

Abstract
A series of experimental studies, along with DFT calculations, are reported that provide a detailed view into the mechanism of Ullmann coupling of phenols with aryl halides in the presence of catalysts generated from Cu(I) and bidentate, anionic ligands. These studies encompass catalysts containing anionic ligands formed by deprotonation of 8-hydroxyquinoline, 2-pyridylmethyl tert-butyl ketone, and 2,2,6,6-tetramethylheptane-3,5-dione. Three-coordinate, heteroleptic species [Cu(LX)OAr]− were shown by experiment and DFT calculations to be the most stable complexes in catalytic systems containing 8-hydroxyquinoline or 2-pyridylmethyl tert-butyl ketone and to be generated reversibly in the system containing 2,2,6,6-tetramethylheptane-3,5-dione. These heteroleptic complexes were characterized by a combination of 19F NMR, 1H NMR, and UV–vis spectroscopy, as well as ESI-MS. The heteroleptic complexes generated in situ react with iodoarenes to form biaryl ethers in high yields without evidence for an aryl radical intermediate. Measurements of 13C/12C isotope effects showed that oxidative addition of the iodoarene occurs irreversibly. This information, in combination with the kinetic data, shows that oxidative addition occurs to the [Cu(LX)OAr]− complexes and is turnover-limiting. A Hammett analysis of the effect of phenoxide electronic properties on the rate of the reaction of [Cu(LX)OAr]− with iodotoluene also is consistent with oxidative addition of the iodoarene to an anionic phenoxide complex. Calculations by DFT suggest that this oxidative addition is followed by dissociation of I− and reductive elimination of the biaryl ether from the resulting neutral Cu(III) complex.
Graphical Abstract
INTRODUCTION
Catalytic arylation of phenols with aryl halides to form biaryl ethers is an important process in organic synthesis because biaryl ether cores are widespread in natural products, molecules of pharmaceutical significance, and agrochemicals.1 Currently, such structural motifs can be generated with palladium catalysts,1 but they are formed more commonly with copper catalysts, and the first synthetic method was developed using a copper reagent. In 1905, Ullmann synthesized biaryl ethers from the reaction of phenols with aryl bromides in the presence of KOH.2 However, the classical Ullmann procedure for biaryl ether formation and other related processes required stoichiometric amounts of copper reagents and high reaction temperatures (typically >160 °C), which severely limited the synthetic scope of this transformation.3 Much effort has been spent recently by many groups to improve the original copper-mediated reaction protocols for the coupling of aryl halides with phenols and other nucleophiles by conducting the reactions with added ancillary ligands (Scheme 1).3a,4 Ancillary ligands used for this chemistry have included phenanthrolines,5 bipyridines,6 dia-mines,7 dipyridylimines,8 amino acids,9 8-hydroxyquinolines,10 diketones,11 pyridylketones,12 and others.13 These widely used ancillary ligands typically fall into two structural types: neutral, bidentate ancillary ligands (LL), such as phenanthrolines and bipyridines, and anionic, bidentate ancillary ligands [H(LX)], such as 8-hydroxyquinolines and diketones.
Scheme 1.

Ullmann Coupling Reactions Catalyzed by Ligated Copper Complexes
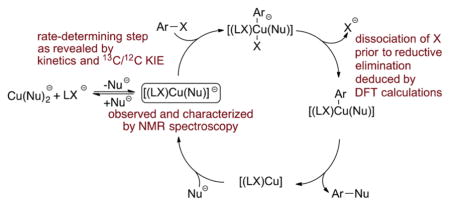
Recently, much effort has been spent by our group and others to elucidate the mechanisms of the coupling processes that form C–N, C–O, and C–C bonds catalyzed by copper complexes of neutral, dative ligand, such as the Ullmann amination,14 the Ullmann biaryl ether formation,15 the Goldberg reaction,16 and the Hurtley reaction.17 The mechanisms of these transformations have long been debated, primarily due to the lack of identification of discrete reactive intermediates involved in the catalytic reactions and experimental studies on the structure, dynamics, and reactivity of these intermediates. Thus, our group synthesized and characterized a series of reactive intermediates containing phenanthrolines, bipyridines, and diamines, and less reactive intermediates lacking ancillary ligands.14–17 Based on kinetic studies and competition experiments on the reactions of these intermediates with aryl iodides, we deduced a mechanism for the catalytic process (Scheme 2) that involved a three-coordinate complex (LL)Cu(Nu) (LL = neutral ancillary ligand, Nu = nucleophile) as the reactive intermediate. Such species have also been prepared in situ and proposed as reactive intermediates based on kinetic studies,14–18 cyclic voltammetric studies,19 and DFT calculations.20 As such, there is a consensus that three-coordinate (LL)Cu(Nu) species form when the catalyst bears neutral bidentate ligands (LL) and that such complexes react with aryl halides to generate the coupled products (Scheme 2).21 Although radical and nonradical pathways have been proposed for the reaction of haloarenes with (LL)Cu(Nu), there is now a consensus based on experimental tests for radical intermediates, as well as DFT calculations, that this oxidative addition occurs without the intermediacy of free aryl radical intermediates.
Scheme 2.
Mechanism of Ullmann Coupling Reactions Catalyzed by Complexes of Netural Bidentate Ancillary Ligands
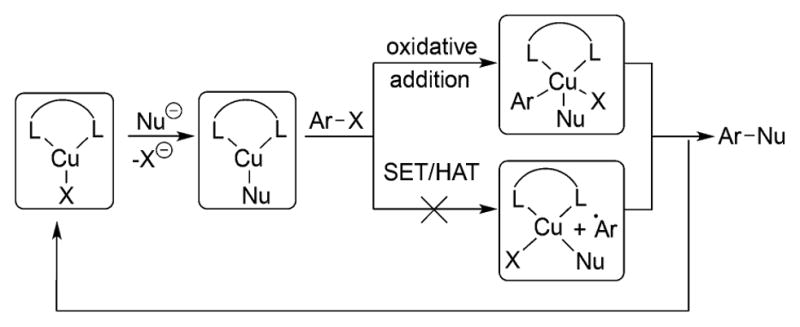
In contrast, existing studies on the mechanism of the coupling of aryl halides with heteroatom nucleophiles catalyzed by copper complexes of anionic ancillary ligands, instead of neutral ancillary ligands, have led to more ambiguous conclusions. If the starting species is a Cu(I) complex with an anionic ligand, then a mechanism involving initial reaction of the nucleophile would generate a three-coordinate anionic copper complex, and such species have not been clearly identified.22
Due to the ambiguity about the formation of such species, mechanisms involving oxidative addition of the aryl halide to a neutral Cu(I) species lacking the nucleophile to form a Cu(III) intermediate have been considered.23 The Cu(III) intermediate would then react with the nucleophile. Experimental22a,24 and theoretical20,22a,24 investigations of the mechanism of coupling reactions catalyzed by CuI ligated to anionic ligands have been reported recently, but no agreement on the identity of the resting state and the rate-determining step was reached, mainly due to the lack of clear experimental characterization of complexes present in the reaction.4a One hydroxo and phenoxo complex ligated by a diketonate ligand were stated to be generated in solution, and a mass corresponding to these compounds was observed by ESI-MS.22a However, the NMR spectra of these complexes are dynamic and are ambiguous or inconsistent with the structures. The reactions of these complexes with haloarenes are limited, and the results did not allow conclusions to be drawn on the chemical or kinetic competence of these complexes to be intermediates in the catalytic cycle. Mechanistic data on the reactions of the complexes of the nucleophiles with haloarenes were gained predominantly by DFT calculations, and the barriers computed do not match well with the barriers of the catalytic reactions. Thus, more detailed studies on a broader range of compounds are needed.
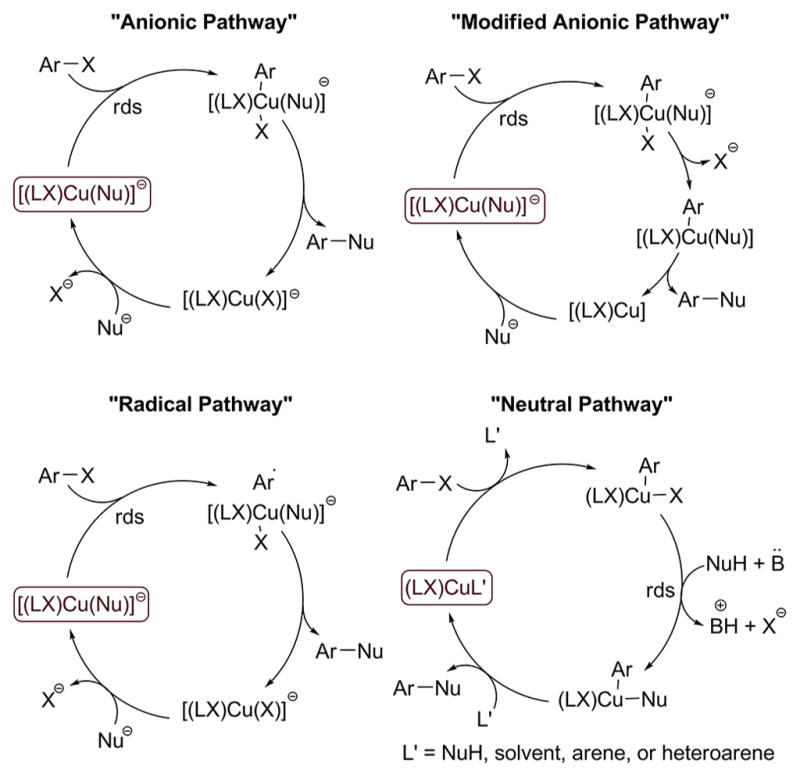
The catalytic cycles that have been proposed, largely on the basis of DFT calculations, for coupling reactions catalyzed by CuI complexes of anionic ancillary ligands are shown in Scheme 3. A detailed rendering of these proposed catalytic cycles to clarify their distinction is also provided in Scheme 4. The first two catalytic cycles, termed “anionic pathways” are analogous to the one proposed for the same transformation catalyzed by copper complexes of neutral ancillary ligands. In these catalytic cycles, [(LX)Cu(Nu)]− reacts with aryl halides in the rate-determining step (RDS). The copper center in the anionic [(LX)Cu(Nu)]− is likely more electron-rich than that in the neutral (LL)Cu(Nu), and [(LX)Cu(Nu)]− could, therefore, react with aryl halides more rapidly than (LL)Cu(Nu). This predicted higher rate is consistent with experimental results on systems in which CuI-catalyzed C–X coupling reactions proceed at lower temperatures in the presence of complexes formed from anionic ancillary ligands than in the presence of complexes formed from neutral ancillary ligands.3a
Scheme 3.
Possible Catalytic Cycles Proposed for Couplings of Aryl Halides with Various Nucleophiles Catalyzed By CuI Coordinated to Anionic Ligands
Scheme 4.

A Series of Reaction Pathways for CuI-Catalyzed Coupling Reactions in the Presence of Anionic Ancillary Ligands
The two anionic pathways in Scheme 3 diverge after the oxidative addition step and are termed the “anionic pathway” and “modified anionic pathway.” In the anionic pathway, reductive elimination occurs to form the ether product from the five-coordinate, anionic copper(III) complex containing one halide, one phenoxide, and one aryl group, in addition to the anionic ancillary ligand. In the modified anionic pathway, reductive elimination is preceded by dissociation of halide from the five-coordinate complex to form a four-coordinate neutral copper-(III) complex containing one phenoxide, one aryl group, and the anionic ligand.
The anionic pathway could be expected to occur because it parallels that for the Ullmann reactions catalyzed by complexes of neutral bidentate ligands.14 The modified anionic pathway was proposed by Jutand and co-workers to occur for reactions of hydroxide or phenoxide anions with iodobenzene in the presence of copper catalysts bearing enolates of diketones as ancillary ligands. These conclusions were based on data gained by cyclic voltammetric studies and DFT calculations,22a,24 but the activation energy computed by DFT (11.3 kcal/mol) is much lower than those corresponding to the experimental reaction rates. The origin of this difference was not addressed.
A third catalytic cycle in Scheme 3, termed the “radical pathway,” occurs by electron transfer. In this proposed mechanism, reaction of the aryl halide with [(LX)Cu(Nu)]− is the rate-determining step20a and could be envisioned to occur because of the high electron density and anionic charge on the copper complex. Such pathways have been proposed for many years,4a and it was proposed recently in one publication reporting DFT calculations.20a However, more recent DFT calculations by Fu predict that this electron-transfer process would have an unreasonably high barrier.20b A radical pathway was also challenged by other researchers based on its inconsistency with the experimental data.4a
A fourth mechanism in Scheme 3, termed the “neutral pathway,” occurs by reaction of the haloarene with the neutral CuI species (LX)Cu(NuH) to form (LX)Cu(X)(Ar) via the neutral (LX)Cu(η2-ArI) complex. In this pathway, the (LX)Cu fragment could be stabilized by a neutral form of the nucleophile. The oxidative addition would be followed by rate-limiting displacement of the halide from the CuIII intermediate by an anionic nucleophile to generate a second CuIII-species, (LX)-Cu(Nu)(Ar). This mechanism proposed by Fu and coworkers20b based on DFT calculations was computed to have the lowest barriers for coupling of amines with iodoarenes in the presence of copper complexes of anionic ancillary ligands and is reminiscent of the well-studied PdII-catalyzed coupling reactions in which oxidative addition of haloarenes is followed by nucleophilic displacement of the halide by nucleophiles.25
Thus, four different mechanisms outlined in Scheme 3 have been proposed in published work for coupling reactions catalyzed by copper complexes ligated by bidentate anionic ligands. These proposals have been made largely or exclusively on the basis of computations by DFT. CuI-species containing anionic ancillary ligands that are potential intermediates or lead to reactive intermediates in the catalytic coupling reactions to form C–O or C–N bonds have not been prepared, clearly characterized by NMR spectroscopy, and evaluated for their competence to be intermediates in the catalytic cycle.4a,22b In the absence of such data, efforts to distinguish between these mechanisms rest on a weak foundation.
Herein we present a combination of detailed experimental and computational studies on the mechanism of the Ullmann arylation of phenols in the presence of anionic ancillary ligands. Three-coordinate CuI complexes of the general formula K[(LX)Cu(OAr)] were prepared in situ, characterized by NMR spectroscopy, UV–vis spectroscopy, and ESI-MS. For the systems containing 8-hydroxyquinoline or 2-pyridylmethyl tert-butyl ketone as the LX ligand, K[(LX)Cu(OAr)] complexes were the most stable copper intermediates, as determined by experimental observations and DFT calculations, implying that these three-coordinate species are the resting states of the catalysts. For 2,2,6,6-tetramethylheptane-3,5-dione as the LX ligand, K[(LX)Cu(OAr)] was found to be generated reversibly in an equilibrium lying at the side of free ligand and the homoleptic cuprates K[Cu(OAr)2]. Kinetic experiments and DFT calculations of catalysts containing each of the three ligands support a pathway by which rate-determining oxidative addition of the iodoarene to the K[(LX)Cu(OAr)] complex occurs and is followed by dissociation of the halide anion and reductive elimination from a neutral CuIII intermediate.
RESULTS AND DISCUSSION
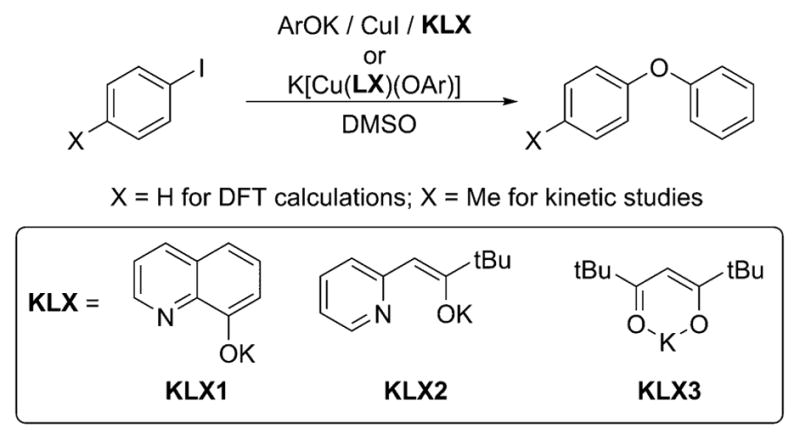
Copper-catalyzed arylation of phenols (Ullmann biaryl ether formation) is typically conducted with a mixture of a phenol, an aryl halide, and a weak base in the presence of catalytic amounts of a Cu(I) salt and either a neutral or an anionic ancillary ligand in DMF, DMSO, or toluene as solvent at 80–110 °C.4a To investigate the mechanism of the Ullmann biaryl synthesis in the presence of anionic ancillary ligands, we have studied the model reaction in Scheme 5 in which KOPh is allowed to react with either iodobenzene (for DFT calculations) or p-iodotoluene (for reactivity and kinetic studies) in the presence of catalytic amounts of CuI and the potassium salts of the anionic ancillary ligands KLX in DMSO.
Scheme 5.
Model Systems Studied in This Work
We have conducted studies with three different types of anionic ancillary ligands, 8-hydroxyquinoline (HLX1), 3,3-dimethyl-1-(pyridin-2-yl)butan-2-one (HLX2), and 2,2,6,6-tetramethylheptane-3,5-dione (HLX3), which are commonly used for the copper-catalyzed arylation of phenols.4a These ligands were chosen for a combination of reasons. First, 8-hydroxyquinoline (HLX1) is a commonly used ligand for copper-catalyzed cross-coupling4a and was even used for couplings on >10 kg scale.26 The pyridyl ketone ligand (HLX2) was chosen because complexes of this ligand were shown to catalyze coupling reactions at room temperature.27 The 1,3-diketone (HLX3) was chosen because it is one of the most commonly used anionic ligands for copper-catalyzed couplings.4a Second, these three ligands comprise O,O and O,N donor groups and the two O,N donor ligands have varying degrees of rigidity. Thus, data on the set of these three ligands would provide a broader view of the mechanism of this reaction than data on just one ligand.
Additionally, the Ullmann etherification reactions are typically conducted under heterogeneous conditions with inorganic bases, such as K2CO3 and Cs2CO3. Because the acidity of phenols and bicarbonate are similar,28 the phenols can be deprotonated in the catalytic system containing carbonate base without assistance by coordination to the copper catalyst. To gain quantitative rate data, we studied a homogeneous reaction system containing potassium salts of phenol and the anionic ancillary ligands.
Synthesis and Characterization of Stable Homoleptic Cuprates Containing Phenoxides and Anionic Ancillary Ligands
Our synthesis of homoleptic cuprates K[Cu(OPh)2] and K[Cu(LX)2] is outlined in Scheme 6. The reaction of copper tert-butoxide (CuOtBu)29 with a solution of a mixture of HOAr and KOAr in THF led to bis-aryloxide cuprate complexes 1a–d. These complexes were characterized by NMR spectroscopy and elemental analysis. The structure of the [Cu(OPh)2] − unit has been deduced by X-ray crystallography previously as the anionic part of a pair of copper ions.15 The reaction of CuOtBu with a mixture of bidentate, anionic ancillary ligands HLX1 and HLX3, and their potassium salts KLX1 and KLX3 in THF (for KLX1) or pentane (for KLX3) furnished the corresponding bis-LX cuprate complexes K[Cu(LX1)2] and K[Cu(LX3)2]. We isolated K[Cu(LX1)2] and K[Cu(LX3)2] in pure form and fully characterized these two complexes, including X-ray diffraction. We were unable to isolate K[Cu(LX2)2] as a pure crystalline material, so K[Cu(LX2)2] was not used in subsequent studies. Both compounds K[Cu(LX1)2] and K[Cu(LX3)2] crystallized as networks (Figures S1 and S2, respectively) in which each individual [Cu(LX)2] − moiety is bridged by a potassium cation. To determine if these homoleptic cuprates were mononuclear in solution, diffusion coefficients of these complexes and their mononuclear structural analogues, Zn-(LX1)2(H2O)2 and Zn(LX3)2,30 were determined by DOSY. We hypothesized that if cuprates K[Cu(LX1)2] and K[Cu(LX3)2] were mononuclear in solution, then their diffusion coefficients should be similar to the diffusion coefficients of their zinc analogues. Indeed, the diffusion coefficient of K[Cu(LX1)2] was similar to that of Zn(LX1)2(H2O)2 (9.29 × 10 −6 and 1.04 × 10−5 cm2 s−1, respectively), and the diffusion coefficient of K[Cu-(LX3)2] was similar to that of Zn(LX3)2 (7.78 × 10 −6 and 9.68 × 10−6 cm2 s−1, respectively). Thus, these results indicate that compounds K[Cu(LX1)2] and K[Cu(LX3)2] are mononuclear in solution.
Scheme 6.
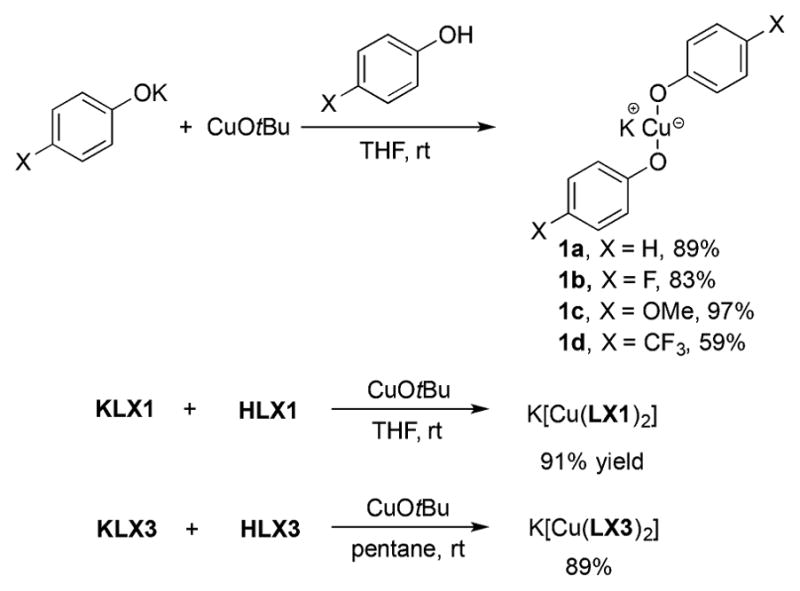
Synthesis of Potassium Cuprates K[Cu(OAr)2] and K[Cu(LX)2]
Reactivity of Bis-phenoxide 1a with Aryl Iodides in the Absence and Presence of Anionic Ancillary Ligands KLX and Cuprate Complexes K[Cu(LX)2]
Reaction of bis-phenoxide 1a with p-iodotoluene at 110 °C over 3 h formed phenyl tolyl ether (2) in a low 17% yield; 83% of complex 1a remained unreacted, based on 1H NMR spectroscopy (Scheme 7). The low reactivity of the ligandless, bis-phenoxide 1a with iodoarenes agrees with similar previous experimental results and theoretical predictions for the reactivity of anionic, two-coordinate CuI-complexes.15 However, the combination of bis-phenoxide 1a and anionic cuprates K[Cu(LX1)2] and K[Cu-(LX3)2] reacted with p-iodotoluene at 80 °C in 3 h to afford the biaryl ether product 2 in 91% and 99% yields, respectively. Moreover, bis-phenoxide 1a reacted with p-iodotoluene in the presence of the potassium salts of the anionic ancillary ligands KLX1–KLX3 at only 50 °C over 1–2 h to form the biaryl ether product 2 in excellent yields (95%, 81%, and 95% with KLX1, KLX2, and KLX3), respectively (Scheme 8). These data provide strong evidence that the anionic ancillary ligands KLX1–KLX3 and bis-phenoxide 1a generate CuI complexes that are kinetically and chemically competent to be intermediates in the catalytic cycle to afford biaryl ether products from reaction with iodoarenes. We conducted detailed spectroscopic studies to deduce the identity of these reactive CuI-intermediates.
Scheme 7.
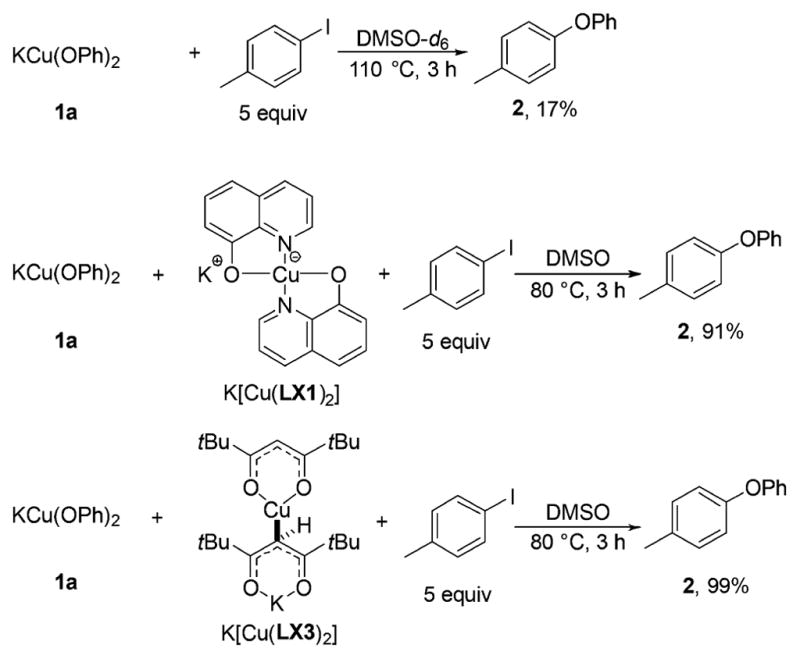
Reactions of 1a with 4-Iodotoluene in the Absence and the Presence of Complexes K[Cu(LX1)2] and K[Cu(LX3)2]a
aYields are reported relative to the number of OPh groups in the starting material.
Scheme 8. Reactions of 1a with 4-Iodotoluene in the Presence of Anionic Ligands KLXa.
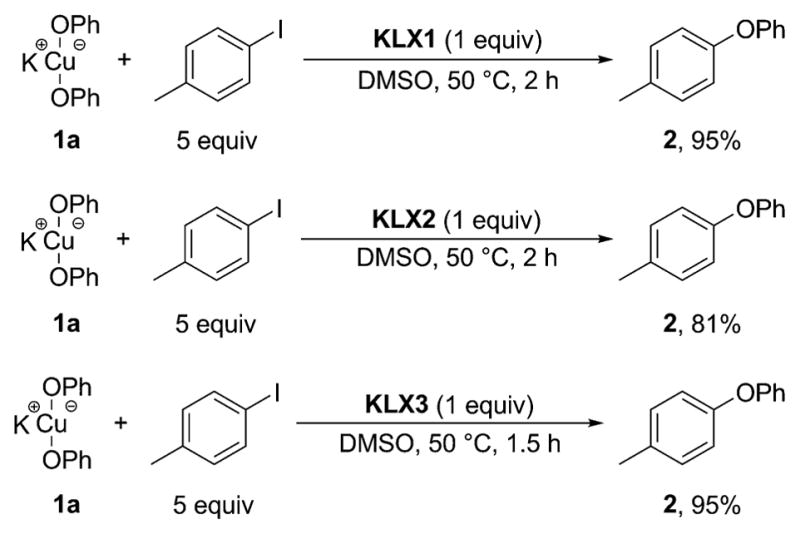
aYields are reported relative to the number of OPh groups in the starting material.
Identity of Catalytic Intermediates
To assess the identity of the species that react with iodoarenes in the experiments depicted in Schemes 7 and 8, we conducted spectroscopic studies of the complexes generated from addition of the bidentate, anionic ancillary ligands to the bis-phenoxides 1. The complexes were characterized by a combination of 1H and 19F NMR spectroscopy, UV–vis spectroscopy, and electrospray ionization mass spectroscopy (ESI-MS).
1H NMR spectra were obtained in DMSO of complex 1a, the potassium salt of each ligand, potassium phenoxide, and combinations of these components. These spectra show clearly that complex 1a reacts with the potassium salt of hydroxyquino-line (KLX1) and the salt of the pyridylketone (KLX2), but not with the anion of the 1,3-diketone. The reactions of 1a with KLX1 and KLX2 consumed 1a, generated free potassium phenoxide, and gave rise to a complex with a new set of phenoxide and ligand signals. These spectra are shown in the Supporting Information.
To observe more directly the phenoxide complexes and to exploit the high resolution of 19F NMR spectroscopy, these experiments were conducted with di(4-fluorophenoxide)cuprate (1b), and the outcomes were monitored by 19F NMR spectroscopy. Figure 1a shows three superimposed individual 19F NMR spectra of 4-fluorophenol (−126.5 ppm), potassium 4-fluorophenoxide (−139.7 ppm), and bis(4-fluorophenoxide)-cuprate (1b, −133.3 and −133.7 ppm) in THF at −100 °C (all referenced internally to fluorobenzene at −113.15 ppm). Figure 1b–e show 19F NMR spectra obtained at room temperature and −100 °C after addition of the anionic (KLX2) or neutral (HLX2) forms of pyridylmethyl tert-butyl ketone to bis-phenoxide 1b. 19F NMR spectra of these samples acquired at room temperature (Figure 1b and d) contained a new broad signal at −136.69 and −131.81 ppm, respectively, due to exchange between the phenoxide in a ligated copper phenoxide with potassium phenoxide or phenol, respectively. The spectra of the sample generated from 1b and KLX2 and the spectra of the sample generated from 1b and HLX2 acquired at −100 °C (Figure 1c and e) each consisted of two sharp resonances. The chemical shift of one of the two resonances in each of the spectra was −135.5 ppm. The identities of these resonances strongly suggest that they correspond to the heteroleptic complex K[Cu(LX2)OAr], which was generated in 65% yield from the reaction of 1b and KLX2 and in 63% yield from the reaction of 1b and HLX2 based on integrations relative to fluorobenzene. The 19F NMR spectrum of the sample generated from KLX2 also contained a signal with a chemical shift near that of the phenoxide, and the spectrum of the sample generated from HLX2 also contained a signal with a chemical shift near that of the phenol. The chemical shift of phenol in this mixture is not identical to that of phenol alone in solution, presumably because of interactions of phenol with ions in solution.
Figure 1.

(a) Superimposed 19F NMR spectra of 4-fluorophenol, bis(4-fluorophenoxide)cuprate (1b), and potassium 4-fluorophenoxide at −100 °C in THF. (b) 19F NMR spectrum of cuprate 1b and KLX2 at room temperature. (c) 19F NMR spectrum of cuprate 1b and KLX2 at −100 °C. (d) 19F NMR spectrum of cuprate 1b and HLX2 at room temperature. (e) 19F NMR spectrum of cuprate 1b and HLX2 at −100 °C. (f) Proposed structure of K[Cu(LX2)OAr] and equilibria to generate the new species observed at −135.5 ppm. All 19F NMR spectra were obtained in THF and are referenced to fluorobenzene at −113.2 ppm.
The 19F NMR spectra showing dynamic processes for complexes generated from bis(4-fluorophenoxide)cuprate (1b) and the enolate of 2,2,6,6-tetramethylheptane-3,5-dione (LX3) are shown in Figure 2. 19F NMR spectra of samples containing 1b and KLX3 or HLX3 acquired at room temperature (Figure 2b and d) contained new broad signals at −134.5 and −130.4 ppm, respectively. However, the 19F NMR spectrum of a sample containing cuprate 1b and KLX3 at −100 °C (Figure 2c) indicated that the potassium salt of the ligand does not signiflcantly displace the phenoxide. The spectrum contained a major signal at −133.8 ppm, which is the same chemical shift as that of cuprate 1b. We do not have a clear explanation for the presence of just one major signal at this chemical shift when KLX3 is added, but this ligand might facilitate exchange between the two forms or conformations of 1b present at −100 °C, and it is clear that the mixed complex K[Cu(LX3)OAr] does not form under these conditions. Only a minor signal was present at the chemical shift corresponding to 4-fluorophenoxide.
Figure 2.

(a) Superimposed 19F NMR spectrum of 4-fluorophenol, bis(4-fluorophenoxide)cuprate (1b), and potassium 4-fluorophenoxide at −100 °C in THF. (b) 19F NMR spectrum of cuprate 1b and KLX3 at room temperature. (c) 19F NMR spectrum of cuprate 1b and KLX3 at −100 °C. (d) 19F NMR spectrum of cuprate 1b and HLX3 at room temperature. (e) 19F NMR spectrum of cuprate 1b and KLX3 at −100 °C. (f) Proposed structure of K[Cu(LX3)OAr] and equilibria to generate the new species observed at −135.1 ppm. All spectra were obtained in THF and are referenced to fluorobenzene at −113.2 ppm.
In contrast to these results, the 19F NMR spectrum at −100 °C of the sample containing β-diketone HLX3 (Figure 2e) indicated that this neutral form of the ligand protonates a bound phenoxide to generate the corresponding heteroleptic compound K[Cu-(LX3)OAr] and phenol. Two resonances were observed at this temperature, one at −135.1 ppm and one at −128.9 ppm. The signal at −135.1 ppm can be assigned to K[Cu(LX3)OAr] because its chemical shift is similar to that of the 19F NMR resonance of heteroleptic complex K[Cu(LX2)OAr] (−135.5 ppm). This complex was formed in 50% yield, as determined by 19F NMR spectroscopy with fluorobenzene as an internal standard. The broad resonance at −128.9 is near that of the free 4-fluorophenol, but, again, is not identical to that of the phenol alone, presumably due to hydrogen bonding with ions at low temperature. Thus, the most stable component in the combination of phenoxy cuprate 1b and anionic diketone ligand under the basic conditions of a catalytic coupling reaction is the unreacted 1b and the free anionic ligand, but the heteroleptic complex can be generated and studied by protonation of a phenoxide from 1b.
19F NMR spectra of dynamic processes observed for complexes generated from bis(4-fluorophenoxide)cuprate (1b) and the anionic or neutral forms of 8-hydroxyquinoline KLX1 and HLX1 are shown in Figure 3. These spectra are similar to those generated from 1b and KLX2 or HLX2 (Figure 1) but contain additional signals besides the heteroleptic compound and the phenol or phenoxide. 19F NMR spectra acquired at room temperature of samples containing bis(4-fluorophenoxide)-cuprate(I) (1b) and KLX1 or HLX1 (Figure 3b and d) contained new broad signals at −136.49 and −130.83 ppm, respectively (Figure 3b). The 19F NMR spectrum obtained at −100 °C of the same sample generated from 1b and KLX1 (Figure 3c) contained resonances that were broader than those from samples containing LX2 and LX3, but the chemical shift of one resonance at −134.8 ppm was similar to those assigned to the heteroleptic complexes containing LX2 and LX3 (−135.5 and −135.1 ppm, respectively) and, therefore, is assigned to the heteroleptic compound K[Cu(LX1)OAr]. This complex formed in 40% yield from the reaction of 1b with KLX1 and in 60% yield from the reaction of 1b and HLX1. The chemical shifts of the other two resonances were near that of the potassium phenoxide. We are uncertain why more than one resonance is observed in the region of the phenoxide in this system. The spectrum of the sample generated from 1b and HLX1 acquired at −100 °C (Figure 3e) contained a sharp resonance with a chemical shift of −134.4 ppm, matching that observed in Figure 3c and corresponding to the heteroleptic compound K[Cu(LX1)OAr]. The other signals in the spectrum correspond to the phenol, again resonating slightly upfield of the phenol alone.
Figure 3.
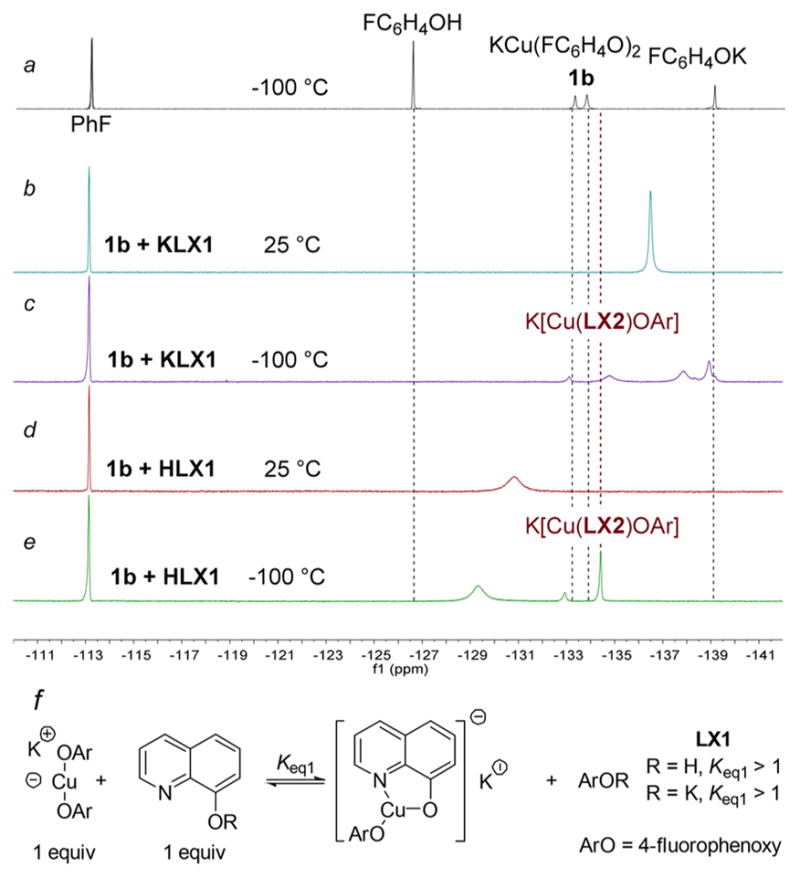
(a) Superimposed 19F NMR spectra of 4-fluorophenol, bis(4-fluorophenoxide)cuprate (1b), and potassium 4-fluorophenoxide at −100 °C in THF. (b) 19F NMR spectrum of cuprate 1b and KLX1 at room temperature. (c) 19F NMR spectrum of cuprate 1b and KLX1 at −100 °C. (d) 19F NMR spectrum of cuprate 1b and HLX1 at room temperature. (e) 19F NMR spectrum of cuprate 1b and HLX1 at −100 °C. (f) Proposed structure of K[Cu(LX1)OAr] and equilibria to generate the new species observed at −134.7 ppm. All spectra were obtained in THF and are referenced to fluorobenzene at −113.2 ppm.
To determine the nuclearity of the K[Cu(LX)OAr] complexes in solution, we attempted to measure the diffusion coefficients of these compounds by DOSY. Again, measurements were obtained by 19F NMR spectroscopy with bis(4-fluoro-phenoxide)cuprate (1b) and an LX ligand at −100 °C in THF. To generate the K[Cu(LX)OAr] compounds in high yield, the ligands HLX1, KLX2, and HLX3 were chosen for these experiments. At −100 °C, we were unable to obtain diffusion coefficients by DOSY for any K[Cu(LX)OAr] compounds due to rapid relaxation (T1 approximately 100 ms and T2 approximately 40 ms) and slow diffusion. As such, significant losses to signal intensity were observed during DOSY measurements. Due to the thermal sensitivity of the K[Cu(LX)OAr] complexes and rapid equilibria between copper-containing starting materials and products, we were unable to perform DOSY experiments at temperatures above −100 °C.
If the slow diffusion of the K[Cu(LX)OAr] compounds resulted from aggregation of the cuprates via bridging potassium cations, then the substitution of potassium for a noncoordinating cation should inhibit aggregation and facilitate diffusion. To test this hypothesis, the K[Cu(LX)OAr] complexes were synthesized in the presence of N(n-butyl)4Cl (1 equiv for each potassium cation) to perform an in situ salt metathesis, and the resulting product was submitted for DOSY measurements. Unfortunately, DOSY measurements still could not be obtained for the prior stated reason. Similarly, attempts to measure diffusion coefficients in a less viscous solvent, 2-MeTHF, were not successful.
Because our attempts to determine the nuclearity of the K[Cu(LX)OAr] complexes by DOSY were not fruitful, we obtained ESI-MS spectra of the K[Cu(LX)OAr] complexes to gain a final set of data on the identity the compounds produced by the addition of LX to bis(4-fluorophenoxide)cuprate (1b). Room-temperature DMF solutions of KLX1, KLX2, or HLX3 were added to cuprate 1b and analyzed by ESI-MS. Due to the air sensitivity of the Cu compounds, the MS source was kept under an atmosphere of nitrogen, and solutions were injected into the spectrometer from a glovebox via a capillary.31 In negative ion mode, Cu-containing species were identified, based on the diagnostic isotope pattern of 63Cu and 65Cu in a ratio of approximately 69:31. Signals with m/z ratios consistent with the formation of mononuclear, three-coordinate Cu complexes comprising a single 4-fluorophenoxide and a single LX bidentate ligand (Chart 1 and Supporting Information) were observed. Signals for the anionic form of 8-hydroxyquinoline LX1− or the homoleptic Cu(I) complexes containing two anionic ligands LX1− or LX3− from 8-hydroxyquinoline and the β-diketone, respectively, were not observed; however, signals for unreacted cuprate 1b were observed in the presence of all ligands KLX. Masses corresponding to oligomeric forms of [Cu(LX)OAr] − were not observed. In positive ion mode, no signals containing copper were detected in the mass spectrum.
Chart 1. ESI-MS Analysis of Solutions Containing Mixtures of 1b and Ligands LXa.
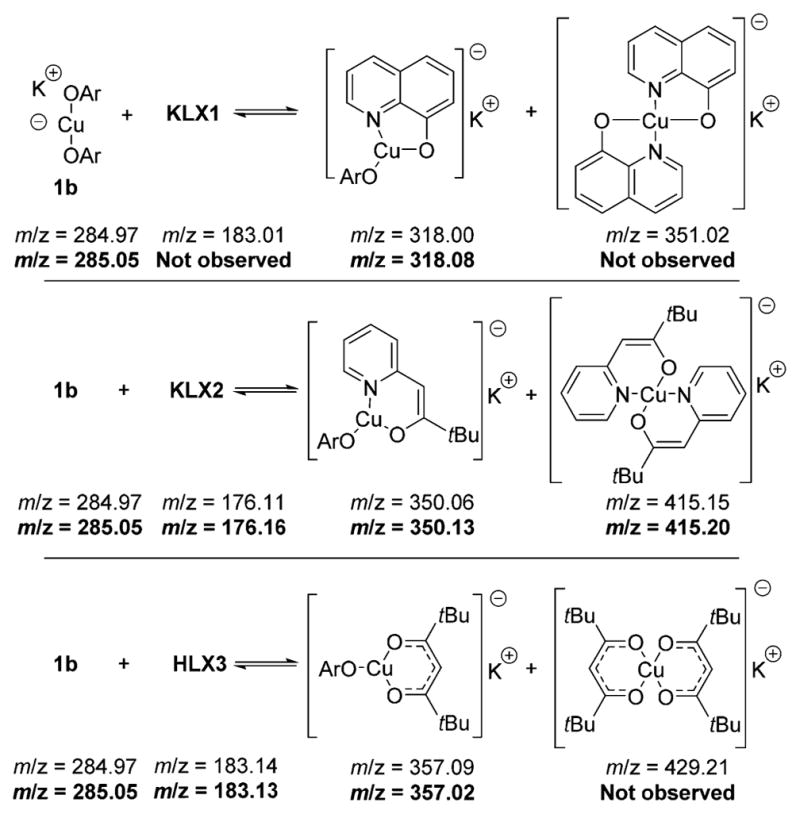
aThe calculated masses are shown without highlighting, and the observed masses are shown in bold. All masses are given for the corresponding anions.
UV–vis studies on the reaction of K[Cu(OPh)2] (1a) with potassium quinolin-8-olate (KLX1) provided additional evidence for formation of the heteroleptic cuprates. The data are provided in the Supporting Information. In brief, a UV–vis spectrum of the solution of KLX1 and complex 1a contained a new absorption band not present in the spectrum of 1a, the spectrum of KLX1, or the spectrum of the homoleptic complex K[Cu(LX1)2]. This band observed in the spectrum of a solution of KLX1 and complex 1a is consistent with the formation of a compound that we propose to be K[Cu(LX1)(OPh)].
In summary, the 19F NMR and 1H NMR spectra, UV–vis spectra, and ESI-MS data together provide strong evidence that the combination of the potassium salts of the anionic ligands LX and cuprate 1b form three-coordinate, mononuclear species of the structure K[Cu(LX)(OAr)] as the most stable structure (for the anions KLX1 and KLX2 derived from hydroxyquinoline and pyridylmethyl tert-butyl ketone) or as a species in equilibrium with the bis-phenoxy cuprate and free potassium salt KLX3 of the β-diketone.
Tests for the Presence of Aryl Radicals during the Ullmann Arylation of Phenols Catalyzed by Complexes of LX1–LX3
After identification of the major species formed from copper phenoxides and LX1–LX3, we sought to determine the mechanism of the reactions of these species with iodoarenes (Scheme 3). First, to distinguish between the anionic and radical pathways of Schemes 3 and 4, the potential formation of aryl radicals was assessed. The intermediacy of an aryl radical was probed by conducting reactions of the copper complexes with an aryl halide linked to an olefin that serves as a radical clock. The aryl radical that would be generated from o-(allyloxy)iodo-benzene 3 is known to cyclize to yield the 2,3-dihydrobenzo-furanylmethyl radical with a rate constant of 9.6 × 109 s−1 in DMSO.32 The resulting primary radical can then abstract a hydrogen atom from the solvent, dimerize, or combine with the phenoxide ligand to form a C–O bond.
A 1:1 mixture of KLX1–KLX3 and K[Cu(OPh)2] (1a) was allowed to react with 3.0 equiv of o-(allyloxy)iodobenzene 3 (1.5 equiv with respect to phenoxide) in DMSO (Scheme 9). After 3 h at 110 °C, the biaryl ether 4 from C–O coupling at the aryl group formed in quantitative yields with all ligands. Comparison of the GC and GC/MS chromatograms with those of an authentic sample of 3-methyl-2,3-dihydrobenzofuran 5 confirmed that no cyclized product 5 was formed. The reactions of KOPh with o-(allyloxy)iodobenzene (3) catalyzed by 10 mol % CuI and 20 mol % KLX1–KLX3 in DMSO at 110 °C formed the biaryl ether 4 from C–O coupling at the aryl group in >99%, >99%, and 89% yields with KLX1, KLX2, and KLX3, respectively, after 24 h (Scheme 10). Again, no products from cyclization were detected. Thus, neither the stoichiometric nor the catalytic reactions yielded the products that one would expect to form from a reaction through a free aryl radical. Therefore, it is likely that the Ullmann coupling to form biaryl ethers catalyzed by CuI-complexes in the presence of anionic ancillary ligands does not involve free aryl radical intermediates. In addition, our results are consistent with recent work to probe the intermediacy of a radical in copper-catalyzed Ullmann coupling.33 Accordingly, the subsequent mechanistic studies, especially the DFT calculations, focused on distinguishing between the nonradical, anionic, and neutral pathways of Schemes 3 and 4.
Scheme 9.
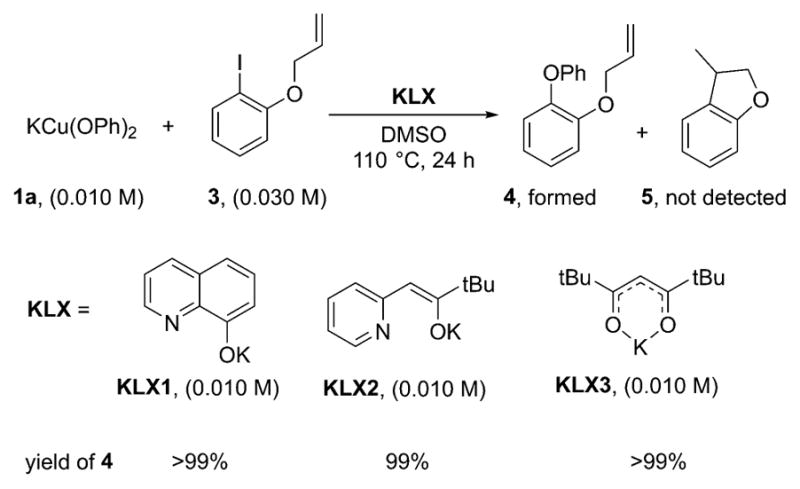
Reactions of 1a with 3 in the Presence of the Ligands KLX
Scheme 10.
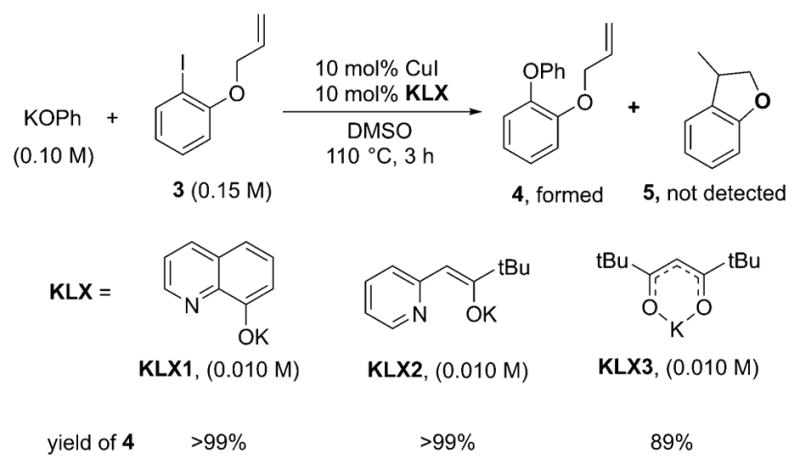
Catalytic Reactions of KOPh with 3 in the Presence of CuI and the Ligands KLX
Kinetic Analysis
Kinetic studies on the stoichiometric reactions of the heteroleptic complexes with iodoarenes distinguished between reaction by the anionic pathways and the neutral pathway. The experiments were performed by generating the K[Cu(LX)(OAr)] complexes in situ by mixing potassium salts of the anionic ligands KLX1–KLX3 with K[Cu(OAr)2] (1) or by mixing a 1:1 ratio of K[Cu(OPh)2] and the cuprate K[Cu(LX3)2] containing the diketonate ligand. The reactions were conducted under pseudo-first-order conditions by allowing the K[Cu(LX)(OAr)] species generated in situ to react at the appropriate temperatures (30 or 50 °C) inside the NMR probe with an excess of the iodoarene. The formation of biaryl ether product 2 was monitored by 1H NMR spectroscopy. To measure the kinetic order in phenoxide, some reactions were conducted in the presence of added PhO− (as opposed to the PhO− solely generated from 1a). Because these reactions with added phenoxide are catalytic (phenoxide and iodoarene are both present in the presence of copper), initial rates of these reactions were measured with varying concentrations of added PhO−.
For the reaction at 30 °C between iodotoluene and K[Cu(LX1)(OPh)] generated from the potassium salt of 8-hydroxyquinoline KLX1 and K[Cu(OPh)2], the concentration of the biaryl ether product increased monoexponentially, indicating a first-order dependence of the rate of the reaction on the concentration of K[Cu(LX1)(OAr)] (Figure 4a). Consistent with this observation, doubling the concentration of KLX1 and K[Cu(OPh)2] did not alter kobs (Table S1). The pseudo-first-order rate constants kobs for these reactions conducted with varying concentrations of iodotoluene between 0.1 and 0.8 M were found to correlate linearly with the concentrations of 4-iodotoluene (Figure 4b), indicating that the reaction is first-order in the iodoarene. The reaction rate was not affected by the amount of added PhO−, indicating a zero-order dependence of the reaction on this component of the catalytic system (Figure 4c).
Figure 4.

(a) Plot of the formation of the product 2 from the reaction of p-iodotoluene (0.20 M) with K[Cu(LX1)OPh] generated in situ from the reaction of KLX1 (0.010 M) with KCu(OPh)2 (1a) (0.010 M) in DMSO-d6 at 30 °C. (b) Plot of kobs versus [p-iodotoluene] for the reaction of p-iodotoluene (0.10–0.80 M) with K[Cu(LX1)OPh] generated in situ from the reaction of KLX1 (0.010 M) with KCu(OPh)2 (1a) (0.010 M) in DMSO-d6 at 30 °C. (c) Plot of the added [KOPh] (0.00–0.080 M) vs the initial rate for the formation of the product 2 from the reaction of p-iodotoluene (0.10 M) with K[Cu(LX1)OPh] generated in situ from the reaction of KLX1 (0.010 M) with KCu(OPh)2 (1a) (0.010 M) in DMSO-d6 at 50°C.
Similar results were obtained for the reaction of 4-iodotoluene with K[Cu(LX2)(OPh)] − generated from KCu(OPh)2 (1a) and the salt KLX2 derived from the pyridylmethyl tert-butyl ketone (Figures S7–S8 of the Supporting Information). The reaction was clearly first-order in the copper complex, based on the monoexponential appearance of product, and doubling the concentration of KCu(OPh)2 (1a) and KLX2 did not affect kobs (Table S3). The reaction was also first-order in iodoarene, as determined from the linear plot of kobs vs [ArI]. In addition, the reaction was clearly zero-order in phenoxide.
The kinetic behavior of the reaction catalyzed by the combination of KCu(OPh)2 (1a) and potassium β-diketonate KLX3 at 50 °C was more complex, but the order in iodoarene, phenoxide, and ligand were clear. The appearance of the biaryl ether product 2 from this reaction was linear (Figure 5a), even in the presence of an excess of the ligand KLX3, and reactions with two different initial concentrations of 1a occurred with the same rate (Figure 5a). The origin of this apparent zero-order dependence of the rate on [Cu] is difficult to pinpoint, but it likely results from participation of copper species lacking phenoxide (such as ligated CuI) generated upon conversion of 1a to the biaryl ether 2. To assess this hypothesis, we studied the effect of added CuI on the rate of the reaction of 1a and 4-iodotoluene in the presence of an excess of KLX3. This result shows that, under otherwise identical conditions, the reaction in the presence of 1 equiv of CuI was faster than that in the absence of CuI (Figure 6).
Figure 5.

(a) Plot of the rise of the biaryl ether product 2 from the reactions of p-iodotoluene (0.10 M) with 0.010 M (blue diamonds) or 0.005 M (red circles) of KCu(OPh)2 (1a) in the presence of KLX3 (0.080 M) in DMSO-d6 at 50 °C. (b) Plot of the [p-iodotoluene] vs the initial rate for the rise of the biaryl ether product 4 from the reaction of p-iodotoluene (0.10–0.40 M) with KCu(OPh)2 (1a, 0.01 M) in the presence of KLX3 (0.02 M) in DMSO-d6 at 50 °C. (c) Plot of the concentration of the ligand KLX3 ([L], 0.02–0.08 M) vs the initial rate for the rise of the biaryl ether product 2 from the reaction of p-iodotoluene (0.10 M) with KCu(OPh)2 (1a, 0.01 M) in DMSO-d6 at 50 °C. (d) Plot of the added [KOPh] (0.010–0.080 M) vs the reciprocal of the initial rate (1/rate) for the rise of the biaryl ether product 4 from the reaction of p-iodotoluene (0.20 M) with KCu(OPh)2 (1a, 0.01 M) in the presence of KLX3 (0.02 M) in DMSO-d6 at 50 °C.
Figure 6.
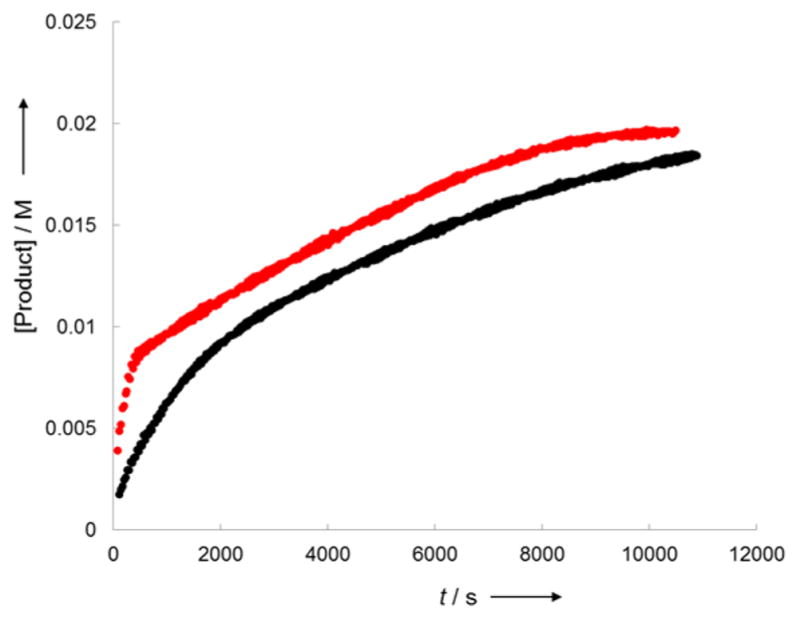
Reaction of 1a (10 mM) with 4-iodotoluene (100 mM) in the presence of KLX3 (80 mM) in the absence (black) or presence of CuI (10 mM, red line) in DMSO at 30 °C.
To determine if the unusual kinetic behavior was inherent to reactions of copper-containing potassium β-diketonate KLX3 or due to the method by which the ligated copper was generated, we conducted reactions of iodotoluene with a mixture of K[Cu-(OPh)2] (1a) and the bis-ligand complex K[Cu(LX3)2]. Under these conditions, the reaction of iodotoluene occurred with a simple monoexponential appearance of the biaryl ether product, as shown in Figure 7a. Moreover, doubling the concentration of K[Cu(OPh)2] (1a) and K[Cu(LX3)2] did not alter kobs (Table S8), consistent with a first-order relationship between total copper concentration and rate.
Figure 7.

(a) Plot of the rise of the product 2 from the reaction of p-iodotoluene (0.10 M) with K[Cu(LX3)OPh] generated in situ from the reaction of K[Cu(LX3)2] (0.010 M) with KCu(OPh)2 (1a) (0.010 M) in DMSO-d6 at 90 °C. (b) Plot of kobs versus [p-iodotoluene] for the reaction of p-iodotoluene (0.10–0.60 M) with in situ generated complex K[Cu(LX3)OPh] from the reaction of K[Cu(LX3)2] (0.010 M) with KCu(OPh)2 (1a) (0.010 M) in DMSO-d6 at 90 °C.
Thus, the orders in iodoarene, phenoxide, and ligand were gained from a combination of reactions of iodoarene with K[Cu(LX3)(OPh)] generated from bis-phenoxide 1a and β-diketonate KLX3, and reactions of iodoarene with a 1:1 mixture of 1a and K[Cu(LX3)2]. Under both conditions, the reaction of the copper complex was first-order in the concentrations of p-iodotoluene (Figure 5b and Figure 7b). Information on the reaction order in phenoxide could not be gained from the mixture of 1a and K[Cu(LX3)2] because the phenoxide would displace the diketonate ligand, as shown by the NMR studies discussed earlier in this paper, but the order in phenoxide was clear from the reactions conducted with K[Cu(LX3)(OPh)] generated reversibly (and endergonically) from 1a and KLX3. Under these conditions, the reaction was clearly inverse first-order in phenoxide (Figure 5d) and first-order in KLX3 (Figure 5c). These reaction orders are consistent with a reversible pre-equilibrium for displacement of one phenoxide in 1a by the diketonate to generate K[Cu(LX3)(OPh)] and reaction of the iodoarene with this heteroleptic complex, as was deduced for reactions of iodoarenes with K[Cu(LX1)(OPh)] and K[Cu-(LX2)(OPh)].
This set of kinetic data on the reactions of iodoarenes with copper complexes containing the three anionic ancillary ligands LX1–LX3 is inconsistent with reaction by the neutral pathway with irreversible, rate-determining oxidative addition of the iodoarene to the neutral complexes [(LX)Cu] or [(LX)Cu-(ArI)]. This pathway would require the dissociation of the phenoxide anion from the more stable [Cu(LX)OPh] − before the rate-determining step, and a reaction by this mechanism would be inverse first-order in PhO−. In contrast to this prediction, the reactions of the iodotoluene with the K[Cu-(LX)OPh] − complexes were zero-order in [PhO−].
Instead, our data are consistent with reaction by one of the anionic pathways with a turnover-limiting oxidative addition or reaction by the neutral pathway with a reversible oxidative addition and an irreversible association of PhO− to the resulting four-coordinate neutral intermediate [Cu(LX)(Ar)I]. Reactions of iodotoluene with K[Cu(LX)OPh] complexes by these two pathways would be zero-order in [PhO−], first-order in [ArI], and first-order in [Cu], and these orders were observed experimentally. Moreover, the reaction of iodotoluene with the combination of K[Cu(OPh)2] and KLX3 by these pathways should be first-order in [ArI], first-order in KLX3, and inverse first-order in phenoxide, and these orders predicted for these two pathways were observed experimentally.
Measurement of 13C/12C Kinetic Isotope Effects
To distinguish between reaction by the anionic pathways with a turnover-limiting oxidative addition and reaction by the neutral pathway with a reversible oxidative addition and irreversible association of PhO− to the resulting four-coordinate neutral intermediate [Cu(LX)(Ar)I], we measured the 13C/12C isotope effect (IE) for the breaking of the C–I bond (at the ipso carbon). We did so following the protocol designed by Singleton.34 We conducted this experiment on the reaction of 4-iodotoluene with potassium 3-methoxyphenoxide (Scheme 11) and compared the experimental values with those computed for the corresponding pathways using Bigeleisen–Mayer theory35 (Table 1). The reactions were conducted at 110 °C in DMSO solvent to 90–97% conversion. The remaining iodoarene was isolated by column chromatography, and the isolated product was analyzed by 13C NMR spectroscopy with gated decoupling to allow accurate integration. The methyl carbon was used as the internal standard, assuming that the 13C/12C isotope effect would be negligible at this position.
Scheme 11. Reaction for Which 13C/12C Isotope Effects (IE) Were Measureda.
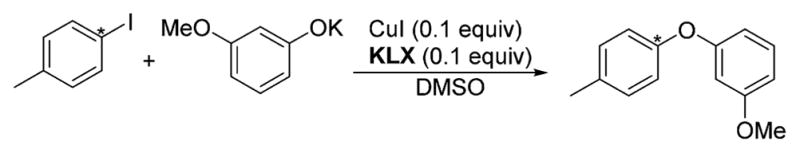
aThe carbon for which the IE was determined is marked with *.
Table 1.
Calculated and Experimental Values of 13C/12C Isotope Effect
| ligand | Calculated Values in DMSO Solvent | Experimental Values | ||
|---|---|---|---|---|
|
|
|
|||
| KIEa | EIEa | exp | σexpb | |
| LX1 | 1.018 | 0.998 | 1.0139 | 0.0046 |
| LX2 | 1.022 | 1.002 | 1.0125 | 0.0049 |
| LX3 | 1.018 | 1.001 | 1.0234 | 0.0045 |
Calculated for DMSO solvent using CPCM.36
Absolute error of determination of the experimental value of IE.
The calculated and experimental values of the 13C/12C isotope effect for the reaction of Scheme 11 are given in Table 1. An isotope effect of unity is predicted for the reaction by the neutral pathway with reversible dissociation of phenoxide, reversible oxidative addition of the C–I bond, and irreversible reaction with phenoxide (calculated equilibrium isotope effect (EIE) values in Table 1). This lack of an isotope effect results from the reversibility of the oxidative addition of the C–I bond and a resulting equilibrium isotope effect that is too small to measure. In contrast, a primary 13C kinetic isotope of about 1.02 is predicted to be observed for the reaction by the anionic pathways because oxidative addition of the C–I bond is irreversible. The measured values for the kinetic isotope effects are clearly outside the range of the calculated equilibrium isotope effects. The experimental values for the reaction in Scheme 11 with the systems containing the ligands LX1 and LX3 matches within experimental error the computed KIE for the oxidative addition of iodobenzene to the K[Cu(LX)(OAr)] complexes. The measured values of the isotope effects for reaction of the complex of LX2 is not as high as that predicted by theory, but it is still outside the range of values predicted for an equilibrium isotope effect. Therefore, these 13C/12C isotope effects are consistent with the predicted primary KIE for turnover-limiting oxidative addition of the aryl iodide in one of the anionic pathways and inconsistent with an EIE value for reversible oxidative addition and irreversible reaction with the phenoxide.
Electronic Effects on the Reactions of K[Cu(LX)(OAr)] Complexes
To gain a final set of data that would help distinguish between the anionic or neutral pathways, we studied the dependence of the kobs values for the reactions of an iodoarene (2-fluoro-4-iodotoluene) with a set of complexes K[Cu(OC4H4X)2] (1a–d) carrying various substituents in the para position of the aryloxide in the presence of KLX2. A linear correlation with a negative value of ρ is expected for each of the anionic pathways (the electron-donating substituents would increase the electron density on copper and thus facilitate the oxidative addition of iodoarene to the corresponding cuprate), whereas no strong correlation would be expected for the neutral pathway because an effect on the dissociation constant to form [Cu(LX)] or [Cu(LX)ArI] from [Cu(LX)OAr] − would be compensated by the opposite effect on the association of phenoxide to [Cu(LX)(Ar)I] to form [Cu(LX)(Ar)I(OPh)]. The plot of kobs vs σ–37 was linear with the ρ value of −0.95 (Figure 8). The substantial accelerating effect of electron-donating groups attached to the phenoxide further supports reaction by an anionic pathway.
Figure 8.
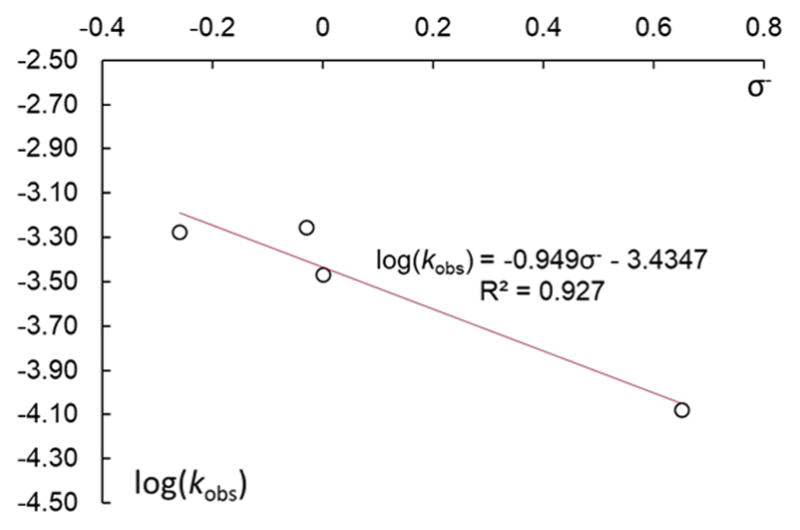
Hammett plot for the reactions of 1a–d (0.05 M) with 2-fluoro-4-iodotoluene (0.5 M) in the presence of KLX2 (0.05 M). The values of kobs were obtained fitting the function Ct = C0(1 − e−kobst) + Cres to the experimental values of the concentration of the corresponding product.
Thus, the series of kinetic data (zero-order in phenoxide, a primary 13C/12C KIE, and a linear Hammett plot with a negative ρ) allow us to conclude that the Ullmann arylation of phenols catalyzed by complexes of anionic LX ligands occurs by an anionic pathway of Scheme 3, not by the neutral pathway.
DFT Calculations
To refine the conclusions drawn from our synthetic and kinetic studies, we have conducted DFT calculations on the structures along the anionic and neutral pathways for the model reaction of phenol with iodobenzene in the presence of ligands LX. These calculations were conducted with the B3LYP functional38 and LANL2TZ39 (Cu,40 I) and 6–311G**41 (all other atoms) basis sets. CPCM36 was used to account for solvation effects in single-point energy calculations.
Figure 9 summarizes the results of our computational analysis. The structures in black correspond to intermediates and transition states that are common to both the anionic and neutral pathways. The structures in red correspond to those lying on the anionic pathway. The structures in blue correspond to those lying on the neutral path. In agreement with our experimental observations, the species [(LX)Cu(OPh)] − (LX-INT-1) was found to lie at the lowest energy among all intermediates lying on the reaction pathway. Dimeric [(LX)Cu-(OPh)]22− compounds containing bridging phenoxides (LX-INT-1-Dimer) were found to be 5–6 kcal/mol in energy above [(LX)Cu(OPh)] − (LX-INT-1). The computed energies of LX-INT-1-Dimer indicate that the monomeric compounds [(LX)-Cu(OPh)] − (LX-INT-1) are more stable than their dimeric forms. The unfavorability of forming LX-INT-1-Dimer likely arises from the association of two anionic compounds. At the same time, the computed energies are accessible under the reaction conditions. Given that we do not observe such dimers experimentally and that our reactions are first-order in copper, we propose that dimers are unlikely to participate in the catalytic cycle, even if these dimers are formed as part of an equilibrium with monomers. The energies of higher order cuprate oligomers were not computed, but we posit that they are less energetically accessible than LX-INT-1-Dimer due to the congregation of multiple anionic species. The complexes containing an η2-bound arene (LX-INT-2a and LX-INT-2b) were computed to lie 17–23 kcal/mol uphill of LX-INT-1, and this large energy difference makes the η2-arene complexes unlikely resting states in the catalytic cycle, as deduced from experiment.
Figure 9.

Computed reaction pathways for Ullmann etherification of aryl iodides in the presence of anionic ligands LX (the numbers correspond to computed values of ΔG/kcal·mol−1). a All attempts to optimize the geometry of this structure were unsuccesful. b LX-INT-3b is 2.5 kcal/mol downhill to LX-TS-1b in gas phase.
The energies of the transition states for oxidative addition to the anionic intermediate are similar to those for oxidative addition to the neutral intermediate. The energies of the transition states for oxidative addition of iodobenzene to the three anionic complexes LX-INT-1 after coordination of the arene to form LX-INT-2a lie 27–29 kcal/mol above the starting complexes and approximately 10–12 kcal/mol above the one anionic η2 arene complex for which we could minimize the structure. The energies of the transition states for oxidative addition to the neutral complexes LX-INT-2b formed after dissociation of the phenoxide and coordination of the arene lie 25–27 kcal/mol above the starting complexes and 4.2–6.5 kcal/mol above the neutral η2 arene complexes. Thus, the overall free energy barriers for the neutral (25–27 kcal/mol) and anionic (27–29 kcal/mol) pathways are too similar to distinguish by computation alone. Our experimental studies did show clearly that the reaction of iodoarene with phenoxide in the presence of the copper complexes ligated by the three bidentate anionic (LX) ligands follows an “anionic” pathway.
On the other hand, the computations clearly show that the barrier to direct reductive elimination from an anionic pentacoordinate species LX-INT-3a is higher than that for reductive elimination from an alternative complex. The energy of the transition state LX-TS-2a for reductive elimination from the anionic, five-coordinate species lies 27–29 kcal/mol above the starting complex. In contrast, dissociation of the iodide anion to generate a neutral tetracoordinate intermediate LX-INT-4 is strongly downhill, and reductive elimination from LX-INT-4 through the transition state LX-TS-2b is only 5–10 kcal/mol above LX-INT-4 and 12–14 kcal/mol uphill from the reactants and the starting anionic phenoxide complex LX-INT-1. Thus, we conclude from these DFT studies that the catalytic reaction occurs by the “modified anionic pathway” during which reductive elimination occurs after dissociation of iodide from the anionic, five-coordinate complex LX-INT-3a. The favorable dissociation of iodide and low barrier for reductive elimination from the resulting four-coordinate neutral species makes the oxidative addition portion of this mechanism, not the reductive elimination portion, rate limiting. These computational results are fully consistent with our kinetic data and are consistent with our deduction from the 13C/12C isotope effect that oxidative addition to the anionic species is irreversible and rate-limiting.
SUMMARY AND CONCLUSIONS
A combination of kinetic and computational studies on the Ullmann biaryl ether formation catalyzed by CuI in the presence of anionic ligands LX provides strong evidence that the reaction occurs by one of the anionic pathways in which the iodoarene reacts with the three-coordinate anionic cuprate complex containing the bidentate anionic ancillary ligand and the reactive phenoxide ligand. Our DFT studies suggest that the reaction occurs by the “modified anionic pathway” of Scheme 3 annotated with the experimental support in Scheme 12 in which reductive elimination occurs after dissociation of the iodide anion. Our variable-temperature NMR data suggest that the resting state of the reaction is the anionic species [Cu(LX)(OAr)] −, and the observed 13C/12C kinetic isotope effects imply that oxidative addition of iodoarenes to these species is the rate-determining step of the catalytic cycle. Many pathways for reaction of nucleophiles with iodoarenes have been proposed, but the combination of characterization of the most stable complex in the system, kinetic data on the reaction of these species with iodoarenes, 13C/12C kinetic isotope effect, and computational information provides strong evidence for reaction of phenoxides by the path in Scheme 12.
Scheme 12.
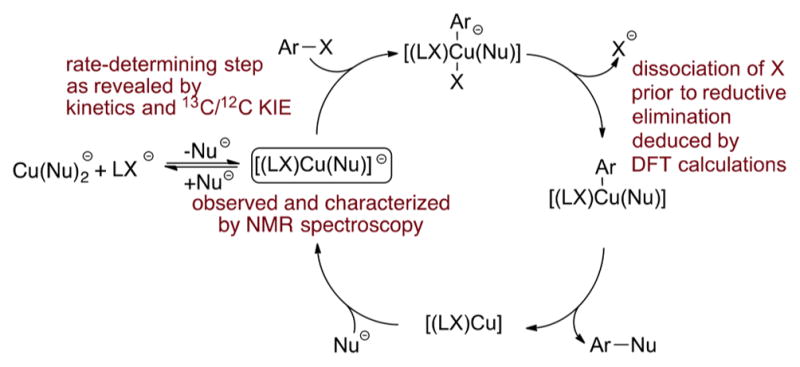
Proposed Catalytic Cycle for the Ullmann Biaryl Ether Formation Catalyzed by CuI in the Presence of Anionic Ligands LX
Although the charges and stability of the anionic phenoxide complex ligated by a bidentate anionic ligand are distinct from those of the neutral phenoxide complexes ligated by a neutral bidentate ligand,15 the pathway for reaction of the anionic species is similar to that for reaction of the neutral species. This mechanism is different from that proposed for the reaction of iodobenzene with the alcohol and with the amine of 5-aminopentanol in the presence of a similar anionic ligand.20 The difference between the mechanisms of the reactions of phenoxides and those of alcohols and amines can be rationalized by the greater stability of the anionic aryloxide-containing complexes [Cu(LX)(OAr)] − in the coupling of phenols than of the anionic alkoxide counterparts computed in prior studies for the reactions of alcohols and amines.20b This greater stability of the phenoxide complexes causes them to be the resting state of the catalyst or to lie only slightly uphill of an alternative resting state of two homoleptic cuprates (the phenoxide and bis-LX complexes) and causes these reactions to occur by the anionic pathway.
Supplementary Material
Acknowledgments
We gratefully acknowledge the NIH NIGMS (5R37-GM055382) for support of this work. We also acknowledge the NIH for support of the Molecular Graphics and Computation Facility at UC Berkeley (NIH S10OD023532). We thank Dr. Jeffrey Pelton and Dr. Hasan Celik for help on the NMR experiments, and we thank Rhonda Stoddard and Scott
McIndoe and the University of Victoria for assistance and facilities to obtain the ESI-MS data.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b11853.
Experimental procedures, characterization, and computational details (PDF)
X-ray data for complex K[Cu(LX1)2] (CIF)
X-ray data for complex K[Cu(LX3)2] (CIF)
References
- 1.Frlan R, Kikelj D. Synthesis. 2006;2006:2271–2285. [Google Scholar]
- 2.Ullmann F, Sponagel P. Ber Dtsch Chem Ges. 1905;38:2211–2212. [Google Scholar]
- 3.(a) Evano G, Blanchard N, Toumi M. Chem Rev. 2008;108:3054–3131. doi: 10.1021/cr8002505. [DOI] [PubMed] [Google Scholar]; (b) Monnier F, Taillefer M. Angew Chem, Int Ed. 2009;48:6954–6971. doi: 10.1002/anie.200804497. [DOI] [PubMed] [Google Scholar]
- 4.(a) Sambiagio C, Marsden SP, Blacker AJ, McGowan PC. Chem Soc Rev. 2014;43:3525–3550. doi: 10.1039/c3cs60289c. [DOI] [PubMed] [Google Scholar]; (b) Ley SV, Thomas AW. Angew Chem, Int Ed. 2003;42:5400–5449. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 5.(a) Kiyomori A, Marcoux JF, Buchwald SL. Tetrahedron Lett. 1999;40:2657–2660. [Google Scholar]; (b) Goodbrand HB, Hu NX. J Org Chem. 1999;64:670–674. [Google Scholar]; (c) Gujadhur RK, Bates CG, Venkataraman D. Org Lett. 2001;3:4315–4317. doi: 10.1021/ol0170105. [DOI] [PubMed] [Google Scholar]
- 6.Weng Z, He W, Chen C, Lee R, Tan D, Lai Z, Kong D, Yuan Y, Huang KW. Angew Chem, Int Ed. 2013;52:1548–1552. doi: 10.1002/anie.201208432. [DOI] [PubMed] [Google Scholar]
- 7.(a) Zanon J, Klapars A, Buchwald SL. J Am Chem Soc. 2003;125:2890–2891. doi: 10.1021/ja0299708. [DOI] [PubMed] [Google Scholar]; (b) Antilla JC, Baskin JM, Barder TE, Buchwald SL. J Org Chem. 2004;69:5578–5587. doi: 10.1021/jo049658b. [DOI] [PubMed] [Google Scholar]; (c) Surry DS, Buchwald SL. Chem Sci. 2010;1:13–31. doi: 10.1039/C0SC00107D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cristau HJ, Cellier PP, Hamada S, Spindler JF, Taillefer M. Org Lett. 2004;6:913–916. doi: 10.1021/ol036290g. [DOI] [PubMed] [Google Scholar]
- 9.Ma D, Cai Q. Org Lett. 2003;5:3799–3802. doi: 10.1021/ol0350947. [DOI] [PubMed] [Google Scholar]
- 10.Fagan PJ, Hauptman E, Shapiro R, Casalnuovo A. J Am Chem Soc. 2000;122:5043–5051. [Google Scholar]
- 11.(a) Buck E, Song ZJ, Tschaen D, Dormer PG, Volante RP, Reider PJ. Org Lett. 2002;4:1623–1626. doi: 10.1021/ol025839t. [DOI] [PubMed] [Google Scholar]; (b) de Lange B, Lambers-Verstappen MH, Schmieder-van de Vondervoort L, Sereinig N, de Rijk R, de Vries AHM, de Vries JG. Synlett. 2006;2006:3105–3109. [Google Scholar]
- 12.Wang D, Cai Q, Ding K. Adv Synth Catal. 2009;351:1722–1726. [Google Scholar]
- 13.(a) Kwong FY, Klapars A, Buchwald SL. Org Lett. 2002;4:581–584. doi: 10.1021/ol0171867. [DOI] [PubMed] [Google Scholar]; (b) Kwong FY, Buchwald SL. Org Lett. 2003;5:793–796. doi: 10.1021/ol0273396. [DOI] [PubMed] [Google Scholar]; (c) Cristau HJ, Cellier PP, Spindler JF, Taillefer M. Chem - Eur J. 2004;10:5607–5622. doi: 10.1002/chem.200400582. [DOI] [PubMed] [Google Scholar]; (d) Rao H, Jin Y, Fu H, Jiang Y, Zhao Y. Chem - Eur J. 2006;12:3636–3646. doi: 10.1002/chem.200501473. [DOI] [PubMed] [Google Scholar]; (e) Zhang Y, Yang X, Yao Q, Ma D. Org Lett. 2012;14:3056–3059. doi: 10.1021/ol301135c. [DOI] [PubMed] [Google Scholar]; (f) Wang Y, Ling J, Zhang Y, Zhang A, Yao Q. Eur J Org Chem. 2015;2015:4153–4161. [Google Scholar]
- 14.Giri R, Hartwig JF. J Am Chem Soc. 2010;132:15860–15863. doi: 10.1021/ja105695s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tye JW, Weng Z, Giri R, Hartwig JF. Angew Chem, Int Ed. 2010;49:2185–2189. doi: 10.1002/anie.200902245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tye JW, Weng Z, Johns AM, Incarvito CD, Hartwig JF. J Am Chem Soc. 2008;130:9971–9983. doi: 10.1021/ja076668w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang Z, Hartwig JF. Angew Chem, Int Ed. 2012;51:1028–1032. doi: 10.1002/anie.201106719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubinina GG, Ogikubo J, Vicic DA. Organometallics. 2008;27:6233–6235. [Google Scholar]
- 19.Lefêvre G, Franc G, Tlili A, Adamo C, Taillefer M, Ciofini I, Jutand A. Organometallics. 2012;31:7694–7707. [Google Scholar]
- 20.(a) Jones GO, Liu P, Houk KN, Buchwald SL. J Am Chem Soc. 2010;132:6205–6213. doi: 10.1021/ja100739h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yu HZ, Jiang YY, Fu Y, Liu L. J Am Chem Soc. 2010;132:18078–18091. doi: 10.1021/ja104264v. [DOI] [PubMed] [Google Scholar]
- 21.Sperotto E, van Klink GPM, van Koten G, de Vries JG. Dalton Trans. 2010;39:10338–10351. doi: 10.1039/c0dt00674b. [DOI] [PubMed] [Google Scholar]
- 22.Lefevre G, Tlili A, Taillefer M, Adamo C, Ciofini I, Jutand A. Dalton Trans. 2013;42:5348–5354. doi: 10.1039/c3dt32948h.(b) A heteroleptic complex [Cu(LX)OPh] − was reported to be characterized by 1H NMR and ESI-MS in ref 22a. This characterization was, however, not supported by comparison with alternative structures (such as homoleptic complexes [Cu(OPh)2] − and [Cu(LX)2] −) which might be in equilibrium with these species and to which the reported resonances could be assigned.
- 23.(a) Xifra R, Ribas X, Llobet A, Poater A, Duran M, Solà M, Stack TDP, Benet-Buchholz J, Donnadieu B, Mahía J, Parella T. Chem - Eur J. 2005;11:5146–5156. doi: 10.1002/chem.200500088. [DOI] [PubMed] [Google Scholar]; (b) Casitas A, Canta M, Solà M, Costas M, Ribas X. J Am Chem Soc. 2011;133:19386–19392. doi: 10.1021/ja2058567. [DOI] [PubMed] [Google Scholar]; (c) Huffman LM, Casitas A, Font M, Canta M, Costas M, Ribas X, Stahl SS. Chem - Eur J. 2011;17:10643–10650. doi: 10.1002/chem.201100608. [DOI] [PubMed] [Google Scholar]; (d) Casitas A, Ribas X. Chem Sci. 2013;4:2301–2318. [Google Scholar]; (e) Rovira M, Soler M, Güell I, Wang MZ, Góomez L, Ribas X. J Org Chem. 2016;81:7315–7325. doi: 10.1021/acs.joc.6b01035. [DOI] [PubMed] [Google Scholar]
- 24.(a) Franc G, Cacciuttolo Q, Lefèvre G, Adamo C, Ciofìni I, Jutand A. ChemCatChem. 2011;3:305–309. [Google Scholar]; (b) Lefèvre G, Franc G, Adamo C, Jutand A, Ciofini I. Organometallics. 2012;31:914–920. [Google Scholar]
- 25.Mann G, Shelby Q, Roy AH, Hartwig JF. Organometallics. 2003;22:2775–2789. [Google Scholar]
- 26.Pu YM, Ku YY, Grieme T, Black LA, Bhatia AV, Cowart M. Org Process Res Dev. 2007;11:1004–1009. [Google Scholar]
- 27.Wang D, Ding K. Chem Commun. 2009:1891–1893. doi: 10.1039/b821212k. [DOI] [PubMed] [Google Scholar]
- 28.Oxtoby DW, Gillis HP, Campion A. Principles of Modern Chemistry. 7. Brooks/Cole; Belmont: 2012. [Google Scholar]
- 29.Sasaki Y, Zhong C, Sawamura M, Ito H. J Am Chem Soc. 2010;132:1226–1227. doi: 10.1021/ja909640b. [DOI] [PubMed] [Google Scholar]
- 30.(a) Cotton FA, Wood JS. Inorg Chem. 1964;3:245–251. [Google Scholar]; (b) Chen ZF, Zhang M, Shi SM, Huang L, Liang H, Zhou ZY. Acta Crystallogr, Sect E: Struct Rep Online. 2003;59:m814–m815. [Google Scholar]
- 31.Lubben AT, McIndoe JS, Weller AS. Organometallics. 2008;27:3303–3306.(b) Despite these precautions, signals with m/z values that are attributed to complexes of Cu were typically accompanied by additional small signals with m/z values 16 and 32 amu greater than the large signal. These additional peaks suggest that a small amount of oxygen was also present.
- 32.Annunziata A, Galli C, Marinelli M, Pau T. Eur J Org Chem. 2001;2001:1323–1329. [Google Scholar]
- 33.Rovira M, Jasikova L, Andris E, Acuna-Pares F, Soler M, Guell I, Wang M-Z, Gomez L, Luis JM, Roithova J, Ribas X. Chem Commun. 2017;53:8786–8789. doi: 10.1039/c7cc04491g. [DOI] [PubMed] [Google Scholar]
- 34.Singleton DA, Thomas AA. J Am Chem Soc. 1995;117:9357–9358. [Google Scholar]
- 35.A computer program called QUIVER was used for these calculations. An optimized version of this program can be downloaded from https://github.com/ekwan/quiver.
- 36.(a) Klamt A, Schuurmann G. J Chem Soc, Perkin Trans. 1993;2:799–805. [Google Scholar]; (b) Andzelm J, Kölmel C, Klamt A. J Chem Phys. 1995;103:9312–9320. [Google Scholar]; (c) Barone V, Cossi M. J Phys Chem A. 1998;102:1995–2001. [Google Scholar]; (d) Cossi M, Rega N, Scalmani G, Barone V. J Comput Chem. 2003;24:669–681. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- 37.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165–195. [Google Scholar]
- 38.(a) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys Rev B: Condens Matter Mater Phys. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 39.(a) Hay PJ, Wadt WR. J Chem Phys. 1985;82:270–283. [Google Scholar]; (b) Wadt WR, Hay PJ. J Chem Phys. 1985;82:284–298. [Google Scholar]; (c) Hay PJ, Wadt WR. J Chem Phys. 1985;82:299–310. [Google Scholar]
- 40.Ehlers AW, Böhme M, Dapprich S, Gobbi A, Höllwarth A, Jonas V, Köhler KF, Stegmann R, Veldkamp A, Frenking G. Chem Phys Lett. 1993;208:111–114. [Google Scholar]
- 41.Krishnan R, Binkley JS, Seeger R, Pople JA. J Chem Phys. 1980;72:650–654. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.