analysis of Kobuvirus in Faecal Samples of European Roller (Coracias Garrulus): For the First Time in a Bird’, Archives of Virology, 160: 345–51. [DOI] [PubMed] [Google Scholar]
- Parker J., Rambaut A., Pybus O. G. (2008) ‘Correlating Viral Phenotypes with Phylogeny: Accounting for Phylogenetic Uncertainty’, Infection, Genetics and Evolution, 8: 239–46. [DOI] [PubMed] [Google Scholar]
- Pham N. T. et al. (2008) ‘Sequence Analysis of the Capsid Gene of Aichi Viruses Detected from Japan, Bangladesh, Thailand, and Vietnam’, Journal of Medical Virology, 80: 1222–7. [DOI] [PubMed] [Google Scholar]
- Phan T. G. et al. (2011) ‘The Fecal Viral Flora of Wild Rodents’, PLoS Pathogens, 7: e1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan P. L. et al. (2013) ‘Bats Are a Major Natural Reservoir for Hepaciviruses and Pegiviruses’, Proceedings of the National Academy of Sciences of the United States of America, 110: 8194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabaa M. A. et al. (2015) ‘The Vietnam Initiative on Zoonotic Infections (VIZIONS): a Strategic Approach to Studying Emerging Zoonotic Infectious Diseases’, EcoHealth, 12: 726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter G. et al. (2008) ‘Candidate New Species of Kobuvirus in Porcine Hosts’, Emerging Infectious Diseases, 14: 1968–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter G., Boros A., Pankovics P. (2011) ‘Kobuviruses - A Comprehensive Review’, Reviews in Medical Virology, 21: 32–41. [DOI] [PubMed] [Google Scholar]
- Reuter G. et al. (2010) ‘Kobuvirus in Domestic Sheep, Hungary’, Emerging Infectious Diseases, 16: 869–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P. (2012) ‘SSE: A Nucleotide and Amino Acid Sequence Analysis Platform’, BMC Research Notes, 5: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits S. L. et al. (2013) ‘Metagenomic Analysis of the Ferret Fecal Viral Flora’, PLoS One, 8: e71595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor L. H., Latham S. M., Woolhouse M. E. (2001) ‘Risk Factors for Human Disease Emergence’, Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 356: 983–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dung N. et al. (2014) ‘Prevalence, Genetic Diversity and Recombination of Species G Enteroviruses Infecting Pigs in Vietnam’, Journal of General Virology, 95: 549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dung N. et al. (2016) ‘Large-Scale Screening and Characterization of Enteroviruses and Kobuviruses Infecting Pigs in Vietnam’, Journal of General Virology, 97: 378–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood D. E., Salzberg S. L. (2014) ‘Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments’, Genome Biology, R46.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse M. E., Gowtage-Sequeria S. (2005) ‘Host Range and Emerging and Reemerging Pathogens’, Emerging Infectious Diseases, 11: 1842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. et al. (2012) ‘Virome Analysis for Identification of Novel Mammalian Viruses in Bat Species from Chinese Provinces’, Journal of Virology, 86: 10999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. et al. (2003) ‘Isolation and Characterization of a New Species of Kobuvirus Associated with Cattle’, The Journal of General Virology, 84: 3069–77. [DOI] [PubMed] [Google Scholar]
- Yamashita T. et al. (1991) ‘Isolation of Cytopathic Small round Viruses with BS-C-1 Cells from Patients with Gastroenteritis’, The Journal of Infectious Diseases, 164: 954–7. [DOI] [PubMed] [Google Scholar]
- Yamashita T. et al. (1998) ‘Complete Nucleotide Sequence and Genetic Organization of Aichi Virus, a Distinct Member of the Picornaviridae Associated with Acute Gastroenteritis in Humans’, Journal of Virology, 72: 8408–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo X., Feschotte C. (2015) ‘Cross-Species Transmission and Differential Fate of an Endogenous Retrovirus in Three Mammal Lineages’, PLoS Pathogens, 11: e1005279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GenBank accession numbers, hosts, and geographic locations of all kobuvirus sequences used in this study are listed in Supplementary Table S2. The tree file representing the time-scale phylogeny of kobuviruses from all hosts based on the VP1 gene (Fig. 1 and Supplementary Fig. S1) is available in Dryad (doi: 10.5061/dryad.98q55).
Conflict of interest: None declared.