

Abstract
The complement cascade is a part of the innate immune system which acts primarily to remove pathogens and injured cells. However, complement activation is also peculiarly associated with tumor progression. Here we report mechanistic insights into this association in multiple immunocompetent orthotopic models of lung cancer. After tumor engraftment, we observed systemic activation of the complement cascade as reflected by elevated levels of the key regulator C3a. Notably, growth of primary tumors and metastases was both strongly inhibited in C3-deficient mice (C3−/− mice), with tumors undetectable in many subjects. Growth inhibition was associated with increased numbers of IFNγ+/TNFα+/IL-10+ CD4+ and CD8+ T cells. Immunodepletion of CD4+ but not CD8+ T cells in tumor-bearing subjects reversed the inhibitory effects of C3 deletion. Similarly, antagonists of the C3a or C5a receptors inhibited tumor growth. Investigations using multiple tumor cell lines in the orthotopic model suggested the involvement of a C3/C3 receptor autocrine signaling loop in regulating tumor growth. Overall, our findings offer functional evidence that complement activation serves as a critical immunomodulator in lung cancer progression, acting to drive immune escape via a C3/C5-dependent pathway.
Keywords: Lung cancer, orthotopic mouse model, complement, CD4+ T Lymphocytes, C3a/C3aR Autocrine Loop
Introduction
Lung cancer remains the leading cause of cancer death in both men and women (1), underscoring the need for new therapeutic approaches. While cancer initiation is mediated by activation of oncogenic drivers and loss of tumor suppressors, tumor progression involves interactions between cancer cells and the tumor microenvironment (TME), and targeting these interactions is an attractive therapeutic strategy. In particular, immune checkpoint inhibitors, such as antibodies blocking the interactions between PD-1 and PD-L1, have shown surprising efficacy in the treatment of multiple cancers, leading to FDA approval of anti-PD-1 antibodies for treatment of non-small cell lung cancer (NSCLC) (2, 3). However, these agents exhibit clinical efficacy only in approximately 25% of unselected patients. Developing novel strategies targeting the immune system and better selection of patients who will benefit from these therapies will require a deeper understanding of the interactions between cancer cells and the TME.
The complement pathway is an important arm of the innate immune system that interfaces to the adaptive immune system. Complement activation can be initiated through the classical, lectin, or alternative pathway. All three pathways share a common endpoint involving covalent fixation and cleavage of complement component (C3) on the surface of targeted cells. Systemic increases in C3 levels have been detected in cancer patients, including lung cancer (4). Although the complement system typically helps in the removal of pathogens and injured cells (5-7), recent studies have revealed that complement activation can in fact promote the spread of tumors (8). These effects are likely mediated through anaphylatoxins (C3a and C5a) produced during complement activation (9). These agents act on immune and inflammatory cells through specific cell surface receptors (C3aR and C5aR).
Our laboratory has developed an orthotopic immunocompetent model of lung cancer, in which murine lung cancer cell lines derived from C57BL/6 mice are implanted directly into the left lobe of the lungs of syngeneic mice (10-12). These cells form a primary tumor, which over a period of 3-4 weeks forms secondary pulmonary tumors in the other lobes and metastases to distant organs, including the liver and brain. This model allows cancer cells to develop in the relevant microenvironment (i.e. in contrast to subcutaneous injection of cancer cells) and in the presence of a complete immune system. The goal of this study was to examine the role of the complement pathway in mediating progression of these tumors. Using multiple murine lung cancer cell lines, we report a critical role for complement on cancer progression through regulation of CD4+ T cells. These studies suggest that inhibiting the complement pathway may represent a rational approach for immunotherapy in the treatment of lung cancer.
Materials and Methods
Cell Lines and Culture Conditions
CMT167 cells (13), originally provided by Dr. Alvin Malkinson (University of Colorado), were stably transfected with firefly luciferase as previously described (12) and maintained in DMEM with 1 g/L glucose containing 10% FBS, 100 U/mL penicillin, 100 μg/ml streptomycin, MEM Non-Essential Amino Acids, 10 mM HEPES, and 500 μg/mL G418 at 37°C in a humidified 5% CO2 atmosphere. Luciferase-expressing Lewis Lung Carcinoma (LLC) cells were purchased from Caliper Life Sciences (LL/2-luc-M38) and maintained in DMEM with 4.5 g/L glucose containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 500 μg/mL G418 at 37°C in a humidified 5% CO2 atmosphere. Both cell lines were periodically tested for mycoplasma infection and were last retested in October 2016. To avoid cross-contamination and phenotypic changes, these cell lines have not been maintained in long-term cultures. All cells used in this study were maintained as frozen stocks and cultured for only 2–4 weeks before use in experiments. Authentication of these cell lines based on morphology, growth curve analysis, and metastatic phenotype was performed regularly, and no phenotypic changes were observed through the duration of the study.
Isolation, Preparation, and Validation of EML4-ALK Murine Cancer Cells
Adenovirus encoding Cas9 and gRNA for the EML4-ALK translocation (30ul 107 PFU/ml, Viraquest, North Liberty, IA) was tracheally instilled into C57BL/6 mice as previously described (14). After 12-20 weeks, animals were euthanized, and visible lung tumors were collected. After mechanically disrupting tumors with razor blades, 50μl of this sample was instilled into the trachea of a recipient C57BL/6 female. Matrigel (15% total v/v) was added to the remaining tumor homogenate and injected into the right flank of the same recipient animal. When flank tumors reached ~1.5 cm (20–30 weeks after transplant), animals were euthanized, secondary lung and flank tumors were collected, processed as described above then plated into RPMI medium with 10% FBS to generate cell lines. DNA was extracted and the EML4-ALK genomic rearrangement was verified by PCR as previously described (14). Sensitivity to crizotinib and TAE-684, an ALK inhibitor was verified in vitro. Orthotopic tumors were then established in C57BL/6 animals using one of these cell lines, designated EA-2. Cells were maintained as frozen stocks and cultured for only 2-4 weeks. Cells were tested for mycoplasma monthly.
Mice
Wild-type C57BL/6J mice and mice globally deficient in complement factor 3 (C3−/−) were obtained from The Jackson Laboratory (Bar Harbor, ME). All mice were bred and maintained in the Center for Comparative Medicine at the University of Colorado Denver. Experiments were done only in 7-10 wk-old male mice. All procedures were performed under protocols approved by the Institutional Animal Care and Use Committee at the University of Colorado Denver.
Orthotopic Mouse Model
Cancer cells (1×105 in 40 μl/injection) were suspended in HBBS containing 1.35 mg/ml Matrigel (BD Biosciences) and injected into the parenchyma of the left lung lobe through the rib cage using a 30-gauge needle, as previously described (10). Mice were sacrificed 2.5–4 weeks after injection. At the time of sacrifice, mice were injected intraperitoneally (i.p.) with 300 mg/kg body weight D- luciferin (Caliper Life Sciences) 10 minutes before euthanasia. Lungs and liver were harvested and immediately imaged for bioluminescence using an IVIS Imaging System 50 Series (Caliper Life Sciences/Xenogen). For metastases, two images were taken per organ (front and back), and the number of metastatic foci was calculated. Primary tumor volume (Length × Width × Height × 0.5) was measured using digital calipers.
Subcutaneous Mouse Model
Cancer cells (2×105) were suspended in HBBS and injected into the flanks of recipients, using a 30-gauge needle. Mice were sacrificed 3–4 weeks after injection. Flank tumor volume (Length × Width × Height × 0.5) was measured using digital calipers.
Plasma Preparation and ELISA for C3a
Plasma samples were prepared in 10 mM EDTA from whole blood collected by terminal cardiac puncture. Cells were removed from plasma by centrifugation. 96-well plates were coated with capture rat anti-mouse C3a antibody (BD Pharmingen, 558250) at 1 μg/µl in pH 6.5 PBS overnight at 4°C. Samples were diluted 1:100 in Reagent Diluent (Reagent Diluent 1, R&D Systems, DY997) and incubated overnight at 4°C. Biotin rat anti-mouse C3a antibody at 0.5 μg/µl (BD Pharmigen, 558251) was used, and analyzed by ELISA using the manufacturer’s protocol.
Immunofluorescence
Tumor specimens and normal lung tissues were harvested and fixed in 4% paraformaldehyde. Paraffin embedded samples were cut into 5 μm sections. Sections were dehydrated, immersed in 0.1% Sudan Black B (Sigma 199664-25G) in 70% ethanol for 20 minutes, washed in TBST, incubated in citrate antigen retrieval solution at 100°C for 2 hours, washed in 0.1M glycine/TBST (Sigma G-8898) for 10 minutes, and placed in 10mg/ml sodium borohydride in Hank’s buffer (Gibco, 14175-095). After blocking with 10% goat serum in equal parts of 5% BSA in TBST and Superblock (ScyTek Laboratories, AAA999) at 4°C overnight, sections were incubated with primary anti-CD3 (Thermo Scientific, MA5-14524) anti-CD4 (eBiosciences, 4SM95), or anti-CD8 (eBiosciences, 4SM15) antibody in equal part solution of 5% BSA in TBST and Superblock for 1 hour at room temperature, washed in TBST, and incubated with secondary goat anti-rabbit IgG Alexa Flour 488 (Invitrogen, A11034) or goat anti-rabbit IgG Alexa Fluor 594 (Invitrogen, A11037). Slides were mounted with Vectashield mounting medium with DAPI (Vector Laboratories, H-1200).
Confocal Microscopy
Lungs were snap frozen in OCT compound (Sakura Finetek). Sagittal sections were cut using a cryostat, warmed to room temperature, and fixed with absolute acetone for 10 minutes. Sections were washed with cold DPBS and non-specific binding was blocked with sterile-filtered 10% heat-inactivated goat serum. Primary antibodies specific for complement C3 (FITC goat IgG to mouse complement C3, MP Biomedicals, #55500, 1:200 dilution), IgM (Cy3 goat anti-mouse IgM, µ-chain specific, Jackson ImmunoResearch, #115-165-020, 1:400 dilution), and complement C4 (Anti-mouse C4, clone 16D2, Hycult, #HM1046, 1:50 dilution) were diluted in sterile-filtered 2% heat-inactivated goat serum in DPBS and incubated overnight. Sections were washed with cold DPBS and when applicable, incubated in secondary antibody (Cy5 goat anti-rat IgG, Jackson ImmunoResearch, #112-175-143, 1:100 dilution). Sections underwent a final series of washes in cold DPBS and then mounted with a 1:1 glycerol/DPBS mounting medium. High-resolution images were obtained with an Olympus FV1000 FCS/RICS confocal microscope (Olympus Scientific Solutions Americas Corp).
Western Blot Analysis
Proteins were isolated from cultured cells in lysis buffer (50 mM β-glycerophosphate, pH 7.2, 0.5% Triton X-100, 5 mM EGTA, 100 μM sodium orthovanadate, 1 mM dithiothreitol, 2 mM MgCl2,) with protease inhibitor cocktail (Sigma). Proteins were fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electrophoretically transferred to PVDF membranes. The anti-C3d antibody was generated as previously described (15).
Lymphocyte Depletion
Mice were injected intraperitoneally (10 mg/kg) with anti-CD4 antibody (Clone GK1.5, BioXcell), anti-CD8 antibody (YTS 169.4, BioXcell), both anti-CD4 and anti-CD8, antibodies, or control IgG (rat IgG2A Clone 2A3 or rat IgG3B Clone LFT-2, BioXcell) twice a week, starting 1 week before CMT-luc tumor implantation.
C3a/C5a Receptor Antagonist Treatment
Mice were intraperitoneally injected daily with PBS or C3aRA, SB290157, trifluoroacetate salt (1mg/kg, Cayman Chemical Company, 15783), starting a day prior to tumor implantation. Mice were subcutaneously injected daily with PMX-control scrambled peptide or C5aRA, PMX-53 Ac-Phe-[Orn-Pro-dCha-Trp-Arg] (1mg/kg), starting a day prior to tumor implantation.
Anchorage Independent Growth
Cell culture plates were coated with base layer containing 1.2% agar in DMEM (#19-017-CV; Corning CellGro) containing 10% fetal bovine serum, penicillin/streptomycin and overlaid with cells suspended in 0.7% agar in DMEM containing 10% fetal bovine serum, penicillin/streptomycin. The plates were incubated at 37°C for 14 days, and size of tumor colonies was determined and counted with ImageJ software.
shRNA Silencing and Proliferation Assay
GIPZ lentiviral shRNA targeting murine C3 (V2LMM_67054 and V3LMM_491809), and the non-silencing lentiviral control shRNA (RHS4349) were purchased from GE Dharmacon, Lafayette, CO, USA. The lentiviral shRNA targeting murine C3a receptor (TRCN0000027362 and TRCN0000027385), murine CIITA (TRCN0000086450), and the negative control (pLKO.1-puro control – SHC001V) shRNAs were prepared at the Functional Genomics Facility, University of Colorado, Denver Anschutz Medical Campus (http://functionalgenomicsfacility.org/). LLC-luc or CMT167-luc cells were transduced with lentiviral particles and stable pool of transfectants were isolated by puromycin (1µg/ml) resistance. The growth rates of the established pools of transfectants were assessed by alive or dead cell count using trypan blue under the hemocytometer.
Preparation of single cell suspension, activation of T lymphyocytes, and analysis by flow cytometry
Mice were sacrificed 10 days after cancer cell injection, the circulation was perfused with PBS/heparin (80 U/ml, Sigma), and left lungs containing tumors were excised. Lung tissues were mechanically dissociated, and incubated at 37°C for 30 minutes with collagenase type II (8480 U/ml, Worthington Biochemical), elastase (7.5mg/ml, Worthington Biochemical), soybean trypsin inhibitor (2mg/ml, Worthington Biochemical) and DNAse I (40 µg/ml, Sigma). Resulting single-cell suspensions were filtered through 70 µm cell strainers (BD), subjected to red blood cells lysis using hypotonic buffer (0.15 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA, pH 7.2), and filtered a second time through 40 µm cell strainers. Cells were stained for 1 hour at 4°C with the following panels of antibodies: Macrophage Panel - CD11b-FITC (Clone M1/70, BD Biosciences), SiglecF-PE (Clone E50-2440, BD Biosciences), Ly6G-PE-Cy7 (Clone 1A8, BD Biosciences), F4/80-APC (Clone CI:A3-1, AbDSerotec), CD11c-APC-Cy7 (Clone HL3, BD Biosciences). T cell Intracellular Staining Panel – IA/IE-PE/Dazzle 594 (Clone M5/114.15.2) CD4-eF450 (Clone RM4-5, eBioscience), CD45-AF700 (Clone 30-F11 eBioscience), and CD8-APCe780 (Clone 53-6.7, eBioscience). Brefeldin A solution (Biolegend), monensin solution (Biolegend), and cell stimulation cocktail (eBioscience) were used at the manufactures’ recommended concentrations for 5 hours at 37°C before the cells were stained with the Intracellular T Cell Panel - IFNγ-AF488 (Clone XMG1.2, eBioscience), IL10-PE (Clone JESS-16E3, eBioscience), TNFα-PerCP-Cy5.5 (Clone MP6-TX22, eBioscience), IL17a-PE-Cy7 (Clone eBio17B7, eBioscience), and FOXp3-eF660 (Clone FJK-16s, eBioscience). T cell PD-1/CD44/CD69 Staining Panel – CD44-FITC (Clone IM7, eBioscience), PD1-PE (Clone J43, eBioscience), CD45-PE-CF594 (Clone 30-F11, eBioscience), TCRB-PE-Cy5 (Clone H57-597, eBioscience), CD69-PE/Cy7 (Clone H1.2F3, eBioscience), FoxP3-APC (Clone FJK-16S, eBioscience), CD8-AF700 (Clone 53-6.7, eBioscience), CD4-APC/Cy7 (Clone GK1.5, eBioscience), and MHCII-eFluor450 (Clone M5/114.15.2, eBioscience). Cells were analyzed at the University of Colorado Cancer Center Flow Cytometry Core using Gallios (Beckman Coulter). The gating strategy involved excluding debris and cell doublets by light scatter, and dead cells by Viability eFluor506 (L34966, Invitrogen). Data were analyzed using Kaluza Software (Beckman Coulter).
Analysis of C3aR Expression
Cells were lifted from flasks by incubating in accutase (Innovative Cell Technologies, #AT104) for 5 minutes at 37°C followed by gentle agitation and brief centrifugation. Cells were re-suspended in primary antibody and incubated for 1 hour on ice. Cells were washed, centrifuged, and if applicable, were incubated in secondary antibody for 1 hour on ice. Following centrifugation, cells were re-suspended in DPBS supplemented with 1% heat-inactivated, sterile-filtered fetal bovine serum and analyzed using a FACSCalibur flow cytometer (BD Biosciences). All antibodies were diluted in DPBS supplemented with 2 mM CaCl2 and 1.5 mM MgCl2 and included the following: C3aR mouse mAb, clone 14D4 (Hycult, #HM1123, 1:50 dilution), purified monoclonal rat IgG2a, clone 54447.11 (R&D Biosystems, #MAB006, 1:100 dilution), Cy5 goat anti-rat IgG H+L (Jackson ImmunoResearch, #112-175-143, 1:200 dilution), FITC goat IgG to mouse Complement C3 (MP Biomedicals, #55500, 1:50 dilution), and FITC whole molecule goat IgG (Jackson ImmunoResearch, #005-090-003, 1:100 dilution). Gating strategy was based on cell viability, which was determined with an Annexin V – Propidium Idodide kit (BD Pharmingen, #556547). Data was analyzed with FlowJo software (FlowJo, LLC).
Analysis of Complement Activation in Human Lung Tumors
A tissue microarray containing sections of 55 human lung adenocarcinomas was obtained from the Lung SPORE Tissue Bank at the University of Colorado Cancer Center. Prior to immunofluorescent staining, human tissue arrays underwent a standard deparaffinization/rehydration protocol followed by heat-induced antigen retrieval. Tissues were blocked for one hour at room temperature with sterile-filtered DPBS supplemented with 1% bovine serum albumin and 2% fetal bovine serum, followed by primary antibody incubation overnight at 4°C. Antibodies used to stain tissues include a DyLight 550-conjugated monoclonal antibody that only recognizes complement fragments C3d and iC3b (mAb 3d29, 18 µg/ml diluted in 1% BSA/2% FBS in DPBS) (15) and DyLight 650 goat anti-human IgM, µ-chain specific (Abcam, #ab96990, diluted to 10 µg/ml in 1% BSA/2% FBS in DPBS). Tissues were incubated in DAPI (Sigma Aldrich, #D9542, diluted 1:6000 in DPBS) for 5 minutes at room temperature, followed by 3 washes in cold DPBS (10 minutes each). Tissues were viewed using the Vectra 3.0 Quantitative Pathology Imaging System (Perkin Elmer). C3d and IgM positivity was determined in a blinded fashion by examination of tissue punches following background subtraction in each individual channel. Tissue punches were considered positive when signal could be observed in ≥ 5% of the total punch. Each adenocarcinoma was represented by 3 randomly placed punches, and was considered positive when signal could be detected in all represented punches.
Recovery of lung cancer cells from tumor-bearing GFP-expressing transgenic mice and transcriptome profiling by RNA sequencing (RNA-seq)
Lung tumors were implanted in GFP-expressing transgenic mice as described above. Three weeks after CMT167 tumor implantation or two weeks after LLC tumor implantation, the pulmonary circulation was perfused with heparinized PBS and tumor-bearing left lungs were isolated. Tissues from four mice were pooled, mechanically dissociated, incubated at 370C for 45 minutes with Collagenase A (1 mg/mL, Roche) and DNAse I (40 μg/mL, Sigma), and processed using a GentleMACS Dissociator (Miltenyi Biotec). Red blood cell lysis was performed for three minutes at room temperature in RBC lysis buffer [0.15 M NH4Cl, 0.1 mM KHCO3, 0.1 mM Na2EDTA (pH 7.2)]. Cell sorting was performed at the University of Colorado Cancer Center Flow Cytometry Shared Resource using an XDP-100 cell sorter (Beckman Coulter). The sorting strategy excluded debris and cell doublets by light scatter and dead cells by DAPI. Lung cancer cells were recovered from tumor-bearing GFP-expressing transgenic mice by sorting for GFP-negative cells. Total RNA was isolated using an RNeasy Plus kit (QIAGEN).
RNA quality and quantity were analyzed using an Agilent 2100 electrophoresis bioanalyzer. RNA-seq library preparation and sequencing were conducted at the University of Colorado Cancer Center Genomics and Microarray Shared Resource. RNA libraries were constructed using an Illumina TruSEQ stranded mRNA Sample Prep Kit (Cat# RS-122-2101). Total RNA was combined with RNA purification beads to bind PolyA RNA to oligodT magnetic beads. mRNA was eluted and converted to double stranded DNA. A-tailing, adapter ligation, and PCR amplification using 15 cycles was performed to complete the library construction. Libraries were quantitated via Qubit, analyzed on a Bioanalyzer Tape Station, and diluted to appropriate concentration to run on an Illumina HiSEQ 2500 High Throughput Flow Cell. RNA-seq reads were obtained using Illumina Hi-seq analysis Pipeline. Reads quality was checked using FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). The median number of reads per condition was 24 million. Reads were then processed and aligned to the UCSC Mus musculus reference genome (build mm10) using TopHat v2 (16). The pre-built Mus musculus UCSC mm10 index was downloaded from the TopHat homepage and used as the reference genome. The default parameters for TopHat were used. The aligned read files were processed by Cufflinks v2.0.2 (17). Reads were assembled into transcripts and their abundance was estimated. Cufflinks uses the normalized RNA-Seq fragment counts to measure the relative abundances of transcripts. The unit of measurement is Fragments Per Kilobase of exon per Million fragments mapped (FPKM). Confidence intervals for FPKM estimates were calculated using a Bayesian inference method (17).
Stimulation of cells in vitro with IFNγ
Cell lines were incubated with recombinant murine IFNγ (Sigma, 100ng/mL) for 48 hours prior to assessments. Total RNA was isolated as previously described, and expression of target genes measured by qRT-PCR.
Statistical Analyses
Data are represented as mean ± SEM. Student’s T test analysis or One-way ANOVA with Tukey post-test was used to evaluate the difference between group(s). P < 0.05 was considered significant.
Results
Complement activation occurs in the presence of orthotopic tumors
We sought to determine if systemic complement activation occurred in the setting of lung tumors in our orthotopic model. We initially used the CMT167 cell line, which is a KRAS mutant lung cancer cell line derived from a spontaneous lung adenocarcinoma in a C57BL/6 mouse (18). Cells were stably transfected with luciferase to allow bioluminescence imaging. Following orthotopic injection of luciferase-expressing CMT167 cells (CMT-luc) into the left lung of C57BL/6 wild type mice, levels of C3a were measured in plasma at 4 weeks. As shown in Fig. 1A, CMT-luc tumor bearing WT mice had elevated levels of C3a compared to naïve WT controls. To determine if these levels represented production by tumor cells or by the host, identical cells were implanted into the lungs of C3-deficient (C3−/−) mice. Levels of C3a were at the limit of detection in naïve C3−/− mice, and there was no increase in the setting of CMT-luc tumors. These data indicate that the presence of tumors in the lung is sufficient to activate C3 systemically in the host animal.
Figure 1. Inhibition of CMT-luc Tumor Growth in C3−/− mice.
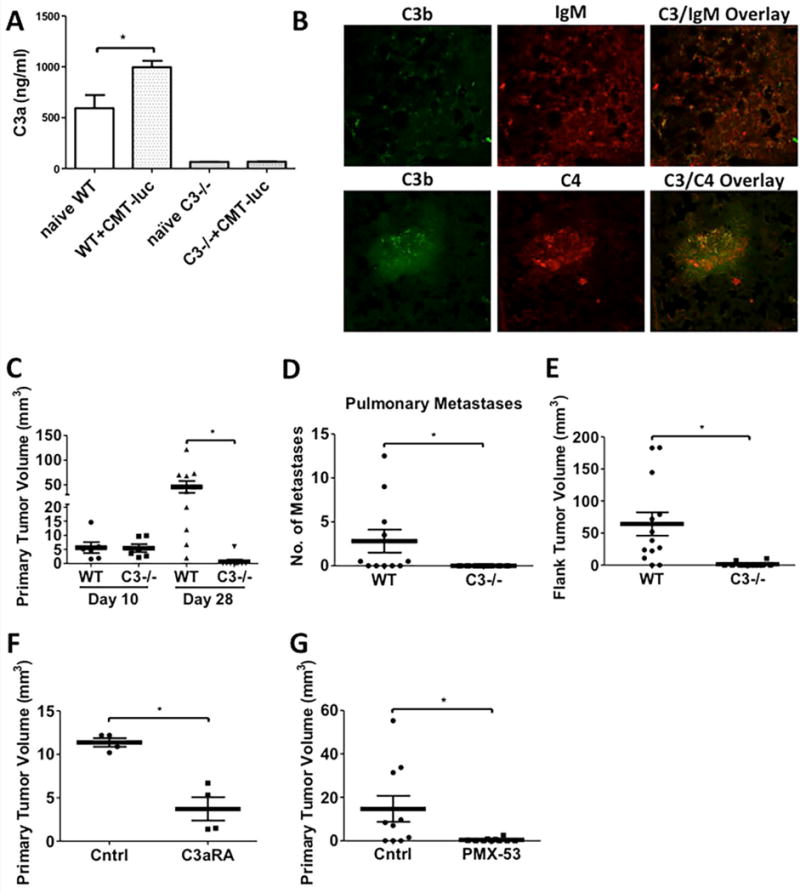
Age matched WT and C3−/− mice were injected with CMT-luc tumors, and the primary tumors, organs, and whole blood were collected at the terminal sacrifice 28 days post-injection. (A) ELISA specific for murine C3a was used to measure C3a in the plasma samples collected from naïve or tumor-bearing WT and C3−/− mice (Naïve WT n = 3; WT+CMT-luc n = 5; naïve C3−/− n = 4; C3−/−+CMT-luc n = 9). (B) Immunofluorescence co-staining for C3 (green) and IgM (red) or C3 (green) and C4 (red) in the primary tumor specimens from WT mice were evaluated by confocal microscopy. (C) Primary tumor volumes 10 or 28 days after CMT-luc implantation in WT or C3−/− mice are shown. (Day 10: WT n = 6 and C3−/− n = 6; Day 28: WT n = 10 and C3−/− n = 9) (D) Number of metastases to the secondary pulmonary space were counted from ex vivo images that captured the luciferase activities of CMT-luc metastases. (WT n = 11; C3−/− n = 9) (E) Flank tumor volumes 28 days after CMT-luc implantation in WT or C3−/− are shown (WT n = 31; Day 28 C3−/− n = 12) (F-G) WT mice are administered with (F) C3a receptor antagonist (C3aRA; SB290157) or (G) PMX-53 (C5a receptor antagonist, C5aRA), starting a day prior to tumor implantation into WT mice. Primary tumor volumes 28 days after tumor implantation in the treated groups and vehicle or control peptide group are shown (F, n = 4 and G, n = 10 each group). *p <0.05. Error bars represent mean ± SEM.
To establish if complement activation occurs locally at the site of the tumor, sections of CMT-luc tumors were stained with antibodies against components of complement activation, C3b and C4, as well as against IgM. Binding of circulating IgM to target antigens initiates the classical pathway of complement activation (5). By immunofluorescence, we observed co-localization of C3b and IgM, as well as co-localization of C3b and C4 in CMT-luc tumors (Fig. 1B). Taken together, our data show that lung cancer cells elicit local complement activation, and this is likely mediated through the classical pathway.
Inhibition of Tumor Growth in C3−/− Mice
To assess the functional role of C3 in the TME, we compared the progression of CMT-luc tumors in WT and C3−/− mice in our orthotopic model. At 10 days after tumor implantation, we observed no significant difference in primary tumor size (Fig. 1C). However, at 4 weeks we saw a dramatic difference in primary tumor size in C3−/− mice (Fig. 1C), with average tumor volume of 45.11 mm3 in WT mice, versus 0.6667 mm3 in C3−/−. This was associated with a complete inhibition of secondary tumor metastases in the other lobes of the lung (Fig 1D). As a second model, cancer cells were implanted subcutaneously into the flanks of C3−/− or WT mice; we observed a similar inhibition of tumor growth (Fig. 1E). To further examine the pathway of complement activation, we compared tumor growth in mice deficient in factor B (fB−/−), a protein necessary for activation of the alternative pathway of complement (7, 19). We observed no significant difference in primary CMT-luc tumor size or pulmonary metastases in fB−/− mice compared to the WT controls (Supplemental Fig. S1A,B) consistent with our staining for IgM indicating that activation in the setting of tumors occurs via the classical pathway.
The pro-tumorigenic effects of complement can be mediated through production of anaphylatoxins (C3a and C5a), which act as pro-inflammatory mediators (9). To test the role of these molecules in our model, we used either a C3a receptor antagonist (C3aRA) (SB290157) (20) or a C5a receptor antagonist, PMX-53 (C5aR) (21). We observed a strong inhibition of CMT-luc tumor growth in mice treated with either the C3aRA (Fig. 1F) or the C5aR (Fig. 1G) compared to vehicle control at day 28, similar to what we observed in C3−/− mice.
Tumor Growth Inhibition in C3−/− Mice is Mediated through CD4+ Lymphocytes
We examined changes in inflammatory and immune populations in tumor-bearing WT and C3−/− mice. Since CMT-luc tumors are virtually undetectable at 4 weeks in C3−/− mice, we harvested animals at 7-10 days, when tumors grown in WT or C3−/− mice were similar in size. T cell populations were analyzed by flow cytometry with the gating strategy and the isotype gating controls shown in Supplemental Fig. S2A and B. We did not detect significant changes in populations of resident alveolar macrophages (MacA; SigF+CD11c+), neutrophils (Ly5G+CD11b+), or recruited monocytes (MacB; CD64highCD11c+) at day 7 (Supplemental Fig. S3A). However, we observed a trend towards higher numbers of both CD4+ and CD8+ T cell populations in tumor bearing lungs of C3−/− mice by flow cytometry (Fig. 2A). The increase in tumor infiltrating lymphocytes (TIL) was confirmed by immunostaining for CD3, CD4 and CD8 (Supplemental Fig. S3B). In addition, we observed higher percentages of IFNγ/TNFα double-positive (Fig. 2B) and IFNγ/TNFα/IL-10 triple-positive, polyfunctional CD4+ and CD8+ T cells in tumor bearing C3−/− mice compared to WT controls in all 3 independent experiments using pools of 3 mice for each experiment (Fig. 2C). Interestingly, abundance of CD4+/FOXP3+ regulatory T cells (Fig 2D) and IL17a+ T cells (Supplemental Fig. S3C) showed no trend. IFNγ, TNFα, or IL-10 single-positive populations of CD4+ and CD8+ T cells showed more modest changes (Supplemental Fig. S3D–F). Examination of activated T cells, by examining CD44/CD69 double-positive cells, also indicated a trend towards increased activation in C3−/− mice compared to the control (Supplemental Fig. S3G). Lastly, the expression of PD1 on CD4 and CD8 T cells showed no consistent difference in either group (Supplemental Fig. S3H).
Figure 2. Tumor Growth Inhibition in C3−/− mice is mediated through CD4+ Lymphocytes.
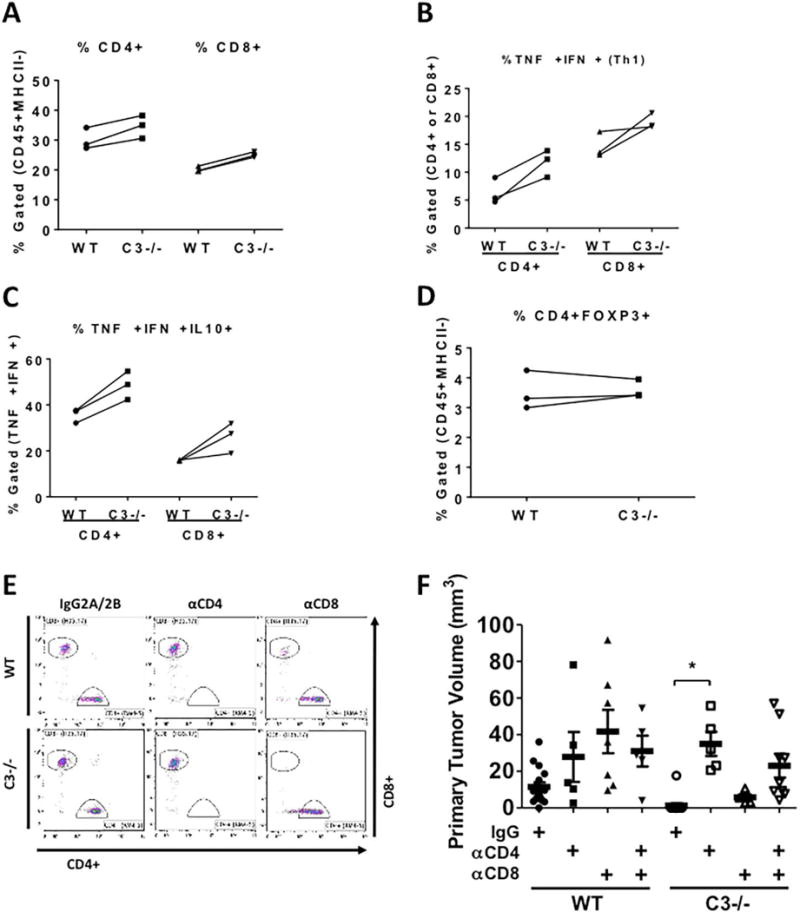
CMT-luc cells were injected into the lungs of WT or C3−/− mice 10 days prior to flow cytometric analysis of the immune populations using the sequential flow cytometry gating strategy shown in Supplemental Figure 2C. (A–D) Three individual experiments using separate isolations of T cells from 3 WT or C3−/− mice are shown. (A) The percentages of CD4+ or CD8+ cells are shown within the tumor-bearing lungs of WT and C3−/− mice. (B) CD4+ and CD8+ populations were analyzed for their ability to make TNFα and IFNγ after in vitro stimulation. (C) The TNFα+ and IFNγ+ CD4+ or CD8+ cells were gated for their ability to produce IL-10. (D) The percentages of CD4+ FOXP3+ (Tregs) are shown in both groups. (E) Representative flow cytometric plots of depletion efficacy measured with CD8 antibody clone (H35.17) and CD4 antibody (RM4-5) are shown (n = 2). The percentages of depleted T cells are shown in Supplementary Table 1. (F) The effects of in vivo depletion of CD4- and CD8- specific T cells on the primary CMT-luc volumes are shown (WT+IgG n = 16; WT+αCD4 n = 5; WT+αCD8 n = 7; WT+αCD4+αCD8 n = 5; C3−/−+IgG n = 14; C3−/−+αCD4 n = 5; C3−/−+αCD8 n = 5; C3−/−+αCD4+αCD8 n = 9). The depletion antibodies were administered starting a week before the tumor implantation, and CMT-luc tumors were harvested 3 weeks after implantation. *p <0.05. Error bars represent mean ± SEM
Previous studies have demonstrated that complement inhibition blocks the growth of TC-1 cells implanted into the flank (8). These investigators demonstrated that the anti-tumor effects of complement inhibition were mediated through CD8+ T cells. In addition, complement acting in an autocrine fashion in CD8+ T cells has been recently shown to inhibit anti-tumor immunity (22). We, therefore, tested the effects of immunodepletion of specific T cell populations on primary tumor growth in our model using CMT-luc tumors. Administration of anti-CD8 or anti-CD4 effectively depleted the respective population, without affecting the other population (Fig. 2E and Supplemental Table 1). Unexpectedly, depletion of CD8+ T cells had a minimal effect on increasing tumor size in C3−/− mice (Fig. 2F). However, immunodepletion of either CD4+ T cells, or double depletion of both CD8+ and CD4+ T cells restored primary CMT-luc tumor size to the level seen in WT mice. These data indicate that the effects of complement depletion are mediated through a CD4 T cell mechanism that is independent of CD8 T cells.
We sought to define the mechanism whereby CD4 cells are inhibiting tumor progression in the setting of complement depletion. We therefore examined gene expression changes in CMT167 cells recovered from in vivo tumors compared to cells grown in vitro, by implanting tumor cells into GFP+ transgenic mice, recovering the GFP-negative populations and performing RNA-seq analysis. Analysis of MHC genes and associated cofactors demonstrated that in vivo, CMT167 cells upregulate MHC class II genes H2-Aa and H2Ab1 and cofactors necessary for MHC class II processing and presentation, such as H2-DM, CIITA, and Ii (Supplemental Fig S4A). This suggests that CD4+ T cells may directly interact with these cancer cells, independent of their role as helper cells. To test this, we silenced expression of CIITA, which is considered the master regulator of MHC Class II in CMT167 cells. Knockdown was validated at both the RNA and protein level (Supplemental Fig S4B,C). To confirm the effects of silencing CIITA, silenced cells and cells transfected with a non-targeting control shRNA were stimulated with IFNγ, and expression of MHC Class II was determined using flow cytometry. CIITA-silenced cells showed a marked impairment in the ability of IFNγ to induce MHC Class II (Supplemental Fig. S4D), consistent with this protein being a critical transcription factor for induction of MHC Class II. To test the role of MHC Class II on the response to complement, cells silenced for CIITA and control cells were implanted subcutaneously into either WT or C3−/− mice. Non-targeting control cells formed tumors in WT mice, but failed to grow in C3−/− mice, similar to parental CMT167 cells. In contrast, CMT167 cells silenced for CIITA formed similar sized tumors in both WT and C3−/− mice (Supplemental Fig. S4E). Based on these data we propose that the mechanism for CD4-mediated inhibition of CMT167 tumors involves direct interactions between MHC Class II expressing cancer cells and CD4 T cells.
Complement Depletion Inhibits Tumor Growth of Multiple Lung Cancer Cell Lines
We examined the effect of C3 depletion using two other murine lung cancer cell lines. We first examined growth of EML4-ALK cells. These cells were generated from mice in which tumor initiation occurred by administration of a CRISPR construct (14) (gift of Dr. Andrea Ventura, Memorial Sloan Kettering Cancer Center). We validated that these cells expressed the fusion kinase and were sensitive to growth inhibition by Crizotinib and TAE684 (Supplemental Fig. S5A, B). EA2 cells were orthotopically implanted into the lungs of WT or C3−/− mice; primary tumor growth for EA2 cells was strongly inhibited in C3−/− mice 3 weeks after implantation, similar to what we observed with CMT-luc tumors with average tumor volume of 8.070 mm3 in WT mice, versus 0.2067 mm3 in C3−/− (p = 0.0065) (Supplemental Fig. S5C).
We also examined luciferase-expressing Lewis Lung Carcinoma cells (LLC-luc), which also express oncogenic KRAS, similar to CMT-luc (23). LLC cells have a G12C mutation, whereas CMT-luc have a G12V mutation. LLC tumor-bearing lungs showed a similar degree of systemic complement activation and C3/C4/IgM deposition as CMT-luc tumors (Fig. 3A, B). However, while growth of primary LLC-luc tumors was significantly inhibited in C3−/− mice (average tumor volume of 482.5 mm3 in WT mice vs 289.8 mm3 in C3−/− mice, p = 0.0081) (Figure 3C), the degree of inhibition was less than seen with CMT-luc tumors. In addition, there was no effect on formation of secondary pulmonary tumors or liver metastases (Fig. 3D, E). LLC-luc tumor growth was unaffected in fB−/− mice (Supplemental Fig. S6A, B). Treatment of tumor-bearing mice with the C3aRA significantly decreased the primary tumor size by approximately 50% (Fig. 3F), and treatment with the C5aR inhibitor showed a trend towards smaller tumor size which did not reach significance (Fig. 3G).
Figure 3. Inhibition of LLC-luc Tumor Growth in C3−/− mice.
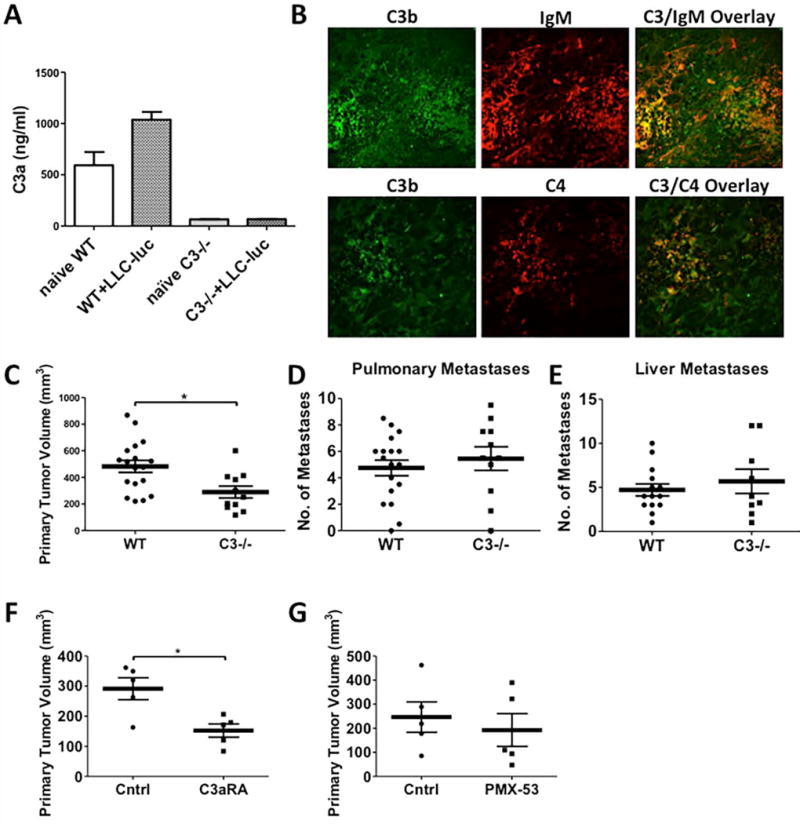
Age and gender-matched WT and C3−/− mice were injected with LLC-luc tumors, and primary tumors, organs, and whole blood were collected at the terminal sacrifice 21 days post-injection. (A) ELISA specific for murine C3a was used to measure C3a in the plasma samples collected from naïve or LLC-luc tumor-bearing WT and C3−/− mice (Naïve WT n = 3; WT+LLC-luc n = 21; naïve C3−/− n = 4; C3−/−+LLC-luc n = 4). Same data from naïve WT and C3−/− mice are used in this graph. (B) Immunofluorescence co-staining for C3 (green) and IgM (red) or C3 (green) and C4 (red) in tumor specimens induced by LLC-luc from C3−/− were evaluated by confocal microscopy. (C) Primary tumor volumes 21 days after LLC-luc implantation in WT or C3−/− mice are shown (WT n = 18; C3−/− n = 11). (D and E) Number of metastases to the secondary pulmonary space and liver were counted from ex vivo images that captured the luciferase activities of LLC-luc metastases (Pulmonary metastasis - WT n = 18; C3−/− n = 11 and Liver metastasis - WT n = 14; C3−/− n = 9). (F-G) WT mice are administered with (F) C3a receptor antagonist (C3aRA; SB290157) or (G) PMX-53 (C5a receptor antagonist, C5aRA), starting a day prior to tumor implantation into WT mice. Primary tumor volumes 21 days after tumor implantation in both treated groups and vehicle or control peptide group are shown (F, n = 5 and G, n = 5 each group). *p <0.05. Error bars represent mean ± SEM.
Previous studies using implantation of ovarian cancer cells demonstrated that these cells expressed C3 protein, which made them insensitive to growth inhibition in C3−/− mice (24). We compared expression of C3 in LLC-luc and CMT-luc cells. Analysis of whole cell lysates showed that LLC-luc, but not CMT-luc express C3/C3b and its degradation products iC3b and C3dg (Supplemental Fig. S7A). The role of cancer-derived C3 was assessed by silencing C3 expression in LLC-luc using two lentiviral shRNA constructs targeting C3 (Fig. 4A). Although the two C3 shRNAs decreased expression of C3, there was no effect on proliferation in vitro (Fig. 4B). However, analysis of anchorage-independent growth showed that both C3-silenced LLC-luc cell lines formed smaller and fewer colonies compared to LLC-luc transfected with a control shRNA (Fig. 4C, D). Next, we examined tumor growth of LLC-luc cells silenced for C3 in vivo. The intermediate knockdown (KD#1) showed no difference in primary tumor size or metastases when implanted into WT mice compared to control cells (Fig. 4E–G). However, there was a greater decrease in both primary tumor and metastases in C3−/− mice. The stronger knockdown of C3 (KD#2) formed smaller primary tumors in WT mice with few detectable pulmonary or liver metastases compared to control cells. In C3−/− mice, both primary tumor growth and metastasis were almost completely inhibited. These observations indicate cooperation between C3 production by the cancer cells and the host.
Figure 4. Silencing C3 Expression in LLC-luc Cells Inhibits Tumor Progression in vivo.
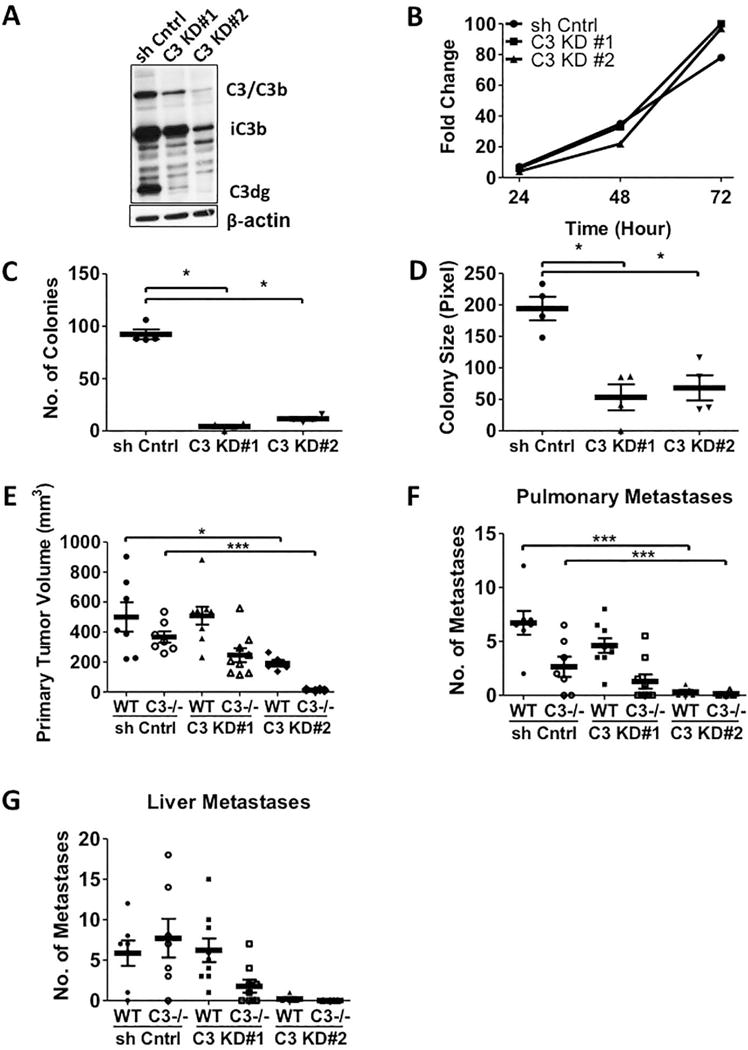
(A) Western blot analysis of the whole cell lysates from LLC-luc cells transduced with two lentiviral shRNA constructs targeting C3 are shown. The anti-C3d antibody that recognizes C3 activation fragments is used. (B) Fold changes in vitro proliferation of two C3 knockdown LLC-luc cell lines are shown. (C and D) Numbers and sizes of C3 KD LLC-luc colonies are shown in the anchorage independent assay (n = 4). (E-G) The primary tumor volumes and number of metastases induced by the two C3 KD LLC-luc cell lines and the control shRNA cell lines are shown. The animals were sacrificed 21 days after the tumor implantation (WT+sh cntrl n = 7; C3−/−+sh cntrl n = 7; WT+C3 KD#1 n = 9; C3−/−+C3 KD#1 n = 9; WT+C3 KD#2 n = 5; C3−/−+C3 KD#2 n =4). * p<0.05 *** p<0.001. Error bars represent mean ± SEM.
We also examined expression of the C3aR in CMT-luc and LLC-luc cells. By flow cytometry, we detected expression of C3aR on the surface of LLC-luc cells, but not CMT-luc cells (Supplemental Fig. S7B). We silenced the expression of C3aR in LLC-luc cells using two independent shRNA constructs and validated knockdown by flow cytometry (Fig. 5A). Similar to what we observed with C3 knockdown, silencing C3aR in LLC-luc cells did not affect cell proliferation, but inhibited anchorage-independent growth (Fig. 5B–D). We implanted the two C3aR knockdown cell lines into WT and C3−/− mice. Although statistically not significant, silencing C3aR expression in LLC-luc led to a trend in smaller primary tumors in WT mice. In C3−/− mice, there was a further inhibition of tumor size (Fig. 5E), similar to what we observed with C3-silenced LLC-luc cells. Interestingly, C3aR silenced cells formed fewer pulmonary metastases than control cells; however, this was similar in WT and C3−/− mice (Fig. 5F). The number of liver metastases detected was not affected by silencing C3aR, and was similar in WT and C3−/− mice (Fig. 5G). While the effects on metastasis are not dramatic, our observations confirm that C3a/C3aR signaling is important in the progression of lung cancer.
Figure 5. Silencing C3a Receptor Expression in LLC-luc Cells Inhibits the Primary Tumor Growth, but not metastasis in vivo.
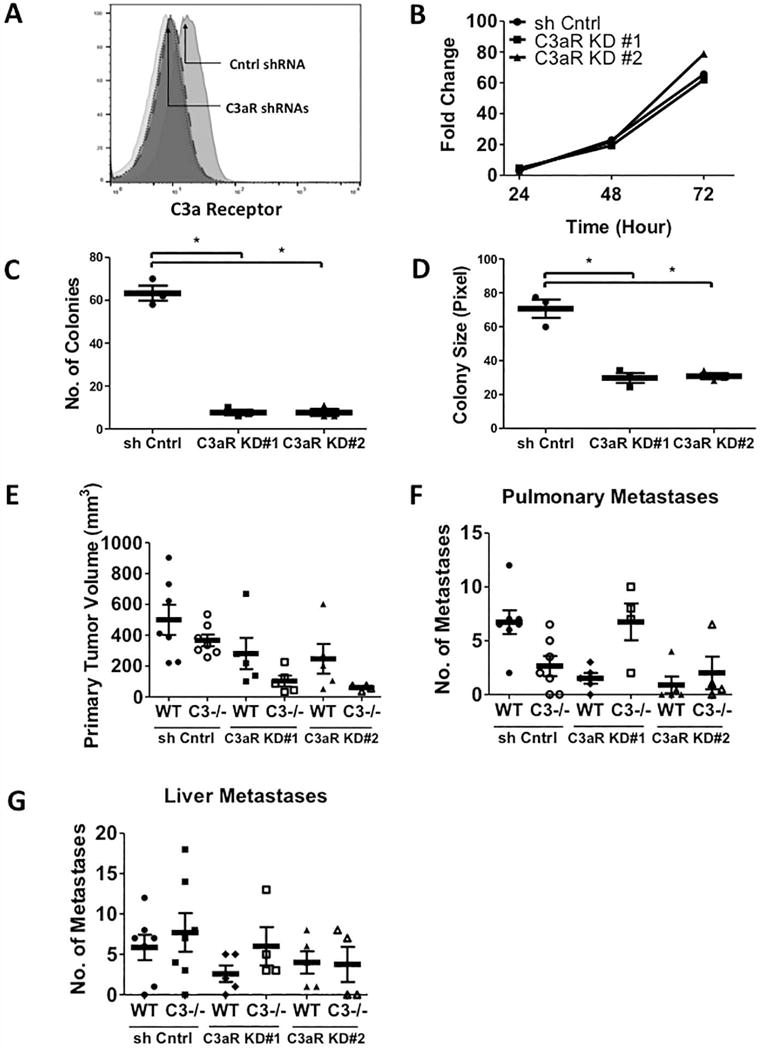
(A) Flow cytometric analysis of cell surface C3a receptor expression on the two C3a receptor knockdown and control LLC-luc cell lines. (Isotype in light grey (median = 9.10); LLC-luc shRNA control on grey (median = 19.8); LLC-luc C3aR KD#1 in dark grey with small dotted line (median = 9.87); LLC-luc C3aR KD#2 in dark grey with big dotted line (median = 10.5)). (B) Fold changes in vitro proliferation of two C3aR knockdown LLC-luc cell lines are shown. (C and D) Numbers and sizes of C3aR KD LLC-luc colonies are shown in the anchorage independent assay. (E-G) The primary tumor volumes (E) and number of pulmonary (F) and liver (G) metastases induced by the two C3aR KD LLC-luc cell lines and the control shRNA cell lines are shown. The animals were sacrificed 21 days after the tumor implantation. (WT+C3aR KD#1 n = 5; C3−/−+C3aR KD#1 n = 5; WT+C3aR KD#2 n = 5; C3−/−+C3aR KD#2 n =4). * p<0.05 Error bars represent mean ± SEM.
Co-Localization of C3d and IgM in Human Lung Tumors
To determine if complement activation occurs in human lung tumors, we immunostained a tissue microarray containing 55 human lung adenocarcinomas with antibodies against C3d and IgM. We observed positive staining for both IgM and C3d in 24 of 55 tumors (43.64%) (Figure 6A). Two examples of tumors positive for both IgM and C3d are shown in Fig. 6B. For these tumors the co-localization of IgM and C3d suggests that complement activation is mediated via the classical pathway, similar to what we observed in our mouse tumors. In addition staining of C3d without detectable IgM staining was observed in an addition 13 tumors. Fig. 6C shows an example of tumors positive for only IgM (top panels) or only C3d (bottom panels).
Figure 6. Immunostaining of Human Lung Tumors.
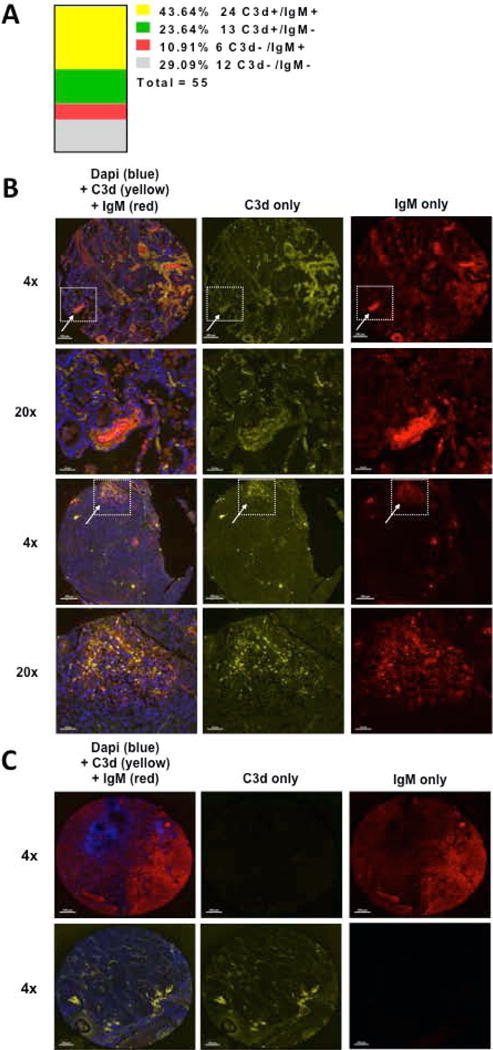
Tissue microarrays containing human lung adenocarcinomas were obtained from the Tissue Bank of the SPORE in Lung Cancer at the University of Colorado and stained for C3d (yellow), IgM (red), and dapi (blue). (A) C3d and IgM positivity was determined in a blinded fashion by examination of tissue punches following background subtraction in each individual channel. Tissue punches were considered positive when signal could be observed in ≥ 5% of the total punch. Each adenocarcinoma was represented by 3 randomly placed punches, and was considered positive when signal could be detected in all represented punches. The histogram shows the frequency and number that were scored double positive (C3d+/IgM+ in yellow), single positive (C3d+/IgM− in green or C3d−/IgM+ in red) or negative both markers (C3d−/IgM− in grey) out of total of 55 patient samples. (B) Two representative staining of C3d+/IgM+ are shown in x4. Areas of IgM and C3d co-deposition within the adenocarcinomas are shown in 20x (arrows). (C) Representative staining of C3d−/IgM+ is shown on the top panels and C3d+/IgM− on the bottom panels. Scale bars = 200 µm.
Discussion
While earlier work suggested that activation of complement would lead to inhibition of tumor growth through direct killing of cancer cells, recent studies have indicated that activation of complement promotes cancer and that complement inhibitors may represent a novel therapeutic strategy for multiple malignancies (8, 25-28). In this study we examined the effects of complement using an orthotopic, immunocompetent model of lung cancer progression. This model recapitulates many of the critical features of human lung cancer; specifically tumors originate in the appropriate microenvironment, and metastasize to distinct organs, including the other lung lobes and liver. Our data support a model in which the presence of a lung tumor results in systemic activation of complement in the host, which is reflected in elevated levels of C3a in the plasma, and deposition of C3 on the surface of cancer cells in the tumor. This appears to be mediated by the classical pathway of complement activation, since we detect IgM, C3b, and C4 co-localizing on cancer cells, and tumor growth is not altered in factor B knockout mice. Importantly, primary tumor growth and distant organ metastasis of 3 independent lung cancer cell lines were inhibited in C3−/− mice, similar to what has been observed in earlier studies in which tumors were grown subcutaneously(29).
Our data support a role for C3a as a major mediator of tumor progression. A C3aR receptor inhibitor blocked growth of both CMT-luc and LLC-luc tumors. The efficacy of this agent was similar to the effect of global C3 deletion. Based on the fact that CMT-luc cells do not express C3 or C3aR, we concluded that C3 does not have cell autonomous effects on CMT-luc cells, and that the tumor inhibitory effects are mediated exclusively through the TME. In contrast, we propose that growth of LLC-luc tumors is mediated, in part, through an autocrine growth loop involving C3 and C3aR. Silencing of either C3 or C3aR expression on these cells inhibited transformed growth in vitro and sensitized tumors to the effects of complement deficiency in the microenvironment. The published studies using ID8-VEGF ovarian cancer cells also suggested autocrine signaling through C3/C3aR (24). Based on the fact that in vivo growth of ID8-VEGF cells was not altered in C3−/− mice, we speculate that LLC-luc cells represent an intermediate state between CMT-luc cells which are wholly dependent on C3 signaling in the TME and ID8-VEGF cells that are independent of C3 proteins made by the host. However, blocking C3aR signaling in LLC-luc, which presumably inhibits both the autocrine loop and effects of C3a on the TME, only partially inhibited tumor growth of LLC-luc tumors. A near-complete inhibition was only observed when cells silenced for C3 were implanted into C3−/− mice. We propose additional downstream pathways mediated by C3 and its activation products contribute to tumor growth of LLC-luc tumors, such as the production of C5a and signaling through C5a receptors in TME.
We observed increases in T cell populations in CMT-luc tumor bearing lungs of C3−/− mice. In particular, polyfunctional activated populations of both CD8+ and CD4+ T cells expressing multiple cytokines were increased in CMT-luc tumor bearing C3−/− mice. Using immunodepleting antibodies we demonstrated a critical role for CD4+ T cells in mediating the effects of complement inhibition. However, in contrast to earlier studies (8, 22, 28), depleting CD8+ T cells had a minimal effect on tumors grown in C3−/− mice, suggesting that the effects of CD4+ cells are independent of their role as helper cells facilitating CD8 T cell function. Several publications have implicated the role of CD4+ T cells in complement-mediated cancer inhibition. Nabizadeh et. al. showed that C3-C3aR signaling modulates the antitumor functions of neutrophils and CD4+ T cells in B16-F0 melanomas (30). C3aR signaling was also shown to negatively regulate induction of Tregs (31). Based on this study we would expect loss of C3R signaling would increase the abundance of Treg, and promote tumor progression. In fact we have not observed any changes in Treg populations in our model. A recent study indicated that production of IL-10 by T cells is controlled through production of C3 and signaling through C3aR or C5aR in an autocrine fashion, and represents a novel immune checkpoint (22). This is consistent with our observation of increased IL-10+ T cells in tumor-bearing C3−/− mice.
It has also been reported that human CD4+ T cells express and activate C3 via cathepsin, leading to regulation of IFNγ secretion (32). We have used RNA-seq to profile cancer cells recovered from in vivo tumors compared to cancer cells grown in vitro to identify pathways that are altered in the setting on in vivo tumors. Interestingly, we detected an interferon-stimulated gene signature and induction of MHC Class II in vivo for CMT-luc cells, but this was not observed in LLC-luc cells. This suggests that CMT-luc cells may be able to present self-antigen(s) through MHC Class II and promote direct interactions with CD4+ T cells, whereas LLC-luc cells would likely not have this type of interaction. Consistent with this model, CMT167 cells silenced for CIITA, showed a blunted induction of MHC Class II, and were able to form tumors in C3-deficient mice. We propose that in the context of CIITA silencing, diminished ability to present self-antigen(s) to CD4 T cells directly, confers an escape from CD4 T cell mediated growth inhibition. There is clear precedent for antitumor effects of CD4 T cells through the production of both IFNγ and TNFα (33). While these data implicate CD4 T cells as a potential direct mediator of antitumor function, restrained by C3, these studies cannot rule out a role for CD4 T cells in also promoting anti-tumor antibody responses. Further studies using models with a defined antigen will be required to discriminate between these mechanisms of action.
Finally, we have observed the markers of classical complement activation in a subset of human lung tumor samples, similar to what we have observed in our mouse model. Further analysis will be required on a larger set of human tumors to identify biomarkers for complement activation. These studies will be critical for patient selection for potential clinical trials using complement inhibitors. In summary, our data indicate that growth of lung tumors in an orthotopic model reproducing the appropriate microenvironment leads to activation of the classical complement pathway, which is critical for tumor progression. Our data firmly suggest that C3 signaling directly or indirectly inhibits the production of multiple cytokine by CD4+ T cells independent of FOXP3+ Tregs, and that CD4+ T cells may be a critical component of the response to complement inhibitors. The role of increased systemic levels of anaphylatoxins, specifically C3a and C5a, on the infiltration of T cells into the tumor suggests that targeting this pathway may represent a novel therapeutic approach to be used in combination with immune checkpoint inhibitors.
Supplementary Material
Acknowledgments
We thank Andrea Ventura for his gift of the adenoviral construct to generate EML4/ALK cell lines. We also thank the Human Immune Monitoring Shared Resource within the University of Colorado Human Immunology and Immunotherapy Initiative for their expert assistance with multispectral imaging on the Vectra 3.0 platform.
Financial Support: This work was supported by the NIH (R01 CA162226 and R01 CA108610 to RA Nemenoff, R01 AI068730 to JD Lambris), Colorado Lung SPORE P50 CA058187 to HY Lee and RA Nemenoff and the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Service (Career Development Award IK2BX001282 to HY Lee). The University of Colorado Cancer Center Flow Cytometry and the Genomics and Microarray Shared Resources is supported by NIH P30CA046934. The University of Colorado Cancer Center Flow Cytometry Core Facility is funded through a support grant from the National Cancer Institute (P30 CA046934). Imaging experiments were performed in the University of Colorado Anschutz Medical Campus Advanced Light Microscopy Core supported in part by NIH/NCATS Colorado CTSI Grant Number UL1 TR001082. J.D.L. is the inventor of patents and/or patent applications that describe the use of complement inhibitors for therapeutic purposes, the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors (i.e. next generation compstatins) for clinical applications, and the inventor of the compstatin technology licensed to Apellis Pharmaceuticals (i.e., 4(1MeW)7W/POT-4/APL-1 and its PEGylated derivatives.
References
- 1.Hollenberg NK. Hypertension and the kidney: determinants of the response to antihypertensive therapy and their implications. Am Heart J. 1993;125:604–8. doi: 10.1016/0002-8703(93)90210-z. [DOI] [PubMed] [Google Scholar]
- 2.Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, Powderly JD, Heist RS, Carvajal RD, Jackman DM, Sequist LV, Smith DC, Leming P, Carbone DP, Pinder-Schenck MC, Topalian SL, Hodi FS, Sosman JA, Sznol M, McDermott DF, Pardoll DM, Sankar V, Ahlers CM, Salvati M, Wigginton JM, Hellmann MD, Kollia GD, Gupta AK, Brahmer JR. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2015;33:2004–12. doi: 10.1200/JCO.2014.58.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sundar R, Soong R, Cho BC, Brahmer JR, Soo RA. Immunotherapy in the treatment of non-small cell lung cancer. Lung Cancer. 2014;85:101–9. doi: 10.1016/j.lungcan.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oner F, Savas I, Numanoglu N. Immunoglobulins and complement components in patients with lung cancer. Tuberk Toraks. 2004;52:19–23. [PubMed] [Google Scholar]
- 5.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nature immunology. 2010;11:785–97. doi: 10.1038/ni.1923. 2924908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 7.Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Parsa AT. Cancer and the complement cascade. Mol Cancer Res. 2010;8:1453–65. doi: 10.1158/1541-7786.MCR-10-0225. [DOI] [PubMed] [Google Scholar]
- 8.Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, Coukos G, Lambris JD. Modulation of the antitumor immune response by complement. Nature immunology. 2008;9:1225–35. doi: 10.1038/ni.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sacks SH. Complement fragments C3a and C5a: the salt and pepper of the immune response. European journal of immunology. 2010;40:668–70. doi: 10.1002/eji.201040355. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Sorenson AL, Poczobutt J, Amin J, Joyal T, Sullivan T, Crossno JT, Jr, Weiser-Evans MC, Nemenoff RA. Activation of PPARgamma in myeloid cells promotes lung cancer progression and metastasis. PLoS One. 2011;6:e28133. doi: 10.1371/journal.pone.0028133. 3228753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poczobutt JM, Nguyen TT, Hanson D, Li H, Sippel TR, Weiser-Evans MC, Gijon M, Murphy RC, Nemenoff RA. Deletion of 5-Lipoxygenase in the Tumor Microenvironment Promotes Lung Cancer Progression and Metastasis through Regulating T Cell Recruitment. J Immunol. 2016;196:891–901. doi: 10.4049/jimmunol.1501648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiser-Evans MC, Wang XQ, Amin J, Van Putten V, Choudhary R, Winn RA, Scheinman R, Simpson P, Geraci MW, Nemenoff RA. Depletion of cytosolic phospholipase A2 in bone marrow-derived macrophages protects against lung cancer progression and metastasis. Cancer Res. 2009;69:1733–8. doi: 10.1158/0008-5472.CAN-08-3766. 2653105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Layton MG, Franks LM. Heterogeneity in a spontaneous mouse lung carcinoma: selection and characterisation of stable metastatic variants. British journal of cancer. 1984;49:415–21. doi: 10.1038/bjc.1984.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, Ogrodowski P, Crippa A, Rekhtman N, de Stanchina E, Lowe SW, Ventura A. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516:423–7. doi: 10.1038/nature13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thurman JM, Kulik L, Orth H, Wong M, Renner B, Sargsyan SA, Mitchell LM, Hourcade DE, Hannan JP, Kovacs JM, Coughlin B, Woodell AS, Pickering MC, Rohrer B, Holers VM. Detection of complement activation using monoclonal antibodies against C3d. J Clin Invest. 2013;123:2218–30. doi: 10.1172/JCI65861. 3635726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franks LM, Carbonell AW, Hemmings VJ, Riddle PN. Metastasizing tumors from serum-supplemented and serum-free cell lines from a C57BL mouse lung tumor. Cancer Res. 1976;36:1049–55. [PubMed] [Google Scholar]
- 19.Matsumoto M, Fukuda W, Circolo A, Goellner J, Strauss-Schoenberger J, Wang X, Fujita S, Hidvegi T, Chaplin DD, Colten HR. Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc Natl Acad Sci U S A. 1997;94:8720–5. doi: 10.1073/pnas.94.16.8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wysoczynski M, Reca R, Lee H, Wu W, Ratajczak J, Ratajczak MZ. Defective engraftment of C3aR−/− hematopoietic stem progenitor cells shows a novel role of the C3a-C3aR axis in bone marrow homing. Leukemia. 2009;23:1455–61. doi: 10.1038/leu.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanian H, Kashem SW, Collington SJ, Qu H, Lambris JD, Ali H. PMX-53 as a dual CD88 antagonist and an agonist for Mas-related gene 2 (MrgX2) in human mast cells. Mol Pharmacol. 2011;79:1005–13. doi: 10.1124/mol.111.071472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Sun SN, Liu Q, Yu YY, Guo J, Wang K, Xing BC, Zheng QF, Campa MJ, Patz EF, Jr, Li SY, He YW. Autocrine Complement Inhibits IL10-Dependent T-cell-Mediated Antitumor Immunity to Promote Tumor Progression. Cancer discovery. 2016;6:1022–35. doi: 10.1158/2159-8290.CD-15-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li HY, McSharry M, Bullock B, Nguyen TT, Kwak J, Poczobutt JM, Sippel TR, Heasley LE, Weiser-Evans MC, Clambey ET, Nemenoff RA. The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade. Cancer Immunol Res. 2017 doi: 10.1158/2326-6066.CIR-16-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho MS, Vasquez HG, Rupaimoole R, Pradeep S, Wu S, Zand B, Han HD, Rodriguez-Aguayo C, Bottsford-Miller J, Huang J, Miyake T, Choi HJ, Dalton HJ, Ivan C, Baggerly K, Lopez-Berestein G, Sood AK, Afshar-Kharghan V. Autocrine effects of tumor-derived complement. Cell reports. 2014;6:1085–95. doi: 10.1016/j.celrep.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, Rouzaut A, Pajares MJ, Montuenga LM, Pio R. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. 2012;189:4674–83. doi: 10.4049/jimmunol.1201654. 3478398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elvington M, Scheiber M, Yang X, Lyons K, Jacqmin D, Wadsworth C, Marshall D, Vanek K, Tomlinson S. Complement-dependent modulation of antitumor immunity following radiation therapy. Cell reports. 2014;8:818–30. doi: 10.1016/j.celrep.2014.06.051. 4137409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pio R, Ajona D, Lambris JD. Complement inhibition in cancer therapy. Seminars in immunology. 2013;25:54–64. doi: 10.1016/j.smim.2013.04.001. 3733085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vadrevu SK, Chintala NK, Sharma SK, Sharma P, Cleveland C, Riediger L, Manne S, Fairlie DP, Gorczyca W, Almanza O, Karbowniczek M, Markiewski MM. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res. 2014;74:3454–65. doi: 10.1158/0008-5472.CAN-14-0157. [DOI] [PubMed] [Google Scholar]
- 29.Afshar-Kharghan V. The role of the complement system in cancer. J Clin Invest. 2017;127:780–9. doi: 10.1172/JCI90962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nabizadeh JA, Manthey HD, Steyn FJ, Chen W, Widiapradja A, Md Akhir FN, Boyle GM, Taylor SM, Woodruff TM, Rolfe BE. The Complement C3a Receptor Contributes to Melanoma Tumorigenesis by Inhibiting Neutrophil and CD4+ T Cell Responses. J Immunol. 2016;196:4783–92. doi: 10.4049/jimmunol.1600210. [DOI] [PubMed] [Google Scholar]
- 31.Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4(+) cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3(+) regulatory T cells. Nature immunology. 2013;14:162–71. doi: 10.1038/ni.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, Subias M, Pickering MC, Drouet C, Meri S, Arstila TP, Pekkarinen PT, Ma M, Cope A, Reinheckel T, Rodriguez de Cordoba S, Afzali B, Atkinson JP, Kemper C. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. 2013;39:1143–57. doi: 10.1016/j.immuni.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, Ranta F, Ullrich S, Mocikat R, Braungart K, Mehra T, Fehrenbacher B, Berdel J, Niessner H, Meier F, van den Broek M, Haring HU, Handgretinger R, Quintanilla-Martinez L, Fend F, Pesic M, Bauer J, Zender L, Schaller M, Schulze-Osthoff K, Rocken M. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494:361–5. doi: 10.1038/nature11824. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.