

Abstract
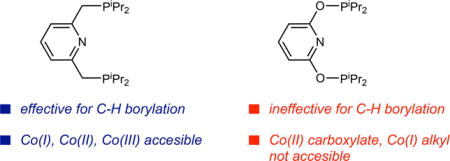
The activation of readily prepared, air-stable cobalt (II) bis(carboxylate) pre-catalysts for the functionalization of C(sp2)–H bonds has been systematically studied. With the pyridine bis(phosphine) chelate, iPrPNP, treatment of 1-(O2CtBu)2 with either B2Pin2 or HBPin generated cobalt boryl products. With the former, reduction to (iPrPNP)CoIBPin was observed while with the latter, oxidation to the cobalt(III) dihydride boryl, trans-(iPrPNP)Co(H)2BPin occurred. The catalytically inactive cobalt complex, Co[PinB(O2CtBu)2]2, accompanied formation of the cobalt-boryl products in both cases. These results demonstrate that the pre-catalyst activation from cobalt(II) bis(carboxylates), although effective and utilizes an air-stable precursor, is less efficient than activation of cobalt(I) alkyl or cobalt(III) dihydride boryl complexes, which are quantitatively converted to the catalytically relevant cobalt(I) boryl. Related cobalt(III) dihydride silyl and cobalt(I) silyl complexes were also synthesized from treatment of trans-(iPrPNP)Co(H)2BPin and (iPrPNP)CoPh with HSi(OEt)3, respectively. No catalytic silylation of arenes was observed with either complex likely due to the kinetic preference for reversible C–H reductive elimination rather than product- forming C–Si bond formation from cobalt(III). Syntheses of the cobalt(II) bis(carboxylate) and cobalt(I) alkyl of iPrPONOP, a pincer where the methylene spacers have been replaced by oxygen atoms, were unsuccessful due to deleterious P–O bond cleavage of the pincer. Despite their structural similarity, the rich catalytic chemistry of iPrPNP was not translated to iPrPONOP due to the inability to access stable cobalt precursors as a result of ligand decomposition via P–O bond cleavage.
Keywords: C–H activation, borylation, cobalt, PNP ligand, PONOP ligand
Graphical abstract
1. Introduction
The selective functionalization of carbon-hydrogen bonds with soluble transition metal complexes is of long-standing interest in organic chemistry and catalysis.[1] Among the many methods now developed, metal-catalyzed borylation of arene C(sp2)–H bonds is among the most powerful owing to orthogonal selectivity to traditional electrophilic aromatic substitution and the synthetic versatility of the resulting arylboronate products.[2,3] Precious metal catalysts, particularly those based on iridium, are the most commonly used.[2] Increased emphasis on sustainable catalytic methods has generated considerable interest in developing C–H borylation catalysts with earth abundant transition metals such as iron[4] and cobalt.[5] Our laboratory has evaluated a variety of cobalt complexes bearing tridentate ligands[5] and discovered that electron-rich bis(phosphino)pyridine (iPrPNP) chelates[6] are the most effective (Scheme 1).[5a,7]
Scheme 1.
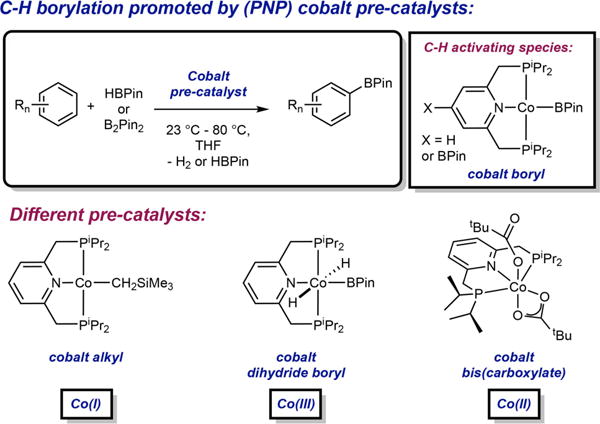
(PNP)cobalt pre-catalysts employed for C–H borylation of arenes. The parent pincer ligand (X=H) is shown for the different catalysts for simplicity.
Detailed mechanistic studies on the [(iPrPNP)Co] family of compounds support a pathway involving the borylated cobalt (I) boryl, 4-BPin-(iPrPNP)CoBPin, as responsible for activation of the C(sp2)–H bond of substituted pyridines and arenes with B2Pin2 (Pin=pinacolate) as the boron source. Modification of the 4-position of the pincer preceded borylation of the substrate.[8] A cobalt(I)-boryl compound was also identified as the C(sp2)–H activating species during the borylation of 5-membered heteroarenes with HBPin but competing borylation of the cobalt pincer complex was not observed.[9] With these heteroarenes and boron source, the catalyst resting state is the cobalt(III) complex, trans-(iPrPNP)Co(H)2BPin.
An important but often overlooked feature of these compounds is that C–H borylation catalysis can be initiated by (iPrPNP)cobalt complexes in different oxidation states. Cobalt(I) alkyl,[5a] cobalt(III) dihydride boryl,[7,8] as well as cobalt(II) bis(carboxylates)[7,10] have all been identified as effective pre-catalysts for this transformation (Scheme 1). Successful catalysis initiated from cobalt precursors across three oxidation states highlights the electronic versatility of first row metals that can be leveraged for distinct reactivity. The air-stability of (iPrPNP)Co(O2CR)2 compounds and their ease of synthesis from commercially available Co(O2CR)2 and the free chelate, without the need for pyrophoric alkyl lithium or highly reactive and reducing main group hydride reagents, makes them attractive for applications in synthesis.
The activation mode of [(iPrPNP)Co(I)] alkyl pre-catalysts is well understood as stoichiometric experiments have established quantitative formation of (iPrPNP)CoBPin (1-BPin) upon treatment with either HBPin or B2Pin2. A sequence involving oxidative addition of a B–H or B–B followed by H–H or H–B reductive elimination, possibly involving phosphine dissociation,[11] generates the intermediate responsible for activation of the C(sp2)–H bond of the substrate (Schemes 2a and 2b).[5a,8,9]
Scheme 2.

Different activation modes of pincer cobalt borylation catalysts.
Despite a mature understanding of how (iPrPNP)cobalt(I) alkyl and cobalt(III) dihydride boryl pre-catalysts enter the C(sp2)–H borylation cycle, little is known about how the related cobalt(II) bis(carboxylates) undergo activation with HBPin and B2Pin2 (Scheme 2c). It is also non-obvious if the same cobalt(I) active species form and if so, how the reduction from Co(II) to Co(I) occurs.[12] Here we describe systematic studies to understand the activation of (iPrPNP)Co(O2CtBu)2 with both B2Pin2 and HBPin. Attempted synthesis of a related cobalt(II) bis(carboxylate) where the methylene subunits of the pincer were replaced with oxygen atoms, (iPrPONOP)Co(O2CtBu)2, were unsuccessful and resulted in P–O bond cleavage of the ligand and extrusion of the carboxylate groups on the cobalt to yield a new four-coordinate modified ligand complex.
2. Results and Discussion
2.1. Reaction of 1-(O2CtBu)2 with B2Pin2 and HBPin
Stoichiometric studies were conducted with the 4-methyl substituted variant of the cobalt(II) pre-catalyst, (4-Me-iPrPNP) Co(O2CtBu)2 (1-O2CtBu)2,[5] because of its established ortho to fluorine selectivity in the C(sp2)–H borylation of fluoroarenes and its resistance to deactivation through catalyst borylation.[8] Addition of ten equiv. of B2Pin2 to a THF-[d8] solution of (1-O2CtBu)2 at 23°C produced no reaction as judged by 1H NMR spectroscopy. Heating the mixture to 80°C for 1 hour resulted in formation of the cobalt(I) boryl dinitrogen complex, (4-Me-iPrPNP)CoBPin(N2)2 1-BPin(N2),[8] Scheme 3a). It is possible that reduction of Co(II) to Co(I) occurs by initial formation of an unobserved cobalt(II) diboryl,[13] which undergoes B–B reductive elimination to Co(0) and subsequent comproportionation to two equivalents of Co(I). If operative, this mode of pre-catalyst activation is analogous to the B2Pin2 mediated reduction of (Cy3P)2Pd(OAc)2 to (Cy3P)2Pd,[14a] although comproportionation was not observed.[14b] Likewise, treatment of a THF-[d8] solution of 1-(O2CtBu)2 with four equiv. of HBPin at 23°C produced 1-(H)2BPin in 68% yield. A paramagnetic cobalt byproduct, assigned as Co[PinB(O2CtBu)2]2, was also observed in 32% yield. This product is analogous to Co[PinB(OAc)2]2,[5d] a byproduct identified following treatment of terpyridine cobalt(II) bis(acetate) complexes with HBPin. These observations demonstrate that both B2Pin2 and HBPin are sufficient activators and formal reductants for generating cobalt(I) boryl complexes from cobalt(II) precursors.
Scheme 3.

Stoichiometric reactions of 1-(O2CtBu)2 with B2Pin2 and HBPin.
2.2. Reactions of 1-(H)2BPin and 1-Ph with HSi(OEt)3
The reactivity of 1-(O2CtBu)2 with silanes was also explored to evaluate the potential of [4-Me-(iPrPNP)Co] compounds for catalytic C(sp2)–H silylation chemistry.[15] Treatment of 1-(O2CtBu)2 with two equivalents of HSi(OEt)3 at 23°C in benzene-[d6] for 48 hours produced an intractable mixture of compounds. An alternative route to a cobalt silyl compound was explored by reversible reductive elimination-oxidative addition from a well-defined cobalt(III) compound. Addition of 1 equivalent of HSi(OEt)3 to a benzene-[d6] solution of 1-(H)2BPin produced a new diamagnetic product identified as the cobalt(III) compound, 1-(H)2Si(OEt)3 (Scheme 4a).[16] A diagnostic triplet centered at −9.02 ppm was observed in the benzene-[d6] 1H NMR spectrum corresponding to the Co–H. A single, sharp 31P resonance was observed at 98.67 ppm, consistent with formation of a six-coordinate [(iPrPNP)Co(III)] compound.[5a,8,17]
Scheme 4.
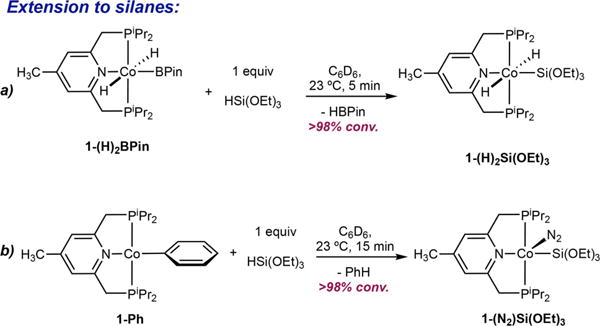
Stoichiometric reactions of 1-(H)2BPin and 1-Ph with HSi (OEt)3.
The synthesis of cobalt-silyl compounds was also explored from other [(iPrPNP)Co(I)] sources. Addition of one equivalent of HSi(OEt)3 to a benzene-[d6] solution of 4-Me(iPrPNP) CoCH3, 1-CH3[8] at 23°C and monitoring the progress of the reaction for 16 hours by 1H and 31P NMR spectroscopies established formation of a new, diamagnetic cobalt compound, tentatively assigned as the cobalt(I) silyl[18] dinitrogen compound, 1-Si(OEt)3(N2). Other unidentified cobalt compounds accompanied formation of 1-Si(OEt)3(N2). The cobalt silyl compound exhibits a 31P NMR resonance at 76.39 ppm, a value typical for Co(I) complexes of this type,[5a,8,17] and a vN2 at 2054 cm−1 in benzene-d6 solution, diagnostic of dinitrogen coordination.[8] 1-Si(OEt)3(N2) was also generated more cleanly from addition of HSi(OEt)3 to 4-Me(iPrPNP)CoPh (1-Ph) at 23°C for 15 minutes (Scheme 4b). Evaluation of 1-(O2CtBu)2, 1-(H)2BPin and 1-CH3 for the catalytic C–H silylation of benzofuran with HSi(OEt)3 at 80°C both in the presence and in the absence of cyclohexene as an acceptor[15] (see SI for details) resulted in no evidence for formation of silylated products with complete recovery of the arene. The observed lack of reactivity may be a result of the kinetic preference of the cobalt(III) intermediate to preferentially undergo reversible C–H reductive elimination rather than product-forming C–Si bond formation. Such a pathway is supported by the synthesis of 1-Si(OEt)3(N2) from 1-Ph where (iPrPNP)Co(Ph)Si(OEt)3H is likely an intermediate that generates (iPrPNP)CoSi(OEt)3 exclusively.
2.3. Chemistry of (iPrPONOP) Cobalt Compounds
The synthesis of cobalt(II) bis(carboxylate) compounds of iPrPONOP,[19] where the [CH2] linkers have been replaced with oxygen atoms, was also explored. This ligand was of interest due to its relative ease of synthesis and potentially distinct electronic properties and coordination preferences as compared to iPrPNP. Previous work from Schrodi[20a] and coworkers have demonstrated that iPrPONOP-supported iron carbonyl complexes contain a more electron deficient metal center relative to the iPrPNP-variant.[20b] If similar properties translated on to cobalt, isolable Co(I) complexes would be expected.
Addition of iPrPONOP to anhydrous cobalt pivalate[21] did not yield the anticipated cobalt(II) bis(carboxylate), (iPrPONOP)Co(O2CtBu)2. Instead, the planar cobalt compound 2 was isolated in 36% yield as orange crystals arising from P–O bond cleavage of the pincer upon metalation (Scheme 5). The fate of the remainder of one iPrPONOP ligands has not been determined. Notably, metalation of iPrPONOP with CoCl2 has been reported to yield (iPrPONOP)CoCl2, 2-Cl2,[22] suggesting that the metal carboxylates play a role in the ligand degradation process. Magnetic susceptibility measurements on 2 are consistent with an S=1/2 ground state (μeff=1.8 μB, 23°C). Cooling a saturated ether solution of 2 at −35 C produced crystals suitable for X-ray diffraction and a solid-state representation of 2 is shown in Figure 1.
Scheme 5.
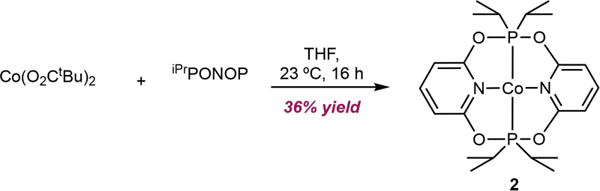
Synthesis of 2.
Figure 1.

Solid state structure of 2 at 30% probability ellipsoids. Hydrogen atoms were omitted for clarity.
Complex 2 was evaluated for the C(sp2)–H borylation of a selection of heteroarenes using HBPin as the boron source. Stirring a mixture of neat benzofuran, HBPin and 5 mol% of 2 at 80°C for 16 hours resulted in complete conversion to the 2-benzofuranyl product (Scheme 6). No catalytic turnover was observed with other five-membered heteroarenes including: substituted furans, benzothiophene, 2-methylthiophene and N-methylindole.
Scheme 6.

Evaluation of 2 for catalytic C–H borylation.
Because the synthesis of the targeted cobalt bis(carboxylate) complex of iPrPONOP was not synthesized through standard methods, preparation of a well-defined cobalt(I) alkyl containing this pincer was explored. Addition of two equivalents of LiCH2SiMe3 to a toluene solution of (iPrPONOP) CoCl2,[22] 2-Cl2, produced an intractable mixture of products arising from P–O bond cleavage within the chelate as evidenced by formation of iPr2PCH2SiMe3 (Scheme 7a). The synthesis and alkylation (iPrPONOP)CoCl was also pursued, owing to the rich chemistry associated with related (iPrPNP) CoR (R=H, alkyl, aryl) compounds.[23] Unfortunately, addition of one equivalent of LiCH2SiMe3 to a toluene solution of (iPrPONOP)CoCl, 2-Cl, also produced an intractable mixture of cobalt products. Similarly, iPr2PCH2SiMe3 was identified by 1H and 31P NMR spectroscopy, consistent with P–O bond cleavage in the chelate (Scheme 7b).[24]
Scheme 7.
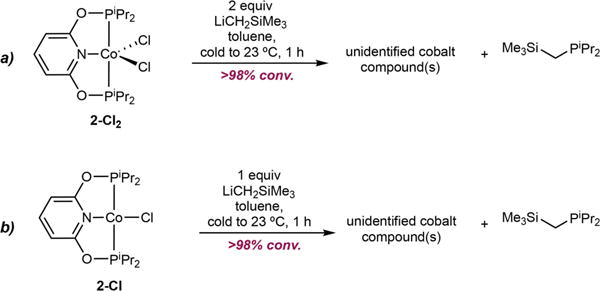
Attempted synthesis of a cobalt alkyl derived from iPrPONOP.
3. Conclusions
Studies on the activation of the readily prepared, air-stable C(sp2)–H arene borylation pre-catalyst, 1-(O2CtBu)2, with both B2Pin2 and HBPin were conducted. With B2Pin2, reduction to the cobalt(I) boryl, 1-BPin, was observed while with HBPin oxidation to the cobalt(III) dihydride boryl, 1-H2 (BPin) occurred. In both cases, formation of the catalytically inactive Co[PinB(O2CtBu)2]2 accompanied generation of the cobalt boryl compounds. Related pincer-ligated cobalt(I) and cobalt(III) silyl complexes have been synthesized from treatment of 1-Ph and 1-(H)2BPin with HSi(OEt)3, respectively. Despite their structural resemblance to the cobalt boryl compounds, no C–H functionalization reactivity was observed. Attempted synthesis of the cobalt(II) bis(carboxylate) with the related pincer, iPrPONOP, resulted in the isolation of complex 2 arising from extrusion of carboxylate ligands on cobalt and P–O bond cleavage of the chelate. Despite the structural similary of iPrPNP and iPrPONOP, the rich catalytic chemistry of iPrPNP cobalt compounds was not observed with iPrPONOP due to the deleterious P–O bond cleavage of the chelate. As a result, stable cobalt pre-catalysts with this pincer have not yet been synthesized.
Supplementary Material
Acknowledgments
J.V.O. acknowledges the 2015 Howard Hughes Medical Institute International Student Research Fellowship and the 2016 Harold W. Dodds Honorific Fellowship (awarded by the Graduate School at Princeton University). We also thank Dr. Jamie Neely for the preparation of 2-Cl. Financial support was provided by NIH (R01 GM121441).
Footnotes
Dedicated to Professor Robert Bergman with congratulations on the Wolf Prize.
Supporting information for this article is available on the WWW under https://doi.org/10.1002/ijch.201700072
References
- 1.a) Godula K, Sames D. Science. 2006;312:67–72. doi: 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]; b) Rouquet G, Chatani N. Angew Chem Int Ed. 2013;52:11726–11743. doi: 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]; c) McMurray L, O’Hara F, Gaunt M. Chem Soc Rev. 2011;40:1885–1898. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]; d) Gutekunst WR, Baran PS. Chem Soc Rev. 2011;40:1976–1991. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]; e) Colby DA, Tsai AS, Bergman RG, Ellman JA. Acc Chem Res. 2012;45:814–825. doi: 10.1021/ar200190g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Iverson CN, Smith MR. J Am Chem Soc. 1999;121:7696–7697. [Google Scholar]; b) Cho JY, Tse MK, Holmes D, Maleczka RE, Smith MR. Science. 2002;295:305–308. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]; c) Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. J Am Chem Soc. 2002;124:390–391. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]; d) Ishiyama T, Takagi J, Hartwig JF, Miyaura N. Angew Chem Int Ed. 2002;41:3056–3058. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 3.Hall DG. Boronic Acids. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 4.a) Mazzacano TJ, Mankad NP. J Am Chem Soc. 2013;135:17258–17261. doi: 10.1021/ja408861p. [DOI] [PubMed] [Google Scholar]; b) Hatanaka T, Ohki Y, Tatsumi K. Chem – Asian J. 2010;5:1657–1666. doi: 10.1002/asia.201000140. [DOI] [PubMed] [Google Scholar]; c) Dombray T, Werncke CG, Jiang S, Grellier M, Vendier L, Bontemps S, Sortais JB, Sabo-Etienne S, Darcel C. J Am Chem Soc. 2015;137:4062–4065. doi: 10.1021/jacs.5b00895. [DOI] [PubMed] [Google Scholar]; d) Yan G, Jiang Y, Kuang C, Wang S, Liu H, Zhang Y, Wang J. Chem Commun. 2010;46:3170–3172. doi: 10.1039/b926945b. [DOI] [PubMed] [Google Scholar]
- 5.a) Obligacion JV, Semproni SP, Chirik PJ. J Am Chem Soc. 2014;136:4133–4136. doi: 10.1021/ja500712z. [DOI] [PubMed] [Google Scholar]; b) Schaefer BA, Margulieux GW, Small BL, Chirik PJ. Organometallics. 2015;34:1307–1320. [Google Scholar]; c) Scheuermann ML, Johnson EJ, Chirik PJ. Org Lett. 2015;17:2716–2719. doi: 10.1021/acs.orglett.5b01135. [DOI] [PubMed] [Google Scholar]; d) Léonard NG, Bezdek MJ, Chirik PJ. Organometallics. 2017;36:142–150. [Google Scholar]
- 6.Khaskin E, Diskin-Posner Y, Weiner L, Leitus G, Milstein D. Chem Commun. 2013;49:2771–2773. doi: 10.1039/c3cc39049g. [DOI] [PubMed] [Google Scholar]
- 7.Obligacion JV, Bezdek MJ, Chirik PJ. J Am Chem Soc. 2017;139:2825–2832. doi: 10.1021/jacs.6b13346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Obligacion JV, Semproni SP, Pappas I, Chirik PJ. J Am Chem Soc. 2016;138:10645–10653. doi: 10.1021/jacs.6b06144. [DOI] [PubMed] [Google Scholar]
- 9.Obligacion JV, Chirik PJ. ACS Catal. 2017;7:4366–4371. doi: 10.1021/acscatal.7b01151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cobalt bis(carboxylate) pre-catalysts have also been utilized in benzylic borylation (reference 10a) and hydrosilation (reference 10b and c):; a) Palmer WN, Obligacion JV, Chirik PJ. J Am Chem Soc. 2016;138:766–769. doi: 10.1021/jacs.5b12249. [DOI] [PubMed] [Google Scholar]; b) Noda D, Tahara A, Sunada Y, Nagashima H. J Am Chem Soc. 2016;138:2480–2483. doi: 10.1021/jacs.5b11311. [DOI] [PubMed] [Google Scholar]; c) Schuster CH, Diao T, Pappas I, Chirik PJ. ACS Catal. 2016;6:2632–2636. [Google Scholar]
- 11.Xu H, Bernskoetter W. J Am Chem Soc. 2011;133:14956–14959. doi: 10.1021/ja2072548. [DOI] [PubMed] [Google Scholar]
- 12.It has been shown that silanes can reduce cobalt(II) amides can be reduced to cobalt(I) amides in alkene hydrosilation, see:; Liu Y, Deng L. J Am Chem Soc. 2017;139:1798–1801. doi: 10.1021/jacs.6b12938. [DOI] [PubMed] [Google Scholar]
- 13.a) Dai C, Stringer G, Corrigan JF, Taylor NJ, Marder TB, Norman NC. J Organomet Chem. 1996;513:273–275. [Google Scholar]; b) Adams CJ, Baber RA, Batsanov AS, Bramham G, Charmant JPH, Haddow MF, Howard JAK, Lam WH, Lin Z, Marder TB, Norman NC, Orpen AG. Dalton Trans. 2006:1370–1373. doi: 10.1039/b516594f. [DOI] [PubMed] [Google Scholar]
- 14.a) Wei CS, Davies GHM, Soltani O, Albrecht J, Gao Q, Pathirana C, Hsiao Y, Eastgate MD. Angew Chem Int Ed. 2013;52 doi: 10.1002/anie.201210252. [DOI] [PubMed] [Google Scholar]; 5822–5826; B2Pin2-mediated formation of a Ni(I) boryl intermediate from NiI2 was also invoked in cross coupling although no nickel intermediates were isolated, see:; b) Xu H, Zhao C, Qian Q, Deng W, Gong H. Chem Sci. 2013;4:4022–4029. [Google Scholar]
- 15.a) Cheng C, Hartwig JF. Science. 2014;343:853–857. doi: 10.1126/science.1248042. [DOI] [PubMed] [Google Scholar]; b) Cheng C, Hartwig JF. J Am Chem Soc. 2015;137:592–595. doi: 10.1021/ja511352u. [DOI] [PubMed] [Google Scholar]
- 16.An analogous (tBuPNP)cobalt(III) dihydride silyl has also been reported, see:; Scheuermann ML, Semproni SP, Pappas I, Chirik PJ. Inorg Chem. 2014;53:9463–9465. doi: 10.1021/ic501901n. [DOI] [PubMed] [Google Scholar]
- 17.Semproni SP, Atienza CCH. Chem Sci. 2014;5:1956–1960. [Google Scholar]
- 18.Other Co(I) silyl compounds have been reported:; a) Aylett BJ, Campbell JM. Chem Commun. 1965;217 [Google Scholar]; b) Mo Z, Xiao J, Gao Y, Deng L. J Am Chem Soc. 2014;136:17414–17417. doi: 10.1021/ja510924v. [DOI] [PubMed] [Google Scholar]
- 19.Salem H, Shimon LJW, Diskin-Posner Y, Leitus G, Ben-David Y, Milstein D. Organometallics. 2009;28:4791–4806. [Google Scholar]
- 20.a) DeRieux WW, Wang A, Schrodi Y. J Organomet Chem. 2014;772:60–67. doi: 10.1016/j.jorganchem.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Trovitch RJ, Lobkovsky E, Chirik PJ. Inorg Chem. 2006;45:7252–7260. doi: 10.1021/ic0608647. [DOI] [PubMed] [Google Scholar]
- 21.Aromi G, Batsanov AS, Christian P, Helliwell M, Parkin A, Parsons S, Smith AA, Timco GA, Winpenny RE. Chem Eur J. 2003;9:5142–5161. doi: 10.1002/chem.200304993. [DOI] [PubMed] [Google Scholar]
- 22.Smith AD, Saini A, Singer LM, Phadke N, Findlater M. Polyhedron. 2016;114:286–291. [Google Scholar]
- 23.a) Semproni SP, Milsmann C, Chirik PJ. J Am Chem Soc. 2014;136:9211–9224. doi: 10.1021/ja504334a. [DOI] [PubMed] [Google Scholar]; b) Neely JM, Bezdek MJ, Chirik PJ. ACS Cent Sci. 2016;2:935–942. doi: 10.1021/acscentsci.6b00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ligand modification via hydride attack at the para position of the ligand in PONOP ligated metal compounds was also observed in the presence of superhydride. Furthermore, P–O bond cleavage was observed in some cases, making the characterization of the (PONOP)M complexes challenging. See; Kundu S, Brennesel WW, Jones WD. Inorg Chem. 2011;50:9443. doi: 10.1021/ic201102v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.