

How does the genome become unstable during aging?
Abstract
The causal relationship between genomic instability and replicative aging is unclear. We reveal here that genomic instability at the budding yeast ribosomal DNA (rDNA) locus increases during aging, potentially due to the reduced cohesion that we uncovered during aging caused by the reduced abundance of multiple cohesin subunits, promoting increased global chromosomal instability. In agreement, cohesion is lost during aging at other chromosomal locations in addition to the rDNA, including centromeres. The genomic instability in old cells is exacerbated by a defect in DNA double-strand break (DSB) repair that we uncovered in old yeast. This was due to limiting levels of key homologous recombination proteins because overexpression of Rad51 or Mre11 reduced the accumulation of DSBs and largely restored DSB repair in old cells. We propose that increased rDNA instability and the reduced DSB repair capacity of old cells contribute to the progressive accumulation of global chromosomal DNA breaks, where exceeding a threshold of genomic DNA damage ends the replicative life span.
INTRODUCTION
Aging is a complex universal biological process characterized by progressive functional decline of biological macromolecules. It serves as the greatest risk factor for numerous diseases including cancer, neurodegenerative disorders, cardiovascular diseases, and many others (1). Understanding the molecular basis of aging is, thus, imperative for the design of therapeutics to delay or prevent age-related disorders. Unfortunately, there are still huge gaps in our understanding of aging.
Budding yeast is one of the most informative eukaryotic model organisms for aging studies because of its short life span and ease of genetic and environmental manipulations (2). Its asymmetric cell division, together with daughter cell rejuvenation, has made it uniquely suitable for accurately measuring replicative life span. However, because the proportion of aged cells in a logarithmically growing yeast culture is negligible, it was a significant challenge to obtain enough old cells for molecular analyses until the development of the “Mother Enrichment Program” (MEP). The MEP enables estradiol-inducible mitotic arrest of only daughter cells (3) and allows the isolation of more, and older, old cells than previously possible, allowing one to gain novel molecular insights into events during replicative aging.
One important hallmark of aging is increased genomic instability (4). In agreement, we previously observed increased levels of translocations, amplification of chromosomal regions, DNA breaks, and retrotransposition during yeast aging (5). What causes increased genomic instability during aging, and whether this genomic instability causes aging, is unknown. Notably, most of the human premature aging syndromes are characterized by defects in genome surveillance mechanisms (6).
Packaging of the genomic DNA with histones into chromatin is critical to regulate all genomic processes, including genomic stability (7). The chromatin structure has been shown to be a critical modulator of aging in yeast and human fibroblasts (1). Previous studies from our laboratory found global loss of histones from all regions of the yeast genome during replicative aging (5). An open chromatin conformation is likely to allow factors to gain inappropriate access to the DNA, compromising gene expression and genomic integrity. Loss of histones during aging causes globally increased gene expression and accumulation of DNA breaks (5). The major pathway to repair DNA breaks accurately is homologous recombination (HR) (8). Defects in HR could account for the translocations, loss of heterozygosity (LOH), amplifications, and elevated levels of DNA breaks that occur in old yeast. Following DNA double-strand break (DSB) formation, repair by the HR pathway involves DNA-end processing/resection by MRN/X (Mre11-Rad50-Nbs1/Xrs2) and the Sae2 protein, followed by replication protein A (RPA) loading onto single-stranded DNA. This is followed by Rad51 nucleoprotein filament formation and strand exchange into the duplex DNA of the sister chromatid, assisted by Rad52, Rad54, and other proteins. Information is copied from the sister chromatid to accurately restore the genomic information (8). Recent studies have indicated that the efficiency of HR is reduced during in vitro senescence of human fibroblasts (9). Whether this is the case in other organisms and whether this contributes to genomic instability during aging are unclear.
While loss of genomic integrity can affect all genomic regions during aging, certain regions of the genome are particularly fragile. The ribosomal DNA (rDNA) locus is one of the most damaged regions of the genome during yeast replicative aging (5). In agreement, the rDNA is the region that mediates age-induced LOH (10). The rDNA locus is highly conserved in eukaryotes, with a large number of tandemly repeated sequences, comprising both genes and intergenic regions with noncoding elements. Repetitive in nature, the rDNA is highly recombinogenic, enabling fluctuations in rDNA copy numbers (11). Although production of extrachromosomal rDNA circles (ERCs) that result from changes in rDNA copy number had been proposed as a cause of aging in yeast (12), the rDNA model of aging (13) suggests that they are by-products of rDNA instability. This rDNA model of aging states that loss of silencing of the noncoding regions within the rDNA, for example, by deletion of SIR2, leads to increased noncoding gene expression, which displaces cohesion and leads to unequal rDNA recombination and rDNA instability. Ganley et al. suggest that the rDNA instability lengthens the cell cycle through unknown signals, eventually inducing cessation of cell division. Notably, dietary restriction and inhibition of target of rapamycin (TOR), two well-known life-extending mechanisms, also reduce rDNA instability (13). Although the available evidence correlates rDNA instability with aging, it is unclear how or whether this instability influences life span.
Using old yeast cells isolated by the MEP, we show that the rDNA locus becomes more recombinogenic during yeast replicative aging, causing increased global genomic instability. We also found that the noncoding RNAs (ncRNAs) reported to be involved in regulating yeast life span (14) are highly overexpressed in old cells, but the transcripts are mostly derived from ERCs, not the chromosomal rDNA locus, in old cells. We observed loss of cohesin and cohesion during aging, not only at the rDNA but also at other locations. However, loss of cohesin occupancy occurred independent of ncRNA transcription from the rDNA locus. Furthermore, HR repair efficiency is severely compromised in aged yeast cells due to less translation of key DNA repair proteins because overexpression of key HR proteins rescued the genomic stability of old cells.
RESULTS
ncRNA transcription is highly induced from the rDNA locus in old cells
The rDNA locus in budding yeast is located in a single cluster at the right arm of chromosome (chr) XII, consisting of approximately 150 tandem repeats of rDNA units (Fig. 1A). Each individual repeat includes 5S and 35S ribosomal RNA (rRNA) genes and nontranscribed spacers 1 and 2 (NTS1 and NTS2). The NTS1 region has a replication fork barrier (RFB) site, which allows replication to proceed in a single direction, to prevent collision with RNA polymerase transcribing the 35S genes. The binding of Fob1 protein to the RFB sites may lead to a replication fork block, resulting in a fragile site that can be accompanied by a DSB. The NTS2 region has an origin of replication or autonomously replicating sequences (ARS) and a cohesin-associating region (CAR) (15, 16), which ensures equal sister chromatid recombination. In our RNA sequencing analysis of replicative aging (5), transcription from NTS1 and NTS2 was the most induced out of all the yeast genes during aging. Given that transcription of the NTS1 and NTS2 ncRNAs had never been reported in wild-type (WT) yeast, we performed validation by reverse transcription polymerase chain reaction (PCR). Aged cells isolated using the MEP (25 or more generations old) showed clear transcriptional induction from NTS1 and NTS2 (fig. S1A) in old cells (Fig. 1B) from both DNA strands, consistent with having bidirectional promoters (17). RNA polymerase I transcription is inversely proportional to ncRNA production at rDNA, suggesting that RNA polymerase I activity may silence ncRNA production at this locus (18). However, the levels of 25S and 18S rRNA as an indication of RNA polymerase I activity are comparable in young and old cells (fig. S1, B and C), indicating that the increased ncRNA during aging is not due to loss of RNA polymerase I–mediated regulation.
Fig. 1. Transcription of ncRNA from the rDNA during aging.
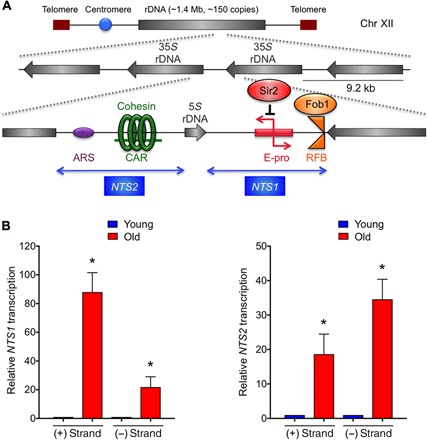
(A) Structure of yeast rDNA locus. (B) Strand-specific reverse transcription quantitative PCR analysis from both the Watson (+) and Crick (−) strands of NTS1/2 during aging. RNA levels were normalized to ACT1 before normalizing to 1 for young cells in each case. The average and SEM of three replicates are plotted. *P < 0.05, as determined by Student’s t test.
Cohesion is lost not only from the rDNA but also globally during aging
Transcription from the bidirectional EXP promoter (E-pro) of NTS1 in yeast deleted for SIR2 has been reported to promote dissociation of cohesin from the rDNA locus to regulate recombination (17) (Fig. 1A). Because we observed increased ncRNA from the rDNA during aging (Fig. 1B), presumably as a consequence of histone loss during aging (5), we asked whether this transcription displaces cohesin from the rDNA. We examined CARs within the rDNA, a centromeric region of chr III and a cohesin-depleted region of chr III (chr III: 309,955 to 310,355) as a negative control. Mcd1 occupancy was significantly lower in aged cells within the rDNA in comparison to young cells, as determined by chromatin immunoprecipitation (ChIP) analysis (Fig. 2A). However, there was a similar reduction of cohesin occupancy outside of the rDNA locus (CARL2) and at the centromeric CAR in old cells, indicating that reduced cohesin occupancy during aging extended beyond the rDNA locus.
Fig. 2. Cohesion is lost during replicative aging.
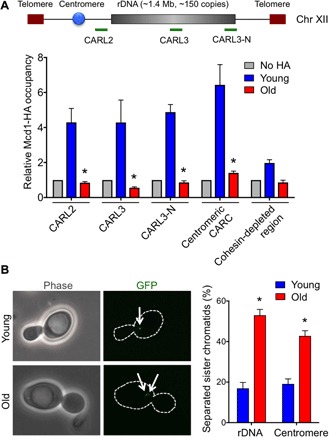
(A) ChIP analysis of hemagglutinin (HA)–tagged Mcd1 in young and old cells. An untagged strain was used as a negative control. The average and SEM of four replicates are plotted. *P < 0.05, as determined by Student’s t test. (B) Loss of cohesion at rDNA and centromeric regions with aging. Representative images are shown on the left. The average and SEM of three replicates are plotted. The arrows indicate one spot or two spots of GFP lac repressor. *P < 0.05, as determined by Student’s t test.
To determine whether the reduction of cohesin occupancy during aging is functionally relevant, we measured cohesion. We visualized cohesion at the rDNA and centromere by observing Lac repressor–green fluorescent protein (GFP) localization in strains where the lacO array was inserted into the rDNA or closer to the centromere (19). We only considered large budded cells where all GFP signal was still in the mother cell to focus on cohesion after replication but before chromosome segregation. Cohesed sister chromatids yield a single GFP spot, whereas cohesion loss yields two GFP spots (Fig. 2B). The percentage of separated sister chromatids was significantly higher at the rDNA and centromere in older cells (Fig. 2B). Hence, there is a significant loss of cohesion with aging at the rDNA and other regions of the genome.
Cohesin displacement from the rDNA is not due to ncRNA
We further examined the relationship between ncRNA expression from the rDNA and cohesion as it relates to longevity in the rDNA theory of aging (13). Previously, when transcription was induced from the galactose-inducible bidirectional Gal1/10 promoter (Gal-pro) inserted in place of the native E-pro promoter in the NTS1 locus (Fig. 3A), it increased rDNA instability and reduced life span (14). In contrast, transcription inhibition from Gal-pro by the addition of glucose reduced rDNA instability and increased life span (14). Using the same strains, we found that in log-phase yeast cultures, neither repression nor induction of NTS1 transcription by Gal-pro had a significant effect on cohesin occupancy at CARL2 (Fig. 3A). Meanwhile, both repression and induction of NTS1 transcription by Gal-pro significantly reduced cohesin occupancy at the intergenic noncoding regions of the rDNA (CARL3 and CARL3-N; Fig. 3A). Notably, repression of NTS1 transcription led to the greatest loss of cohesin occupancy at these sites (Fig. 3A). Hence, although NTS1 transcription may limit replicative life span (14), this is independent of its effect on cohesin occupancy at the rDNA. In agreement, deletion of FOB1, which strongly reduced NTS1/2 transcription (Fig. 3B), has little effect on cohesin occupancy at either E-pro or Gal-pro (fig. S2A). Our findings indicate that the cohesin removal that occurs upon Sir2 loss (accompanied by NTS1 gene expression) (17) should not be extrapolated to suggest that increased expression of NTS1 displaces cohesin in WT cells because that is not the case (Fig. 3A). Together, our data show that the loss of cohesion in old cells is not due to increased NTS1 transcription but, instead, is due to reduced total levels of cohesin protein.
Fig. 3. NTS1/2 transcription does not directly correlate with cohesin association.
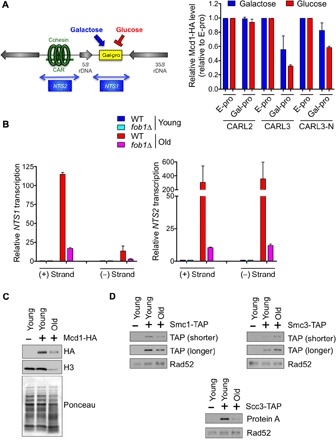
(A) ChIP analysis of Mcd1 occupancy in log-phase cultures. Data shown were normalized to a positive control region and then to E-pro. The average and SEM of two replicates are plotted. (B) Strand-specific qRT-PCR analysis, as in Fig. 1B, in WT and fob1Δ strains. (C) Western analysis of HA-tagged Mcd1 and (D) tandem affinity purification (TAP)–tagged Smc1, Smc3, and Scc3 with untagged strain as a control. Histone H3 protein level is known to be decreased in old cells (52), whereas Rad52 protein level remains constant during aging (this study). Antibodies against TAP or protein A were used, as indicated.
The majority of the ncRNA from the rDNA in old cells arises from ERCs
Because we found no correlation between NTS1 transcription and cohesin occupancy (Fig. 3A), we asked whether the ncRNAs produced during aging even come from the rDNA on chr XII. We compared the ncRNA levels in WT yeast and a fob1Δ mutant, where ERCs accumulate to a much lower degree during aging in fob1Δ cells (20). Because the fob1Δ mutant cells live longer, we isolated cells with a mean age of ~30 replicative divisions and found that deletion of FOB1 caused an eightfold reduction in NTS1 transcription, and a 30-fold reduction in NTS2 transcription, in the old cells (Fig. 3B). Hence, we conclude that the majority of NTS1/2 transcription in old cells comes from the ERCs and not the rDNA locus on chr XII.
Given that loss of cohesion during aging is independent of ncRNA transcription from the rDNA, we asked whether the abundance of cohesin was reduced during aging. The levels of the Mcd1 subunit of cohesin were drastically reduced during aging (Fig. 3C and fig. S2B). We also observed reduced levels of Smc1 and Scc3, but not Smc3, cohesin subunits during aging (Fig. 3D). We conclude that lower protein levels of multiple cohesion subunits are the likely explanation for the cohesion defect in old cells.
The rDNA region and chr XII become unstable during aging
The loss of cohesion at the rDNA during aging (Fig. 2) prompted us to ask whether there is unequal recombination in old cells resulting in rDNA repeat expansion/contraction. We compared the length of chr XII in young and old cells via pulse-field gel electrophoresis (PFGE) followed by Southern hybridization with an rDNA-specific probe (Fig. 4A). Whereas young cells showed one band for chr XII, indicating uniform rDNA locus length, aged cells showed variability in its size with two main populations of chr XII, which is apparent as early as middle age (~14 to 15 divisions) but increases with age (fig. S3). Notably, while the rDNA probe displayed chr XII specificity in young cells, it additionally hybridized to all other chromosomes in old cells (Fig. 4A and fig. S3). Together with our previous observation of rDNA sequences fused to other chromosomal sequences in old cells (5), these data suggest that small rDNA repeat fragments insert into other yeast chromosomes during aging.
Fig. 4. The rDNA locus and chr XII become increasingly unstable with age.
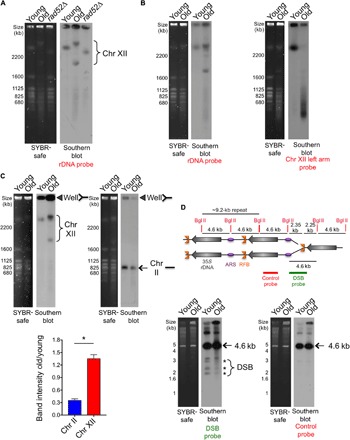
(A) Analysis of yeast chromosomes by PFGE. Left panel: SYBR-safe staining. Right panel: Southern hybridization using a chr XII (rDNA) probe. The rad52Δ mutant was used as a control with a different rDNA copy number. (B) As in (A), probes to the rDNA or the left arm of chr XII on the same samples. (C) Left panel: SYBR-safe staining of same number of cells analyzed by PFGE. Right panel: Southern blot using the indicated probe. Closed triangles indicate stuck DNA in the well. Quantification of the relative intensity of stuck DNA in old cells compared to young cells is shown below with the average and SEM of three replicates. *P < 0.05, significant change, as determined by Student’s t test. (D) The DSB at the RFB is not more abundant in old cells. Schematic showing use of Bgl II digestion to detect DSB at RFB. Southern detection using either the DSB probe or the control probe is shown. The asterisk indicates DNA fragments generated from DSBs near the RFB, whereas the arrows indicate fragments from restriction digestion with no DSB at RFB.
To understand why the rDNA probe always detected chr XII with a longer and a shorter size in old cells, we asked whether these were both intact chr XII. Strikingly, we consistently observed that a chr XII left arm probe (centered 84 kb from the left telomere) did not hybridize to either of the presumed chr XII bands in old cells despite being clearly apparent in the total DNA stain (Fig. 4B). Instead, the left arm of chr XII underwent severe fragmentation in old cells, as seen by the smear of lower-molecular-weight bands (Fig. 4B). This indicates that the two bands that we had assumed were chr XII in old cells have lost at least some of their left arm. However, the bands were still very large, so we asked whether the rDNA array had expanded during aging. To determine the size of the rDNA array during aging, we digested with Bam HI, which cuts at sites throughout the genome but not within the rDNA array (21). We successfully liberated the rDNA array from young cells but not old cells (fig. S4). Using triple the amount of zymolyase enzyme, it was apparent that Bam HI entered the cells because the other chromosomes in the old cells became highly fragmented (fig. S4). However, both chr XII bands remained intact in the old cells. Given that there are Bam HI sites throughout the remainder of chr XII outside of the rDNA, a rational explanation of these results is that the two bands that we had assumed were chr XII in old cells are likely to be hugely expanded rDNA arrays.
We asked whether we could find direct evidence of rDNA recombination actively occurring in old cells. Recombination intermediates are unable to enter PFGE gels, leading to their being stuck in the wells. An analysis of “stuck DNA” in the wells has been used previously to show that there are more rDNA recombination intermediates in mother cells (~3 to 4 generations) compared to daughter cells (~0 to 1 generations), in conjunction with sir2 and fob1 mutant cells showing increased and decreased rDNA recombination intermediates, respectively (22). By comparing daughters to much older mothers (25 or more generations), we found that chr XII detected by the rDNA probe was four times more likely to be stuck in the well compared to chr II in old mothers (Fig. 4C), indicating that old cells have more rDNA recombination intermediates. Next, we asked whether old yeast cells have more DSBs within the rDNA locus. Bgl II digestion releases a 4.6-kb fragment when the RFB is intact and yields smaller fragments when there is a DSB at the RFB, as detected by Southern blotting. We found no significant increase in DSB formation around the RFB within the rDNA in aged cells (Fig. 4D). All the evidence suggests that DSBs at the RFB within the rDNA occur with the same frequency in old and young cells but that old cells are more likely to repair these DSBs using unequal sister recombination or fusion to other chromosomes.
Global chromosomal instability in old cells is caused by rDNA instability
We noted clear evidence of chromosomal fragmentation in old cells, apparent from the smearing of the chromosomal signals for all old chromosomes in the total DNA stain of all our PFGE analyses. If the aging genome experienced random DNA breakage, then the loss of intactness of the chromosomes would be inversely proportional to chromosome length, with the longest chromosomes becoming least intact with age. Quantitation of total DNA staining showed that the intactness of chromosomes in old cells was inversely proportional to their length (fig. S5A). To more accurately examine this relationship, we performed PFGE followed by Southern analysis of the second longest chromosome (chr II), a middle-sized chromosome (chr X), and the smallest chromosome (chr I). The smallest chromosome had the least loss of intactness in old cells, whereas the longest chromosome had the greatest loss of intactness during aging (Fig. 5A). These data indicate that there is DSB accumulation along the length of chromosomes in old cells.
Fig. 5. The aging genome accumulates random damage, whereas rDNA instability appears to promote global genomic instability.
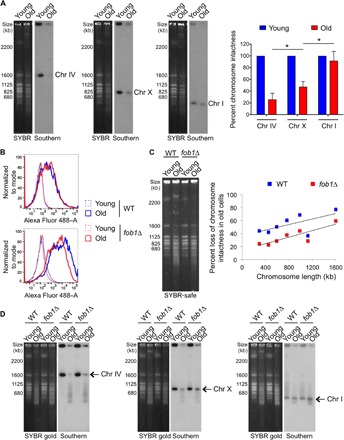
(A) As in Fig. 4A using the same numbers of young and old WT yeast cells probed for three chromosomes of different sizes: chr IV (long), chr X (middle-sized), and chr I (short). Left panels: SYBR-safe staining. Right panels: Southern blots. The graph shows percent loss of band intensity in aged cells measured from Southern blots, following normalizing signal intensity of young cells to 100% for each chromosome. The average and SEM of three replicates are plotted. The asterisk indicates significant change in chromosomal band intensities between chromosomes of different lengths in old cells (P < 0.05), as determined by Student’s t test. (B) Old cells have more global DNA damage than young cells, and this is partially restored in FOB1 deleted cells, as determined by flow cytometry analysis. Top panel: Levels of fluorescein-labeled dUTP in the absence of TdT indicating the background incorporation. Bottom panel: Levels of fluorescein-labeled dUTP in the presence of TdT that indicates levels of DNA damage. For both panels, dotted lines indicate young cells, and solid lines indicate old cells. (C) PFGE analysis from same numbers of young and old yeast cells from both WT and fob1Δ strains. The net percent loss of chromosomal band intensities during aging was measured from quantifying band intensities of all chromosomes from SYBR-safe stained agarose gels. The averages of two replicates are plotted here, along with the best-fit line for each group. The R2 value for WT is 0.39911, and for fob1Δ, the value is 0.72577. Individual experimental data are shown in fig. S6. (D) Similar analysis as in (A), with cells from both WT and fob1Δ strains. In each case, the left panel shows SYBR gold staining of chromosomes, and right panel shows Southern hybridization using the indicated probe.
To obtain a more quantitative measurement of the extent of damage in old cells, we measured DSBs using terminal deoxynucleotidyl transferase (TdT) to incorporate fluorescein-labeled deoxyuridine triphosphate (dUTP) onto DNA breaks, followed by flow cytometry analysis. Whereas young cells had very low levels of DSBs, the old cells had significantly more DSBs (Fig. 5B). To determine whether global chromosomal instability was related to rDNA instability, we compared the level of DSBs between WT yeast and a fob1Δ mutant, which has much less DSBs at the RFB within the rDNA (20). We found that old fob1Δ cells had reduced global levels of DSBs (Fig. 5B), consistent with global DNA damage correlating with levels of rDNA instability/ERCs. To verify whether the damage was chromosomal, we examined the intactness of chromosomes during aging between WT yeast and a fob1Δ mutant. With the fob1Δ mutant, we observed a similar trend of chromosomal intactness inversely proportional to chromosome length in old cells by total DNA staining, but the general chromosomal intactness was greater in old fob1Δ mutants in comparison to WT cells (Fig. 5C and fig. S5, B and C). In agreement, Southern analysis showed that the chromosomal intactness was better maintained in the fob1Δ mutant when compared to WT for every chromosome and that the loss of chromosome intactness in old cells was more pronounced in longer chromosomes than in smaller ones (Fig. 5D). These data indicate that rDNA instability is proportional to, and likely responsible for, loss of global chromosomal stability in old cells.
DNA repair is impaired in aged cells
We observed previously that replicatively aged cells accumulate DSBs (5) and that these DSBs randomly accumulate across the genome (Fig. 5). To examine whether DSB repair is defective in old yeast cells, we analyzed their ability to repair DSBs induced globally by the alkylating agent methyl methane sulfonate (MMS) by resolving intact yeast chromosomes using PFGE. In young cells, MMS treatment induced smearing of the chromosome bands indicative of DSBs, and repair was completed 6 hours after removal of MMS from the media (Fig. 6A). In contrast, there was a clear defect in old cells in the ability to reestablish intact chromosomes following MMS removal (Fig. 6A). The presence of DNA breaks causes cells to accumulate in the G2/M phase (23). During the addition of MMS and washing out the DNA damaging agent, it was clear that young cells accumulated in the G2/M phase following DNA damage and subsequently reenter the cell cycle following DNA repair (fig. S6A). By contrast, the old cells were already predominantly in the G2/M phase before inducing damage (fig. S6A). The reason why old cells accumulated in the G2/M phase is unclear. One possibility is the activation of cell cycle checkpoints, such as the spindle assembly or DNA damage checkpoint. We asked whether the elevated level of DNA damage in old cells resulted in persistent DNA damage checkpoint activity by measuring Rad53 phosphorylation as an indicator of the activated checkpoint. We could not detect Rad53 phosphorylation in old cells in the absence of exogenous damage induction (fig. S6B), but induction of DNA damage led to an equivalent degree of Rad53 phosphorylation in young and old cells (fig. S6B), indicating that old cells have an intact checkpoint response.
Fig. 6. Aged cells have impaired DSB repair due to reduced repair protein levels.
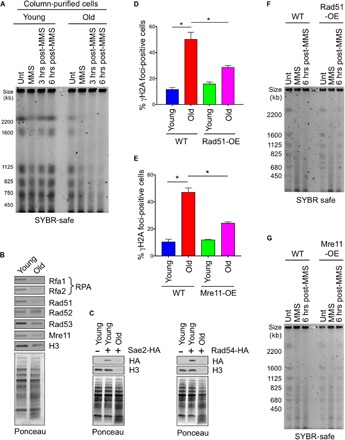
(A) PFGE analysis of chromosomes from the same numbers of young and old WT yeast cells. Samples were collected before damage induction (Unt), after MMS treatment (MMS), and after MMS removal and recovery at 3 and 6 hours, respectively. (B and C) Western blot analysis to measure key DNA repair protein levels during aging. Samples are loaded according to equal cell numbers. (B) Proteins using commercial antibodies. (C) HA-tagged proteins, with untagged strain as additional control. Ponceau staining is shown to show equal loading. (D) Quantification of percentage of young and old cells with detectable γH2A foci in both WT and single-copy Rad51-overexpression (OE) strains. The average and SEM of three replicates are plotted. *P < 0.05, as determined by Student’s t test. (E) As in (D), but for Mre11-OE. (F) As for (A) in old WT and old Rad51-overexpression yeast cells. (G) As in (F), but for Mre11-OE.
Most DSB repair in yeast is performed by HR (8). We found that the levels of several key proteins involved in different stages of HR were severely reduced during aging, as compared to young cells (Fig. 6, B and C). These included Mre11 and Sae2, RPA (Rfa1 and Rfa2), Rad51, Rad53, and Rad54. The only HR protein that did not appear to have significantly reduced levels during yeast aging was Rad52.
Overexpression of Rad51 and Mre11 in old cells restores DSB repair efficiency
To investigate whether the reduced levels of HR proteins in old cells are responsible for the profound defect in DSB repair (Fig. 6A), we overexpressed several of the key HR proteins and asked whether they could restore DSB repair. Aged cells show increased accumulation of γH2A foci, indicative of increased DNA damage during aging (5). When we introduced an extra copy of RAD51, we found a significant reduction in γH2A foci in old cells (Fig. 6D and fig. S7A). Similarly, an extra copy of MRE11 also led to a significant reduction in γH2A foci in old cells (Fig. 6E and fig. S7B). In agreement, overexpression of Rad51 or Mre11 each partially rescued the DSB repair defect in old cells (Fig. 6, F and G). Given that single-copy overexpression of Rad51 extends replicative life span in yeast (24), these data demonstrate that the reduced DSB repair in old cells is a cause of aging because partially fixing the DSB repair defect in old cells extends replicative life span (fig. S8).
DISCUSSION
We have uncovered a global loss of cohesion in replicatively old cells. This is the likely cause of the increased rDNA instability and extrachromosomal rDNA circle (ERC) accumulation that promotes global genomic instability in replicatively old cells. Furthermore, we have discovered a profound defect in DSB repair in replicatively aged yeast due to limiting levels of key components of the HR machinery. This DSB repair defect in old cells limits the replicative life span because restoration of DSB repair by overexpressing HR proteins extends life span (see model in fig. S8).
Loss of cohesion during replicative aging
Our findings necessitate updating the rDNA theory of aging. While we discovered that there is a massive increase in ncRNA transcription from the rDNA locus in old yeast cells (Fig. 1) and a loss of cohesion from the rDNA locus in old cells (Fig. 2), the loss of rDNA cohesion is unlikely to depend on the increased ncRNA transcription in old cells (Fig. 3). Furthermore, even forced transcriptional induction of ncRNAs from the rDNA locus in log-phase cell populations, which are predominantly young, was not sufficient to markedly displace cohesin from the rDNA (Fig. 3A). Instead, both repression and expression of NTS1 under the control of the GAL1 promoter led to reduced cohesin occupancy at the rDNA (Fig. 3A). The reason for this is unclear, but perhaps, the replacement of E-pro with Gal-pro changed the DNA sequences necessary for efficient cohesin recruitment. Rather than NTS1 transcription leading to cohesin displacement in old cells, the reason for the loss of cohesion during aging is reduced levels of multiple cohesion subunits (Fig. 3, C and D), which is most likely due to reduced protein synthesis of these proteins during aging.
Strikingly, we observed loss of cohesion during aging not only at the rDNA but also at other genomic locations (Fig. 2). The potential implications of this loss of cohesion for life span and human diseases of aging, including cancer, is profound. Loss of cohesion at centromeres would lead to chromosome loss and gain during mitotic aging. Minichromosome loss from mother cells during yeast replicative aging is a widely observed phenomenon and is likely due to the loss of cohesion in old cells. Functionally, cohesion loss during meiotic aging in mammals is a leading cause for missegregation of chromosomes associated with advanced maternal age, causing birth defects and developmental abnormalities (25, 26). Furthermore, aneuploidy has been shown to shorten the replicative life span of yeast (27). In addition to causing aneuploidy during mitotic aging, loss of cohesion during mitotic aging would also contribute to increased genomic instability given that cohesin is recruited to DSBs to promote accurate DSB repair (28). Loss of cohesion would also lead to unequal sister chromatid recombination and ERC accumulation.
The accumulation of ERCs has long been known to occur during yeast aging and has been proposed to be a causative reason for yeast aging (12). Indirect evidence of enlarged and fragmented nucleoli in old yeast suggested that ERC accumulation occurs in the nucleoli of old yeast cells (12). Similarly, yeast strains carrying high-copy plasmids bearing the rDNA also display punctate/fragmented nucleolar structure (29) similar to the nucleolar phenotypes observed in old cells. During the rejuvenation that occurs when yeast undergo meiosis or upon experimental expression of the meiotic transcriptional activator Ndt80, the only age-related phenotype that disappeared was nucleolar damage (30). Hence, nucleolar changes, likely due to ERC accumulation, may be a primary force behind the aging process.
The molecular reason for ERC accumulation during aging was unclear. ERCs form during aging as a consequence of unequal sister chromatid recombination between rDNA repeats (31). The loss of cohesion during aging that we have uncovered here (Fig. 2) is possibly the cause for the accumulation of ERCs during aging because cohesion is essential for equal sister chromatid recombination between the rDNA repeats. We also found ERCs to be the source of the majority of the ncRNAs from the rDNA in old cells (Fig. 3B). Whether ERCs can also contribute to the generation of active rRNAs in old cells is yet to be determined. However, given the regulation of rRNA transcription is not dependent on whether the rDNA repeats are on chr XII or on a plasmid structure similar to ERCs (29), it is plausible that ERCs also generate rRNAs in the nucleolus. However, because the levels of rRNAs remain unaltered during aging (fig. S1, B and C), we expect that the rRNAs encoded by the ERCs can be functional within the ribosome. Our study also provides evidence in support of extensive increases in the size of the rDNA array in old cells, coupled with rearrangements or deletions of other regions of chr XII (Fig. 4B). Although future analyses are required to understand the mechanistic basis for these changes, we speculate that loss of cohesion during aging is involved.
ERCs are not unique to yeast aging. Extrachromosomal circular DNAs similar to ERCs arise during Drosophila aging (32). Extrachromosomal circular DNAs also appear during both in vitro and in vivo aging of mammalian cells (33, 34). We predict that cohesion is also lost during aging of metazoans including mammals and is responsible for the circular DNA accumulation seen in these systems. Given that cancer is a disease of aging, it is interesting that rDNA instability has been shown to occur in some adult solid tumors (35). Given that a previous study found an inverse correlation between the levels of ncRNA from the rDNA and replicative life span in yeast (14), it is relevant that the majority of the ncRNA transcription during aging is coming from the ERCs (Fig. 3B). This places ERCs at center stage again for contributing to perturbing cellular homeostasis.
Instability of the rDNA and its influence on global chromosomal stability during aging
Our data demonstrate that repair efficiency declines with old age, with DSBs accumulating along the chromosomes. There is a direct correlation between rDNA instability/ERC levels and accumulation of global chromosomal damage in old cells (Fig. 5). This is likely more than mere correlation because the global damage accumulation in old cells is less in the fob1 mutant, where the rDNA is less recombinogenic and has less ERCs. It is probable that an unstable rDNA locus and/or accumulated ERCs in the nucleolus titrates key limiting factors, which would otherwise be available to maintain genomic integrity, as previously suggested (12, 36). Notably, others have shown that deletion of FOB1 improved instability at the rDNA locus, without improving global genomic instability (37). These studies were in log-phase cultures, which are predominantly young cells. Similarly, when we examined intactness of chromosomes in young cells, we saw no difference between WT and fob1Δ strains (fig. S5B). This is consistent with the protective effect of Fob1 on global genomic integrity only occurring in old cells, suggesting that a threshold of rDNA instability and/or ERCs has to be reached before it can influence global genomic integrity.
Another study using the MEP, albeit analyzing much younger old cells (9 divisions old compared to 25 or more divisions old in our work), found reduced pausing of the replication machinery at the RFB within the rDNA in older cells (38). The molecular reason for this is currently unknown. Regardless, this result is consistent with the fact that we find similar levels of DSBs close to the RFB in old and young cells (Fig. 4D), whereas the efficiency of DSB repair is greatly reduced in old cells (Fig. 6). Hence, it is possible that old cells have reduced generation of DSBs near the RFB due to reduced replication fork pausing, but in old cells, these DSBs are not effectively repaired, leading to equivalent steady-state levels of DSBs at the RFB in young and old cells.
Reduced HR in yeast limits the replicative life span
Our studies reveal a defect in DSB repair, presumably via HR in replicatively aged yeast (Fig. 6), that limits replicative life span (24). The reason for the HR repair defect in replicatively old yeast is reduced levels of key HR proteins in old cells (Fig. 6, B and C), which was possibly due to a defect in their protein synthesis in old cells. In agreement with our observations in yeast, a decline in HR efficiency occurs during replicative senescence of human fibroblast cells in vitro and also during meiotic aging in germ cells of old flies (9, 39). Replicative senescence of human fibroblasts was accompanied by reduced levels of key HR proteins, including Rad51, Rad52, NBS1, and SIRT6 (9), demonstrating that reduced levels of key HR proteins are a common feature of replicative aging yeast and human cells. In human fibroblasts, however, the HR repair defect during replicative senescence is due to reduced levels of the histone deacetylase SIRT6, as SIRT6 overexpression reestablished DSB repair (9). The molecular reason why Rad52 protein levels remain constant through aging, while most DSB repair proteins become less abundant, is not clear. This is unlikely to be related to their relative protein half-lives because Rad52 has a short half-life compared to the long half-life of Rad51, at least in young cells (40). We have shown previously that all transcripts become more abundant during aging (5), but whether there are global decreases in protein stability and/or protein synthesis during yeast replicative aging, and how Rad52 escapes these age-induced changes, remains to be determined.
The optimal level of the HR protein Rad51 seems extremely important in maintaining genomic integrity. While deficiency of Rad51 causes increased sensitivity to DNA damaging agents in budding and fission yeast (41, 42), Rad51 disruption results in the accumulation of chromosomal aberrations leading to cell death in vertebrate cells and cell lethality in mouse embryonic stem cells (43, 44). Rad51 overexpression also negatively influences HR in yeast and promotes chromosomal instability in mammalian cells (45, 46). However, the addition of an extra copy of RAD51 in yeast is better tolerated and is beneficial for longevity, as illustrated by moderate life span extension in yeast (24). This is likely due to the partial restoration of DSB repair in old cells that resulted from introduction of an extra copy of RAD51 (Fig. 6, D and F). Reintroduction of an extra copy of MRE11 also partially fixed the DSB repair defect in old cells (Fig. 6, E and G). Together, these results indicate that DSBs accumulate in old cells due to the DSB repair defect and that this is a cause of replicative aging.
Aging is a very complex multifactorial process, and replicative aging in yeast is accompanied by a myriad of altered cellular events. Our work suggests that although rDNA instability, including the accumulation of ERCs, contributes to life span regulation, it is not the only factor promoting genomic instability during aging. During mitotic aging, cellular resources become limiting, and other problems including defective cohesion, defective DSB repair, and inaccurate repair of DSB lesions within the rDNA arise, as we have uncovered here. We also propose that the limiting levels of repair factors and cohesin in old cells lead to the accumulation of genomic damage, chromosomal rearrangements, and potentially chromosome loss. Presumably, eventually, a threshold of genomic damage that is sensed by the cell to cause cell division to halt is crossed, marking the end of the replicative life cycle (fig. S8). Future work should aim at understanding the contribution of these events in the regulation of life span in greater detail.
MATERIALS AND METHODS
Yeast strains and media
All the yeast strains used in this study are listed in table S1. YEP (1% yeast extract and 2% peptone) media and SC (synthetic complete) media were used with either glucose, galactose, or raffinose added to 2%.
Purification of old cells
Isolation of aged cells was performed using the MEP, as previously described (3, 5). Briefly, 2 × 107 cells from YEPD (1% yeast extract, 2% peptone, and 2% dextrose) mid–log-phase culture were collected and washed twice in 1× phosphate-buffered saline (PBS), followed by resuspension in PBS containing Ez-Link Sulfo-NHS-LC-LC-Biotin (3 mg/ml; Thermo Scientific, P121338). Cells were incubated at room temperature for 30 min and washed twice using YEPD. These cells were used to seed culture at a density of 2 ×104 biotinylated cells/ml in YEPD. 17β-Estradiol (Sigma, E8875) was added to a final concentration of 1 μM to initiate the MEP, and cells were cultured at 30°C for 30 hours to obtain cells of desired age. At the collection time, cells were collected via centrifugation and washed twice in PBS, resuspended at a density of 5 × 108 cells/ml in 1 ml of PBS, and incubated for 30 min at room temperature with 50 μl of streptavidin-coated magnetic beads (Miltenyi Biotec, NC9821945). After streptavidin incubation, cells were washed in PBS, resuspended in 8 ml of PBS, and loaded onto an LS MACS column (Miltenyi Biotec, NC9777034) equilibrated with 5 ml of PBS. After gravity flow-through of unlabeled cells and debris, columns were washed thrice with 8 ml of PBS. The columns were then removed from the magnetic field, and aged cells were eluted by gravity flow with 8 ml of PBS and processed accordingly for the subsequent experimental purpose.
RNA isolation and cDNA synthesis
Total RNA isolation was performed using the MasterPure Yeast RNA Isolation Kit (Epicentre Biotechnologies, MPY03100) following the manufacturer’s protocols using the same number of cells for each sample. The deoxyribinuclease I digestion steps were included in our RNA isolation to obtain a purer sample for subsequent analyses. The quality of the RNA was confirmed by running RNA samples in formaldehyde gels. cDNA synthesis was performed using the Transcriptor First Strand cDNA Synthesis Kit (Roche, 04379012001), to be analyzed by subsequent qPCR analyses using LightCycler 480 SYBR Green I Master Mix (Roche, 04887352001) using the manufacturer’s instructions in a LightCycler 480 II PCR machine (Roche). The average and SEM of three independent experiments are plotted in Figs. 1A and 3B. P values were determined using Student’s t test using Prism 6 software (GraphPad Software Inc.).
Pulse-field gel electrophoresis
Analyses of intact yeast chromosomes were performed using a CHEF-DR II system (Bio-Rad), followed by Southern hybridization using probes against different chromosomes. Yeast cells grown in YEPD or YEPR (1% yeast extract, 2% peptone, and 2% raffinose) were isolated at different time points using the MEP, as indicated in the figure or figure legends. Generally 2 × 107 cells were used in each well to prepare agarose gel molds following the manufacturer’s instructions (Bio-Rad). The electrophoretic conditions used for Figs. 4 and 5 were 3 V/cm for 72 hours at 14°C with a 300- to 900-s pulse time and a 1% agarose gel with 0.5× tris-borate EDTA (TBE), and for Fig. 6, the electrophoretic conditions used were 6 V/cm for 24 hours at 14°C with a 60- to 120-s pulse time and a 0.5% agarose gel with 0.5× TBE.
Southern blot
Intact chromosomes resolved in a CHEF agarose gel were transferred to a nylon membrane via the capillary method, and specific chromosomes were detected using probes labeled by random prime labeling with 32P (Rediprime II Random Prime Labelling system, GE Healthcare Life Sciences, RPN1633) following the manufacturer’s protocol. The primer sequences used for amplifying the probes are listed in the table S2.
Analysis of branched recombination intermediates (stuck DNA analysis)
Branched recombination intermediates that were not resolved by CHEF gels remained in the plug well (stuck DNA). The analysis of branched recombination intermediate formation was performed for two chromosomes, chr II and chr XII (rDNA), in both young and old samples. Relative proportions of stuck DNA in the well during aging was measured by quantifying the signal from the Southern blot using probes against respective chromosomes. P values were determined using Student’s t test using Prism 6 software (GraphPad Software Inc.).
DSB analysis
Measurement of DSB formation at rDNA (using restriction digestion by Bgl II) was performed following the procedure described earlier with small modifications (47). The procedure of embedding the cells in agarose plugs followed by different enzymatic digestions was performed similarly as described, and the gel plug was loaded into 1% agarose gel to run in TBE at 30 V for 14 hours. The gel was stained in SYBR safe to be used for visualization of DNA and subsequent Southern blot using specific probes.
Chromatin immunoprecipitation
Samples for ChIP were collected at different time points of MEP, as indicated in the figure legends. Briefly, cells were cross-linked with formaldehyde, followed by shearing of the chromatin by sonication and immunoprecipitation using specific antibody (48). Quantitation of the DNA sequences in the input samples and immunoprecipitates was performed via real-time PCR analysis. A polyclonal antibody against rabbit HA (Abcam, ab9110) was used to measure the occupancy of Mcd1, a cohesion subunit, at different regions around the rDNA locus and control regions.
Western blot
Total protein extracts were isolated by boiling samples in 2X sample buffer [0.06 M tris-HCl (pH 6.8), 10% glycerol, 2% SDS, 5% 2-mercaptoethanol, 0.0025% bromophenol blue] before loading gels. Samples were collected according to equal cell numbers. Antibodies used were HA (Abcam, ab9110), H3 (Abcam, ab1791), Rad51 (Abcam, ab63798), Rad52 (Santa Cruz, sc-50445), Rad53 (49) Rfa1 and Rfa2 (50), Mre11 (LSBio, LS-C155765), α-tubulin (AbD Serotec, MCA78G), γ-tubulin (Abcam, ab27074), phospho-H2A S129 (Abcam, ab15083), and TAP tag antibody (Thermo Fisher Scientific, CAB1001). Secondary antibodies were anti-rabbit immunoglobulin G (IgG) (H + L), Alexa Fluor 594 (Invitrogen, A-11037), IRDye 680RD goat anti-rabbit IgG (H + L) (LICOR, 925–68071), IRDye 800CW goat anti-mouse IgG (H + L) (LICOR, 925–32210), anti-mouse horseradish peroxidase (HRP) (Promega, W4021), anti-rabbit HRP (Promega, W4011), and anti-rat HRP (Sigma, A5795).
Cohesion assay (GFP two-spot analysis)
To investigate sister chromatid cohesion, we used strains where a LacO array is inserted into rDNA or centromere expressing LacI-GFP fusion protein (19). Aged cells were collected following two rounds of purification. Exponential growth of the culture was maintained by limiting the cells to go through ~6 doublings before each round of sorting. Cells surface-labeled with Ez-Link Sulfo-NHS-LC-LC-Biotin (Thermo Scientific) were grown exponentially in YPD (yeast extract, peptone, and dextrose) media and affinity-purified following incubation with streptavidin-coated magnetic beads (Miltenyi Biotec, NC9821945) using an LS MACS column (Miltenyi Biotec) and the procedure as previously described (51). Cells at the G2/M phase were scored for a single fluorescent GFP dot (maintenance of cohesion) versus two separated fluorescent GFP dots (loss of cohesion), as visualized by fluorescence microscopy. More than 100 cells were counted for each sample. P values were determined using Student’s t test using Prism 6 software (GraphPad Software Inc.).
Repair assay using MMS
For the repair assay, biotinylated cells were grown in YEPD media according to the MEP method, and samples were collected before inducing any damage. For damage induction for Fig. 6A, 0.07% MMS (Sigma-Aldrich, 129925-5G) was used for 30 min followed by washing the cells in fresh YEPD media thrice. To analyze postdamage repair kinetics, samples were collected 3 and 6 hours after washing with MMS. All the samples collected at different time points went through magnetic biotin-streptavidin purification in a cold room as described previously. For old samples, damage induction with MMS was started at 22 hours into growing the cells following the MEP procedure so that the 6-hour repair time course was finished within 30 hours of MEP. For each time point, 2 × 107 cells were collected and processed for making DNA plugs in agarose for subsequent PFGE analysis. The condition that was used to induce damage for Fig. 6 (F and G) was 0.5% MMS for 5 min, with all other procedures performed similarly as previously described.
Immunofluorescence
Samples for immunofluorescence were collected following the MEP procedure. Cells were fixed with 4% formaldehyde followed by spheroplasting using Zymolyase 100T (10 mg/ml; United States Biological, Z1004) for 40 to 60 min. Spheroplasting was confirmed visually under the microscope. Spheroplasts were harvested and applied to a polylysine-coated microscope slide. The cells were permeabilized using methanol and acetone followed by blocking with 3% PBS–bovine serum albumin and incubation with primary and appropriate fluorescent secondary antibodies.
Flow cytometry analysis using TdT
Young and old cells were fixed with 3.7% methanol-free paraformaldehyde (EM-grade) for 20 min, followed by washing with 1X PBS and resuspension of the cell pellet in SPM buffer [1.2 M sorbitol, 50 mM KHPO4 (pH 7.4), 1 mM MgCl2]. The yeast cell wall was digested with Zymolyase 100T (10 mg/ml), followed by washing the cells with SPM buffer three times. Permeabilization was performed using permeabilization buffer (0.1% Triton X-100 and 0.1% Na-citrate) for 2 min on ice, followed by washing twice with 1X PBS. The samples were stained using a TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) staining kit (Roche, cat. no. 11684795810) following the manufacturer’s protocol and resuspended in 500 μl of 1X PBS to be analyzed using a BD LSR II flow cytometer.
Supplementary Material
Acknowledgments
We thank J. Han, R. Kang, and J.-H. Seol for assistance during this study. We are grateful to W. Dang, P. McGee, X. Zhao, and T. Kobayashi for providing yeast strains and plasmids. We thank J. Petrini for the use of his CHEF DR III apparatus, G. Brush for the Rfa1 and Rfa2 antibodies, and A. Pellicioli for the Rad53 antibody. S.P. was a recipient of the Hearst Foundation Student Research and Education Fellowship and the Marilyn and Frederick R. Lummis, Jr., M.D., Fellowship in Biomedical Sciences. Funding: This work was supported by NIH grants R01 CA95641 and AG050660 to J.K.T. Author contributions: J.K.T. guided the work, oversaw the project, and edited the manuscript. S.P. performed the experiments in Figs. 1 to 6 and fig. S1 to S5, S6A, S7, and S8. M.C. performed the experiment in fig. S6B. S.D.P. helped with some related experiments. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions of this paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/eaaq0236/DC1
fig. S1. No change in rRNA levels during aging.
fig. S2. Cohesin occupancy is not affected by NTS1 transcription, but cohesins are reduced during aging.
fig. S3. Appearance of two major chromosomal bands containing rDNA during aging.
fig. S4. The two major chromosomal bands containing rDNA appear to comprise mainly rDNA repeats in old cells.
fig. S5. Increased chromosomal instability proportional to chromosome length and rDNA instability is observed during aging in WT cells.
fig. S6. The DNA damage checkpoint is intact in old cells.
fig. S7. Rad51 and Mre11 overexpression.
fig. S8. Model for rDNA instability and reduced HR causing global genomic instability to limit replicative life span.
table S1. Yeast strains used in this study.
table S2. Primers used in this study.
Reference (53)
REFERENCES AND NOTES
- 1.Pal S., Tyler J. K., Epigenetics and aging. Sci. Adv. 2, e1600584 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Longo V. D., Shadel G. S., Kaeberlein M., Kennedy B., Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 16, 18–31 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindstrom D. L., Gottschling D. E., The mother enrichment program: A genetic system for facile replicative life span analysis in Saccharomyces cerevisiae. Genetics 183, 413–422 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez-Otin C., Blasco M. A., Partridge L., Serrano M., Kroemer G., The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu Z., Chen K., Xia Z., Chavez M., Pal S., Seol J.-H., Chen C.-C., Li W., Tyler J. K., Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 28, 396–408 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorbunova V., Seluanov A., DNA double strand break repair, aging and the chromatin connection. Mutat. Res. 788, 2–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feser J., Tyler J., Chromatin structure as a mediator of aging. FEBS Lett. 585, 2041–2048 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jasin M., Rothstein R., Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 5, a012740 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mao Z., Tian X., Van Meter M., Ke Z., Gorbunova V., Seluanov A., Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc. Natl. Acad. Sci. U.S.A. 109, 11800–11805 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindstrom D. L., Leverich C. K., Henderson K. A., Gottschling D. E., Replicative age induces mitotic recombination in the ribosomal RNA gene cluster of Saccharomyces cerevisiae. PLOS Genet. 7, e1002015 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi T., A new role of the rDNA and nucleolus in the nucleus—rDNA instability maintains genome integrity. Bioessays 30, 267–272 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Sinclair D. A., Guarente L., Extrachromosomal rDNA circles—A cause of aging in yeast. Cell 91, 1033–1042 (1997). [DOI] [PubMed] [Google Scholar]
- 13.Ganley A. R. D., Kobayashi T., Ribosomal DNA and cellular senescence: New evidence supporting the connection between rDNA and aging. FEMS Yeast Res. 14, 49–59 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Saka K., Ide S., Ganley A. R., Kobayashi T., Cellular senescence in yeast is regulated by rDNA noncoding transcription. Curr. Biol. 23, 1794–1798 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi T., Strategies to maintain the stability of the ribosomal RNA gene repeats—Collaboration of recombination, cohesion, and condensation. Genes Genet. Syst. 81, 155–161 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi T., Regulation of ribosomal RNA gene copy number and its role in modulating genome integrity and evolutionary adaptability in yeast. Cell. Mol. Life Sci. 68, 1395–1403 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi T., Ganley A. R. D., Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science 309, 1581–1584 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Cesarini E., Mariotti F. R., Cioci F., Camilloni G., RNA polymerase I transcription silences noncoding RNAs at the ribosomal DNA locus in Saccharomyces cerevisiae. Eukaryot. Cell 9, 325–335 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ide S., Miyazaki T., Maki H., Kobayashi T., Abundance of ribosomal RNA gene copies maintains genome integrity. Science 327, 693–696 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Defossez P. A., Prusty R., Kaeberlein M., Lin S. J., Ferrigno P., Silver P. A., Keil R. L., Guarente L., Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol. Cell 3, 447–455 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Johzuka K., Terasawa M., Ogawa H., Ogawa T., Horiuchi T., Condensin loaded onto the replication fork barrier site in the rRNA gene repeats during S phase in a FOB1-dependent fashion to prevent contraction of a long repetitive array in Saccharomyces cerevisiae. Mol. Cell. Biol. 26, 2226–2236 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganley A. R. D., Ide S., Saka K., Kobayashi T., The effect of replication initiation on gene amplification in the rDNA and its relationship to aging. Mol. Cell 35, 683–693 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Weinert T. A., Hartwell L. H., The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 241, 317–322 (1988). [DOI] [PubMed] [Google Scholar]
- 24.Dang W., Sutphin G. L., Dorsey J. A., Otte G. L., Cao K., Perry R. M., Wanat J. J., Saviolaki D., Murakami C. J., Tsuchiyama S., Robison B., Gregory B. D., Vermeulen M., Shiekhattar R., Johnson F. B., Kennedy B. K., Kaeberlein M., Berger S. L., Inactivation of yeast Isw2 chromatin remodeling enzyme mimics longevity effect of calorie restriction via induction of genotoxic stress response. Cell Metab. 19, 952–966 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lister L. M., Kouznetsova A., Hyslop L. A., Kalleas D., Pace S. L., Barel J. C., Nathan A., Floros V., Adelfalk C., Watanabe Y., Jessberger R., Kirkwood T. B., Höög C., Herbert M., Age-related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr. Biol. 20, 1511–1521 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Chiang T., Duncan F. E., Schindler K., Schultz R. M., Lampson M. A., Evidence that weakened centromere cohesion is a leading cause of age-related aneuploidy in oocytes. Curr. Biol. 20, 1522–1528 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sunshine A. B., Ong G. T., Nickerson D. P., Carr D., Murakami C. J., Wasko B. M., Shemorry A., Merz A. J., Kaeberlein M., Dunham M. J., Aneuploidy shortens replicative lifespan in Saccharomyces cerevisiae. Aging Cell 15, 317–324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ünal E., Heidinger-Pauli J. M., Koshland D., DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7). Science 317, 245–248 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Nierras C. R., Liebman S. W., Warner J. R., Does Saccharomyces need an organized nucleolus? Chromosoma 105, 444–451 (1997). [PubMed] [Google Scholar]
- 30.Ünal E., Kinde B., Amon A., Gametogenesis eliminates age-induced cellular damage and resets life span in yeast. Science 332, 1554–1557 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinclair D. A., Mills K., Guarente L., Molecular mechanisms of yeast aging. Trends Biochem. Sci. 23, 131–134 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Larson K., Yan S.-J., Tsurumi A., Liu J., Zhou J., Gaur K., Guo D., Eickbush T. H., Li W. X., Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLOS Genet. 8, e1002473 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kunisada T., Yamagishi H., Ogita Z.-i., Kirakawa T., Mitsui Y., Appearance of extrachromosomal circular DNAs during in vivo and in vitro ageing of mammalian cells. Mech. Ageing Dev. 29, 89–99 (1985). [DOI] [PubMed] [Google Scholar]
- 34.Cohen Z., Lavi S., Replication independent formation of extrachromosomal circular DNA in mammalian cell-free system. PLOS ONE 4, e6126 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stults D. M., Killen M. W., Williamson E. P., Hourigan J. S., Vargas H. D., Arnold S. M., Moscow J. A., Pierce A. J., Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 69, 9096–9104 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Oberdoerffer P., Michan S., McVay M., Mostoslavsky R., Vann J., Park S.-K., Hartlerode A., Stegmuller J., Hafner A., Loerch P., Wright S. M., Mills K. D., Bonni A., Yankner B. A., Scully R., Prolla T. A., Alt F. W., Sinclair D. A., SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menzel J., Malo M. E., Chan C., Prusinkiewicz M., Arnason T. G., Harkness T. A. A., The anaphase promoting complex regulates yeast lifespan and rDNA stability by targeting Fob1 for degradation. Genetics 196, 693–709 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cabral M., Cheng X., Singh S., Ivessa A. S., Absence of non-histone protein complexes at natural chromosomal pause sites results in reduced replication pausing in aging yeast cells. Cell Rep. 17, 1747–1754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delabaere L., Ertl H. A., Massey D. J., Hofley C. M., Sohail F., Bienenstock E. J., Sebastian H., Chiolo I., LaRocque J. R., Aging impairs double-strand break repair by homologous recombination in Drosophila germ cells. Aging Cell 16, 320–328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asleson E. N., Livingston D. M., Investigation of the stability of yeast rad52 mutant proteins uncovers post-translational and transcriptional regulation of Rad52p. Genetics 163, 91–101 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saeki T., Machida I., Nakai S., Genetic control of diploid recovery after gamma-irradiation in the yeast Saccharomyces cerevisiae. Mutat. Res. 73, 251–265 (1980). [DOI] [PubMed] [Google Scholar]
- 42.Muris D. F., Vreeken K., Carr A. M., Broughton B. C., Lehmann A. R., Lohman P. H., Pastink A., Cloning the RAD51 homologue of Schizosaccharomyces pombe. Nucleic Acids Res. 21, 4586–4591 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sonoda E., Sasaki M. S., Buerstedde J.-M., Bezzubova O., Shinohara A., Ogawa H., Takata M., Yamaguchi-Iwai Y., Takeda S., Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 17, 598–608 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsuzuki T., Fujii Y., Sakumi K., Tominaga Y., Nakao K., Sekiguchi M., Matsushiro A., Yoshimura Y., Morita T., Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. U.S.A. 93, 6236–6240 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richardson C., Stark J. M., Ommundsen M., Jasin M., Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene 23, 546–553 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Paffett K. S., Clikeman J. A., Palmer S., Nickoloff J. A., Overexpression of Rad51 inhibits double-strand break-induced homologous recombination but does not affect gene conversion tract lengths. DNA Repair (Amst.) 4, 687–698 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Weitao T., Budd M., Hoopes L. L., Campbell J. L., Dna2 helicase/nuclease causes replicative fork stalling and double-strand breaks in the ribosomal DNA of Saccharomyces cerevisiae. J. Biol. Chem. 278, 22513–22522 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Chen C. C., Carson J. J., Feser J., Tamburini B., Zabaronick S., Linger J., Tyler J. K., Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 134, 231–243 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fiorani S., Mimun G., Caleca L., Piccini D., Pellicioli A., Characterization of the activation domain of the Rad53 checkpoint kinase. Cell Cycle 7, 493–499 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Brush G. S., Kelly T. J., Phosphorylation of the replication protein A large subunit in the Saccharomyces cerevisiae checkpoint response. Nucleic Acids Res. 28, 3725–3732 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park P. U., McVey M., Guarente L., Separation of mother and daughter cells. Methods Enzymol. 351, 468–477 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Feser J., Truong D., Das C., Carson J. J., Kieft J., Harkness T., Tyler J. K., Elevated histone expression promotes life span extension. Mol. Cell 39, 724–735 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., Arkin A. P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., Brachat S., Campanaro S., Curtiss M., Davis K., Deutschbauer A., Entian K.-D., Flaherty P., Foury F., Garfinkel D. J., Gerstein M., Gotte D., Güldener U., Hegemann J. H., Hempel S., Herman Z., Jaramillo D. F., Kelly D. E., Kelly S. L., Kötter P., LaBonte D., Lamb D. C., Lan N., Liang H., Liao H., Liu L., Luo C., Lussier M., Mao R., Menard P., Ooi S. L., Revuelta J. L., Roberts C. J., Rose M., Ross-Macdonald P., Scherens B., Schimmack G., Shafer B., Shoemaker D. D., Sookhai-Mahadeo S., Storms R. K., Strathern J. N., Valle G., Voet M., Volckaert G., Wang C. Y., Ward T. R., Wilhelmy J., Winzeler E. A., Yang Y., Yen G., Youngman E., Yu K., Bussey H., Boeke J. D., Snyder M., Philippsen P., Davis R. W., Johnston M., Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/eaaq0236/DC1
fig. S1. No change in rRNA levels during aging.
fig. S2. Cohesin occupancy is not affected by NTS1 transcription, but cohesins are reduced during aging.
fig. S3. Appearance of two major chromosomal bands containing rDNA during aging.
fig. S4. The two major chromosomal bands containing rDNA appear to comprise mainly rDNA repeats in old cells.
fig. S5. Increased chromosomal instability proportional to chromosome length and rDNA instability is observed during aging in WT cells.
fig. S6. The DNA damage checkpoint is intact in old cells.
fig. S7. Rad51 and Mre11 overexpression.
fig. S8. Model for rDNA instability and reduced HR causing global genomic instability to limit replicative life span.
table S1. Yeast strains used in this study.
table S2. Primers used in this study.
Reference (53)