

Abstract
The catalytic transformation of a C(sp3)–H bond to a C(sp3)–C bond via an iron carbene intermediate represents a long-standing challenge. Despite the success of enzymatic and small molecule iron catalysts mediating challenging C(sp3)–H oxidations and aminations via high-valent iron oxos and nitrenes, C(sp3)–H alkylations via isoelectronic iron carbene intermediates have thus far been unsuccessful. Iron carbenes have been inert, or shown to favor olefin cyclopropanation and heteroatom-hydrogen insertion. Herein we report an iron phthalocyanine-catalyzed alkylation of allylic and benzylic C(sp3)–H bonds. Mechanistic investigations support that an electrophilic iron carbene mediates homolytic C–H cleavage and rebounds from the resulting organoiron intermediate to form the C–C bond; both steps are tunable via catalyst modifications. These studies suggest that for iron carbenes, distinct from other late metal carbenes, C–H cleavage is partially rate-determining and must be promoted to effect reactivity.
Graphical abstract
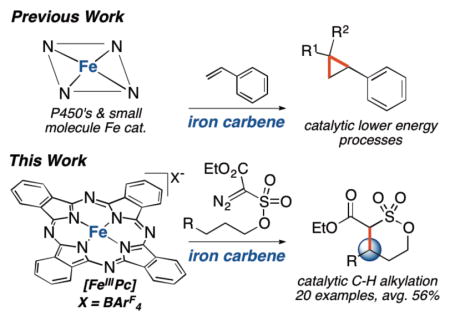
Iron is one of the most abundant elements, totaling one-third of the Earth’s mass, and is emerging as an important metal for homogenous catalysis1a–f. Iron small molecule catalysts have been demonstrated to catalyze challenging C–H oxidation and amination processes via high-valent metal oxos and nitrenes1b,1c,1d,1f,. In contrast, no iron catalyst has been demonstrated to alkylate C(sp3)–H bonds via an isoelectronic metallocarbene intermediate.
Naturally occurring and engineered P450 enzymes form only inert carbenes or carbenes active for lower energy processes than C–H alkylation (e.g. cyclopropanations, heteroatom-hydrogen bond insertions)2 (Fig. 1). Small molecule iron catalysts capable of forming carbenes also fail to catalyze C–H alkylation, favoring alternative reaction pathways3. Curiously, nearly all other late metals, including copper4a,4b, cobalt4c, silver4a, palladium4d, rhodium4e, and ruthenium4f, form metallocarbenes catalytically active for C–H alkylation. Herein we report an iron-catalyzed alkylation of allylic and benzylic C(sp3)–H bonds and provide evidence of an iron carbene intermediate. Distinct from other late metal carbenes, C–H cleavage is partially rate-determining and tunable via catalyst modifications.
Figure 1.
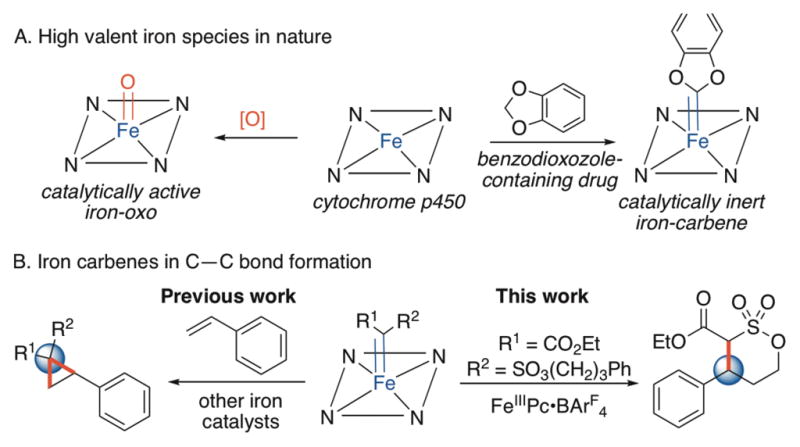
Challenges of iron carbene C–H activation.
Iron carbene complexes have been generated stoichiometrically at low temperatures, isolated5a, and/or demonstrated to undergo C–H insertion in a separate step at elevated temperatures (e.g. 80°C)5b,c. We hypothesized that one reason these reactions are not catalytic is due to the distinct energetic requirements for each step. At the elevated temperatures needed for C–H insertion, thermal decomposition of the diazoester or metal carbene into a free carbene may occur. Literature reports claiming iron-catalyzed C–H alkylation with methyl phenyldiazoacetate at 80°C are ambiguous6a because this diazoester is reported and observed by us (Supplementary Information, SI) to show significant non-metal mediated alkylation reactivity at this temperature6b.
We hypothesized that with iron, there is a higher kinetic barrier to C–H insertion than in rhodium and copper systems where metallocarbene formation is rate determining4a,7,8. This difference may explain the predominance of lower energy pathways in iron carbene reactions, such as competitive dimerization to furnish olefins7. Reactivity and selectivity of metal carbenes is highly tunable4e,9a; therefore, we aimed to electronically and sterically tune the catalyst and carbene precursor to form a metallocarbene intermediate reactive enough to insert into C(sp3)–H bonds at temperatures that mitigate deleterious pathways and do not decompose the diazoprecursor.
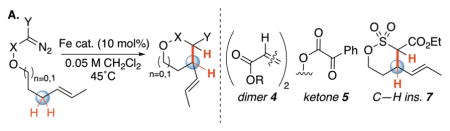
We first evaluated iron catalysts used for metallocarbene-mediated cyclopropanations and heteroatom-hydrogen insertions for the intramolecular alkylation of allylic C–H bonds (Table 1A)3,5b,10. We examined a series of substituted diazo compounds (acceptor 1, donor-acceptor 2, acceptor-acceptor 3) with varying degrees of electrophilicity and steric bulk9a. With iron porphyrin and phthalocyanine catalysts, diazoester 1 converted to olefin dimer (4, 93% and 84%, respectively), suggestive of iron carbene formation9a. Bulkier disubstituted carbene precursor 2 disfavored dimerization with all catalysts, but favored ketone product 5 over alkylation9a.
Table 1.
Reaction optimization.
| A. | ||||
|---|---|---|---|---|
| ||||
|
| ||||
| Catalyst: | Acceptor: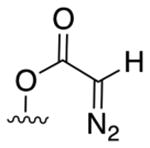 1a |
Donor/acceptor: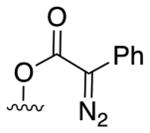 2a |
Acceptor/acceptor: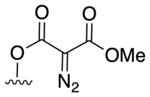 3a |
Acceptor/acceptor: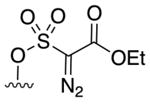 6b |
|
|
||||
| FeIIICl3·PyBOX | 99% rsm | 90% rsm | 97% rsm | 98% rsm |
| FeIII(TTP)CI | 93% dimer 4 | 4% ketone 5 95% rsm |
96% rsm | 98% rsm |
| [FeIIIPc]CI | 84% dimer 4 | 12% ketone 5 58% rsm |
95% rsm | 3% C—H ins. 7 97% rsm |
|
| ||||
| B. | ||||
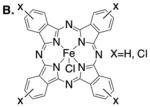
|
Catalysts: | Acceptor/acceptor 6: | ||
| [FeIIIPc]CI, NaBArF4 | 48% (53%c) C—H ins. 7 | |||
| [FeIIICl8Pc]CI, NaBArF4 | 25% C—H ins. 7 | |||
| [FeIIICl16Pc]CI, NaBArF4 | 38% C—H ins. 7 | |||
R = CH2CH2CH=CHCH3. Reaction stirred for 24 hours unless otherwise noted. Pc = phthalocyanine; TPP = tetraphenylporphyrin; PyBOX = 2,6-bis[(4R)-4-phenyl-2-oxazolinyl]pyridine; NaBArF4= Sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate. Isolated yields are average of two runs. rsm = recovered starting material.
n = 0.
n = 1.
Run with a 1 h slow addition of substrate.
We hypothesized that augmenting the electrophilicity of the disubstituted diazo compound would increase its reactivity by producing a strongly electrophilic metallocarbene that could more readily engage in higher energy C–H insertion pathways. Under rhodium catalysis, such electrophilic diazo compounds require very active catalysts to form the metallocarbene; however, once formed, the resulting carbene is more reactive towards C–H insertion9a. Acceptor-acceptor diazoester 3 resulted in predominately recovered starting material. Upon examination of the 13C shifts of the α-carbon of various acceptor-acceptor diazo precursors, we observed that sulfonate esters appeared to be the most electrophilic and thus investigated their diazoesters (6) for C–H alkylation reactivity9b. The catalyst with the greatest π-accepting character, iron phthalocyanine chloride ([FeIIIPc]Cl), formed C–H alkylated δ-sultone product 7 in low yield (3%) but with excellent selectivity (97% rsm).
Examination of non-coordinating counterions to render the iron catalyst more electrophilic led to a significant increase in yield with both AgSbF6 (45%, SI) and NaBArF4 (48%) (Table 1B). Adding the substrate to [FeIIIPc] over an hour further increased the yield (53%). Catalysts with halogenated phthalocyanine ligand frameworks formed C–H alkylated product in lower yield than the unsubstituted, commercial catalyst (e.g. [(FeIIICl8Pc)Cl]). Iron (II) phthalocyanine and iron porphyrin complexes gave very little or no C–H alkylation (see SI), highlighting the significance of the FeIII oxidation state and the ligand framework10. NaBArF4 alone gave no product (see SI).
Allylic C–H alkylations are rare under both noble and base metal catalysis. Specifically, under rhodium and copper catalysis, chemoselectivity issues arise wherein cyclopropanation of the olefin competes with C-H insertion4a,4e,9. We investigated the scope of this iron-catalyzed reaction across a range of allylic diazosulfonate esters (Table 2A). Bulky trisubstituted olefins, olefins with proximal, protected oxygen functionality, and styrenyl substrates all undergo alkylation in preparative yields (8–13). Consistent with an electrophilic metallocarbene intermediate, [FeIIIPc]-catalyzed C–H alkylation is sensitive to substrate electronics (13 vs. 10)4c,11. This contrasts observations with metallo-radical intermediates invoked in cobalt catalysis, where substrate electronics do not affect reactivity.4c Chemoselectivity for iron-catalyzed allylic C–H alkylation is maintained with more proximal olefins where cyclopropanation would form the geometrically preferred 6-membered sultone9b (14–15). In contrast, rhodium catalysis shows poor chemoselectivity for C–H insertion, forming cyclopropanated products. We also demonstrate one example of ethereal C—H alkylation (16).
Table 2.
Substrate scope.

|
lsolated yields are the average of three runs. Products yields are based on a mixture of diastereomers.
d.r. = 11:1, relative stereochemistry not assigned.
d.r. = 10:1, relative stereochemistry not assigned.
For all other compounds, diastereomeric ratios range from 2:1 to 1:1.3 syn:anti. ins. = C—H insertion; cyclopro. = cyclopropanation; Rh2(OAc)4 = rhodium acetate dimer; TBDPS = tert-butyldiphenylsilyl; SO2Ph = phenyl sulfonyl.
Benzylic C(sp3)–H bonds were evaluated under these reaction conditions and shown to readily undergo alkylation (Table 2B). Substrates containing electron rich aryl rings are alkylated in high yield (17–18), with no cyclopropanation observed even for a naphthalene-containing substrate (19). [FeIIIPc] promotes C–H alkylation adjacent to chromene and indole heterocycles (20–21), and tolerates lactam and thiophene motifs (23–24). A substrate containing a benzodioxole moiety is readily alkylated (22), despite the activated methylenedioxy functionality2a (Fig. 1A).
We investigated the application of this reaction to the late-stage functionalization of δ-tocopherol, a chromane antioxidant (Table 2C). Tocopherol derivative 25 was subjected to [FeIIIPc]-catalyzed C–H alkylation and furnished 26 in 78% yield. The δ-sulfone motif has been demonstrated to be readily derivitized9b. Accordingly, a three step elaboration of the δ-sultone via nucleophilic displacement yielded 27, bearing an unsymmetrical tertiary center with two new functional group handles, in 44% overall yield.
We next sought to investigate the mechanism and the involvement of an iron carbene intermediate in this reaction. We hypothesized that C–H alkylation occurs via an iron-bound carbene intermediate that, analogous to iron oxos1b,1f and iron nitrenes1c,1d, promotes homolytic cleavage of the C–H bond followed by recombination with the resulting carbon-centered radical to form the new C–C bond (Fig. 2A). Alternatively, [FeIIIPc] could serve as a Lewis acid to decompose the diazosulfonate ester to a free carbene capable of inserting into proximal C–H bonds. We investigated unligated iron salts and UV light with no added catalyst, conditions demonstrated to form reactive free carbenes from acceptor-acceptor diazo species12. FeCl3 and FeCl2 resulted in predominately recovered starting material (Fig. 2B), whereas irradiation of 6 with UV light (254 nm) gave compound 28, generated from C–H insertion of the free carbene into the dichloromethane solvent (Fig. 2B). Product 28 is not observed under [FeIIIPc]-catalysis, which is also run in dichloromethane. These divergent outcomes support the hypothesis that [FeIIIPc]-catalyzed C–H alkylation does not proceed via free carbene intermediates.
Figure 2.

Mechanistic studies.
We next sought to probe the involvement of an iron-bound carbene intermediate in both the C–H bond cleavage and C–C bond formation steps (Fig. 2C, 2D). In rhodium-catalysis proceeding via metallocarbene intermediates, varying the carboxylate ligands has been shown to influence both the intramolecular kinetic isotope effect (KIE) of C–H cleavage 7,11,12b and the selectivity of C–C bond formation4e,9a,13. We observed a change in the KIE with varied ligand electronics: [FeIIIPc] gave the highest KIE (5.0), followed by [FeIIICl8Pc] (4.8) and [FeIIICl16Pc] (4.5). These data support involvement of the iron complex in the C–H cleavage step. The larger KIE values for iron versus rhodium (1.8), support the proposed stepwise versus concerted mechanism (Fig. 2C)7. A stepwise mechanism accounts for the improved chemoselectivity of iron relative to rhodium catalysts for C–H insertion over cyclopropanation; a stabilized allylic radical is preferred over the secondary aliphatic radical formed during stepwise olefin oxidation processes1c,4c.
We also probed the effect of ligand electronics on the C-C bond forming step. We performed a study on Z-olefin substrate 32 to determine if scrambling of the double bond geometry occurred during allylic C–H alkylation (Fig. 2D). Under Rh2(OAc)4 catalysis, no isomerization of the olefin was observed, consistent with a concerted mechanism of C–H insertion. In contrast, under [FeIIIPc] catalysis we observed scrambling of olefin geometry, consistent with a stabilized carbon radical intermediate. The extent of olefin isomerization is dependent on the electronic substitution of the ligand, with the electron deficient chlorinated iron catalysts affording products with less isomerization than the unsubstituted phthalocyanine (10:1 vs 3:1). Under cobalt porphyrin catalysis, in the absence of a chiral pocket, isomerization during functionalization of Z-olefins occurs to a greater extent than in the iron system4c. This suggests that C–H alkylation with iron proceeds with less free radical character than cobalt4c. Electron withdrawing ligands may destabilize an iron-alkyl species prompting recombination at a faster rate.
We hypothesized that C–H insertion is rate-determining for iron-catalyzed alkylation, unlike rhodium and copper catalysis 4a,7,8. Intermolecular KIE studies that measured initial rates on parallel reactions with benzylic substrate 30 and 30-d2 revealed a primary KIE of 3.1 under [FeIIIPc] catalysis (Fig. 2C). This is consistent with C–H cleavage being part of the rate-determining step of the reaction. Initial rate measurements with [FeIIICl16Pc] showed a lower KIE of 1.4 suggesting that with a more electron deficient iron catalyst, formation of the metallocarbene, which requires donation of electron density from the metal center to extrude nitrogen, competes energetically with C–H cleavage7.
We report an iron-catalyzed C(sp3)–H alkylation via a metallocarbene intermediate. [FeIIIPc] alkylates allylic and benzylic C(sp3)–H bonds with broad scope. Mechanistic studies demonstrate the ability to exert catalyst control on the reactivity and selectivity during C–H cleavage and functionalization. Future studies will be aimed at elucidating the nature of the iron carbene intermediate, as well as further development of this highly tunable species to access stronger aliphatic C(sp3)–H bond types and intermolecular C–H alkylations.
Supplementary Material
Acknowledgments
Financial support provided by the NIGMS MIRA (R35 GM122525). J.R.G is a National Science Foundation and Springborn Graduate Fellow. C.I.W. is an Illinois Distinguished Graduate Fellow. We thank L. Zhu for assistance with NMR and Dr. J.R. Clark for checking our experimental procedure.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website at DOI:
Experimental details and characterization data (PDF) Spectral data (PDF)
References
- 1.(a) Enthaler S, Junge K, Beller M. Angew Chem Int Ed. 2008;47:3317–3321. doi: 10.1002/anie.200800012. [DOI] [PubMed] [Google Scholar]; (b) Chen MS, White MC. Science. 2007;318:783–787. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; (c) Paradine SM, White MC. J Am Chem Soc. 2012;134:2036–2039. doi: 10.1021/ja211600g. [DOI] [PubMed] [Google Scholar]; (d) Hennessy ET, Betley TA. Science. 2013;340:591–595. doi: 10.1126/science.1233701. [DOI] [PubMed] [Google Scholar]; (e) Shang R, Ilies L, Nakamura E. Chem Rev. 2017;117:9086–9139. doi: 10.1021/acs.chemrev.6b00772. [DOI] [PubMed] [Google Scholar]; (f) Oloo WN, Que L. Acc Chem Res. 2015;48:2612–2621. doi: 10.1021/acs.accounts.5b00053. [DOI] [PubMed] [Google Scholar]
- 2.(a) Stepan AF, Mascitti V, Beaumont K, Kalgutkar AS. Med Chem Comm. 2013;4:631–652. [Google Scholar]; (b) Coelho PS, Brustad EM, Kannan A, Arnold FH. Science. 2013;339:307–310. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]; (c) Kan SBJ, Lewis RD, Chen K, Arnold FH. Science. 2016;354:1048–1051. doi: 10.1126/science.aah6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Shen JJ, Zhu SF, Cai Y, Xu H, Xie XL, Zhou QL. Angew Chem Int Ed. 2014;53:13188–13191. doi: 10.1002/anie.201406853. [DOI] [PubMed] [Google Scholar]; (b) Zhu SF, Zhou QL. Acc Chem Res. 2012;45:1365–1377. doi: 10.1021/ar300051u. [DOI] [PubMed] [Google Scholar]
- 4.(a) Caballero A, Diaz-Requejo MM, Fructos MR, Olmos A, Urbano J, Perez PJ. Dalton Trans. 2015;44:20295–20307. doi: 10.1039/c5dt03450g. [DOI] [PubMed] [Google Scholar]; (b) Flynn CJ, Elcoate CJ, Lawrence SE, Maguire AR. J Am Chem Soc. 2010;132:1184–1185. doi: 10.1021/ja909713a. [DOI] [PubMed] [Google Scholar]; (c) Cui X, Xu X, Jin LM, Wojtas L, Zhang XP. Chem Sci. 2015;6:1219–1224. doi: 10.1039/c4sc02610a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gutierrez-Bonet A, Julia-Hernandez F, de Luis B, Martin R. J Am Chem Soc. 2016;138:6384–6387. doi: 10.1021/jacs.6b02867. [DOI] [PubMed] [Google Scholar]; (e) Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem Rev. 2010;110:704–724. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]; (f) Reddy AR, Zhou CY, Guo Z, Wei JH, Che CM. Angew Chem Int Ed. 2014;53:14175–14180. doi: 10.1002/anie.201408102. [DOI] [PubMed] [Google Scholar]
- 5.(a) Russell SK, Hoyt JM, Bart SC, Milsmann C, Stieber SCE, Semproni SP, DeBeer S, Chirik PJ. Chem Sci. 2014;5:1168–1174. [Google Scholar]; (b) Li Y, Huang JS, Zhou ZY, Che CM, You XZ. J Am Chem Soc. 2002;124:13185–13193. doi: 10.1021/ja020391c. [DOI] [PubMed] [Google Scholar]; (c) Zhao SK, Knors C, Helquist P. J Am Chem Soc. 1989;111:8527–8528. [Google Scholar]
- 6.(a) Mbuvi HM, Woo LK. Organometallics. 2008;27:637–645. [Google Scholar]; (b) Tortoreto C, Rackl D, Davies HM. Org Lett. 2017;19:770–773. doi: 10.1021/acs.orglett.6b03681. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura E, Yoshikai N, Yamanaka M. J Am Chem Soc. 2002;124:7181–7192. doi: 10.1021/ja017823o. [DOI] [PubMed] [Google Scholar]
- 8.Pirrung MC, Morehead AT. J Am Chem Soc. 1996;118:8162–8163. [Google Scholar]
- 9.(a) Davies HML, Beckwith REJ. Chem Rev. 2003;103:2861–2903. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; (b) Wolckenhauer SA, Devlin AS, Du Bois J. Org Lett. 2007;9:4363–4366. doi: 10.1021/ol701950d. [DOI] [PubMed] [Google Scholar]
- 10.Sharma VB, Jain SL, Sain B. Catal Lett. 2009;94:57–59. [Google Scholar]
- 11.Wang P, Adams J. J Am Chem Soc. 1994;116:3296–3305. [Google Scholar]
- 12.(a) Yang JM, Cai Y, Zhu SF, Zhou QL. Org Biomol Chem. 2016;14:5516–5519. doi: 10.1039/c5ob02418h. [DOI] [PubMed] [Google Scholar]; (b) Demonceau A, Noels AF, Costa JL, Hubert AJ. J Mol Catal. 1990;58:21–26. [Google Scholar]
- 13.(a) Wang JB, Chen B, Bao J. J Org Chem. 1998;63:1853–1862. [Google Scholar]; (b) Taber DF, Joshi PV. J Org Chem. 2004;69:4276–4278. doi: 10.1021/jo0303766. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.