

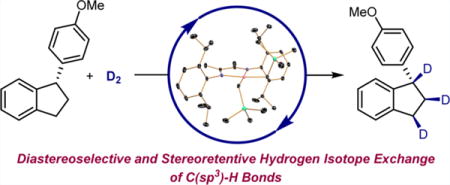
Abstract
Cobalt dialkyl complexes bearing α-diimine ligands proved to be active precatalysts for the nondirected, C(sp3)−H selective hydrogen isotope exchange (HIE) of alkylarenes using D2 gas as the deuterium source. Alkylarenes with a variety of substitution patterns and heteroatom substituents on the arene ring were successfully labeled, enabling high levels of incorporation into primary, secondary, and tertiary benzylic C(sp3)−H bonds. In some cases, the HIE proceeded with high diastereoselectivity and application of the cobalt-catalyzed method to enantioenriched substrates with benzylic stereocenters provided enantioretentive hydrogen isotope exchange at tertiary carbons.
Keywords: cobalt, deuterium, α-diimine, hydrogen isotope exchange, isotopic labeling, C–H activation
Graphical abstract
Hydrogen isotope exchange (HIE) of C–H bonds promoted by transition-metal catalysts is a valuable tool for the preparation of isotopically labeled organic molecules. HIE is particularly useful for studying mechanisms of C–H bond activation1 and for late-stage deuterium (2H, or D) or tritium (3H, or T) labeling of pharmaceuticals.2 The selective incorporation of deuterium into an active pharmaceutical ingredient (API) can significantly alter its metabolism2d,3 and is a common strategy for the design of new therapeutics.2d,3b,c,4 The radiolabeling of APIs with tritium is essential in preclinical adsorption, distribution, metabolism, and excretion (ADME) studies,2a,c and preferred conditions generally use T2 gas at low pressures rather than T2O in order to facilitate safe handling by minimizing radioactivity-to-volume ratio and production of radioactive waste.2a,c
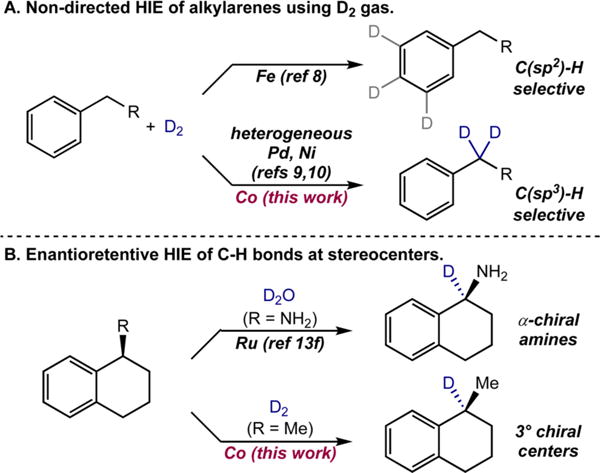
Most transition-metal-catalyzed HIE methods using D2 or T2 gas are selective for C(sp2)−H bonds of arenes and heteroarenes,5 owing to the established oxidative addition preferences of reduced metal complexes.6 The most commonly used catalysts for HIE are homogeneous iridium complexes that typically rely on interaction with directing groups in the substrate to promote selective C(sp2)−H HIE.7 Our laboratory recently reported a bis(N-heterocyclic-carbene)pyridine pincer iron-catalyst that exhibited highly predictable, sterically driven C(sp2)−H site selective HIE that is orthogonal to that observed with iridium (Scheme 1A).8
Scheme 1.
C–H Bond Selectivity in Nondirected Transition-Metal-Catalyzed HIE and Methods for Enantioretentive HIE of C(sp3)−H Bonds
Selective C(sp3)−H HIE in the presence of C(sp2)−H bonds but in the absence of directing groups is rare and presents an unmet need for labeling of drug-like molecules. Precedent exists with heterogeneous Pd,9 Pt, Rh, Ni,10 and Co11 catalysts but these methods often require the use of D2O or other deuterated solvents.6 HIE of benzylic C–H bonds of essentially unfunctionalized alkylarenes has been reported using Pd/C6,9 or Raney Ni6,10 with D2 gas (Scheme 1A), but the process can require multiple iterations of catalyst pretreatment with D2 in order to obtain optimal isotopic enrichment.12
Few examples of stereoretentive or enantioretentive C(sp3)−H isotope exchange have been reported.13 The deuteration of α-C–H bonds of β-chiral alcohols by ruthenium was reported to proceed without loss of enantiopurity.13a Stereoretentive HIE β- to primary amines in phenylalanine derivatives was also described using Pd/C and D2 gas.13b In both of these examples, increased reaction temperature resulted in epimerization. Raney Ni has been used to deuterate carbohydrates without epimerization using D2O under microwave radiation.13c Enantioretentive HIE of α-C–H bonds of chiral primary amines was recently demonstrated with heterogeneous ruthenium catalysts13d,e and D2 gas or homogeneous ruthenium catalysts and D2O (Scheme 1B).13f Nondirected, stereo-retentive labeling of C–H bonds at tertiary carbon centers, however, has not been demonstrated. Here we present a method that uses an α-diimine cobalt(II) dialkyl precatalyst to promote preferential C(sp3)−H HIE in the presence of C(sp2)−H sites using D2 gas. The earth abundant metal catalyst is also active for tertiary centers (Scheme 1B), enabling enantioretentive reactions and making it appealing for late-stage isotopic labeling of APIs.
Our laboratory recently reported α-diimine cobalt14 and nickel15 dialkyl and bis(carboxylate) complexes that are active for the polyborylation of C(sp3)−H bonds in alkylarenes, a shift from the sterically driven C(sp2)−H selectivity more commonly observed with iridium16 and other cobalt borylation catalysts.17 These observations motivated exploration of these catalysts in HIE reactions not only for applications in late stage deuteration and tritiations but also to gain mechanistic insight into the origin of selectivity toward C(sp3)−H functionalization. The N-alkyl-substituted α-diimine cobalt dialkyl complex, (CyADI)Co(CH2SiMe3)2 (1, Scheme 2A), was selected for initial HIE reactions due to its superior performance in catalytic benzylic C(sp3)−H borylation.14 Stirring a dodecane solution of toluene (4) with 5 mol % of 1 under 4 atm of D2 for 24 h at 80 °C resulted in deuterium labeling at multiple C–H positions (Scheme 2B). The highest degree of incorporation occurred at the benzylic C–H bonds (>95%), but the m- and p- positions were also deuterated (11% and 14%, respectively). Arene reduction (12%) to methylcyclohexane was also observed. From these data, it is evident that 1 preferentially activates and functionalizes benzylic C(sp3)−H sites, consistent with observations from C–H borylation, but also accesses C-(sp2)−H positions, a mode of reactivity not previously observed during C–B bond forming catalysis with this precatalyst.14
Scheme 2. α-Diimine Cobalt Complexes Used in This Work and Their Performance in Catalytic HIE of Toluene Using D2.
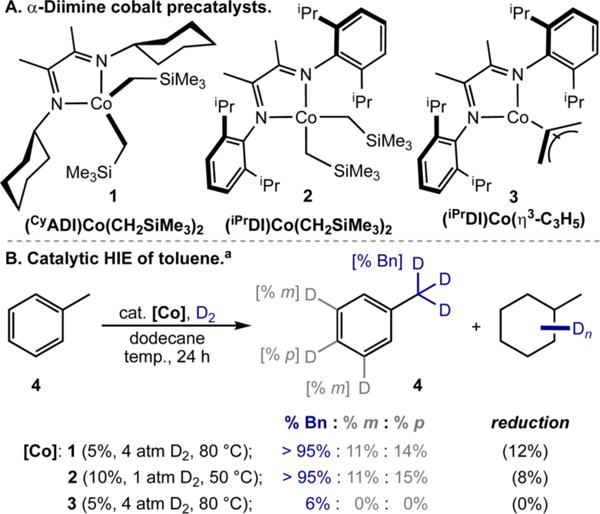
aReactions conducted with 0.55 mmol of toluene in 0.55 mL of dodecane for 24 h. Catalyst loading, pressure of D2 gas, and reaction temperature included in parentheses. See SI for details.
Because arene reduction could be deleterious in the context of late-stage HIE, the precatalyst and reaction conditions were modified to minimize this pathway (see SI, Table S1). Optimal conditions were identified that employed 10 mol % of the N-aryl substituted α-diimine cobalt dialkyl complex, (iPrDI)Co-(CH2SiMe3)2 (2, Scheme 2A),14 at 50 °C and 1 atm of D2 (Scheme 2B). Under these conditions, high levels (>95%) of deuterium incorporation into the benzylic C–H bonds was observed with less arene reduction (8%). The related cobalt η3-allyl complex, (iPrDI)Co(η3-C3H5) (3, Scheme 2A),18 was also evaluated but only provided trace levels of deuterium incorporation (Scheme 2B), highlighting the importance of the cobalt(II) dialkyl precatalyst for entry into the catalytic HIE manifold.
With optimized conditions using cobalt precatalyst 2 established, the generality of the method was investigated with a series of substituted methylarenes. The HIE protocol was selective for the benzylic C–H bonds of p-, m-, o-, and polysubstituted methylarenes (Scheme 3A) and proved tolerant of heteroatom substituents on the arene, including fluoro (5), methoxy (6, 12), and dimethylamino (7) functional groups. For m-tolylboronic acid pinacol ester (8), an elevated temperature (80 °C, 4 atm D2) was required to observe deuterium incorporation, but the reaction also provided a large degree of C(sp2)−H labeling and a significant amount (53%) of reduced arene.19
Scheme 3. HIE of Alkylarenes Using Pre-Catalyst 2 and D2a.
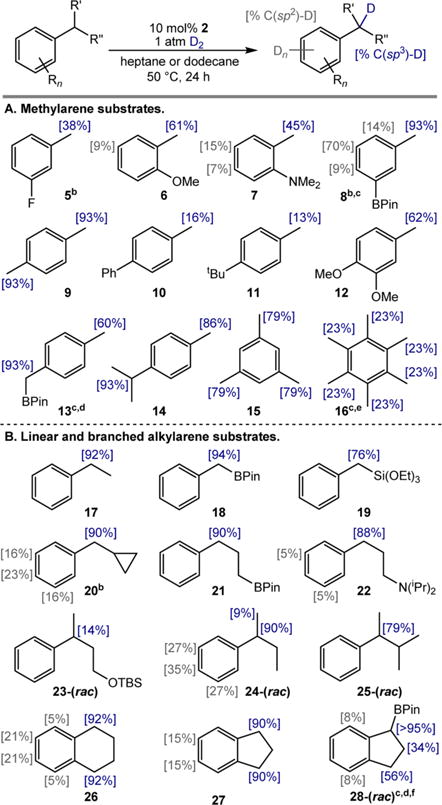
aReactions conducted with 0.55 mmol of alkylarene, 10 mol % of 2, and 1 atm of D2 in 0.55 mL of heptane or dodecane solvent at 50 °C for 24 h. Percent deuterium incorporation at the carbon position indicated in brackets. See SI for details. bArene reduction observed. See SI for details. cReaction carried out at 80 °C. dFive mol % of 2 used. eCPME used as the solvent. fRun with 0.34 mmol of substrate.
Methylarenes with substituents in the 4-position, including p-xylene (9), 4-phenyltoluene (10), and 4-tert-butyltoluene (11), also underwent cobalt-catalyzed C(sp3)−H HIE, although the latter two exhibited comparatively low levels of deuterium incorporation, possibly due to steric effects disfavoring coordination of the substrate to the cobalt catalyst prior to C–H activation. With p-cymene (14), incorporation at the tertiary benzylic C–H position (93%) was only slightly higher than at the primary methyl group (86%). At elevated temperature and pressure (80 °C, 4 atm D2), use of 5 mol % of 2 to label 4-methyl-benzylboronic acid pinacol ester (13) provided a higher degree of deuterium incorporation α- to boron (93%) than in the methyl position (60%). This result is consistent with the apparent activating effect of a [BPin] substituent toward C–H functionalization previously observed in the context of benzylic C–H borylation14,15,20 and with the formation of stabilized α-boryl organometallic intermediates previously invoked in transition-metal catalysis.21 This effect implies that the increased acidity of C–H bonds α- to boron substituents22 coincides with increased rates of C–H functionalization by transition metals.
Cobalt-catalyzed HIE was also applied to arenes with additional methyl groups. A high degree of deuterium incorporation was observed in the benzylic C–H positions of mesitylene (15). At elevated temperatures and pressures (80 °C, 4 atm D2) in cyclopentylmethyl ether (CPME), isotopic exchange was even successful with hexamethylbenzene (16), providing a modest amount of deuterium incorporation (23%) after 24 h and demonstrating compatibility of the method with ethereal solvents.
Substrates with secondary and tertiary benzylic C–H bonds also highlight the unique selectivity of the cobalt-catalyzed method (Scheme 3B). Selective incorporation of deuterium into the secondary benzylic C–H bonds of ethylbenzene (17), benzylboronic acid pinacol ester (18), and benzyltriethoxysilane (19) was observed using standard conditions. In the isotope exchange reaction with benzylcyclopropane (20), incorporation was observed in the benzylic as well as the m-and p-positions. Importantly, no evidence for cyclopropane ring-opening was observed. This result is consistent with either a nonradical mechanism for C–H activation, or a benzylic hydrogen atom abstraction and rapid radical rebound to a cobalt intermediate occurring at a rate greater than the rate of cyclopropane ring-opening.23
Alkylarenes with longer alkyl chains terminated by boronate (21) or amine (22) substituents were also preferentially deuterated in the benzylic position. Branched alkylarenes exhibited high selectivity for tertiary benzylic C(sp3)−H bonds under standard catalytic conditions. With racemic tert-butyldimethylsilyl (TBS) protected 3-phenylbutanol (23-(rac)) and 2-phenyl-3-methylbutane (25-(rac)), the benzylic C–H bonds were exclusively labeled. With sec-butylbenzene (24-(rac)), 9% of the homobenzylic methyl group was deuterated.
Bicyclic alkylarenes were also subjected to cobalt-catalyzed HIE. With tetralin (26) and indan (27), the benzylic methylene positions were selectively labeled and accompanied by a small degree of incorporation into the arene C(sp2)−H bonds. These substrates have proven to be challenging for benzylic C–H borylation with both α-diimine cobalt14 and nickel15 catalysts. These HIE results demonstrate that C(sp3)−H functionalization of these substrates is possible in a similar catalytic regime, suggesting that C–B bond formation may be the challenging step that limits substrate scope in those protocols. The pinacol ester of 1-indanylboronic acid (28-(rac)) underwent deuteration at elevated temperature and pressure (80 °C, 4 atm D2) using 5 mol % of 2, resulting in preferential incorporation into the α-boryl benzylic position (>95%) than the benzylic methylene site (56%), again consistent with an apparent [BPin] activating effect.14,15,20
The deuteration of tertiary benzylic C(sp3)−H bonds inspired exploration of HIE in enantioenriched substrates containing tertiary benzylic stereocenters. Initial experiments were conducted with racemates to verify the applicability of the method to this class of substrates. With 1 atm of D2 and 10 mol % of 2 at 50 °C, racemic 1-(4′-methoxyphenyl)indan (29-(rac)) was successfully labeled (Scheme 4A). Incorporation was preferred on the face of the saturated ring anti- to the aryl substituent, consistent with a mechanism in which selectivity is controlled by precoordination of the arene to the cobalt catalyst intermediate resulting in diastereoselective C(sp3)−H activation.
Scheme 4. Diastereoselective and Enantioretentive HIE of Benzylic C–H Bonds Using Precatalyst 2 and D2a,b.
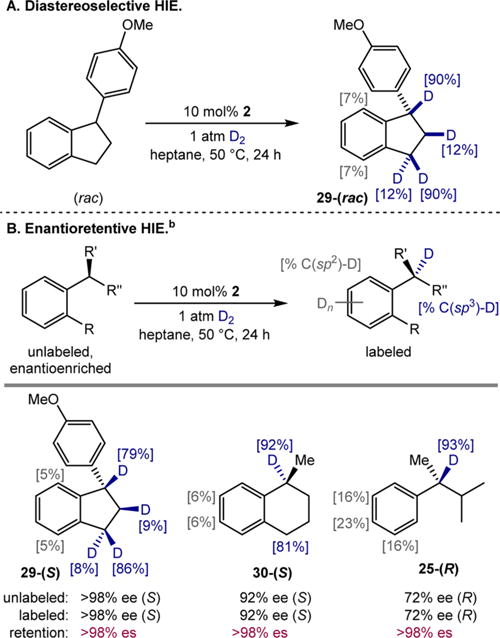
aReactions conducted with 0.55 mmol of alkylarene, 10 mol % of 2, and 1 atm of D2 in 0.55 mL of heptane solvent at 50 °C for 24 h. Percent deuterium incorporation at the carbon position indicated in brackets. See SI for details. bEnantiomeric excess (ee) determined by chiral gas chromatography. See SI for details.
Repeating the procedure with enantiopure arylindan (>98% ee (S)) replicated the diastereoselectivity observed with the racemic substrate, and the enantiopurity of the starting material was preserved in the deuterated product (>98% ee (S), 29-(S), Scheme 4B), demonstrating that this HIE protocol proceeds with high levels of enantiospecificity (es).24 With (S)-1-methyltetralin (92% ee (S), 30-(S)), stereochemical integrity was again conserved (>98% es) with high levels of deuterium incorporation into the tertiary benzylic stereocenter (92%), as well as the benzylic methylene (81%). With the acyclic substrate (R)-2-phenyl-3-methylbutane (72% ee (R), 25-(R)), enantiospecific HIE (>98% es) was achieved with a high degree (93%) of deuterium labeling of the tertiary benzylic C–H bond.
In summary, a cobalt-catalyzed method for the selective C(sp3)−H hydrogen isotope exchange of alkylarenes with D2 gas has been discovered that proceeds with stereoretention when applied to enantioenriched substrates. The enhanced scope and functional group tolerance of this HIE method compared to the previous C(sp3)−H borylation method with the same catalysts14 implies that the latter protocol is limited not by C–H activation but rather by C–B bond formation. Studies to improve the scope of C(sp3)−H HIE and borylation and to develop alternative C(sp3)−H functionalization methods are currently in progress.
Supplementary Material
Acknowledgments
An NSF GOALI (CHE-1564379) is acknowledged for financial support. WNP also acknowledges Eli Lilly for partial support through a graduate fellowship.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b02051.
Complete experimental details including precatalyst optimization studies and characterization data for deuterated products (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.(a) Burwell RL., Jr Acc Chem Res. 1969;2:289–296. [Google Scholar]; (b) Labinger JA, Bercaw JE. Nature. 2002;417:507–514. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; (c) Jones WD. Acc Chem Res. 2003;36:140–146. doi: 10.1021/ar020148i. [DOI] [PubMed] [Google Scholar]; (d) Lersch M, Tilset M. Chem Rev. 2005;105:2471–2526. doi: 10.1021/cr030710y. [DOI] [PubMed] [Google Scholar]
- 2.(a) Voges R, Heys JR, Moenius T. Preparation of Compounds Labeled with Tritium and Carbon-14. John Wiley and Sons, Inc.; New York: 2009. [Google Scholar]; (b) Isin EM, Elmore CS, Nilsson GN, Thompson RA, Weidolf L. Chem Res Toxicol. 2012;25:532–542. doi: 10.1021/tx2005212. [DOI] [PubMed] [Google Scholar]; (c) Lockley WJS, McEwen A, Cooke R. J Labelled Compd Radiopharm. 2012;55:235–257. [Google Scholar]; (d) Gant TG. J Med Chem. 2014;57:3595–3611. doi: 10.1021/jm4007998. [DOI] [PubMed] [Google Scholar]
- 3.(a) Belleau B, Burba J, Pindell M, Reiffenstein J. Science. 1961;133:102–104. doi: 10.1126/science.133.3446.102. [DOI] [PubMed] [Google Scholar]; (b) Meanwell NA. J Med Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]; (c) Katsnelson A. Nat Med. 2013;19:656. doi: 10.1038/nm0613-656. [DOI] [PubMed] [Google Scholar]
- 4.(a) Timmins GS. Expert Opin Ther Pat. 2014;24:1067–1075. doi: 10.1517/13543776.2014.943184. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Halford B. Chem Eng News. 2016;94:32–36. [Google Scholar]
- 5.(a) Junk T, Catallo WJ. Chem Soc Rev. 1997;26:401–406. [Google Scholar]; (b) Atzrodt J, Derdau V, Fey T, Zimmermann J. Angew Chem, Int Ed. 2007;46:7744–7765. doi: 10.1002/anie.200700039. [DOI] [PubMed] [Google Scholar]; (c) Lockley WJS, Heys JR. J Labelled Compd Radiopharm. 2010;53:635–644. [Google Scholar]
- 6.(a) Jones WD, Feher FJ. Acc Chem Res. 1989;22:91–100. [Google Scholar]; (b) Xue XS, Ji P, Zhou B, Cheng JP. Chem Rev. 2017;117:8622–8648. doi: 10.1021/acs.chemrev.6b00664. [DOI] [PubMed] [Google Scholar]
- 7.(a) Hesk D, Das PR, Evans B. J Labelled Compd Radiopharm. 1995;36:497–502. [Google Scholar]; (b) Herbert JM. J Labelled Compd Radiopharm. 2010;53:658–661. [Google Scholar]; (c) Nilsson GN, Kerr W. J Labelled Compd Radiopharm. 2010;53:662–667. [Google Scholar]; (d) Brown JA, Cochrane AR, Irvine S, Kerr WJ, Mondal B, Parkinson JA, Paterson LC, Reid M, Tuttle T, Andersson S, Nilsson GN. Adv Synth Catal. 2014;356:3551–3562. [Google Scholar]
- 8.Yu RP, Hesk D, Rivera N, Pelczer I, Chirik PJ. Nature. 2016;529:195–199. doi: 10.1038/nature16464. [DOI] [PubMed] [Google Scholar]
- 9.Atkinson JG, Luke MO, Stuart RS. Can J Chem. 1967;45:1511–1518. [Google Scholar]
- 10.Heys JR. J Labelled Compd Radiopharm. 2010;53:716–721. [Google Scholar]
- 11.Tsukinoki T, Ishimoto K, Mukumoto M, Suzuki M, Kawaji T, Nagano Y, Tsuzuki H, Mataka S, Tashiro M. J Chem Res Synop. 1996;1:66–67. [Google Scholar]
- 12.Azran J, Shimoni M, Buchman O. J Catal. 1994;148:648–653. [Google Scholar]
- 13.(a) Takahashi M, Oshima K, Matsubara S. Chem Lett. 2005;34:192–193. [Google Scholar]; (b) Maegawa T, Akashi A, Esaki H, Aoki F, Sajiki H, Hirota K. Synlett. 2005:0845–0847. [Google Scholar]; (c) Cioffi EA, Bell RH, Le B. Tetrahedron: Asymmetry. 2005;16:471–475. [Google Scholar]; (d) Taglang C, Martínez-Prieto LM, del Rosal I, Maron L, Poteau R, Philippot K, Chaudret B, Perato S, Lone AS, Puente C, Dugave C, Rousseau B, Pieters G. Angew Chem, Int Ed. 2015;54:10474–10477. doi: 10.1002/anie.201504554. [DOI] [PubMed] [Google Scholar]; (e) Jere FT, Miller DJ, Jackson JE. Org Lett. 2003;5:527–530. doi: 10.1021/ol0274211. [DOI] [PubMed] [Google Scholar]; (f) Hale LVA, Szymczak NK. J Am Chem Soc. 2016;138:13489–13492. doi: 10.1021/jacs.6b07879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palmer WN, Obligacion JV, Pappas I, Chirik PJ. J Am Chem Soc. 2016;138:766–769. doi: 10.1021/jacs.5b12249. [DOI] [PubMed] [Google Scholar]
- 15.Palmer WN, Zarate C, Chirik PJ. J Am Chem Soc. 2017;139:2589–2592. doi: 10.1021/jacs.6b12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Ishiyama T, Miyaura N. Pure Appl Chem. 2006;78:1369–1375. [Google Scholar]; Ishiyama T, Miyaura N. J Organomet Chem. 2003;680:3–11. [Google Scholar]; (c) Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; (d) Hartwig JF. Acc Chem Res. 2012;45:864–873. doi: 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
- 17.(a) Obligacion JV, Semproni S, Chirik PJ. J Am Chem Soc. 2014;136:4133–4136. doi: 10.1021/ja500712z. [DOI] [PubMed] [Google Scholar]; (b) Schaefer BA, Margulieux GW, Small BL, Chirik PJ. Organometallics. 2015;34:1307–1320. [Google Scholar]; (c) Obligacion JV, Semproni SP, Pappas I, Chirik PJ. J Am Chem Soc. 2016;138:10645–10653. doi: 10.1021/jacs.6b06144. [DOI] [PubMed] [Google Scholar]; (d) Léonard NG, Bezdek MJ, Chirik PJ. Organometallics. 2017;36:142–150. [Google Scholar]; (e) Obligacion JV, Bezdek MJ, Chirik PJ. J Am Chem Soc. 2017;139:2825–2823. doi: 10.1021/jacs.6b13346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palmer WN, Diao T, Pappas I, Chirik PJ. ACS Catal. 2015;5:622–626. [Google Scholar]
- 19.The electron-deficient nature of the substrate may favor arene reduction compared to toluene under identical reaction conditions (20%, see SI, Table S1) and may also account for a lack of HIE at 50 °C due to less reversible substrate coordination to the catalyst.
- 20.Shimada S, Batsanov AS, Howard JAK, Marder TB. Angew Chem, Int Ed. 2001;40:2168–2171. doi: 10.1002/1521-3773(20010601)40:11<2168::AID-ANIE2168>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 21.(a) Pereira S, Srebnik M. Tetrahedron Lett. 1995;36:1805–1808. [Google Scholar]; (b) Joannou MV, Moyer BS, Meek SJ. J Am Chem Soc. 2015;137:6176–6179. doi: 10.1021/jacs.5b03477. [DOI] [PubMed] [Google Scholar]; (c) Joannou MV, Moyer BS, Goldfogel MJ, Meek SJ. Angew Chem, Int Ed. 2015;54:14141–14145. doi: 10.1002/anie.201507171. [DOI] [PubMed] [Google Scholar]; (d) Endo K, Ohkubo T, Hirokami M, Shibata T. J Am Chem Soc. 2010;132:11033–11035. doi: 10.1021/ja105176v. [DOI] [PubMed] [Google Scholar]; (e) Sun C, Potter B, Morken JP. J Am Chem Soc. 2014;136:6534–6537. doi: 10.1021/ja500029w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Rathke MW, Kow R. J Am Chem Soc. 1972;94:6854–6856. [Google Scholar]; (b) Matteson DS, Moody RJ. J Am Chem Soc. 1977;99:3196–3197. [Google Scholar]; (c) Matteson DS, Moody RJ. Organometallics. 1982;1:20–28. [Google Scholar]
- 23.The rate constant of cyclopropane ring-opening of the corresponding cyclopropylbenzyl radical was previously determined to be 2.7 × 105 s−1 at 22 °C in hexane solution:; Masnovi J, Samsel EG, Bullock RM. J Chem Soc, Chem Commun. 1989:1044, 1045. [Google Scholar]
- 24.Percent “es”, or enantiospecificity, indicates the percentage of the reaction that proceeds with enantioretention and is defined as: % es = (% ee of product)/(% ee of starting material) * 100.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.