

Abstract
Protein–protein interactions drive every aspect of cell signaling, yet only a few small-molecule inhibitors of these interactions exist. Despite our ability to identify critical residues known as hot spots, little is known about how to effectively engage them to disrupt protein–protein interactions. Here, we take advantage of the ease of preparation and stability of pyrrolinone 1, a small-molecule inhibitor of the tight interaction between the urokinase receptor (uPAR) and its binding partner, the urokinase-type plasminogen activator uPA, to synthesize more than 40 derivatives and explore their effect on the protein–protein interaction. We report the crystal structure of uPAR bound to previously discovered pyrazole 3 and to pyrrolinone 12. While both 3 and 12 bind to uPAR and compete with a fluorescently labeled peptide probe, only 12 and its derivatives inhibit the full uPAR·uPA interaction. Compounds 3 and 12 mimic and engage different hot-spot residues on uPA and uPAR, respectively. Interestingly, 12 is involved in a π–cation interaction with Arg-53, which is not considered a hot spot. Explicit-solvent molecular dynamics simulations reveal that 3 and 12 exhibit dramatically different correlations of motion with residues on uPAR. Free energy calculations for the wild-type and mutant uPAR bound to uPA or 12 show that Arg-53 interacts with uPA or with 12 in a highly cooperative manner, thereby altering the contributions of hot spots to uPAR binding. The direct engagement of peripheral residues not considered hot spots through π–cation or salt-bridge interactions could provide new opportunities for enhanced small-molecule engagement of hot spots to disrupt challenging protein–protein interactions.
Graphical abstract
It is estimated that there are more than 200000 protein–protein interactions in a cell. These interactions drive nearly every aspect of cellular function. Small molecules offer an opportunity to explore these interactions in normal and pathological processes. Protein–protein interactions exhibit a wide range of binding thermodynamics and kinetics from weak interactions that occur over small interfaces to tight and stable protein–protein interactions that occur over large interfaces (>1000 Å2). Although the design of small molecules to disrupt weak interactions is relatively straightforward, inhibiting tight interactions is much more challenging. To date, while numerous small molecules have been reported to inhibit protein–protein interactions (reviewed in refs 1 and 2), there are only a few examples of small molecules that disrupt tight protein–protein interactions (Kd = 1–100 nM); examples include antagonists of Bcl-xL3 and IL-2R.4 The ability to disrupt these tight interactions that occur over large surfaces with a small molecule that has a much smaller footprint is attributed to the presence of residues that contribute disproportionately to the binding affinity, also known as hot spots.5–9
The urokinase receptor (uPAR) is a cell surface glycophosphatidylinositol (GPI)-anchored receptor that is part of an extensive network of protein–protein interactions. Its binding partners include serine proteinase urokinase-type plasminogen activator uPA10 and the glycoprotein vitronectin.11–13 The uPAR·uPA protein–protein interaction is a high-affinity (KD = 1 nM) and stable (koff = 10−4 s−1) interaction.14 Crystal structures of the uPAR·uPA complex show that uPA binds to uPAR along a well-defined binding site that is part of a >1000 Å2 interface.15–18 The interaction is mediated by a 25-residue growth factor-like domain (GFD), and residues from a kringle-like domain of uPA. A comprehensive alanine scanning study at the uPAR·uPA interface identified several hot-spot residues that reduced the uPAR·uPA binding affinity by >1 kcal mol−1.14,18,19 In the uPAR·uPA complex, there are hot spots both on uPA (Trp-30, Phe-25, Tyr-24, and Ile-28)14 and on the large binding cleft of uPAR that accommodates the GFD domain (Leu-150, Leu-55, Leu-66, Tyr-57, and Asp-140).18
Previously, we performed a computational screen of commercial libraries to identify small molecules that bind to uPAR and disrupt the uPAR·uPA protein–protein interaction.18,21,22 Compound 1 was discovered following a substructure search using pyrazole, piperidinone, and pyrrolydi-none compounds as templates.23 These compounds were shown to bind to uPAR and displace a fluorescently labeled peptide. In addition to 1, another strategy that consisted of docking chemical libraries to an ensemble of uPAR structures sampled from explicit-solvent molecular dynamics simulations led to 2 (IPR-803).24 Compound 2 binds to uPAR with submicromolar affinity and inhibits the uPAR·uPA complex with single-digit micromolar IC50 values.24 Structure–activity relationships revealed the critical nature of a benzoic acid moiety that was attributed to a salt-bridge interaction with the guanidinium group of Arg-53. We showed that this residue became exposed following explicit-solvent molecular dynamics simulations.16 Our predicted binding mode and interaction with Arg-53 was recently independently confirmed by a crystal structure of an analogue of 2 bound to uPAR.25 To the best of our knowledge, our work with 2 is the first study that led to small-molecule protein–protein antagonists using protein structures sampled from molecular dynamics simulations. The work highlights the importance of considering molecular dynamics of a receptor for the design of small molecules that disrupt tight interactions (Scheme 1).
Scheme 1.
Analysis of three-dimensional structures of protein–protein complexes reveals that hot spots are generally found at the protein–protein interface of tight protein–protein interactions.5,26,27 They can be located either on the ligand or on the receptor that harbors a binding site. It has been suggested that small molecules that bind to hot spots can disrupt tight protein–protein interactions despite their smaller footprint. Early work targeting the IL-2·IL-2Rα complex confirmed that small molecules that inhibited the interaction directly bind to hot spots,28 but the researchers also found that neighboring residues that promote IL-2·IL-2Rα enhanced binding of small molecules. These counterintuitive results can be explained by dynamical effects likely arising from the cooperativity of residues at the interface.29 This cooperativity has led to the suggestion that hot-spot clusters can form hot regions.30 Cooperativity makes it more difficult to select the appropriate hot spots for the rational design of small-molecule inhibitors of protein–protein interactions. The mere binding to hot spots may not necessarily result in more potent inhibition of the protein–protein interaction. A deeper understanding of (i) the interaction energies between small molecules and individual residues and (ii) the dynamical changes that occur upon binding of a small molecule could lead to more effective strategies for the rational design of small-molecule protein–protein interaction inhibitors.
Here, we take advantage of the straightforward synthesis of 1 to prepare 46 derivatives of the compound to gain insight into the forces that lead to small-molecule inhibition of the uPAR·uPA protein–protein interaction. Their activity was measured using fluorescence polarization with a labeled α-helical peptide and an enzyme-linked immunosorbent assay (ELISA) that uses uPAATF, which includes the entire binding interface of the uPAR·uPA interface. We determine the crystal structure of uPAR bound to two small molecules, namely, pyrrolinone 21 (IPR-1175) and pyrrazole 3 (IPR-737), providing a structural basis for the activity of 1 and its derivatives. To gain deeper insight into the basis for the activity of 1 and its derivatives, we conducted explicit-solvent molecular dynamics simulations of uPAR in complex with uPA, 1, 3, and several other derivatives. The resulting structures were used for free energy calculations using molecular mechanics (MM), generalized Born (GB), and solvent-accessible surface (SA) also known as MM-GBSA. Decomposition energy calculations, which correspond to the MM-GBSA free energy between a ligand and individual residues on uPAR, provided deeper insight into the individual contributions of each amino acid to the protein–protein and protein–compound interactions. Finally, we compare the dynamics of the uPAR·uPA interaction with the dynamics of uPAR in complex with small molecules.
MATERIALS AND METHODS
Protein Expression and Purification
suPAR was obtained by a one-step purification process as previously described.31
Crystallization and Structure Determination of uPAR in Complex with Its Inhibitor, 3 (IPR-737), and 12 (IPR-1175)
A stabilized form of human soluble uPAR (H47C/N259C) (denoted suPARcc) was used to facilitate crystallization of suPAR in complex with the inhibitor. suPARcc was expressed and purified as previously described32 and concentrated to 16 mg mL−1. Crystals were formed using 1.96 M ammonium sulfate, 100 mM HEPES (pH 7.5), and 2% (w/v) polyethylene glycol 400 as the precipitant solution by the sitting-drop vapor diffusion method. Crystals were soaked into 40% PEG4000 and 100 mM HEPES (pH 7.5) with 1 mM 3 (IPR-737) or 12 (IPR-1175) for ∼2 days. X-ray diffraction data was collected under cryogenic conditions at 100 K. Crystals were collected on beamline BL17U at the Shanghai Synchrotron Radiation Facility (SSRF) and were processed, integrated, and scaled together with HKL2000.33 The crystal structures of the suPARcc·3 (IPR-737) complex and suPARcc·12 (IPR-1175) complex were determined by the molecular replacement program MOLREP34 of the CCP4 package using Protein Data Bank (PDB) entry 3U74 as a search model. After molecular replacement, inhibitor 3 (IPR-737) or 12 (IPR-1175) was built into the model on the basis of a 2Fobs –Fcalc σ-weighted composite omit map, and then iterative manual model building and model refinement were performed with COOT35 and REFMAC, yielding a final R value of 0.2119 and an Rfree value of 0.2576 in the suPARcc·3 (IPR-737) complex (Table S2), while the values were 0.2349 and 0.2613, respectively, in the suPARcc·12 (IPR-1175) complex (Table S1). These structures were analyzed by PyMOL.4
Fluorescence Polarization
Polarized fluorescence intensities were measured using an EnVision Multilabel plate reader (PerkinElmer) with excitation and emission wavelengths of 485 and 530 nm, respectively.24 A Thermo Scientific Nunc 384-well black microplate was used to prepare samples with a final volume of 50 μL in duplicate. First, the compounds were serially diluted in dimethyl sulfoxide (DMSO) and further diluted in 1× PBS buffer with 0.01% Triton X-100 to yield a final concentration from 100 to 0.046 μM. Triton X-100 was added in the buffer to avoid compound aggregation; 35 μL of the compound solution and 10 μL of PBS containing uPAR were added to the wells, and the mixture was incubated for at least 15 min to allow the compound to bind to the protein. Finally, 5 μL of fluorescent AE147-FAM peptide was added for a total volume of 50 μL in each well, resulting in final uPAR and peptide concentrations of 320 and 100 nM, respectively. The final DMSO concentration was 2%, which had no effect on the binding of the peptide. Controls included wells containing only the peptide and wells containing both protein and peptide each in quadruplicate to ensure the validity of the reaction assay. A unit of millipolarization (mP) was used to calculate the percent inhibition of the compounds. Inhibition constants were determined using the Ki calculator available at http://sw16.im.med.umich.edu/software/calc_ki/.
Microtiter-Based ELISA for uPAR·uPA
High-binding microplates (Greiner Bio-One) were incubated for 2 h at 4 °C with 100 μL of 2 μg mL−1 uPAATF in PBS. Each plate was washed with 0.05% Tween 20 in PBS buffer between each step. A 1:1 mixture of Superblock buffer in PBS (Thermo Fisher Scientific, Inc., Waltham, MA) with 0.04 M NaH2PO4 and 0.3 M NaCl buffer was used for blocking at room temperature for 1 h; 100 μL of 75 nM uPAR in PBS with 0.025% Triton X-100 was added with the indicated concentrations of compounds. Compounds were screened initially at 50 μM. For concentration-dependent studies, a range of compound concentrations from 100 to 0.4 μM were used. The final DMSO concentration was 1%. Following incubation for 30 min and subsequent washing steps, the human uPAR biotinylated antibody (1:3000 dilution of 0.2 mg mL−1 BAF807, R&D Systems, Minneapolis, MN) in PBS containing 1% BSA was added to the wells (100 μL/well) and incubated for 1 h to allow for the detection of bound uPAR. Following washing, 100 μL of streptavidin-bound horseradish peroxidase (84 ng mL−1) in PBS containing 1% BSA was added for 20 min. The signal obtained in the presence of TMB in phosphate-citrate buffer (pH 5) and hydrogen peroxide was stopped by adding a H2SO4 solution and detected using a SpectraMax M5e instrument (Molecular Devices, Sunnyvale, CA).
Molecular Docking
Small molecules were prepared for molecular docking using Maestro (version 9.4, Schrödinger, LLC, New York, NY). Compounds were first processed with LigPrep (version 2.6, Schrödinger, LLC). The receptor protein structure (PDB entry 3BT1 and crystal structures of the uPAR·3 and uPAR·12 complexes) was prepared using the Protein Preparation Wizard workflow in Maestro. Bond orders were assigned, hydrogen atoms added, and disulfide bonds created. For the 3BT1 structure, vitronectin (chain B) was removed and the missing loop at residues Arg-83 and Ala-84 introduced using the Prime36 module in Schrödinger. The missing loops from Ser-81 to Ser-90 (uPAR·3) and from Ile-129 to Lys-139 (uPAR·3 and uPAR·12) were reconstructed using the loop-model class in MODELER (version 9.13).37 Five initial models were constructed and refined using the “fast” molecular dynamics (MD) annealing function. Five additional loop models were constructed for each initial model and similarly refined. A loop model was visually selected from the 25 loop models that best matched the existing loop in the 3BT1 structure. Finally, structures were protonated at pH 7.0 using PROPKA.38
The binding poses of select derivatives of 1 (IPR-1110) were generated using the cocrystal structure of uPAR in complex with 3 (IPR-1175) as a guide in Maestro. The derivatives were docked in a 21 Å box centered on the complexed ligand using Glide39 (Schrödinger, LLC) in standard precision (SP) mode. The common core structure of the analogues found in Table 1 was used to restrict the binding poses of the derivative compounds. All other parameters were set to default values.
Table 1.
Synthesized Derivatives
| Compounds | R1 | R2 | R3 | IC50(μM) | Ki(μM) | MWa | AlogP | HBAb | HBDc | RBd |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 (IPR1110) |
|
|
|
18.4 | 0.7 | 499.7 | 5.4 | 2 | 0 | 4 |
| 4 (IPR1201) |
|
|
|
16.3±1.9 | 1.0±0.2 | 513.8 | 5.8 | 2 | 0 | 4 |
| 5 (IPR1200) |
|
|
|
27.4±2.1 | 2.7±0.4 | 521.4 | 6.1 | 2 | 0 | 5 |
| 6 (IPR1181) |
|
|
|
30.2±2.0 | 3.8±0.4 | 465.3 | 4.7 | 2 | 0 | 4 |
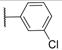
| 7 (IPR1178) |
|
|
|
20.2±1.9 | 1.1±0.2 | 516.2 | 5.8 | 2 | 0 | 4 |
| 8 (IPR1187) |
|
|
|
26.4±1.6 | 2.5±0.5 | 509.8 | 6.1 | 2 | 0 | 5 |
| 9 (IPR1185) |
|
|
|
35.6±5.6 | 1.7±0.1 | 518.7 | 5.2 | 3 | 0 | 4 |
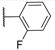
| 10 (IPR1192) |
|
|
|
30.1±2.6 | 1.9±0.4 | 499.7 | 5.4 | 2 | 0 | 4 |
| 11 (IPR1193) |
|
|
|
57.8±1.9 | 1.7±0.1 | 525.8 | 4.9 | 4 | 0 | 4 |
| 12 (IPR1175) |
|
|
|
45.2±2.1 | 2.0±0.5 | 448.9 | 5.3 | 2 | 0 | 5 |
| 13 (IPR1171) |
|
|
|
23.0±2.3 | 1.3±0.1 | 546.7 | 5.2 | 2 | 0 | 4 |
| 14 (IPR1194) |
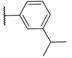
|
|
|
66.7±7.2 | 4.5±0.9 | 448.9 | 5.3 | 2 | 0 | 5 |
| 15 (IPR1195) |
|
|
|
61.7±5.1 | 1.6±0.3 | 462.9 | 5.5 | 2 | 0 | 5 |
| 16 (IPR1186) |
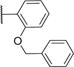
|
|
|
35.3±2.0 | 2.1±0.4 | 512.9 | 5.7 | 3 | 0 | 7 |
| 17 (IPR1182) |
|
|
|
37.1±2.9 | 1.3±0.2 | 471.3 | 4.8 | 3 | 0 | 5 |
| 18 (IPR1196) |
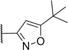
|
|
|
47.3±2.4 | 3.6±0.8 | 453.9 | 4.4 | 4 | 0 | 5 |
| 19 (IPR1152) |
|
|
|
ND | 95.2±1.6 | 450.9 | 3.8 | 3 | 1 | 6 |
| 20 (IPR1119) |
|
|
|
66.5±2.4 | 8.6±2.1 | 489.9 | 5.2 | 2 | 0 | 5 |
| 21 (IPR1183) |
|
|
|
91.1±3.2 | 13.3±2.9 | 477.3 | 4.5 | 3 | 0 | 5 |
| 22 (IPR1176) |
|
|
|
53.8±6.5 | 1.9±0.4 | 507.0 | 6.4 | 3 | 0 | 10 |
| 23 (IPR1174) |
|
|
|
18.4±4.9 | 1.2±0.2 | 521.0 | 6.8 | 3 | 0 | 10 |
| 24 (IPR1179) |
|
|
|
ND | 15.9±2.8 | 537.9 | 6.6 | 2 | 0 | 5 |
| 25 (IPR1177) |
|
|
|
ND | ND | 430.9 | 3.6 | 3 | 1 | 9 |
| 26 (IPR1188) |
|
|
|
ND | 2.0±0.6 | 452.9 | 4.7 | 2 | 0 | 6 |
| 27 (IPR1189) |
|
|
|
ND | 11.8±1.8 | 499.7 | 5.4 | 2 | 0 | 4 |
| 28 (IPR1190) |
|
|
|
21.0±0.7 | 2.6±0.2 | 529.8 | 5.3 | 3 | 0 | 5 |
| 29 (IPR1191) |
|
|
|
29.5±3.3 | 2.1±0.2 | 544.2 | 5.4 | 2 | 0 | 4 |
| 30 (IPR1150) |
|
|
|
ND | ND | 485.7 | 4.9 | 2 | 0 | 4 |
| 31 (IPR1151) |
|
|
|
69.0±1.2 | 31.7±2.6 | 441.3 | 4.8 | 2 | 0 | 4 |
| 32 (IPR1153) |
|
|
|
46.9±3.3 | 4.5±1.4 | 464.9 | 4.4 | 3 | 0 | 7 |
| 33 (IPR1157) |
|
|
|
ND | 17.6±1.6 | 495.8 | 5.6 | 2 | 0 | 4 |
| 34 (IPR1158) |
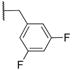
|
|
|
ND | 2.3±0.3 | 456.8 | 4.5 | 2 | 0 | 5 |
| 35 (IPR1159) |
|
|
|
ND | ND | 511.8 | 5.1 | 3 | 0 | 5 |
| 36 (IPR1160) |
|
|
|
ND | 1.9±0.3 | 456.9 | 5.0 | 2 | 0 | 4 |
| 37 (IPR1161) |
|
|
|
87.5±6.1 | 18.6±1.8 | 504.8 | 5.1 | 3 | 0 | 6 |
| 38 (IPR1167) |
|
|
|
ND | 2.3±0.2 | 503.8 | 5.8 | 2 | 0 | 6 |
| 39 (IPR1168) |
|
|
|
23.1±1.6 | 0.9±0.0 | 503.8 | 5.8 | 2 | 0 | 6 |
| 40 (IPR1172) |
|
|
|
93.3±1.6 | 3.7±0.5 | 490.8 | 6.2 | 3 | 0 | 6 |
| 41 (IPR1173) |
|
|
|
97.8±1.0 | 7.7±0.8 | 464.8 | 4.0 | 4 | 0 | 6 |
| 42 (IPR1197) |
|
|
|
77.1±3.4 | 5.5±0.6 | 434.9 | 5.1 | 2 | 0 | 5 |
| 43 (IPR1198) |
|
|
|
ND | 55.3±2.0 | 411.8 | 2.9 | 4 | 0 | 4 |
| 44 (IPR1607) |
|
|
|
24.0±3.2 | 0.8±0.1 | 482.9 | 5.6 | 2 | 0 | 5 |
| 45 (IPR2260) |
|
|
|
15.7±1.3 | 0.4±0.1 | 569.0 | 5.8 | 4 | 0 | 9 |
| 46(IPR1609)XHS12 |
|
|
|
21.6±1.6 | 0.6±0.1 | 540.9 | 5.5 | 4 | 0 | 7 |
| Compounds | R1 | R2 | R3 | R4 | IC50(μM) | Ki (μM) | MWa | AlogP | HBAb | HBDc | RBd |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 47 (IPR1154) |
|
|
|
Me | ND | 33.5±2.2 | 514.8 | 6.1 | 3 | 0 | 5 |
| 48 (IPR1155) |
|
|
|
iPr | ND | ND | 542.8 | 6.8 | 3 | 0 | 6 |
Molecular weight
Hydrogen bond acceptor
Hydrogen bond donor
Rotatable bonds.
Molecular Dynamics Simulations
The binding poses were used to run MD simulations using the AMBER14 and AmberTools15 packages.40 Each compound was assigned AM1-BCC41 charges and gaff42 atom types using the antechamber program.43 Complexes were immersed in a box of TIP3P44 water molecules. No atom on the complex was within 14 Å of any side of the box. The solvated box was further neutralized with Na+ or Cl− counterions using the tleap program.
Simulations were performed using the GPU accelerated version of the pmemd program with ff12SB45 and gaff42 force fields under periodic boundary conditions. All bonds involving hydrogen atoms were constrained by using the SHAKE algorithm,46 and a 2 fs time step was used in the simulation. The particle mesh Ewald47 (PME) method was used to treat long-range electrostatics. Simulations were run at 298 K under 1 atm in the NPT ensemble employing a Langevin thermostat and a Berendsen barostat. Water molecules were first energy-minimized and equilibrated by running a short simulation with the complex fixed using Cartesian restraints. This was followed by a series of energy minimizations in which the Cartesian restraints were gradually relaxed from 500 to 0 kcal Å−2, and the system was subsequently gradually heated to 298 K via a 48 ps MD run. Via assignment of different initial velocities, 10 independent simulations that are 10 ns in length each were performed for the protein–compound structures.
Free Energy Calculations
In each of the 10 trajectories (10 ns in length), the first 2 ns was discarded for equilibration. MD snapshots were saved every 1 ps, yielding 8000 structures per trajectory. A total of 80000 snapshots were generated per 100 ns of simulation; 1000 snapshots were selected at regular intervals from the 80000 snapshots for free energy calculations using the cpptraj program.48 The molecular mechanics-generalized Born surface area (MM-GBSA)49 method was used to calculate the free energy using the MMPBSA.py script50 and Onufriev’s GB model.51,52 Solvent-accessible surface area (SASA) calculations were switched to the ICOSA method, where surface areas are computed by recursively approximating a sphere around an atom, starting from an icosahedron. Salt concentrations were set to 0.1 M. The entropy was determined by normal mode calculations53 with the nmode module from 100 of the 1000 snapshots used in the free energy calculations. The maximal number of cycles of minimization was set to 10000. The convergence criterion for the energy gradient to stop minimization was 0.5.
The MM-GBSA binding free energy is expressed as
where ΔEGBTOT is the combined internal and solvation energies, T is the temperature, and ΔSNM is the entropy determined by normal mode calculation. The solvation energy is determined using generalized Born (GB) solvation models (ΔEGBSOL) (igb = 2):
where ΔEGBSOLV is the solvation free energy and ΔEGAS is the molecular mechanical energies (gas phase). The gas-phase energies are composed of two components:
where ΔEELE is the nonpolar electrostatic energy and ΔEVDW is the polar van der Waals energy. The GB solvation free energy is expressed by
where ΔESURF and ΔEGB are the nonpolar and polar contributions to the solvation free energy, respectively. All the binding energies are determined by
where ECOM, EREC, and ELIG are total energies corresponding to the complex, receptor, and ligand, respectively.
Decomposition Energy
The decomposition energy used in this work was determined using the MMPBSA.py script50 available in AMBER14. The script provides several schemes for decomposing calculated free energies into specific residue contributions using either GB or PB implicit-solvent models. These schemes were developed by Gohlke and co-workers.54 The per-residue decomposition scheme was used in this work. Similar to the free energy calculations, we use the GB solvation model from Onufriev and co-workers.51,52 The energy terms are decomposed according to the scheme outlined in the AMBER14 manual using the following equation:
where the first and second terms represent the average contribution over snapshots i from the MD simulation in residues j on the receptor and ligand, respectively. The term E(i,j) corresponds to the contribution of the gas-phase and solvation energies, that is
where EVDW and EELE are the van der Waals and electrostatic energies in the gas phase (EGAS), respectively. EGB and ESURF are the polar and nonpolar contributions to the solvation free energy by the GB solvation model (EGBSOLV), respectively. Entropy is not included in the decomposition method.
The GB model is described in detail by Onufriev and associates55 and in the AMBER14 manual. The GB model we selected (igb = 2, GBOBC model I) approximates the solvation electrostatic EGB by an analytical formula:
where rij is the distance between atoms i and j, Ri and Rj are the effective Born radii of atoms i and j, respectively, K is the Debye–Hückel screening parameter, ε is the dielectric constant, and f GB is a smooth function. Each atom in the GB model is represented as a sphere with radius ρi with charge qi. The f GB function is expressed as
and is used to describe the distance between two atoms and their effective Born radii.
The nonpolar contribution to the solvation free energy is calculated by approximating the total SASA of the molecule:
where γ and β are the surface tension and offset terms, respectively. The ICOSA method is used to determine SASA.40,54 In this method, surface areas are computed by recursively approximating a sphere around an atom. The first sphere is modeled as an icosahedron. In each subsequent step, the faces of the polyhedron are divided into four equal-sized triangles to better approximate the sphere.
Chemistry
All chemicals were purchased from commercially available sources and used as received. Column chromatography was performed with silica gel (25–63 μm). High-resolution mass spectra were recorded on an Agilent 6520 Accurate Mass Q-TOF instrument. 1H nuclear magnetic resonance was recorded in CDCl3 or DMSO on a Bruker 500 MHz spectrometer. Reverse-phase liquid chromatography and mass spectrometry were performed on an Agilent 1100 LC/MSD instrument fitted with an Eclipse XBD-C18 (4.6 mm × 150 mm) column eluting at 1.0 mL min−1 employing an (acetonitrile/methanol)/water gradient (each containing 5 mM NH4OAc) from 70 to 100% acetonitrile/methanol over 15 min and holding at 100% acetonitrile/methanol for 2 min. Chemical shifts are reported in parts per million using either residual CHCl3 or DMSO as an internal reference. All compounds are >95% pure unless otherwise stated. Syntheses of 3-(hexyloxy)-aniline and 3-(hexyloxy)-4-methylaniline were performed using a protocol described by Marco and co-workers.56 β-Diketoesters were synthesized with modification according to Milagre and co-workers.57 Derivatives of 1 were synthesized by a modified procedure of Rose and co-workers.58 Full compound characterization is provided in the Supporting Information.
RESULTS
Synthesis of Pyrrolinones, Stability, Reactivity, and Selectivity Studies
The ease of synthesis of 1 (IPR-1110) prompted us to prepare 46 derivatives to explore the uPAR·uPA binding interface (Table 1). 1 (IPR-1110) was modified at three sites, R1–R3. Most of the structure–activity exploration was focused on R1 and R2. The synthesis of these compounds was straightforward (Figure S1) as described previously.23
The pyrrolinone core structure of 1 suggests that nucleophilic residues within a protein could potentially form covalent adducts through nucleophilic attack at the carbon bearing the hydroxyl group of the core five-membered ring. To explore this possibility, we employed electrospray ionization (ESI) mass spectrometry. As shown in Figure S2, there was no adduct formation when uPAR was incubated with the compound, ruling out nonspecific covalent bond formation as the mechanism by which the pyrrolinone compounds antagonize uPAR–protein interactions. Next, we investigated the stability of the compound in buffer (PBS), methanol, and uPAR using high-performance liquid chromatography (Figure S3). The UV spectra for all three conditions were identical (Figure S3a), and the mass spectra corresponding to the major peaks were also identical (Figure S3b). This suggests that the compound is stable both in buffer and in the presence of uPAR.
Finally, we investigated the selectivity of the pyrrolinone compounds by testing them for inhibition of an unrelated protein–protein interaction between the α and β subunits of the CaV2.2 calcium channel (Figure S4). The CaVαβ interaction is mediated by a tight single-digit nanomolar interaction that occurs over a large interface consisting primarily of an α helix of the α subunit binding to a large well-defined pocket on the β subunit (Figure S4). Compound 1 (Figure S4) showed no inhibition of this interaction up to 25 μM. The effect of 1 on the CaVαβ interaction at 50 and 100 μM can be attributed to aggregation considering the sharp rise in activity from nearly 0% inhibition at 25 μM to more than 75% inhibition at 50 μM. This was not observed for uPAR, whereby 1 exhibited a gradual increase in its level of inhibition of binding of AE-147 to uPAR (Figure S4). These studies confirm that the compounds bind to uPAR in a selective manner as evidenced by the lack of activity against another unrelated protein–protein interaction. It is possible that compound 1 inhibits interactions that are similar to the uPAR·uPA interaction. Future studies that explore compounds in cell culture should explore these compounds for inhibition of interactions that are similar to uPAR·uPA interactions.
Crystal Structures of Compounds Bound to uPAR
The crystal structure of uPAR was previously determined in complex with ATF (PDB entry 2FD6) (Figure 1a) or α-helical peptide AE-147 (PDB entry 1YWH) (Figure 1b). These structures revealed a large interface between uPAR and uPA that contains several hot spots, including Leu-55, Tyr-57, Leu-66, Asp-140, and Leu-150. All the hot spots on uPAR at the uPAR·uPA interaction are listed in Table S1. We hypothesized that compounds that disrupt the protein–protein interaction between uPAR and uPA likely directly engage these hot spots. To test this hypothesis, we resorted to X-ray crystallography to determine the structure of 1 (IPR-1110) and derivatives in complex with uPAR. We also attempted to determine the structures of pyrazole, piperidinone, pyrrolidinone, and butan-amine compounds that we had previously shown to bind to uPAR.21 We obtained structures for two compounds bound to uPAR, namely, pyrrolinone 12 (IPR-1175) (Figure 2 and Table S2) and pyrazole 3 (IPR-737) (Figure 3 and Table S3).
Figure 1.
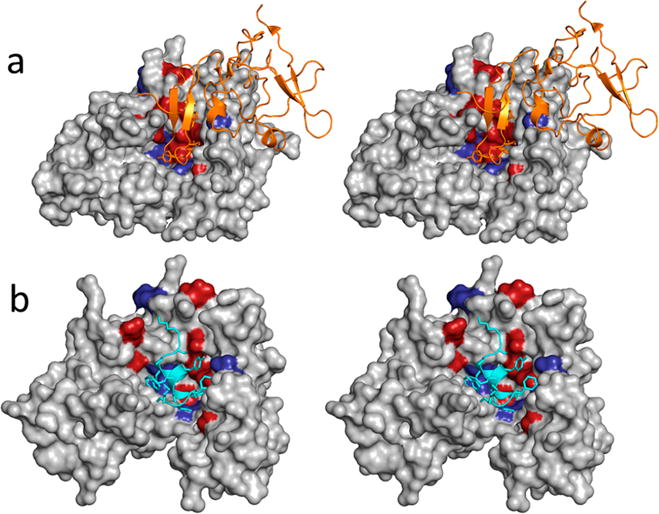
(a) Three-dimensional structure of the uPAR·uPAATF complex (PDB entry 2FD6). uPAR is shown as a gray solvent-accessible surface. Residues whose mutations to alanine result in changes in binding affinity of ≥1 kcal mol−1 are colored red, while those that lead to changes between 0.5 and 1 kcal mol−1 are colored purple. uPAATF is shown as an orange cartoon. (b) Three-dimensional structure of uPAR in complex with the AE-157 peptide (PDB entry 1YWH). The peptide is shown as a cyan cartoon, and side chains that come in contact with uPAR are shown as capped sticks. uPAR is rendered and color-coded in a manner similar to that of panel a.
Figure 2.
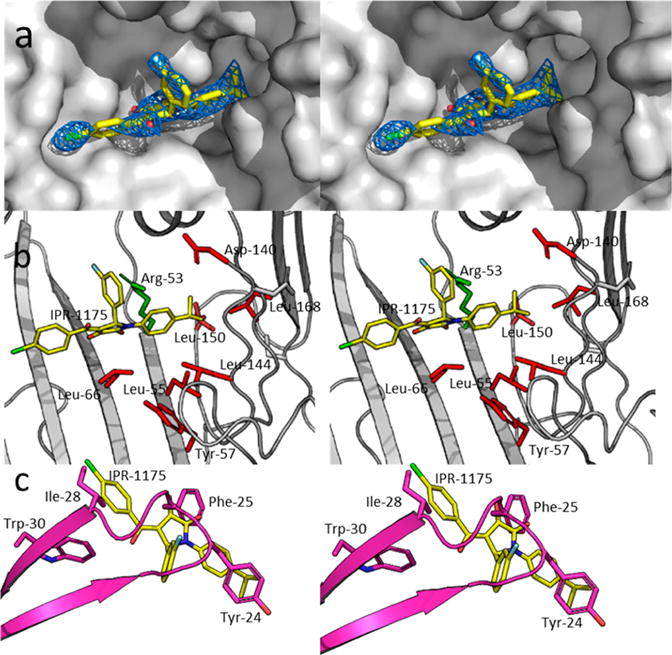
Mode of binding of 12 (IPR-1175) upon the central ligand binding cavity of suPARcc. suPARcc is shown as a gray solvent-accessible surface. 12 (IPR-1175) is shown as capped sticks. Atoms are color-coded by atom type with N, C, O, Cl, and F colored blue, yellow, red, green, and light blue, respectively. The 2Fo – Fc electron density map of 12 (IPR-1175) at 1σ is colored blue. (b) Crystal structure of 12 (IPR-1175) bound to uPAR except that uPAR is shown as capped sticks to highly hot-spot residues located within the uPAR cavity. Hot spots are shown as red capped sticks. Arg-53, which is not considered a hot spot, is colored green. (c) Crystal structure of the uPAR·12 (IPR-1175) complex superimposed on the structure of the uPAR·uPAATF complex to illustrate the overlap between hot spots on uPA and substituents on 12 (IPR-1175).
Figure 3.
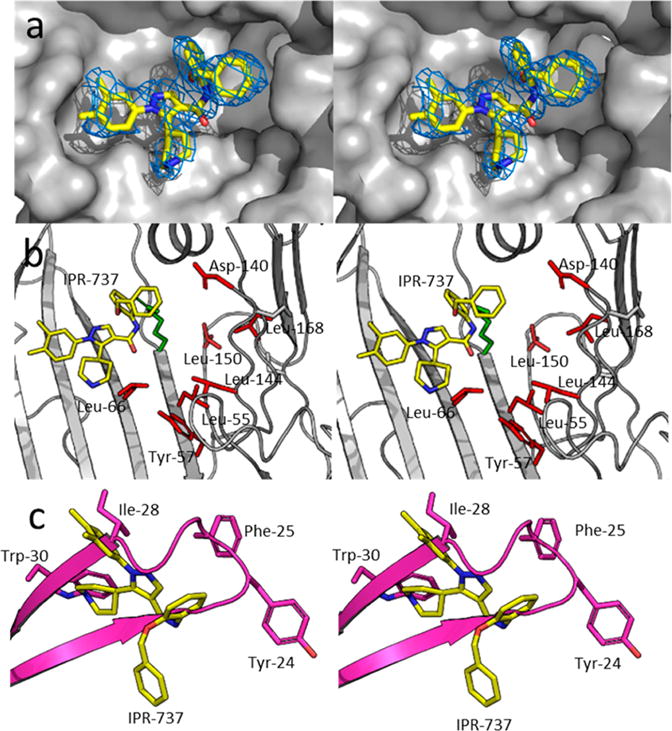
Mode of binding of 3 (IPR-737) upon the central ligand binding cavity of suPARcc. suPARcc is shown as a gray solvent-accessible surface. 3 (IPR-737) is shown as capped sticks. Atoms are color-coded by atom type with N, C, O, Cl, and F colored blue, yellow, red, green, and light blue, respectively. A 2Fo – Fc electron density map of 3 (IPR-737) at 1σ is colored blue. (b) Crystal structure of 3 (IPR-737) bound to uPAR except that uPAR is shown as capped sticks to highly hot-spot residues located within the uPAR cavity. Hot spots are shown as red capped sticks. (c) Crystal structure of the uPAR·3 (IPR-737) complex superimposed on the structure of the uPAR·uPAATF complex to illustrate the overlap between hot spots on uPA and substituents on 3 (IPR-737).
The structure of uPAR in complex with 12 reveals that the compound is ensconced deeply in the pocket that is occupied by the growth factor-like domain of uPA in its complex with uPAR (Figure 2a). The R1 isopropylphenyl group of the compound points toward the interior of the hydrophobic pocket of uPAR. This pocket accommodates the residues located on the loop of the β turn of the GFD domain of uPA. The substituent makes direct contacts with Leu-150, Leu-168, Val-125, Leu-55, and Arg-53 (Figure 2b). Most of these interactions are hydrophobic except for the cation–π interaction with Arg-53. Cation–π interactions are commonly observed in protein–compound structures and can contribute up to 1 kcal mol−1 to the free energy of binding.59,60 Here, the benzene ring of the R1 group of 12 (IPR-1175) directly faces one of the Nω atoms of the guanidinium ion of Arg-53. The Nω atom is 2.5 Å from one of the ortho carbon atoms of the benzene ring. Unlike R1, the chlorobenzene R3 group of 12 (IPR-1175) points in the opposite direction and occupies a pocket surrounded by residues Thr-27, Val-29, Arg-142, and Leu-40 on uPAR. The R3 group is more exposed to solvent than R1 and does not occupy a pocket composed entirely of hydrophobic residues as evidenced by the presence of Arg-142. Finally, the R2 group of 12 does not occupy a well-defined pocket on uPAR, but it is worth noting that this group is also involved in a cation−π interaction with Arg-53 of uPAR (Figure 2b). Interestingly, the R1 and R3 groups of 12 occupy the same position as uPA hot spots Ile-28 and Tyr-24, respectively (Figure 2c).
3 (IPR-737) adopts a binding mode different from that of 12 (IPR-1175) (Figure 3). 3 (IPR-737) binds outside the hydrophobic pocket of uPAR that is occupied by side chains of the β-turn loop of the GFD domain of uPA (Figure 3a). Unlike 12 (IPR-1175), the compound engages fewer hot spots on uPAR (Figure 3b). Only one of the uPAR hot-spot residues (Leu-66) comes in contact with 3 (IPR-737). Interestingly, analysis of the crystal structure of uPAR in complex with the AE-147 peptide shows that two hydrophobic residues on the peptide (Phe-5 and Tyr-8) come in direct contact with Ile-66 of uPAR (Figure 1b). Hence, interaction with Ile-66 may explain why 3 (IPR-737) can disrupt binding of AE-147 to uPAR. Engagement of Ile-66, however, is not sufficient to disrupt the full uPAR·uPAATF interaction. Ile-66 of 3 (IPR-737) binds to uPAR at a site that is occupied by Ile-28 and Trp-30 of uPA. These two residues are located on the β strands of the β hairpin of the GFD domain of uPA (Figure 3c). This is in contrast to 12 (IPR-1175), which overlaps with amino acids located on the loop region of the β hairpin (Figure 2c).
Structure–Activity Relationship
We prepared 46 derivatives of 1 (IPR-1110) to explore the uPAR binding site at the uPAR·uPA interface. Substituents at R1–R3 of 1 (IPR-1110) were explored. The binding mode of 12 (IPR-1175) (Figure 2a,b) shows that R1 groups point toward a large hydrophobic cavity occupied by several uPA hot-spot residues. An aromatic group at R1 was generally required for inhibition of uPAR·uPA interaction. This is evidenced by complete loss of activity of 25 (IPR-1177), which lacks an aromatic ring at R1. There are six compounds without an aromatic ring directly attached to the central pyrrolidone ring: 26, 32, 34, 38, 39, and 46; in these compounds, a methylene (34) or an ethylene (26, 32, 38, 39, and 46) group separates the central pyrrolinone ring with a benzene ring. Except for 26, 34, and 38, the compounds retained the ability to inhibit the uPAR·uPA protein–protein interaction with IC50 values similar to that of 1 (IPR-1110) for 39 and 46. All three compounds that did not inhibit uPAR·uPA interaction have fluorine atoms on the aromatic ring at R1, suggesting that highly polar groups may not be suitable for the binding site on uPAR. Interestingly, the compounds bind to uPAR with a Ki of 1–2 μM. Replacing the bromine atom at R1 with an iodine such as in 13 (IPR-1171) had no effect on the IC50 but gave a 2-fold reduction in binding affinity. Removal of the bromine group at the meta position and replacing the methyl group with an isopropoxy group in 12 (IPR-1175) or a tert-butyl group such as in 15 (IPR-1195) led to a 2–3-fold increase in the IC50 and a nearly 2-fold increase in Ki. A benzyloxy group in 16 (IPR-1186) increased the IC50 and Ki by 3-fold versus those of 1 (IPR-1110). Generally, replacing the hydrogen atoms at the meta position of the aromatic ring directly attached to the central pyrrolidone ring reduced activity substantially as evidenced by a 5-fold increase in IC50 and a 20-fold increase in Ki for 37. Larger groups at one of the meta positions were also generally undesirable as most compounds had pronounced increases in IC50 such as 36 and 41. For these compounds, it is interesting that 36 retained almost all of its binding affinity as evidenced by a Ki of 1.9 μM that was 2-fold higher than that of 1 (IPR-1110). Compound 41 had reduced affinity, but its Ki value was within 10 μM. Introduction of various substituents at the para position generally weakened the ability to disrupt the uPAR·uPA protein–protein interaction (e.g., 19 and 36), except for 44 (IPR-1607) and 45 (IPR-2260). Both of these compounds had IC50 values similar to that of 1 (IPR-1110). Interestingly, the compound showed a nearly 2-fold increase in its binding affinity for uPAR as evidenced by a Ki of 0.4 μM compared with a Ki of 0.9 μM for 1 (IPR-1110).
Despite the highly favorable cation−π interaction of the R2 group with Arg-53, the substituent has significant exposure to solvent. The addition of a methoxy group at the meta position of the aromatic ring of R2 (21 and 28) reduced the binding affinity and weakened the potency for inhibition of uPAR·uPA interaction. Replacing the fluorine on 4 (IPR-1201) with a chlorine on 7 (IPR-1178), however, does not have much impact on the binding affinity or inhibition of uPAR·uPA. Moving the fluorine from the meta to ortho position on 10 reduces the Ki and IC50 by 2-fold. It is interesting to note that a methoxy group at the meta position of 21 has a much more significant impact on both binding with a 10-fold reduction in Ki and inhibition of the protein–protein interaction as evidenced by a nearly 5-fold increase in IC50. Replacing the hydrogen atom with a fluorine atom of this compound to generate 28 improves the Ki and IC50 by 5-fold versus those of compound 21. In compound 24, we introduced a tert-butyl group at the para position of the aromatic ring at R2, which led to complete abrogation of the inhibition of uPAR·uPA interaction (IC50 not determined), although direct binding to uPAR was still detected but its level was reduced by more than an order of magnitude (Ki = 15.9 ± 2.8 μM). A nearly similar effect was observed for 33 (IPR-1157), and an even more dramatic effect when a methoxy group was introduced at the para position of R2 with no inhibition or binding detected within the 100 μM range.
Molecular Dynamics Simulations and Free Energy Calculations
To further explore the interaction between compounds and individual amino acids at the uPAR·uPA interface, we resorted to explicit-solvent molecular dynamics simulations and free energy calculations. We also performed free energy decomposition calculations to investigate the interaction of each amino acid with the compounds. First, we generated a model for the structure of 12 derivatives of 1 (IPR-1110) bound to uPAR using the binding mode of 12 (IPR-1175). These compounds were selected to ensure a uniform distribution across the range of binding affinities. Along with 3 (IPR-737) and 12 (IPR-1175), these complexes were subjected to 100 ns explicit-solvent molecular dynamics simulations. Snapshots were collected at regular intervals, and free energy calculations using the MM-GBSA approach were conducted for each of the compounds as shown in Figure 4a and Table S4. It is worth noting that end-point free energy calculations cannot accurately reproduce the absolute value of the free energy of binding. These calculations, however, have been shown in numerous studies, including ours, to accurately rank-order protein–compound complexes. The MM-GBSA free energy consists of five components, namely, the nonpolar and polar potential energies, the polar and nonpolar solvation energies, and the entropy (Table S4). The calculated MM-GBSA free energy correlated positively with the experimentally determined Ki values with a Pearson’s r of 0.8, a Spearman’s ρ of 0.41, and a Kendall’s τ of 0.29 (Figure 4a).
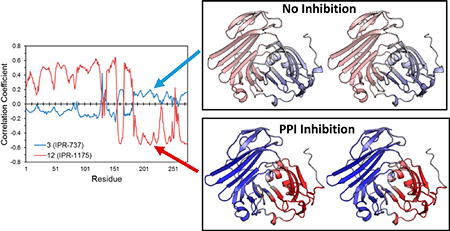
Figure 4.
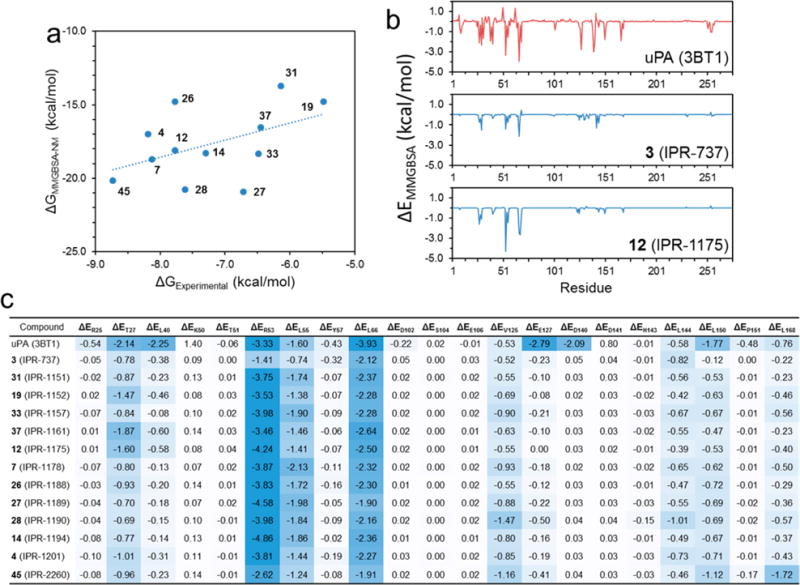
(a) Plot of the MM-GBSA free energy vs inhibition constant Ki that was measured using the fluorescence polarization assay. The correlation coefficients of all 12 selected derivatives are as follows: r = 0.48, ρ = 0.41, and τ = 0.29. (b) Decomposition of the free energy of binding for uPA, 12 (IPR-1175), and 3 (IPR-737). The decomposition energy consists of the interaction energy between the ligand and each residue on uPAR determined for a collection of snapshots that were obtained from molecular dynamics simulations. (c) Decomposition energies for a select number of hot-spot and non-hot-spot residues for a set of 1 (IPR-1110) derivatives.
To gain deeper insight into the interaction of the small molecules with individual residues on uPAR, a decomposition energy calculation was performed for each compound (Figure 4b). The decomposition energy consists of polar and nonpolar interaction potential energy, and the polar and nonpolar solvation energies. These calculations were also performed for the uPAR·uPA complex. On uPAR, mutations at 21 residues at the uPAR·uPA interface significantly impaired the interaction.18 Among these residues, only a subset is considered hot spots by the traditional definition (ΔΔG > 1 kcal mol−1). Additionally, not all hot spots are included in this list of residues. We examine the local interaction between uPA and compounds with uPAR at these residues (Figure 4c). As expected, uPA strongly engaged these residues. The change in free energy from the experimental alanine scan correlated with the per-residue decomposition (r = 0.38, ρ = 0.17, and τ = 0.01). When only hot-spot residues are considered, the correlation decreases to r = 0.18, ρ = 0.11, and τ = 0.10. At the 21 residues mentioned above, the correlation is r = 0.11, ρ = 0.28, and τ = 0.19. Comparison of the decomposition energies of 3 (IPR-737) and 12 (IPR-1175) provides insight into the residues that contribute to the ability of 12 to disrupt the protein–protein interaction. Compound 3 (IPR-737) decomposition interaction energies were much weaker than those observed for uPA. In contrast, 12 (IPR-1175) showed much more favorable interaction energies that were comparable to those of uPA, particularly to Thr-27, Leu-55, Leu-66, and Arg-53. It is interesting to note that among all the 21 residues considered, the experimental binding affinity of compounds correlated most strongly with their interaction energies with Leu-150 and Leu-168 with Pearson correlation coefficients of 0.57 and 0.52, respectively (Table S5). In addition, the experimental kinetic rate constants were also compared to the residue decomposition energies of these derivatives (Table S6).
Arg-53 Enhances Binding through Cooperativity
Arg-53 is not considered a hot spot, because alanine scanning studies revealed that it contributes only 0.7 kcal mol−1,18 but it appears to play a critical role in the binding of small molecules as we have demonstrated previously for 2. Considering the cation−π interaction of 12 with Arg-53, we hypothesized that the residue also plays an essential role in the activity of the compound. We explored the possibility that Arg-53 may be enhancing interaction of the compound with hot spots through cooperative binding. MM-GBSA free energy calculations for double mutants that include Arg-53 were also conducted along with separate MM-GBSA calculations for the individual residues. We ran explicit-solvent molecular dynamics simulations and free energy calculations for both the single- and double-mutation states of the uPAR·uPA (Figure 5a) and uPAR·12 (Figure 5b) complexes (Table S7).
Figure 5.
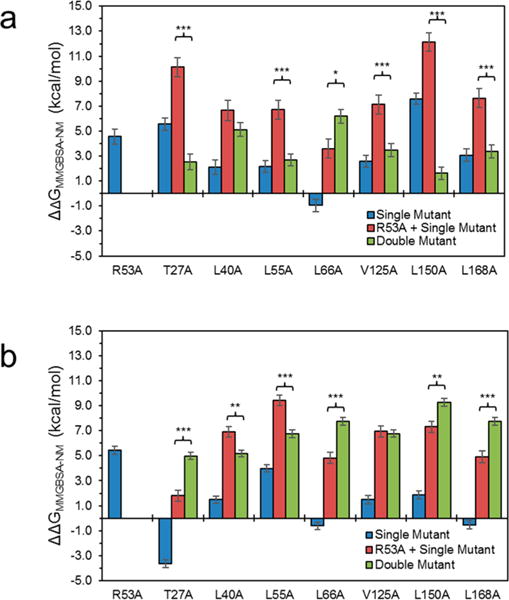
Cooperativity of singly and doubly mutated (a) uPAR·uPA and (b) uPAR·12 complexes. The difference and standard error between the free energy of each mutant with the respective wild-type complex (blue), the sum of the ΔΔG of the Arg-53-Ala mutant and the single mutant (red), and the double mutant (green). Statistical significance (independent two-sample t test): *p < 0.01, **p < 0.001, and ***p < 0.0001.
For the uPAR·uPA complex, mutation of all residues except for one (Leu-66) resulted in a penalty in the MM-GBSA free energy consistent with experimental alanine scanning data for these residues (Figure 5a). The sum of the MM-GBSA free energy change as a result of mutation of Arg-53 and each of the amino acids was nearly always unfavorable as shown in Figure 5a. However, it is interesting that when both amino acids (Arg-53 and one of the amino acids in Figure 5) were simultaneously mutated, the resulting change in the MM-GBSA free energy was nearly always different than the sum of the individual mutations (Figure 5a). For example, the sum of the individual mutations of Leu-150 and Arg-53 was 12 kcal mol−1, yet the Leu-150-Ala/Arg-153-Ala double mutation resulted in an only 2 kcal mol−1 change in the free energy of binding. Another interesting example is that of Leu-66, for which the sum of individual mutations of this residue and Arg-153 to alanine was 4 kcal mol−1, yet the double mutant was much less favorable at nearly 7 kcal mol−1. These results strongly suggest cooperativity between Arg-53 and hot-spot residues toward the binding of uPA to uPAR. For the uPAR·uPA complex, it appears that Arg-53 significantly reduces the size of the contributions of hot spots to the binding of uPA to uPAR.
We explored whether Arg-53 and other residues bind to 12 in a cooperative manner. In the uPAR·12 complex, mutation of Thr-27, Leu-66, and Leu-168 resulted in more favorable binding, while mutation of the other residues resulted in a loss of affinity of 12 for uPAR (Figure 5b). Mutation of Arg-53 to alanine, as expected, resulted in the most pronounced effect on the binding of 12 to uPAR. The sum of the contributions of Arg-53 and other residues was unfavorable in nearly every case. Interestingly, double mutants also consistently exhibited an unfavorable effect that was different from the effect of the sum of each mutation. For example, in the case of Thr-27, mutation of this residue along with Arg-53 resulted in a 5 kcal mol−1 increase in the free energy of binding compared with only 2 kcal mol−1 for the sum of the free energy change of individual mutations. A similar increase was found for Leu-66 and Leu-168. For the other residues, the double mutant showed a change in the free energy less pronounced than the sum of the free energy change for the individual mutations. For example, mutation of Leu-55 and Arg-53 to alanine resulted in a combined free energy change of 9 kcal mol−1, while the Leu-55-Ala/Arg-53-Ala double mutation resulted in a 7 kcal mol−1 less favorable interaction. Other than for Val-125, Arg-53 exhibited cooperativity with each of the residues that were considered. Interestingly, double mutants with Arg-53 and another residue all resulted in unfavorable energy for both uPAR·uPA and uPAR·12 complexes. Interestingly, for two of the hot spots, Leu-66 and Leu-150, Arg-53 enhances the interaction of 12 with uPAR by a combined 5 kcal mol−1, while Arg-53 weakens the interaction of 12 with hot-spot Leu-55 by 2 kcal mol−1.
Small-Molecule uPAR·uPA Inhibitors Alter uPAR Dynamics
The cooperativity among residues prompted us to explore the effect of the two classes of compounds on the dynamics of uPAR. We investigated the correlation of the motion of 3, 12, and several other compounds to each residue on uPAR (Figure 6a). Visual inspection of the color-coded map in Figure 6a reveals a dramatic difference in the correlation profile of compounds. For example, pyrrolinone 12 and derivatives generally shows strong correlated motion with residues 1–60 on uPAR, while pyrazole 3 shows no correlation. To compare the effect of 3 and 12 on the dynamics of uPAR with uPA, we generated a cross-correlated map of the motion of uPAR in the presence of uPA (Figure 6b). Interestingly, the effects of 12 on the dynamics of uPAR were remarkably similar to those of uPA on uPAR (Figure 6c,e), while 3 (Figure 6d) showed little correlation. These results suggest small molecules that replicate the dynamics of the native ligand in a protein–protein interaction are more likely to disrupt the interaction.
Figure 6.
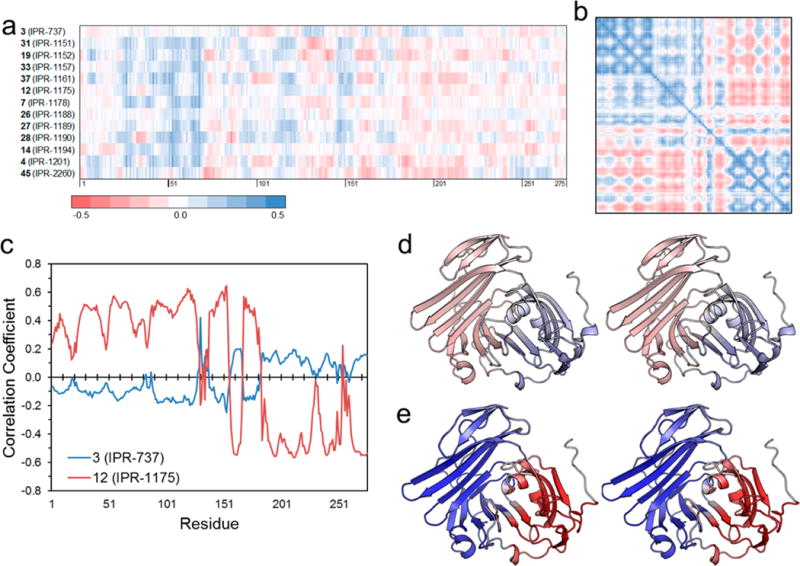
(a) Dynamic cross-correlation matrix (DCCM) cross section of 3 and select derivatives of 1 with uPAR. (b) DCCM of uPAR in the uPAR·uPA complex, colored using the same scheme used for panel a. (c) Pearson’s correlation between the cross-correlation of uPAR·3 and uPAR·12 complexes and the cross-correlation of individual residues of uPAR in the uPAR·uPA complex. Stereoviews of three-dimensional structures of uPAR in the (d) uPAR·3 and (e) uPAR·12 complexes, colored on the basis of the correlation coefficient in panel c from positive correlation (blue) to no correlation (white) to negative correlation (red).
DISCUSSION
Protein–protein interactions range from transient to tight. Transient interactions are weaker micromolar-affinity interactions that typically involve simpler binding epitopes such as a linear peptide bound to a cavity.61,62 Tight interactions occur between proteins with single- or double-digit nanomolar-range binding constants. Tight interactions involve larger interfaces (1000–3000 Å2) with binding epitopes consisting of a single secondary structure such as an α helix, or a more complex binding epitope that spans multiple secondary structures.1,2,63 While there are numerous examples of small molecules that disrupt weak protein–protein interactions, only a few tight interactions have been successfully inhibited with small molecules.1 Despite the smaller footprint of small molecules compared with the large protein–protein interface of tight interactions with complex epitopes, it has been suggested that small molecules disrupt these interactions by engaging hot spots at the interface.9,26,27 Hot spots can be located on either the ligand or the receptor binding cleft.5,8,64
Here, we explore small-molecule antagonists of the uPAR·uPA interaction, a tight and stable protein–protein interaction. The uPAR·uPA interface has a well-defined binding cavity that contains multiple hot-spot residues that contribute ≥1 kcal mol−1 to the binding affinity. These hot spots are located on both uPAR and its ligand, uPA. Small molecules that (i) mimic the position of hot spots on the ligand (uPA) and (ii) engage hot spots on the receptor (uPAR) are expected to disrupt this interaction. Previously, we had identified two classes of compounds that disrupt the protein–protein interaction. The first compound, 2, was identified by docking a commercial library to an ensemble of structures of uPAR collected from molecular dynamics simulations.24 The compound showed binding affinity in the submicromolar range and an IC50 for disruption of uPAR·uPA interaction in the single-digit micro-molar range. Interestingly, the predicted mode of binding of 2 to a structure of uPAR sampled from molecular dynamics simulation revealed a salt bridge between the benzoic acid moiety of 2 and Arg-53.24 A recent crystal structure of an analogue of 2 bound to uPAR confirmed our predicted binding mode of the compound and supported the existence of a salt-bridge interaction with Arg-53.20 Arg-53 is buried in the crystal structure of the uPAR·uPA complex. The snapshot of 2 bound to uPAR that emerged from our molecular dynamics simulations revealed an exposed Arg-53. Interestingly, the apo structure of uPAR, which was determined after we discovered 2, reveals an exposed Arg-53.65 This supported our rationale for using molecular dynamics simulations to sample conformations that differ from the structure of uPAR in its complex with uPA. The second class of uPAR·uPA antagonists that we discovered were pyrrolinone compounds.23 Interestingly, these compounds were identified in a ligand-based approach using the structure of pyrazole, pyrrolodinone, piperidinone, and butan-amine compounds that were identified by structure-based virtual screening using the structure of uPAR from the uPAR· uPA complex.21 The carbon that bears the hydroxyl group of the pyrrolinone compounds considered in this work may be prone to attack by nucleophiles. However, the OH group makes a poor leaving group, making covalent adduct formation between the compound and protein unlikely. We confirmed this with mass spectrometry. Further evidence that argues against the formation of a covalent adduct is the fact that replacement of OH with a methoxy or isopropoxy, each of which is a much better leaving group than OH is, resulted in lower-affinity compounds. Also, the lack of exposed nucleophiles such as cysteine residues on uPAR makes it highly unlikely that 1 and derivatives covalently attach to uPAR. In addition, we show that the compound is stable in buffer, methanol, and uPAR.
We synthesize nearly 50 derivatives of pyrollinone 1 to explore the uPAR·uPA protein–protein interaction. We determined the crystal structure of a derivative of 1, namely 12 (IPR-1175), and a pyrazole compound 3 (IPR-737) that binds to uPAR but does not disrupt the uPAR·uPA interface. The structures reveal that the pyrazole compounds bind to a site on uPAR that is occupied by a uPA side chain located on the β strands of the β turn of the GFD domain of uPA. The 2-pyrrolinone compound, however, adopts a different binding mode. In addition, the compound comes in direct contact with several hot spots. This explains why both compounds bind to uPAR but only the 2-pyrrolone inhibits uPAR·uPA interaction within the 100 μM concentration range considered in this work. It was interesting that two of the aromatic rings of 12 engaged Arg-53, directly forming highly favorable cation−π interactions. Cation−π interactions are electrostatic interactions that are considered to be among the strongest noncovalent interactions in protein structures,66 while mutation of this residue seems to be a common feature of small molecules that we have found to inhibit the uPAR·uPA protein–protein interaction. Previously, we found that a salt-bridge interaction between a benzoic acid moiety on 2 (IPR-803) and the guanidinium ion of Arg-53 was the critical factor that led to inhibition of uPAR·uPA interaction.24 Removal of the benzoic acid led to loss of inhibition of the protein–protein interaction. The use of hot spots on uPAR and uPA in future studies is expected to lead to derivatives that are substantially more potent than the parent compound. This could be achieved by enhancing the affinity of compounds to individual hot-spot residues or by mimicking critical hot spots located on uPA, such as Phe-25 or Trp-30.
While Arg-53 is not considered a hot spot, its interaction with small molecules appears to be essential for successful inhibition of the uPAR·uPA interaction by small molecules. Interestingly, molecular dynamics simulation-based free energy decomposition reveals that Arg-53 makes the strongest interaction with uPA compared with other uPA residues that come in contact with uPAR. Despite the strong interaction, alanine scanning studies show that Arg-53 only moderately contributes to the binding affinity (0.7 kcal mol−111). This can be explained by the fact that decomposition energies do not take into consideration the configurational entropy contributions to the binding, in contrast to the alanine scanning that used surface plasmon resonance, which includes both enthalpy and entropy. Considering the position of Arg-53 within the core structure of uPAR, it is possible that mutation of Arg-53 may have destabilized uPAR and hence enhanced entropy contributions to the binding affinity, despite the significant loss of enthalpy. Small-molecule inhibitors are expected to disrupt only the interaction of residues and not their contributions to the entropy of binding. This suggests that residues that are not considered hot spots should not be ignored in the design of small-molecule protein–protein interaction inhibitors.
The discovery of hot spots at protein–protein interfaces was a significant breakthrough in the field of protein–protein interaction drug discovery. The primary tools to identify hot spots is to systematically mutate each residue to alanine and measure the effect on the binding thermodynamics and kinetics. This process is known as alanine scanning. One limitation of these alanine scanning experiments is that they do not consider the effects of neighboring residues, which may enhance or weaken the contributions of individual residues.67,68 How to take advantage of hot spots for small-molecule drug discovery is still unclear. To gain deeper insight into the role of Arg-53 in the binding of the small-molecule inhibitors, we explored whether any cooperativity between the residue and other residues that come in contact with 1 exists. Interestingly, we found that the independent mutation of Arg-53 and other hot spots resulted in changes in the predicted binding affinity that were different from those measured when both Arg-53 and hot spots were mutated. For uPA, mutation of hot spots resulted in a less favorable MM-GBSA free energy for six of the seven mutations, yet mutations of Arg-53 and each of these residues resulted in dramatically different free energy changes. For example, the mutation of Arg-53 and Leu-150 led to a combined 13 kcal mol−1 MM-GBSA free energy change, yet the combined mutation of these two residues resulted in a change of only 2 kcal mol−1, a dramatic difference. For compound 12, mutation of Arg-53 and Leu-150 led to MM-GBSA free energy changes of 6 and 2 kcal mol−1, respectively. This is in contrast to the dual mutation of both residues that resulted in an unfavorable change of >9 kcal mol−1.
In summary, our work provides insight into the forces that drive small molecules to disrupt tight and stable protein–protein interactions. We show that mere binding is not a sufficient requirement for disrupting the uPAR·uPA protein–protein interaction. Even binding to hot-spot residues is not a guarantee of disruption of the protein–protein interaction. We find that residues that are not considered hot spots in alanine scanning experiments such as Arg-53 in uPAR can provide a strong anchor for small molecules to enhance their binding affinity. These anchors can also strengthen binding of small molecules to other hot spots through cooperativity. Also, salt-bridge and π–cation interactions appear to be critical for disruption of the uPAR·uPA interaction. A previously discovered uPAR antagonist, IPR-803, formed a salt-bridge interaction that was essential for inhibition of uPAR·uPA interaction,24 and compounds reported in this work bind to Arg-53 through π–cation interactions. These types of interactions are likely to be favorable at protein–protein interfaces considering the shallow nature of the pockets and the high degree of solvent exposure. These interactions may be a straightforward strategy for enhancing the inhibition potency of compounds that bind to the target. Another important finding of this work is the importance of molecular dynamics in the inhibition of tight PPIs by small molecules. We discovered that small molecules that inhibit uPAR·uPA interaction alter the dynamics of uPAR in a manner similar to that of uPA. This suggests that drug discovery efforts targeting protein–protein interactions should also consider the effect of compounds on the dynamics of the receptor as opposed to merely focusing on the interaction energy. Small molecules that bind tightly to hot spots and also modulate the dynamics of the receptor in a manner similar to that of the native tight ligand are expected to lead to more effective inhibitors. These findings can help guide the design of derivatives of IPR-1110 through modification of the core structure of the compound or by the introduction of new substituents.
Supplementary Material
Acknowledgments
Funding
The research was supported by the National Institutes of Health (CA135380) (S.O.M.), American Cancer Society Research Scholar Grant RSG-12-092-01-CDD (S.O.M.), and the 100 Voices of Hope (S.O.M.). Computer time on the Big Red II and Karst supercomputer at Indiana University (S.O.M.) is supported in part by Lilly Endowment, Inc., through its support for the Indiana University Pervasive Technology Institute, and in part by the Indiana METACyt Initiative.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.6b01039.
Chemical structures of compounds that emerged from virtual screening against the uPAR crystal structure and multiple conformers, hot spots on uPAR of the uPAR·uPA interaction, adapted from ref 18 (Table S1), X-ray data collection and refinement statistics of the suPARcc·12 (IPR-1175) complex (Table S2), X-ray data collection and refinement statistics of the suPARcc·3 (IPR-737) complex (Table S3), calculated free energies ± the standard error of 3 and select derivatives of 1, showing the individual free energy terms (Table S4), correlation coefficients of the individual free energy terms and correlation to select residues of uPAR to illustrate the relationship between the decomposition energy and inhibition constant Ki for select derivatives of 1 from Figure 4c (Table S5), correlation coefficients to illustrate the relationship between the decomposition energy of select residues from Figure 4c and kinetic rate constants for select derivatives of 1 (Table S6), and calculated free energies ± the standard error of single and double mutants in the uPAR·uPA and uPAR·12 complexes (Table S7) (PDF)
ORCID
Mingdong Huang: 0000-0002-1377-6786
Samy O. Meroueh: 0000-0002-3948-7499
Author Contributions
D.L. and D.X. contributed equally to this work.
Notes
The authors declare no competing financial interest.
References
- 1.Arkin MR, Tang Y, Wells JA. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing Toward the Reality. Chem Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watkins AM, Arora PS. Structure-based inhibition of protein-protein interactions. Eur J Med Chem. 2015;94:480–488. doi: 10.1016/j.ejmech.2014.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 4.Wilson CG, Arkin MR. Probing structural adaptivity at PPI interfaces with small molecules. Drug Discovery Today: Technol. 2013;10:e501–508. doi: 10.1016/j.ddtec.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 5.Cukuroglu E, Engin HB, Gursoy A, Keskin O. Hot spots in protein-protein interfaces: towards drug discovery. Prog Biophys Mol Biol. 2014;116:165–173. doi: 10.1016/j.pbiomolbio.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci U S A. 2002;99:14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clackson T, Wells JA. A Hot Spot of Binding Energy in a Hormone-Receptor Interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 8.Keskin O, Ma B, Nussinov R. Hot regions in protein–protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol. 2005;345:1281–1294. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 9.Bogan AA, Thorn KS. Anatomy of Hot Spots in Protein Interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 10.Magdolen V, Rettenberger P, Koppitz M, Goretzkt L, Kessler H, Weidle UH, Konig B, Graeff H, Schmitt M, Wilhelm O. Systematic Mutational Analysis of the Receptor-Binding Region of the Human Urokinase-Type Plasminogen Activator. Eur J Biochem. 1996;237:743–751. doi: 10.1111/j.1432-1033.1996.0743p.x. [DOI] [PubMed] [Google Scholar]
- 11.Gardsvoll H, Ploug M. Mapping of the vitronectin-binding site on the urokinase receptor: involvement of a coherent receptor interface consisting of residues from both domain I and the flanking interdomain linker region. J Biol Chem. 2007;282:13561–13572. doi: 10.1074/jbc.M610184200. [DOI] [PubMed] [Google Scholar]
- 12.Madsen CD, Ferraris GM, Andolfo A, Cunningham O, Sidenius N. uPAR-induced cell adhesion and migration: vitronectin provides the key. J Cell Biol. 2007;177:927–939. doi: 10.1083/jcb.200612058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huai Q, Zhou A, Lin L, Mazar AP, Parry GC, Callahan J, Shaw DE, Furie B, Furie BC, Huang M. Crystal structures of two human vitronectin, urokinase and urokinase receptor complexes. Nat Struct Mol Biol. 2008;15:422–423. doi: 10.1038/nsmb.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin L, Gardsvoll H, Huai Q, Huang M, Ploug M. Structure-based engineering of species selectivity in the interaction between urokinase and its receptor: implication for preclinical cancer therapy. J Biol Chem. 2010;285:10982–10992. doi: 10.1074/jbc.M109.093492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardsvoll H, Dano K, Ploug M. Mapping part of the functional epitope for ligand binding on the receptor for urokinase-type plasminogen activator by site-directed mutagenesis. J Biol Chem. 1999;274:37995–38003. doi: 10.1074/jbc.274.53.37995. [DOI] [PubMed] [Google Scholar]
- 16.Huai Q, Mazar AP, Kuo A, Parry GC, Shaw DE, Callahan J, Li Y, Yuan C, Bian C, Chen L. Structure of human urokinase plasminogen activator in complex with its receptor. Science. 2006;311:656–659. doi: 10.1126/science.1121143. [DOI] [PubMed] [Google Scholar]
- 17.Llinas P, Helénè Le Du M, Gardsvoll H, Dano K, Ploug M, Gilquin B, Stura EA, Menez A. Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J. 2005;24:1655–1663. doi: 10.1038/sj.emboj.7600635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardsvoll H, Gilquin B, Le Du MH, Menez A, Jorgensen TJ, Ploug M. Characterization of the functional epitope on the urokinase receptor. Complete alanine scanning mutagenesis supplemented by chemical cross-linking. J Biol Chem. 2006;281:19260–19272. doi: 10.1074/jbc.M513583200. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z, Lin L, Huai Q, Huang M. Challenges for drug discovery-a case study of urokinase receptor inhibition. Comb Chem High Throughput Screening. 2009;12:961. doi: 10.2174/138620709789824727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mani T, Wang F, Knabe WE, Sinn AL, Khanna M, Jo I, Sandusky GE, Sledge GW, Jr, Jones DR, Khanna R, Pollok KE, Meroueh SO. Small-molecule inhibition of the uPAR.uPA interaction: synthesis, biochemical, cellular, in vivo pharmacokinetics and efficacy studies in breast cancer metastasis. Bioorg Med Chem. 2013;21:2145–2155. doi: 10.1016/j.bmc.2012.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mani T, Liu D, Zhou D, Li L, Knabe WE, Wang F, Oh K, Meroueh SO. Probing binding and cellular activity of pyrrolidinone and piperidinone small molecules targeting the urokinase receptor. ChemMedChem. 2013;8:1963–1977. doi: 10.1002/cmdc.201300340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang F, Eric Knabe W, Li L, Jo I, Mani T, Roehm H, Oh K, Li J, Khanna M, Meroueh SO. Design, synthesis, biochemical studies, cellular characterization, and structure-based computational studies of small molecules targeting the urokinase receptor. Bioorg Med Chem. 2012;20:4760–4773. doi: 10.1016/j.bmc.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu D, Zhou D, Wang B, Knabe WE, Meroueh SO. A New Class of Orthosteric uPAR.uPA Small-Molecule Antagonists Are Allosteric Inhibitors of the uPAR.Vitronectin Interaction. ACS Chem Biol. 2015;10:1521–1534. doi: 10.1021/cb500832q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khanna M, Wang F, Jo I, Knabe WE, Wilson SM, Li L, Bum-Erdene K, Li J, Sledge GW, Khanna R, Meroueh SO. Targeting multiple conformations leads to small molecule inhibitors of the uPAR.uPA protein-protein interaction that block cancer cell invasion. ACS Chem Biol. 2011;6:1232–1243. doi: 10.1021/cb200180m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rullo AF, Fitzgerald KJ, Muthusamy V, Liu M, Yuan C, Huang M, Kim M, Cho AE, Spiegel DA. Re-engineering the Immune Response to Metastatic Cancer: Antibody-Recruiting Small Molecules Targeting the Urokinase Receptor. Angew Chem, Int Ed. 2016;55:3642. doi: 10.1002/anie.201510866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci U S A. 2002;99:14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 28.Thanos CD, DeLano WL, Wells JA. Hot-Spot Mimicry of a Cytokine Receptor by a Small Molecule. Proc Natl Acad Sci U S A. 2006;103:15422–15427. doi: 10.1073/pnas.0607058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hyde J, Braisted AC, Randal M, Arkin MR. Discovery and characterization of cooperative ligand binding in the adaptive region of interleukin-2. Biochemistry. 2003;42:6475–6483. doi: 10.1021/bi034138g. [DOI] [PubMed] [Google Scholar]
- 30.Keskin Z, Gursoy A, Ma B, Nussinov R. Principles of protein-protein interactions: What are the preferred ways for proteins to interact? Chem Rev. 2008;108:1225–1244. doi: 10.1021/cr040409x. [DOI] [PubMed] [Google Scholar]
- 31.Jacobsen B, Gårdsvoll H, Juhl Funch G, Ostergaard S, Barkholt V, Ploug M. One-step affinity purification of recombinant urokinase-type plasminogen activator receptor using a synthetic peptide developed by combinatorial chemistry. Protein Expression Purif. 2007;52:286–296. doi: 10.1016/j.pep.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 32.Xu X, Gardsvoll H, Yuan C, Lin L, Ploug M, Huang M. Crystal structure of the urokinase receptor in a ligand-free form. J Mol Biol. 2012;416:629–641. doi: 10.1016/j.jmb.2011.12.058. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.Vagin A, Teplyakov A. MOLREP: an Automated Program for Molecular Replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 35.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins: Struct, Funct, Genet. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 37.Sali A, Blundell TL. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 38.Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 39.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2 Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 40.Case DA, Berryman JT, Betz RM, Cerutti DS, Cheatham TE, III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Luchko T, Luo R, Madej B, Merz KM, Monard G, Needham P, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Salomon-Ferrer R, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, York DM, Kollman PA. AMBER 2015. University of California; San Francisco: 2015. [Google Scholar]
- 41.Jakalian A, Jack DB, Bayly CI. Fast, Efficient Generation of High-quality Atomic Charges. AM1-BCC Model: II Parameterization and Validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and Testing of a General Amber Force Field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Wang W, Kollman PA, Case DA. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J Mol Graphics Modell. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 44.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 45.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ryckaert JP, Ciccotti G, Berendsen JJC. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 47.Darden T, York D, Pedersen L. Particle Mesh Ewald: An N × log(N) Method for Ewald Sums in Large Systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 48.Roe DR, Cheatham TE., 3rd PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 49.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
- 50.Miller BR, 3rd, McGee TD, Jr, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J Chem Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 51.Onufriev A, Bashford D, Case DA. Exploring Protein Native States and Large-scale Conformational Changes with a Modified Generalized Born Model. Proteins: Struct, Funct, Genet. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 52.Feig M, Onufriev A, Lee MS, Im W, Case DA, Brooks CL., 3rd Performance Comparison of Generalized Born and Poisson Methods in the Calculation of Electrostatic Solvation Energies for Protein Structures. J Comput Chem. 2004;25:265–284. doi: 10.1002/jcc.10378. [DOI] [PubMed] [Google Scholar]
- 53.Brooks B, Karplus M. Harmonic Dynamics of Proteins: Normal Modes and Fluctuations in Bovine Pancreatic Trypsin Inhibitor. Proc Natl Acad Sci U S A. 1983;80:6571–6575. doi: 10.1073/pnas.80.21.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gohlke H, Kiel C, Case DA. Insights into Protein–Protein Binding by Binding Free Energy Calculation and Free Energy Decomposition for the Ras–Raf and Ras–RalGDS Complexes. J Mol Biol. 2003;330:891–913. doi: 10.1016/s0022-2836(03)00610-7. [DOI] [PubMed] [Google Scholar]
- 55.Onufriev A, Bashford D, Case DA. Exploring Protein Native States and Large-scale Conformational Changes with a Modified Generalized Born Model. Proteins: Struct, Funct, Genet. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 56.De Marco A, De Candia M, Carotti A, Cellamare S, De Candia E, Altomare C. Lipophilicity-related inhibition of blood platelet aggregation by nipecotic acid anilides. Eur J Pharm Sci. 2004;22:153–164. doi: 10.1016/j.ejps.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 57.Milagre CD, Milagre HM, Moran PJ, Rodrigues JA. Chemoenzymatic synthesis of alpha-hydroxy-beta-methyl-gamma-hydroxy esters: role of the keto-enol equilibrium to control the stereoselective hydrogenation in a key step. J Org Chem. 2010;75:1410–1418. doi: 10.1021/jo902227f. [DOI] [PubMed] [Google Scholar]
- 58.Richter A, Rose R, Hedberg C, Waldmann H, Ottmann C. An optimized small-molecule stabiliser of the 14–3-3-PMA2 protein-protein interaction. Chem - Eur J. 2012;18:6520–6527. doi: 10.1002/chem.201103761. [DOI] [PubMed] [Google Scholar]
- 59.Mahadevi AS, Sastry GN. Cation-pi interaction: its role and relevance in chemistry, biology, and material science. Chem Rev. 2013;113:2100–2138. doi: 10.1021/cr300222d. [DOI] [PubMed] [Google Scholar]
- 60.Mecozzi S, West AP, Dougherty DA. Cation-pi interactions in aromatics of biological and medicinal interest: electrostatic potential surfaces as a useful qualitative guide. Proc Natl Acad Sci U S A. 1996;93:10566–10571. doi: 10.1073/pnas.93.20.10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nooren IMA, Thornton JM. Structural characterisation and functional significance of transient protein-protein interactions. J Mol Biol. 2003;325:991–1018. doi: 10.1016/s0022-2836(02)01281-0. [DOI] [PubMed] [Google Scholar]
- 62.Acuner Ozbabacan SE, Engin HB, Gursoy A, Keskin O. Transient protein–protein interactions. Protein Eng, Des Sel. 2011;24:635–648. doi: 10.1093/protein/gzr025. [DOI] [PubMed] [Google Scholar]
- 63.Pelay-Gimeno M, Glas A, Koch O, Grossmann TN. Structure-Based Design of Inhibitors of Protein–Protein Interactions: Mimicking Peptide Binding Epitopes. Angew Chem, Int Ed. 2015;54:8896–8927. doi: 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cukuroglu E, Gursoy A, Keskin O. HotRegion: a database of predicted hot spot clusters. Nucleic Acids Res. 2012;40:D829–833. doi: 10.1093/nar/gkr929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu X, Gårdsvoll H, Yuan C, Lin L, Ploug M, Huang M. Crystal structure of the urokinase receptor in a ligand-free form. J Mol Biol. 2012;416:629–641. doi: 10.1016/j.jmb.2011.12.058. [DOI] [PubMed] [Google Scholar]
- 66.Dougherty DA. The cation− π interaction. Acc Chem Res. 2013;46:885–893. doi: 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vaughan CK, Buckle AM, Fersht AR. Structural response to mutation at a protein-protein interface. J Mol Biol. 1999;286:1487–1506. doi: 10.1006/jmbi.1998.2559. [DOI] [PubMed] [Google Scholar]
- 68.Schreiber G, Fersht AR. Energetics of protein-protein interactions: analysis of the barnase-barstar interface by single mutations and double mutant cycles. J Mol Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.