

Abstract
Upwards of 90% of individuals with Bardet-Biedl syndrome (BBS) display rod-cone dystrophy with early macular involvement. BBS is an autosomal recessive, genetically heterogeneous, pleiotropic ciliopathy for which 21 causative genes have been discovered to date. In addition to retinal degeneration, the cardinal features of BBS include obesity, cognitive impairment, renal anomalies, polydactyly, and hypogonadism. Here, we review the genes, proteins, and protein complexes involved in BBS and the BBS model organisms available for the study of retinal degeneration. We include comprehensive lists for all known BBS genes, their known phenotypes, and the model organisms available. We also review the molecular mechanisms believed to lead to retinal degeneration. We provide an overview of the mode of inheritance and describe the relationships between BBS genes and Joubert syndrome, Leber Congenital Amaurosis, Senior-Løken syndrome, and non-syndromic retinitis pigmentosa. Finally, we propose ways that new advances in technology will allow us to better understand the role of different BBS genes in retinal formation and function.
Keywords: Bardet-Biedl syndrome, eye, retinal degeneration, ciliopathy, blindness, BBSome, photoreceptor, nyctalopia, Joubert syndrome, Leber Congenital Amaurosis, Senior-Løken syndrome
1. Bardet-Biedl Syndrome
Bardet-Biedl syndrome (BBS) is an autosomal recessive, genetically heterogeneous, pleiotropic disorder (1–3) caused by mutations in 21 genes (Table 1) that are involved in proper function of the primary cilium. Eight of these proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9, and BBS18) form a complex, the BBSome, which is involved in protein trafficking within cilia, as well as intracellular trafficking (4–13). BBS6, BBS10, and BBS12 are involved in a chaperonin complex required for BBSome assembly (11, 14–16). BBS3, BBS14, and BBS17 are responsible for proper trafficking of the BBSome to and within cilia (17–21). Other BBS genes are not as closely associated with the BBSome (BBS11, BBS13, BBS15–16, and BBS19–21) (11, 22–29) and their role in cilia has not yet been fully characterized. BBS proteins are also thought to play a role in protein trafficking in the photoreceptor connecting cilium and outer segment (OS), which have similarities to the primary cilium (30, 31).
Table 1.
Summary of BBS gene HGNC names, classification, and models of study.
| BBS Gene | HGNC | Classification | Support (Ref # are PMID) |
|---|---|---|---|
| BBS1 | BBS1 | BBSome | Mouse (15322545, 18032602, 23160237) Zebrafish (24069149) Cell Culture (22500027) |
| BBS2 | BBS2 | BBSome | Mouse (15539463) Cell Culture (22500027) |
| BBS3 | ARL6 | BBSome Associated | Mouse (22139371) Zebrafish (21282186) Cell Culture (22139371) |
| BBS4 | BBS4 | BBSome | Mouse (16794820, 15173597) Zebrafish (24681783) Cell Culture (22500027, 24681783) |
| BBS5 | BBS5 | BBSome | Mouse (25849460) Zebrafish (16399798) Cell Culture (22500027) |
| BBS6 | MKKS | BBSome Chaperonin |
Mouse (15772095) Cell Culture (20080638) |
| BBS7 | BBS7 | BBSome | Mouse (23572516) Zebrafish (24938409) Cell Culture (22500027) |
| BBS8 | TTC8 | BBSome | Mouse (21646512) Zebrafish (20643117) Cell Culture (22500027) |
| BBS9 | PTHB1 | BBSome | Zebrafish (22479622) Cell Culture (22500027) |
| BBS10 | BBS10 | BBSome Chaperonin |
Mouse (26273430) Zebrafish (16582908) Cell Culture (20080638) |
| BBS11 | TRIM32 | Non-BBSome | Mouse (19155210, 21775502) Zebrafish (16606853) Cell Culture (28498859) |
| BBS12 | BBS12 | BBSome Chaperonin |
Mouse (22958920) Zebrafish (17160889) Cell Culture (20080638) |
| BBS13 | MKS1 | Non-BBSome | Mouse (19776033, 21045211) Zebrafish (18327255) |
| BBS14 | CEP290 | BBSome Associated |
Mouse: (16632484, 21623382) Zebrafish (18327255) Cell Culture (25552655) |
| BBS15 | WDPCP | Non-BBSome | Mouse (24302887) Zebrafish (24302887) Cell Culture (20671153) |
| BBS16 | SDCCAG8 | Non-BBSome | Mouse (24722439, 24302887) Zebrafish (20835237) Cell Culture (20835237, 24722439) |
| BBS17 | LZTFL1 | BBSome Associated |
Mouse (27312011, 26216965) Cell Culture (22510444, 26216965) |
| BBS18 | BBIP1 | BBSome | Mouse (24316073) Cell Culture (24316073) |
| BBS19 | IFT27 | Non-BBSome | Mouse (25446516) Cell Culture (25446516) |
| BBS20 | IFT74 | Non-BBSome | Zebrafish (27486776) |
| BBS21 | C8ORF37 | Non-BBSome | Mouse (32) Zebrafish (27008867) |
1.1. Clinical Diagnosis
The clinical diagnosis of BBS is typically defined as having either four primary features or three primary and two secondary features (33). Primary features include retinal degeneration, postaxial polydactyly, truncal obesity, cognitive impairment, hypogonadism, and renal anomalies. Secondary features include speech delay, developmental delay, behavioral abnormalities, orodental abnormalities, brachydactyly/syndactyly, ataxia/poor conditioning, mild hypertonia, diabetes mellitus, cardiovascular anomalies, hepatic involvement, craniofacial dysmorphism, Hirschsprung disease, anosmia, and additional eye abnormalities such as strabismus, cataracts, and astigmatism (33).
Obesity occurs in 72–92% of patients beginning in early childhood and worsening with age (34). Polydactyly is present in almost 95% of BBS patients, primarily with a postaxial presentation (69% of BBS cases) (35). Many BBS patients suffer from hypogonadism and genital anomalies such as hypoplastic fallopian tubes and uterus (36). Abnormal spermatozoa have been found in male patients. However, patients of both sexes have been reported to have successfully reproduced (2, 37). Renal failure may not present until late childhood and onward but is a major cause of morbidity and mortality in this disorder (38). Cognitive impairment is the most variable primary phenotype: 29% of BBS patients display severe intellectual disability, 38% display moderately reduced intelligence, and 29% display average intelligence. Some patients even display intellectual performance in the higher range of abilities within the normal population (4%) (35). The role of BBS genes in obesity (39, 40), renal failure (41, 42), and the cilium (42, 43) have been comprehensively reviewed elsewhere; here, we review the role of BBS genes in syndromic and non-syndromic retinal degeneration.
1.2. Mode of Inheritance
The autosomal recessive nature of BBS has been clearly established by segregation analysis of large pedigrees and consanguineous families. The prevalence of BBS is population dependent with an estimated incidence of 1:160,000 in Switzerland and Tunisia, 1:18,000 in Newfoundland, and 1:36,000 in the mixed Arab populations in Kuwait (37, 44–46). It has been reported that homozygous or compound heterozygous mutations in 5 BBS genes (BBS1, BBS10, BBS2, BBS9, and MKKS (BBS6)) cause the majority of BBS cases (33, 46). However, there is some controversy about how frequently BBS displays a “triallelic” inheritance pattern caused by three pathogenic alleles at two different loci (47). This hypothesis is based on BBS patients identified with three putative mutant alleles in two different BBS genes (BBS2 and BBS6) and unaffected individuals carrying two BBS2 mutations. The same group reported that BBS1 has a complex inheritance pattern (48). The group identified asymptomatic individuals with homozygous mutations for BBS1. In addition, these researchers reported a higher prevalence of the M390R allele in a control cohort compared to the number of affected M390R individuals, suggesting an oligogenic model of BBS for this allele (48). Upon further investigation using the exome aggregation consortium data, the minor allele frequency for the M390R allele is known to be three times lower than previously reported by the group and thus does not seem to follow an oligogenic model of inheritance.
In addition, studies by other groups have not supported a triallelic inheritance pattern, and the vast majority of BBS patients reported to date display classic Mendelian autosomal recessive inheritance (49–51). For example, Biesecker et al. describe an Amish family with three children affected by homozygous mutations in BBS6 (50). The father is a carrier and the mother has a non-penetrant homozygous BBS mutation. Sequencing of eight additional BBS genes in this family did not support triallelic inheritance. Another study of eight BBS genes found no families with involvement of two BBS loci consistent with triallelic inheritance (49). Later studies involving 14 BBS genes also failed to detect evidence of a triallelic model of BBS (51). Autosomal recessive inheritance is also consistent with data from multiple BBS mouse models (30, 31, 52–58). One important ramification of the triallelic model of inheritance is the counseling of parents; the 25% recurrence risk of an autosomal recessive disorder differs from the lower than 25% recurrence risk associated with an oligogenic inheritance pattern.
It has also been proposed that a third mutation in a different BBS gene can result in a more severe phenotype in patients with homozygous or compound heterozygous mutations in a BBS gene (59). The ability of BBS genes to cause other ciliopathies and non-syndromic retinal degeneration provides support for the presence of modifier genes, as do data from animal models. For example, the BBS14 (CEP290) homozygous mutant mouse displays increased obesity and an accelerated rate of retinal degeneration when only a single copy of BBS4 is present (60). In one family with two affected individuals homozygous for BBS7 mutations, the individual with a third mutation in BBS4 had a more severe phenotype (61). In theory, genetic modifiers may act on homozygous BBS gene mutations to affect the phenotypic presentation of BBS. It is important to note that many of the genes reported to cause non-syndromic retinal degeneration code for ciliary proteins and hence, they potentially interact with each other (62).
1.3. Retinal Degeneration
Retinal degeneration is the most highly penetrant feature of BBS, making this disorder the second most common syndromic retinal degeneration behind Usher syndrome (46). In a large systematic study of ophthalmic features in BBS adult patients, 60 of 62 patients had clinically evident retinopathy and the other 2 had evidence of retinopathy using electrophysiologic testing (63). BBS patients typically develop symptoms of retinitis pigmentosa (RP) in the first decade of life and often reach legal blindness between the second and third decades of life (64). Night blindness (nyctalopia) is the most common initial visual symptom and is usually first noted around 8.5 years of age (2, 65).
RP causes decreased peripheral vision, which presents as a constricted visual field (65, 66). During this progressive loss of the rods, the patient also loses some cone photoreceptors, which causes loss of visual acuity and a diminution of color discrimination (67). Electrophysiologically, BBS patients are usually found to have a rod-cone dystrophy with early macular involvement (Figure 1) (46, 65–68).
Figure 1.
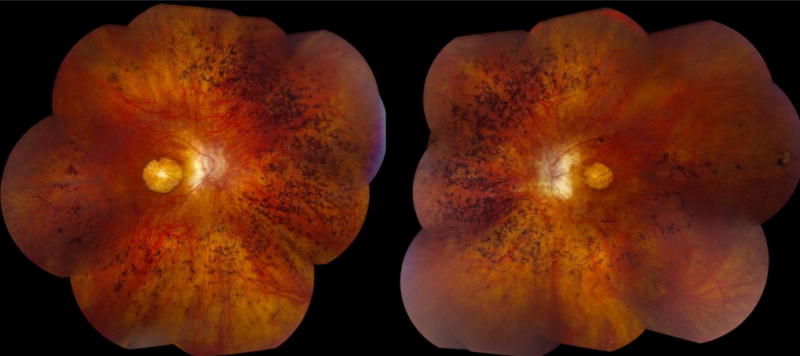
Right (20/100 best corrected visual acuity (BCVA)) and left (20/80 BCVA) fundus photographs of a 47-year-old male with BBS caused by a homozygous M390R mutation in BBS1 showing the characteristic macular atrophy and peripheral bone spicule pigmentation. This patient has a history of postaxial polydactyly of the lower extremities and first noted visual symptoms at age 14.
2. Molecular Mechanisms
2.1. Animal Models of BBS
Investigating the cause of retinal degeneration has been a focus in many BBS animal models. With the advent and increased availability of knockout, knock-in, and transgenic mice, mouse models for a large number of BBS genes have become available to study the role of BBS proteins in the development and maintenance of the mammalian retina (24, 30, 31, 52–55, 57, 58, 69–81). Zebrafish, an organism with a rapidly developing eye, has also been used to investigate how loss of BBS genes affects eye development and maintenance, as well as the effect on vision (70).
BBS can be caused by mutations in two groups of genes: BBSome/BBSome-related genes and non-BBSome genes. Mutations in BBSome genes are more common in humans, which resulted in earlier identification of these genes compared to most of the non-BBSome genes (1, 4–10, 17). The recent advent of next-generation sequencing methods has allowed for the identification of several non-BBSome genes in smaller pedigrees or isolated cases (11, 26). While most BBS genes now have a mouse model available, the retinal phenotype is most well studied in the BBSome (models available in 7 of the 8 genes and a zebrafish model available for the remaining BBSome gene) (30, 54, 58, 69, 70, 73), chaperonin complex genes (3 of 3 mouse models available) (57, 71, 72), or BBSome-interacting genes (3 of 3) (Table 1) (31, 52, 76, 77, 80). While mouse models of the non-BBSome or BBSome-interacting genes Bbs11, Bbs13, Bbs15, Bbs16, Bbs19, and Bbs21 are available (24, 32, 55, 78, 79, 81), with a zebrafish model in bbs20 available (28), retinal phenotypes in the Bbs19 mouse and bbs20 zebrafish are unreported and knockout mice for Bbs11 and Bbs13 lack retinal BBS phenotypic features (24, 78). In addition, the Bbs15 mouse model has anophthalmia, a severe phenotype that is not consistent with patient phenotypes (81).
2.2. Molecular Mechanism of BBS in Retinal Degeneration
The majority of BBS model organisms with retinal phenotypes display rhodopsin localization defects, suggesting a common mechanism of disease. Under normal conditions, rhodopsin is localized to the outer segment (OS) of rods and performs the first step of phototransduction. In BBS mutant mice, rhodopsin is mislocalized to the inner segment (IS) and cell bodies of the outer nuclear layer (ONL) (30, 54, 55, 73) diminishing its presence in the OS (57). Apoptosis of photoreceptor cells has also been noted (54, 82). In addition to rhodopsin mislocalization, BBS mouse models display a disorganization of the OS (30, 31) and progressive loss of OS, IS, and ONL with preservation of the inner retinal layers (53, 57). This phenotype is also observed in the zebrafish knockdown model of bbs9, which shows a complete loss of the OS (70).
Most of the research pertaining to BBS mouse models has focused on changes to photoreceptor structure and localization of specific proteins like rhodopsin, transducin, and arrestin. However, recent work in the Bbs17 mouse model has identified global changes to the OS proteome in mutant mice. Datta et al. identified abnormal enrichment of 138 proteins in the OS of Bbs17 mutant mice, whereas only 8 proteins showed decreased OS localization. Many of these enriched proteins were absent from the WT OS prep. Accumulation of non-OS proteins in mutants was concomitant with a loss of these proteins in the cell body (IS, ONL, synaptic terminal) where they are normally required for regulation of retinal function. One such set of mislocalized IS proteins was the SNARE complex, which mediates membrane fusion events. STX3, a component of the SNARE complex has been linked to rhodopsin trafficking in photoreceptors, is mislocalized to the OS in multiple BBS mouse models (31). As such, the authors suggest that BBS proteins are directly or indirectly required for proper OS protein trafficking and that photoreceptor cell death in BBS mutants may be due to the combination of insufficient protein functions in the cell body and aberrant accumulation of proteins in the OS (Figure 2).
Figure 2.
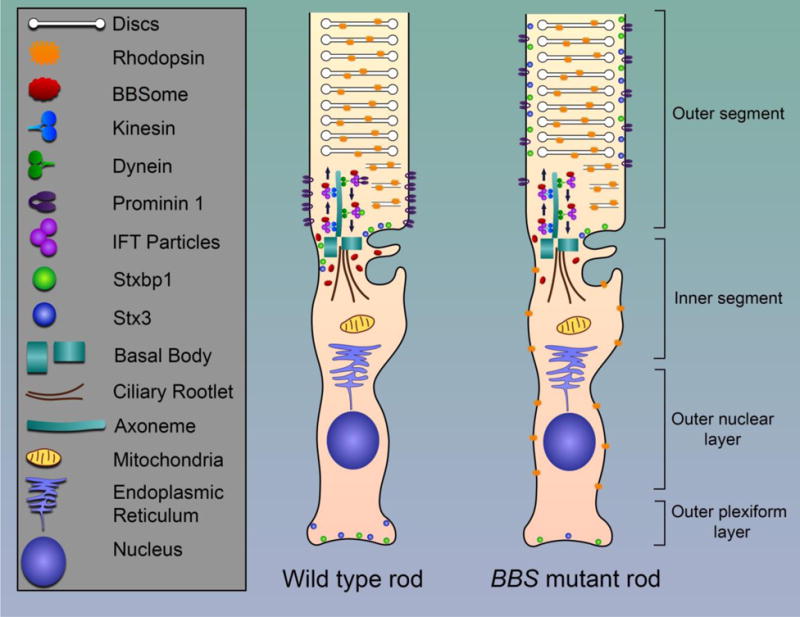
Schematic illustration of the rod photoreceptor for wild type mice and BBS mutant mice. The BBSome is involved in protein trafficking along the axoneme. In Bbs8 and Bbs17 mutant mice, proteins are mislocalized. In a wild type mouse, STX3 and STXBP1 are located at the synapse and base of the cilium. Rhodopsin is localized to the discs. In addition, Prominin 1 is located at the base of the outer segment. In BBS mutant mice, STX3 and STXBP1 are found throughout the outer segment. Rhodopsin is found at the inner segment and outer nuclear layer. In addition, Prominin 1 is found throughout the outer segment.
2.3. In vitro Molecular Mechanisms of BBS
The connecting cilium of photo-receptors is the analogous structure of the transition zone of the primary cilium of non-photoreceptor cell types (83). The connecting cilium derives its name from the fact that it connects the inner segment, which contains the metabolic and trafficking components of photoreceptors, with the outer segment, which contains the photo-transduction apparatus. Like the primary cilium, the connecting cilium consists of an axoneme that is comprised of nine microtubule doublets. There is trafficking of proteins and membrane components through the connecting cilium by a process known as intraflagellar transport (IFT), a process requiring BBS proteins (84, 85).
The BBSome plays a role in transport within the cell, including the known effects of BBSome associated proteins on the leptin receptor (86) and the insulin receptor (87), though the mechanism for this trafficking is not well understood. In addition to intracellular trafficking, in the presence of BBS mutations, an array of ciliary receptors are mislocalized, including: the smoothened receptor (19), somatostatin receptor type 3 (88), melanin-concentrating hormone receptor 1 (88), Neuropeptide Y Family Receptors (89), OSM-9 (90), and ODR-10 (90). The majority of these receptor trafficking defects involve G-protein coupled receptors, which would affect cAMP signaling and phosphatidylinositol signaling (91). Due to the shared structure of the connecting cilium and the transition zone of the primary cilium, the retinal degeneration phenotype is likely due to changes in ciliary receptor trafficking and not generalized intracellular trafficking. Additional work is needed to further define the role and interacting partners of the BBSome in photoreceptor cells.
2.4. Transcriptional Variation
Consistent with the role of BBS proteins in retinal development and maintenance, retina-specific isoforms have been identified in BBS3, BBS5, and BBS8 (77, 92, 93). Loss of the bbs3 retina-specific isoform (bbs3l) in zebrafish causes vision loss that cannot be rescued by the canonical isoform of Bbs3. Loss of the Bbs3l isoform in the mouse causes disorganization of the IS but does not lead to severe retinal dysmorphology. However, green cone opsin mislocalization can be rescued with Bbs3l, suggesting an important role for this isoform specific to eye morphology and physiology. The retinal isoform of BBS5, BBS5L, has not been studied at a functional level in the eye, but is known to localize to the axonemal structure of the photoreceptor and is found at the boundary of the IS and OS, in the IS and OS, the inner plexiform layer, and ganglion cell layer but not in the connecting cilium (92). Based on current patient information and known mutations, no specific pathology can currently be ascribed to BBS5L (92). Interestingly, a mutation that results in the skipping of the BBS8 retina-specific isoform has been identified in patients with non-syndromic RP, supporting the role of this isoform in retinal pathology (93).
The presence of these splice variants highlights the unique characteristics of the retina and confirm there is still much to be learned about the retina, its function, and the role of BBS genes in eye development and function. To date, approximately one quarter of retinal genes have been found to express alternate transcripts (94). It is possible that other BBS genes have unidentified retinal specific splice variants that may account for additional cases of non-syndromic RP or may modify the eye phenotype of BBS patients. Future research could pursue identification of additional splice variants and further investigation of the role of known isoforms like BBS8 and BBS5L.
3. Other Disorders Attributed to BBS Genes
Retinal degeneration is a common feature of many ciliopathies. Mutations in genes that usually cause BBS have currently been identified in three additional ciliopathies that present with retinal degeneration as well as in non-syndromic retinal degeneration (Table 2). Here, we examine the similarities and differences between these disorders.
Table 2.
Summary of BBS genes, references in which they were classified as causative genes of BBS, and BBS genes that cause other ciliopathies (Ref # are PMID).
| Gene | Bardet-Biedl Syndrome | Non-Syndromic Retinitis Pigmentosa | Leber Congenital Amaurosis | Senior-Løken Syndrome | Joubert Syndrome | Meckel-Gruber Syndrome |
|---|---|---|---|---|---|---|
| BBS1 | 12118255 | 23143442 | ||||
| BBS2 | 11285252 | 25541840 | ||||
| ARL6 (BBS3) | 15314642 | 19956407 | ||||
| BBS4 | 11381270 | |||||
| BBS5 | 15137946 | |||||
| MKKS (BBS6) | 10973238 | |||||
| BBS7 | 12567324 | |||||
| TTC8 (BBS8) | 14520415 | 20451172 | ||||
| PTHB1 (BBS9) | 16380913 | |||||
| BBS10 | 20805367 | |||||
| TRIM32 (BBS11) | 16606853 | |||||
| BBS12 | 17160889 | |||||
| MKS1 (BBS13) | 18327255 | 24886560 | 16415886 | |||
| CEP290 (BBS14) | 18327255 | 16909394 | 16682973 | 16682973 | 17564974 | |
| WDPCP (BBS15) | 20671153 | |||||
| SDCCAG8 (BBS16) | 20835237 | 20835237 | ||||
| LZTFL1 (BBS17) | 22510444 | |||||
| BBIP1 (BBS18) | 24026985 | |||||
| IFT27 (BBS19) | 24488770 | |||||
| IFT74 (BBS20) | 27486776 | |||||
| C8ORF37 (BBS21) | 27008867 | 22177090 |
3.1. Leber Congenital Amaurosis
Leber congenital amaurosis (LCA) is the most severe retinal degeneration of the ciliopathies reviewed here. LCA is usually apparent shortly after birth and is characterized by severe dysfunction of both photoreceptor types, nystagmus, reduced pupillary response, keratoconus, and hyperopia (95–97). LCA patients commonly display excessive eye poking and rubbing (98). This behavior is known as Franceschetti’s oculo-digital sign and contributes to the development of keratoconus in these patients (95, 98) Currently, there have been at least 17 genes implicated in the development of LCA including CEP290 (BBS14)(99).
3.2. Joubert Syndrome
Joubert syndrome is a rare disorder that affects many organ systems. A cardinal feature of Joubert syndrome is the “molar tooth sign” resulting from abnormal development of the mid-brain, which is identified using cranial magnetic resonance imaging (100). Other common characteristics include hypotonia, abnormal eye movements, ataxia, irregular breathing patterns, and mild to severe developmental delay (101, 102). Joubert syndrome may also present with retinal degeneration, cystic kidney disease, nephronophthisis, polydactyly, encephalocele, and endocrine defects (103–107). The age of onset of retinal degeneration is variable, and was seen to only occur in approximately 30% of patients. It can be as severe as neonatal onset of congenital blindness, but it may also present as a stable loss of vision (108). Currently, there are at least 34 genes associated with Joubert syndrome including MKS1 (BBS13) and CEP290 (BBS14)(109).
3.3. Senior-Løken Syndrome
Senior-Løken syndrome (SLS), first reported in 1961, is a rare autosomal recessive disease characterized by having both nephronophthisis and LCA (110, 111). Nephronophthisis is characterized by the development of fluid-filled cysts known as cystic dilation within the kidney leading to polyuria, polydipsia, weakness, fatigue, and eventually renal failure (98, 112–115). SLS is classified as infantile, juvenile, or adolescent depending on the manifestation of renal failure. RP and the subsequent blindness that follows may occur as early as birth or it can manifest as a progressive loss of vision during childhood (114–116). Usually all SLS patients have been diagnosed with alterations to their eyes by 10 years of age. Patients with juvenile renal failure, which is linked to mutations in NPHP5 and CEP290, have a greater propensity for RP (116, 117). Milder RP has been observed in patients with mutations in other NPHP genes even though disease symptoms often present during the first two years of life with almost all SLS patients diagnosed with eye changes by 10 years of age (118, 119). Presently, at least 9 genes are associated with SLS including CEP290 (BBS14) and SDCCAG8 (BBS16) (26, 116, 120–123).
3.4. Non-Syndromic Retinal Degeneration
The clinical findings in patients with syndromic and non-syndromic BBS-associated retinal degeneration are very similar; both groups experience early central vision loss coupled with a progressive loss of peripheral vision which can ultimately result in complete blindness in some patients. Mutations in five different BBS genes have been reported to cause non-syndromic RP. While the BBS1 M390R mutation causes the greatest percentage of clinically diagnosed BBS, Estrada-Cuzcano et al. identified two non-syndromic RP patients that were homozygous for the M390R mutation (124). Shevach identified a set of 4 missense mutations in BBS2 that cosegrated with non-syndromic RP in five independent families (125). A homozygous mutation in ARL6 (BBS3) and a novel splice site mutation in TTC8 (BBS8) have been identified in patients with non-syndromic RP (93, 126). Finally, Estrada-Cuzcano et al. reported that homozygous splice and homozygous missense mutations in C8orf37 (BBS21) are associated with non-syndromic autosomal-recessive cone-rod dystrophy (CRD) and RP (127). The combination of low disease prevalence, plus gene- and mutation-level heterogeneity makes the inference of mutation specificity for BBS versus non-syndromic retinal degeneration challenging.
4. Future Directions
In order to accurately distinguish between variants that cause BBS, other ciliopathies, and non-syndromic retinal degeneration, more work is required to understand the many different BBS proteins and their functions. This will include the use of transcriptomics, proteomics, animal studies, and cellular studies using patient-derived induced pluripotent stem cells to provide a comprehensive analysis of each gene and its role in the retina and beyond.
The availability of numerous animal models will allow for further in-depth investigation into the development of BBS retinopathy. For example, it is currently unknown whether accumulation of one specific protein in the OS is sufficient to induce photoreceptor degeneration or whether degeneration is caused by a cumulative stress on the OS trafficking system. It would be interesting to compare the OS proteome of BBS17 mutant mice with proteomes from other BBS models, including BBS models that don’t directly interact with the BBSome, to determine whether certain proteins remain consistently enriched in the OS. Additional data could be obtained utilizing RNA sequencing on similar samples to identified transcription differences within the eye. These data could provide vital clues in identifying which proteins require precise localization or splice variants to prevent retinal degeneration and may provide a shared mechanism of OS degeneration between BBSome and non-BBSome genes. Protein mislocalization could also be used to identify novel BBS or retinal ciliopathy genes.
However, developing mouse models of every specific patient mutation is time consuming, expensive, and impractical. As such, other methods will need to be utilized to assess the pathogenicity of these mutations. Recent efforts to characterize the three-dimensional structure of the BBSome as well as in silico modeling of selected mutations observed in patients (128) could be used to predict the effect of novel mutations on protein structure and BBSome assembly. The functional effects of patient mutations could also be assessed in vitro. The CRISPR/Cas9 system greatly facilitates the creation of cell lines harboring patient mutations for further study. Additionally, induced pluripotent stem cells (iPSCs) derived from patient fibroblasts could be differentiated into photoreceptor precursor cells (129). These cells would allow researchers to study the effect of patient mutations in a disease-relevant cell type in the context of the patient’s specific genetic background. Of course, the ultimate goal is to be able to treat patients with BBS. Patients in the early stages of disease can potentially be treated with gene therapy (130), whereas patients with advanced disease could be treated with photoreceptor precursor cells derived from autologous patient derived iPSCs. Investigative studies of such work is underway (131).
Some patients with the clinical diagnosis of BBS have not yet had the specific molecular cause of their disease identified. It is possible that causal mutations lie in regulatory or non-exonic regions, as well as in alternatively spliced isoforms of known BBS genes; additional sequencing and molecular characterization will be required to identify these variants. It is also possible that these patients harbor mutations in genes that cause BBS that have yet to be identified as such. It may be worth cataloging interactors of the BBSome to identify potential sequencing targets for patients. Future work could involve identifying cargos of the BBSome, other interactors, or other genes with ciliary function in order to determine their likelihood of causing BBS.
5. Conclusions
In summary, we have discussed the diagnostic criteria for BBS, the mode of inheritance of this disorder, and the primary phenotype seen in BBS patients, retinal degeneration. We also reviewed the molecular mechanisms that lead to retinal degeneration in these patients. Animal models have proven to be essential for the study of the outer segment and the role of protein mislocalization in the development of this phenotype. In addition to the animal models, in vitro studies have isolated specific receptors in the cilium that are affected by changes to BBS genes, suggesting a possible role of BBS on their ciliary localization and ability to respond normally to extracellular signaling. Retina-specific transcriptional isoforms have been identified in three BBS genes, with two showing an association to retinal degeneration. We have also reviewed the role of mutations in BBS genes in associated but clinically distinct ciliopathies such as Leber Congenital Amaurosis, Joubert syndrome, Senior-Løken syndrome, and non-syndromic retinitis pigmentosa. Furthermore, we have discussed potential approaches to answer current gaps in knowledge surrounding BBSome-associated and non-BBSome associated genes and how these two separate pools of genes can lead to shared phenotypes. As we continue to study this disease and its current proteins, we will gain a deeper understanding of the molecular mechanisms, potentially identify additional genes that cause this disorder, and allow for the development of therapeutic treatments to better the vision quality in BBS patients.
Acknowledgments
We would like to thank Armin Sokocevic for his help with the illustration of Figure 2.
This work was supported by NIH grants R01 EY-011298 and RO1 EY-017168 to VCS and by the Roy J. Carver Charitable Trust to VCS and EMS.
References
- 1.Sheffield VC, Carmi R, Kwitek-Black A, Rokhlina T, Nishimura D, Duyk GM, et al. Identification of a Bardet-Biedl syndrome locus on chromosome 3 and evaluation of an efficient approach to homozygosity mapping. Hum Mol Genet. 1994;3(8):1331–5. doi: 10.1093/hmg/3.8.1331. [DOI] [PubMed] [Google Scholar]
- 2.Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437–46. [PMC free article] [PubMed] [Google Scholar]
- 3.Sheffield V, Zhang Q, Heon E, Drack AV, Stone EM, Carmi R. The Bardet-Biedl Syndrome. In: Erickson RP, Wynshaw-Boris AJ, editors. Epstein’s inborn errors of development : the molecular basis of clinical disorders of morphogenesis. Third. p. xlvii. 1498 pages. [Google Scholar]
- 4.Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425(6958):628–33. doi: 10.1038/nature02030. [DOI] [PubMed] [Google Scholar]
- 5.Badano JL, Ansley SJ, Leitch CC, Lewis RA, Lupski JR, Katsanis N. Identification of a novel Bardet-Biedl syndrome protein, BBS7, that shares structural features with BBS1 and BBS2. Am J Hum Genet. 2003;72(3):650–8. doi: 10.1086/368204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li JB, Gerdes JM, Haycraft CJ, Fan Y, Teslovich TM, May-Simera H, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117(4):541–52. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- 7.Mykytyn K, Braun T, Carmi R, Haider NB, Searby CC, Shastri M, et al. Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat Genet. 2001;28(2):188–91. doi: 10.1038/88925. [DOI] [PubMed] [Google Scholar]
- 8.Mykytyn K, Nishimura DY, Searby CC, Shastri M, Yen HJ, Beck JS, et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet. 2002;31(4):435–8. doi: 10.1038/ng935. [DOI] [PubMed] [Google Scholar]
- 9.Nishimura DY, Searby CC, Carmi R, Elbedour K, Van Maldergem L, Fulton AB, et al. Positional cloning of a novel gene on chromosome 16q causing Bardet-Biedl syndrome (BBS2) Hum Mol Genet. 2001;10(8):865–74. doi: 10.1093/hmg/10.8.865. [DOI] [PubMed] [Google Scholar]
- 10.Nishimura DY, Swiderski RE, Searby CC, Berg EM, Ferguson AL, Hennekam R, et al. Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. Am J Hum Genet. 2005;77(6):1021–33. doi: 10.1086/498323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheidecker S, Etard C, Pierce NW, Geoffroy V, Schaefer E, Muller J, et al. Exome sequencing of Bardet-Biedl syndrome patient identifies a null mutation in the BBSome subunit BBIP1 (BBS18) J Med Genet. 2014;51(2):132–6. doi: 10.1136/jmedgenet-2013-101785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, et al. The Conserved Bardet-Biedl Syndrome Proteins Assemble a Coat that Traffics Membrane Proteins to Cilia. Cell. 2010;141(7):1208–19. doi: 10.1016/j.cell.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Q, Yu D, Seo S, Stone EM, Sheffield VC. Intrinsic protein-protein interaction-mediated and chaperonin- assisted sequential assembly of stable bardet-biedl syndrome protein complex, the BBSome. The Journal of biological chemistry. 2012;287(24):20625–35. doi: 10.1074/jbc.M112.341487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slavotinek AM, Stone EM, Mykytyn K, Heckenlively JR, Green JS, Heon E, et al. Mutations in MKKS cause Bardet-Biedl syndrome. Nat Genet. 2000;26(1):15–6. doi: 10.1038/79116. [DOI] [PubMed] [Google Scholar]
- 15.Stoetzel C, Muller J, Laurier V, Davis EE, Zaghloul NA, Vicaire S, et al. Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am J Hum Genet. 2007;80(1):1–11. doi: 10.1086/510256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seo S, Baye LM, Schulz NP, Beck JS, Zhang Q, Slusarski DC, et al. BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proceedings of the National Academy of Sciences. 2010;107(4):1488–93. doi: 10.1073/pnas.0910268107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan Y, Esmail MA, Ansley SJ, Blacque OE, Boroevich K, Ross AJ, et al. Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat Genet. 2004;36(9):989–93. doi: 10.1038/ng1414. [DOI] [PubMed] [Google Scholar]
- 18.Marion V, Stutzmann F, Gerard M, De Melo C, Schaefer E, Claussmann A, et al. Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet–Biedl syndrome with situs inversus and insertional polydactyly. J Med Genet. 2012;49(5):317–21. doi: 10.1136/jmedgenet-2012-100737. [DOI] [PubMed] [Google Scholar]
- 19.Seo S, Zhang Q, Bugge K, Breslow DK, Searby CC, Nachury MV, et al. A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened. PLoS genetics. 2011;7(11):e1002358. doi: 10.1371/journal.pgen.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stowe TR, Wilkinson CJ, Iqbal A, Stearns T. The centriolar satellite proteins Cep72 and Cep290 interact and are required for recruitment of BBS proteins to the cilium. Mol Biol Cell. 2012;23(17):3322–35. doi: 10.1091/mbc.E12-02-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbelanne M, Hossain D, Chan DP, Peranen J, Tsang WY. Nephrocystin proteins NPHP5 and Cep290 regulate BBSome integrity, ciliary trafficking and cargo delivery. Hum Mol Genet. 2015;24(8):2185–200. doi: 10.1093/hmg/ddu738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiang AP, Beck JS, Yen HJ, Tayeh MK, Scheetz TE, Swiderski RE, et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11) Proc Natl Acad Sci U S A. 2006;103(16):6287–92. doi: 10.1073/pnas.0600158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xing D-J, Zhang H-X, Huang N, Wu K-C, Huang X-F, Huang F, et al. Comprehensive Molecular Diagnosis of Bardet-Biedl Syndrome by High-Throughput Targeted Exome Sequencing. PLOS ONE. 2014;9(3):e90599. doi: 10.1371/journal.pone.0090599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008;40(4):443–8. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 25.Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, et al. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science (New York, NY) 2010;329(5997):1337–40. doi: 10.1126/science.1191184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otto EA, Hurd TW, Airik R, Chaki M, Zhou W, Stoetzel C, et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet. 2010;42(10):840–50. doi: 10.1038/ng.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aldahmesh MA, Li Y, Alhashem A, Anazi S, Alkuraya H, Hashem M, et al. IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet-Biedl syndrome. Hum Mol Genet. 2014;23(12):3307–15. doi: 10.1093/hmg/ddu044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindstrand A, Frangakis S, Carvalho CM, Richardson EB, McFadden KA, Willer JR, et al. Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome. Am J Hum Genet. 2016;99(2):318–36. doi: 10.1016/j.ajhg.2015.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heon E, Kim G, Qin S, Garrison JE, Tavares E, Vincent A, et al. Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21) Hum Mol Genet. 2016;25(11):2283–94. doi: 10.1093/hmg/ddw096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis RE, Swiderski RE, Rahmouni K, Nishimura DY, Mullins RF, Agassandian K, et al. A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc Natl Acad Sci U S A. 2007;104(49):19422–7. doi: 10.1073/pnas.0708571104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Datta P, Allamargot C, Hudson JS, Andersen EK, Bhattarai S, Drack AV, et al. Accumulation of non-outer segment proteins in the outer segment underlies photoreceptor degeneration in Bardet-Biedl syndrome. Proc Natl Acad Sci U S A. 2015;112(32):E4400–9. doi: 10.1073/pnas.1510111112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharif AS, Zhou J, Chen Q, Zhang W, Loertscher S, Nguyen K, et al., editors. The Association for Research in Vision and Ophthalmology. Seattle, WA2016: 2016. May, C8orf37 knockout mice display abnormal photoreceptor outer segment morphogenesis and progressive photoreceptor degeneration. [Google Scholar]
- 33.Forsythe E, Beales PL. Bardet-Biedl Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 34.Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013;21(1):8–13. doi: 10.1038/ejhg.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mockel A, Perdomo Y, Stutzmann F, Letsch J, Marion V, Dollfus H. Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies. Prog Retin Eye Res. 2011;30(4):258–74. doi: 10.1016/j.preteyeres.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 36.Stoler JM, Herrin JT, Holmes LB. Genital abnormalities in females with Bardet-Biedl syndrome. Am J Med Genet. 1995;55(3):276–8. doi: 10.1002/ajmg.1320550306. [DOI] [PubMed] [Google Scholar]
- 37.Klein D, Ammann F. The syndrome of Laurence-Moon-Bardet-Biedl and allied diseases in Switzerland. Clinical, genetic and epidemiological studies. J Neurol Sci. 1969;9(3):479–513. doi: 10.1016/0022-510x(69)90091-4. [DOI] [PubMed] [Google Scholar]
- 38.Imhoff O, Marion V, Stoetzel C, Durand M, Holder M, Sigaudy S, et al. Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin J Am Soc Nephrol. 2011;6(1):22–9. doi: 10.2215/CJN.03320410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheffield VC. The blind leading the obese: the molecular pathophysiology of a human obesity syndrome. Trans Am Clin Climatol Assoc. 2010;121:172–81. discussion 81-2. [PMC free article] [PubMed] [Google Scholar]
- 40.Guo DF, Rahmouni K. Molecular basis of the obesity associated with Bardet-Biedl syndrome. Trends Endocrinol Metab. 2011;22(7):286–93. doi: 10.1016/j.tem.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Putoux A, Attie-Bitach T, Martinovic J, Gubler MC. Phenotypic variability of Bardet-Biedl syndrome: focusing on the kidney. Pediatr Nephrol. 2012;27(1):7–15. doi: 10.1007/s00467-010-1751-3. [DOI] [PubMed] [Google Scholar]
- 42.Zaghloul NA, Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest. 2009;119(3):428–37. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Novas R, Cardenas-Rodriguez M, Irigoin F, Badano JL. Bardet-Biedl syndrome: Is it only cilia dysfunction? FEBS Lett. 2015;589(22):3479–91. doi: 10.1016/j.febslet.2015.07.031. [DOI] [PubMed] [Google Scholar]
- 44.Farag TI, Teebi AS. High incidence of Bardet Biedl syndrome among the Bedouin. Clinical Genetics. 1989;36(6):463–4. doi: 10.1111/j.1399-0004.1989.tb03378.x. [DOI] [PubMed] [Google Scholar]
- 45.M’Hamdi O, Ouertani I, Maazoul F, Chaabouni-Bouhamed H. Prevalence of Bardet-Biedl syndrome in Tunisia. Journal of community genetics. 2011;2(2):97–9. doi: 10.1007/s12687-011-0040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stone EM, Andorf JL, Whitmore SS, DeLuca AP, Giacalone JC, Streb LM, et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology. 2017 doi: 10.1016/j.ophtha.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, Hoskins BE, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science (New York, NY) 2001;293(5538):2256–9. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- 48.Beales PL, Badano JL, Ross AJ, Ansley SJ, Hoskins BE, Kirsten B, et al. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am J Hum Genet. 2003;72(5):1187–99. doi: 10.1086/375178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hichri H, Stoetzel C, Laurier V, Caron S, Sigaudy S, Sarda P, et al. Testing for triallelism: analysis of six BBS genes in a Bardet-Biedl syndrome family cohort. Eur J Hum Genet. 2005;13(5):607–16. doi: 10.1038/sj.ejhg.5201372. [DOI] [PubMed] [Google Scholar]
- 50.Nakane T, Biesecker LG. No evidence for triallelic inheritance of MKKS/BBS loci in Amish Mckusick-Kaufman syndrome. American journal of medical genetics Part A. 2005;138(1):32–4. doi: 10.1002/ajmg.a.30593. [DOI] [PubMed] [Google Scholar]
- 51.Abu-Safieh L, Al-Anazi S, Al-Abdi L, Hashem M, Alkuraya H, Alamr M, et al. In search of triallelism in Bardet-Biedl syndrome. Eur J Hum Genet. 2012;20(4):420–7. doi: 10.1038/ejhg.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Q, Nishimura D, Seo S, Vogel T, Morgan DA, Searby C, et al. Bardet-Biedl syndrome 3 (Bbs3) knockout mouse model reveals common BBS-associated phenotypes and Bbs3 unique phenotypes. Proc Natl Acad Sci U S A. 2011;108(51):20678–83. doi: 10.1073/pnas.1113220108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Q, Nishimura D, Vogel T, Shao J, Swiderski R, Yin T, et al. BBS7 is required for BBSome formation and its absence in mice results in Bardet-Biedl syndrome phenotypes and selective abnormalities in membrane protein trafficking. Journal of Cell Science. 2013;126(11):2372–80. doi: 10.1242/jcs.111740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(47):16588–93. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Airik R, Slaats GG, Guo Z, Weiss AC, Khan N, Ghosh A, et al. Renal-retinal ciliopathy gene Sdccag8 regulates DNA damage response signaling. Journal of the American Society of Nephrology : JASN. 2014;25(11):2573–83. doi: 10.1681/ASN.2013050565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Insolera R, Shao W, Airik R, Hildebrandt F, Shi SH. SDCCAG8 regulates pericentriolar material recruitment and neuronal migration in the developing cortex. Neuron. 2014;83(4):805–22. doi: 10.1016/j.neuron.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cognard N, Scerbo MJ, Obringer C, Yu X, Costa F, Haser E, et al. Comparing the Bbs10 complete knockout phenotype with a specific renal epithelial knockout one highlights the link between renal defects and systemic inactivation in mice. Cilia. 2015;4(1):10. doi: 10.1186/s13630-015-0019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mykytyn K, Mullins RF, Andrews M, Chiang AP, Swiderski RE, Yang B, et al. Bardet–Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(23):8664–9. doi: 10.1073/pnas.0402354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Badano JL, Kim JC, Hoskins BE, Lewis RA, Ansley SJ, Cutler DJ, et al. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum Mol Genet. 2003;12(14):1651–9. doi: 10.1093/hmg/ddg188. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y, Seo S, Bhattarai S, Bugge K, Searby CC, Zhang Q, et al. BBS mutations modify phenotypic expression of CEP290-related ciliopathies. Hum Mol Genet. 2014;23(1):40–51. doi: 10.1093/hmg/ddt394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bin J, Madhavan J, Ferrini W, Mok CA, Billingsley G, Heon E. BBS7 and TTC8 (BBS8) mutations play a minor role in the mutational load of Bardet-Biedl syndrome in a multiethnic population. Hum Mutat. 2009;30(7):E737–46. doi: 10.1002/humu.21040. [DOI] [PubMed] [Google Scholar]
- 62.Estrada-Cuzcano A, Roepman R, Cremers FP, den Hollander AI, Mans DA. Non-syndromic retinal ciliopathies: translating gene discovery into therapy. Hum Mol Genet. 2012;21(R1):R111–24. doi: 10.1093/hmg/dds298. [DOI] [PubMed] [Google Scholar]
- 63.Denniston AK, Beales PL, Tomlins PJ, Good P, Langford M, Foggensteiner L, et al. Evaluation of visual function and needs in adult patients with bardet-biedl syndrome. Retina (Philadelphia, Pa) 2014;34(11):2282–9. doi: 10.1097/IAE.0000000000000222. [DOI] [PubMed] [Google Scholar]
- 64.Adams NA, Awadein A, Toma HS. The retinal ciliopathies. Ophthalmic Genet. 2007;28(3):113–25. doi: 10.1080/13816810701537424. [DOI] [PubMed] [Google Scholar]
- 65.Berezovsky A, Rocha DM, Sacai PY, Watanabe SS, Cavascan NN, Salomao SR. Visual acuity and retinal function in patients with Bardet-Biedl syndrome. Clinics (Sao Paulo, Brazil) 2012;67(2):145–9. doi: 10.6061/clinics/2012(02)09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fulton AB, Hansen RM, Glynn RJ. Natural course of visual functions in the Bardet-Biedl syndrome. Arch Ophthalmol. 1993;111(11):1500–6. doi: 10.1001/archopht.1993.01090110066026. [DOI] [PubMed] [Google Scholar]
- 67.Hamel CP. Cone rod dystrophies. Orphanet J Rare Dis. 2007;2:7. doi: 10.1186/1750-1172-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Azari AA, Aleman TS, Cideciyan AV, Schwartz SB, Windsor EA, Sumaroka A, et al. Retinal disease expression in Bardet-Biedl syndrome-1 (BBS1) is a spectrum from maculopathy to retina-wide degeneration. Invest Ophthalmol Vis Sci. 2006;47(11):5004–10. doi: 10.1167/iovs.06-0517. [DOI] [PubMed] [Google Scholar]
- 69.Tadenev AL, Kulaga HM, May-Simera HL, Kelley MW, Katsanis N, Reed RR. Loss of Bardet-Biedl syndrome protein-8 (BBS8) perturbs olfactory function, protein localization, and axon targeting. Proc Natl Acad Sci U S A. 2011;108(25):10320–5. doi: 10.1073/pnas.1016531108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Veleri S, Bishop K, Dalle Nogare DE, English MA, Foskett TJ, Chitnis A, et al. Knockdown of Bardet-Biedl Syndrome Gene BBS9/PTHB1 Leads to Cilia Defects. PLOS ONE. 2012;7(3):e34389. doi: 10.1371/journal.pone.0034389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marion V, Mockel A, De Melo C, Obringer C, Claussmann A, Simon A, et al. BBS-Induced Ciliary Defect Enhances Adipogenesis, Causing Paradoxical Higher-Insulin Sensitivity, Glucose Usage, and Decreased Inflammatory Response. Cell Metabolism. 2012;16(3):363–77. doi: 10.1016/j.cmet.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 72.Fath MA, Mullins RF, Searby C, Nishimura DY, Wei J, Rahmouni K, et al. Mkks-null mice have a phenotype resembling Bardet-Biedl syndrome. Hum Mol Genet. 2005;14(9):1109–18. doi: 10.1093/hmg/ddi123. [DOI] [PubMed] [Google Scholar]
- 73.Abd-El-Barr MM, Sykoudis K, Andrabi S, Eichers ER, Pennesi ME, Tan PL, et al. Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet–Biedl syndrome. Vision Research. 2007;47(27):3394–407. doi: 10.1016/j.visres.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mokrzan EM, Lewis JS, Mykytyn K. Differences in renal tubule primary cilia length in a mouse model of Bardet-Biedl syndrome. Nephron Exp Nephrol. 2007;106(3):e88–96. doi: 10.1159/000103021. [DOI] [PubMed] [Google Scholar]
- 75.Eichers ER, Abd-El-Barr MM, Paylor R, Lewis RA, Bi W, Lin X, et al. Phenotypic characterization of Bbs4 null mice reveals age-dependent penetrance and variable expressivity. Hum Genet. 2006;120(2):211–26. doi: 10.1007/s00439-006-0197-y. [DOI] [PubMed] [Google Scholar]
- 76.Jiang J, Promchan K, Jiang H, Awasthi P, Marshall H, Harned A, et al. Depletion of BBS Protein LZTFL1 Affects Growth and Causes Retinal Degeneration in Mice. Journal of Genetics and Genomics. 2016;43(6):381–91. doi: 10.1016/j.jgg.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pretorius PR, Baye LM, Nishimura DY, Searby CC, Bugge K, Yang B, et al. Identification and functional analysis of the vision-specific BBS3 (ARL6) long isoform. PLoS genetics. 2010;6(3):e1000884. doi: 10.1371/journal.pgen.1000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kudryashova E, Wu J, Havton LA, Spencer MJ. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Human Molecular Genetics. 2009;18(7):1353–67. doi: 10.1093/hmg/ddp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wheway G, Abdelhamed Z, Natarajan S, Toomes C, Inglehearn C, Johnson CA. Aberrant Wnt signalling and cellular over-proliferation in a novel mouse model of Meckel–Gruber syndrome. Developmental Biology. 2013;377(1):55–66. doi: 10.1016/j.ydbio.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 80.Chang B, Khanna H, Hawes N, Jimeno D, He S, Lillo C, et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum Mol Genet. 2006;15(11):1847–57. doi: 10.1093/hmg/ddl107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cui C, Chatterjee B, Lozito TP, Zhang Z, Francis RJ, Yagi H, et al. Wdpcp, a PCP Protein Required for Ciliogenesis, Regulates Directional Cell Migration and Cell Polarity by Direct Modulation of the Actin Cytoskeleton. PLOS Biology. 2013;11(11):e1001720. doi: 10.1371/journal.pbio.1001720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Swiderski RE, Nishimura DY, Mullins RF, Olvera MA, Ross JL, Huang J, et al. Gene expression analysis of photoreceptor cell loss in bbs4-knockout mice reveals an early stress gene response and photoreceptor cell damage. Invest Ophthalmol Vis Sci. 2007;48(7):3329–40. doi: 10.1167/iovs.06-1477. [DOI] [PubMed] [Google Scholar]
- 83.Horst CJ, Johnson LV, Besharse JC. Transmembrane assemblage of the photoreceptor connecting cilium and motile cilium transition zone contain a common immunologic epitope. Cell Motil Cytoskeleton. 1990;17(4):329–44. doi: 10.1002/cm.970170408. [DOI] [PubMed] [Google Scholar]
- 84.Yen HJ, Tayeh MK, Mullins RF, Stone EM, Sheffield VC, Slusarski DC. Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer’s vesicle cilia function. Hum Mol Genet. 2006;15(5):667–77. doi: 10.1093/hmg/ddi468. [DOI] [PubMed] [Google Scholar]
- 85.Wei Q, Zhang Y, Li Y, Zhang Q, Ling K, Hu J. The BBSome controls IFT assembly and turnaround in cilia. Nat Cell Biol. 2012;14(9):950–7. doi: 10.1038/ncb2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seo S, Guo DF, Bugge K, Morgan DA, Rahmouni K, Sheffield VC. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet. 2009;18(7):1323–31. doi: 10.1093/hmg/ddp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Starks RD, Beyer AM, Guo DF, Boland L, Zhang Q, Sheffield VC, et al. Regulation of Insulin Receptor Trafficking by Bardet Biedl Syndrome Proteins. PLoS genetics. 2015;11(6):e1005311. doi: 10.1371/journal.pgen.1005311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci U S A. 2008;105(11):4242–6. doi: 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Loktev AV, Jackson PK. Neuropeptide Y family receptors traffic via the Bardet-Biedl syndrome pathway to signal in neuronal primary cilia. Cell Rep. 2013;5(5):1316–29. doi: 10.1016/j.celrep.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 90.Xu Q, Zhang Y, Wei Q, Huang Y, Li Y, Ling K, et al. BBS4 and BBS5 show functional redundancy in the BBSome to regulate the degradative sorting of ciliary sensory receptors. Sci Rep. 2015;5:11855. doi: 10.1038/srep11855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–49. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 92.Bolch SN, Dugger DR, Chong T, McDowell JH, Smith WC. A Splice Variant of Bardet-Biedl Syndrome 5 (BBS5) Protein that Is Selectively Expressed in Retina. PLoS One. 2016;11(2):e0148773. doi: 10.1371/journal.pone.0148773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Riazuddin SA, Iqbal M, Wang Y, Masuda T, Chen Y, Bowne S, et al. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am J Hum Genet. 2010;86(5):805–12. doi: 10.1016/j.ajhg.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Farkas MH, Grant GR, White JA, Sousa ME, Consugar MB, Pierce EA. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genomics. 2013;14(1):486. doi: 10.1186/1471-2164-14-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chung DC, Traboulsi EI. Leber congenital amaurosis: clinical correlations with genotypes, gene therapy trials update, and future directions. J AAPOS. 2009;13(6):587–92. doi: 10.1016/j.jaapos.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 96.Heher KL, Traboulsi EI, Maumenee IH. The natural history of Leber’s congenital amaurosis. Age-related findings in 35 patients. Ophthalmology. 1992;99(2):241–5. doi: 10.1016/s0161-6420(92)31985-2. [DOI] [PubMed] [Google Scholar]
- 97.Koenekoop RK. An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv Ophthalmol. 2004;49(4):379–98. doi: 10.1016/j.survophthal.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 98.Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol. 2011;26(7):1039–56. doi: 10.1007/s00467-010-1731-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Weleber RG, Francis PJ, Trzupek KM, Beattie C. Leber Congenital Amaurosis. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 100.Maria BL, Boltshauser E, Palmer SC, Tran TX. Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol. 1999;14(9):583–90. doi: 10.1177/088307389901400906. discussion 90-1. [DOI] [PubMed] [Google Scholar]
- 101.Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19(9):813–25. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- 102.Parisi M, Glass I. Joubert Syndrome and Related Disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 103.Doherty D. Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin Pediatr Neurol. 2009;16(3):143–54. doi: 10.1016/j.spen.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hildebrandt F, Nothwang HG, Vossmerbaumer U, Springer C, Strahm B, Hoppe B, et al. Lack of large, homozygous deletions of the nephronophthisis 1 region in Joubert syndrome type B. APN Study Group. Arbeitsgemeinschaft fur Padiatrische Nephrologie. Pediatr Nephrol. 1998;12(1):16–9. doi: 10.1007/s004670050394. [DOI] [PubMed] [Google Scholar]
- 105.Tusa RJ, Hove MT. Ocular and oculomotor signs in Joubert syndrome. J Child Neurol. 1999;14(10):621–7. doi: 10.1177/088307389901401001. [DOI] [PubMed] [Google Scholar]
- 106.van Dorp DB, Palan A, Kwee ML, Barth PG, van der Harten JJ. Joubert syndrome: a clinical and pathological description of an affected male and a female fetus from the same sibship. Am J Med Genet. 1991;40(1):100–4. doi: 10.1002/ajmg.1320400121. [DOI] [PubMed] [Google Scholar]
- 107.Saraiva JM, Baraitser M. Joubert syndrome: a review. Am J Med Genet. 1992;43(4):726–31. doi: 10.1002/ajmg.1320430415. [DOI] [PubMed] [Google Scholar]
- 108.Chatterjee S, Lufkin T. The Sound of Silence: Mouse Models for Hearing Loss. Genetics Research International. 2011;2011:9. doi: 10.4061/2011/416450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Parisi M, Glass I. Joubert Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 110.Loken AC, Hanssen O, Halvorsen S, Jolster NJ. Hereditary renal dysplasia and blindness. Acta Paediatr. 1961;50:177–84. doi: 10.1111/j.1651-2227.1961.tb08037.x. [DOI] [PubMed] [Google Scholar]
- 111.Senior B, Friedmann AI, Braudo JL. Juvenile familial nephropathy with tapetoretinal degeneration. A new oculorenal dystrophy. Am J Ophthalmol. 1961;52:625–33. doi: 10.1016/0002-9394(61)90147-7. [DOI] [PubMed] [Google Scholar]
- 112.Mongeau JG, Worthen HG. Nephronophthisis and medullary cystic disease. Am J Med. 1967;43(3):345–55. doi: 10.1016/0002-9343(67)90191-x. [DOI] [PubMed] [Google Scholar]
- 113.Strauss MB, Sommers SC. Medullary cystic disease and familial juvenile nephronophthisis. N Engl J Med. 1967;277(16):863–4. doi: 10.1056/NEJM196710192771606. [DOI] [PubMed] [Google Scholar]
- 114.Dekaban AS. Familial occurrence of congenital retinal blindness and developmental renal lesions. J Genet Hum. 1969;17(3):289–96. [PubMed] [Google Scholar]
- 115.Schimke RN. Hereditary renal-retinal dysplasia. Ann Intern Med. 1969;70(4):735–44. doi: 10.7326/0003-4819-70-4-735. [DOI] [PubMed] [Google Scholar]
- 116.Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005;37(3):282–8. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- 117.Perrault I, Delphin N, Hanein S, Gerber S, Dufier JL, Roche O, et al. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat. 2007;28(4):416. doi: 10.1002/humu.9485. [DOI] [PubMed] [Google Scholar]
- 118.Medhioub M, Cherif D, Benessy F, Silbermann F, Gubler MC, Le Paslier D, et al. Refined mapping of a gene (NPH1) causing familial juvenile nephronophthisis and evidence for genetic heterogeneity. Genomics. 1994;22(2):296–301. doi: 10.1006/geno.1994.1387. [DOI] [PubMed] [Google Scholar]
- 119.Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol. 2017;9(3) doi: 10.1101/cshperspect.a028191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38(6):674–81. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 121.Coussa RG, Otto EA, Gee HY, Arthurs P, Ren H, Lopez I, et al. WDR19: an ancient, retrograde, intraflagellar ciliary protein is mutated in autosomal recessive retinitis pigmentosa and in Senior-Loken syndrome. Clin Genet. 2013;84(2):150–9. doi: 10.1111/cge.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet. 2013;132(8):865–84. doi: 10.1007/s00439-013-1297-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bizet AA, Becker-Heck A, Ryan R, Weber K, Filhol E, Krug P, et al. Mutations in TRAF3IP1/IFT54 reveal a new role for IFT proteins in microtubule stabilization. Nat Commun. 2015;6:8666. doi: 10.1038/ncomms9666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Estrada-Cuzcano A, Koenekoop RK, Senechal A, De Baere EB, de Ravel T, Banfi S, et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Arch Ophthalmol. 2012;130(11):1425–32. doi: 10.1001/archophthalmol.2012.2434. [DOI] [PubMed] [Google Scholar]
- 125.Shevach E, Ali M, Mizrahi-Meissonnier L, McKibbin M, El-Asrag M, Watson CM, et al. Association between missense mutations in the BBS2 gene and nonsyndromic retinitis pigmentosa. JAMA Ophthalmol. 2015;133(3):312–8. doi: 10.1001/jamaophthalmol.2014.5251. [DOI] [PubMed] [Google Scholar]
- 126.Aldahmesh MA, Safieh LA, Alkuraya H, Al-Rajhi A, Shamseldin H, Hashem M, et al. Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol Vis. 2009;15:2464–9. [PMC free article] [PubMed] [Google Scholar]
- 127.Estrada-Cuzcano A, Neveling K, Kohl S, Banin E, Rotenstreich Y, Sharon D, et al. Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am J Hum Genet. 2012;90(1):102–9. doi: 10.1016/j.ajhg.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fenn TD, Schnieders MJ, Brunger AT, Pande VS. Polarizable atomic multipole x-ray refinement: hydration geometry and application to macromolecules. Biophys J. 2010;98(12):2984–92. doi: 10.1016/j.bpj.2010.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wiley LA, Burnight ER, DeLuca AP, Anfinson KR, Cranston CM, Kaalberg EE, et al. cGMP production of patient-specific iPSCs and photoreceptor precursor cells to treat retinal degenerative blindness. Sci Rep. 2016;6:30742. doi: 10.1038/srep30742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Seo S, Mullins RF, Dumitrescu AV, Bhattarai S, Gratie D, Wang K, et al. Subretinal Gene Therapy of Mice With Bardet-Biedl Syndrome Type 1Subretinal Gene Therapy of Mice With BBS1. Investigative Ophthalmology & Visual Science. 2013;54(9):6118–32. doi: 10.1167/iovs.13-11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tucker BA, Mullins RF, Stone EM. Stem cells for investigation and treatment of inherited retinal disease. Hum Mol Genet. 2014;23(R1):R9–R16. doi: 10.1093/hmg/ddu124. [DOI] [PMC free article] [PubMed] [Google Scholar]