

Abstract
DNA methylation by de novo DNA methyltransferases 3A (DNMT3A) and 3B (DNMT3B) is essential for genome regulation and development1, 2. Dysregulation of this process is implicated in various diseases, notably cancer. However, the mechanisms underlying DNMT3 substrate recognition and enzymatic specificity remain elusive. Here we report a 2.65-Å crystal structure of the DNMT3A-DNMT3L-DNA complex where two DNMT3A monomers simultaneously attack two CpG dinucleotides, with the target sites separated by fourteen base pairs within the same DNA duplex. The DNMT3A–DNA interaction involves a target recognition domain (TRD), a catalytic loop and DNMT3A homodimeric interface. A TRD residue Arg836 makes crucial contacts with CpG, ensuring DNMT3A enzymatic preference towards CpG sites in cells. Hematological cancer-associated somatic mutations of the substrate-binding residues decrease DNMT3A activity, induce CpG hypomethylation, and promote transformation of hematopoietic cells. Together, our study reveals the mechanistic basis for DNMT3A-mediated DNA methylation and establishes its etiologic link to human disease.
Mammalian DNA methylation is an important epigenetic mechanism crucial for gene silencing and imprinting, X-inactivation, genome stability, and cell fate determination3. It is established mainly at CpG dinucleotides by de novo methyltransferases DNMT3A and DNMT3B1, 2, and subsequently maintained by DNA methyltransferase 1 (DNMT1) in a replication-dependent manner4. The enzymatic function of DNMT3A and DNMT3B is further regulated by DNMT3-like protein (DNMT3L) in germ and embryonic stem cells (ESCs)5–7. Deregulation of DNMT3A and DNMT3B is associated with various human diseases including hematological cancer8–10. However, the molecular mechanisms underpinning DNMT3A-mediated methylation, especially substrate recognition and catalytic preference towards CpG, remain elusive. Here we generated a productive DNMT3A–DNMT3L–DNA complex using the C-terminal domains of DNMT3A and DNMT3L (Fig. 1a). The DNA molecule consists of a 10-mer, central CpG-containing DNA strand annealed to an 11-mer, 2′-deoxy-Zebularine (dZ)-containing strand (target strand), which results in a (CpG) • (dZpG) sequence context and permits formation of stable, covalent DNMT3A–DNA complexes (Extended Data Fig. 1a,b). The crystal structure of the DNMT3A–DNMT3L–10/11-mer DNA complex, bound to cofactor byproduct S-Adenosyl-L-homocysteine (AdoHcy), was subsequently determined at 3.1 Å resolution (Extended Data Fig. 1c).
Figure 1. Structure of the DNMT3A–DNMT3L tetramer in complex with a 25-mer DNA duplex containing two CpG sites.
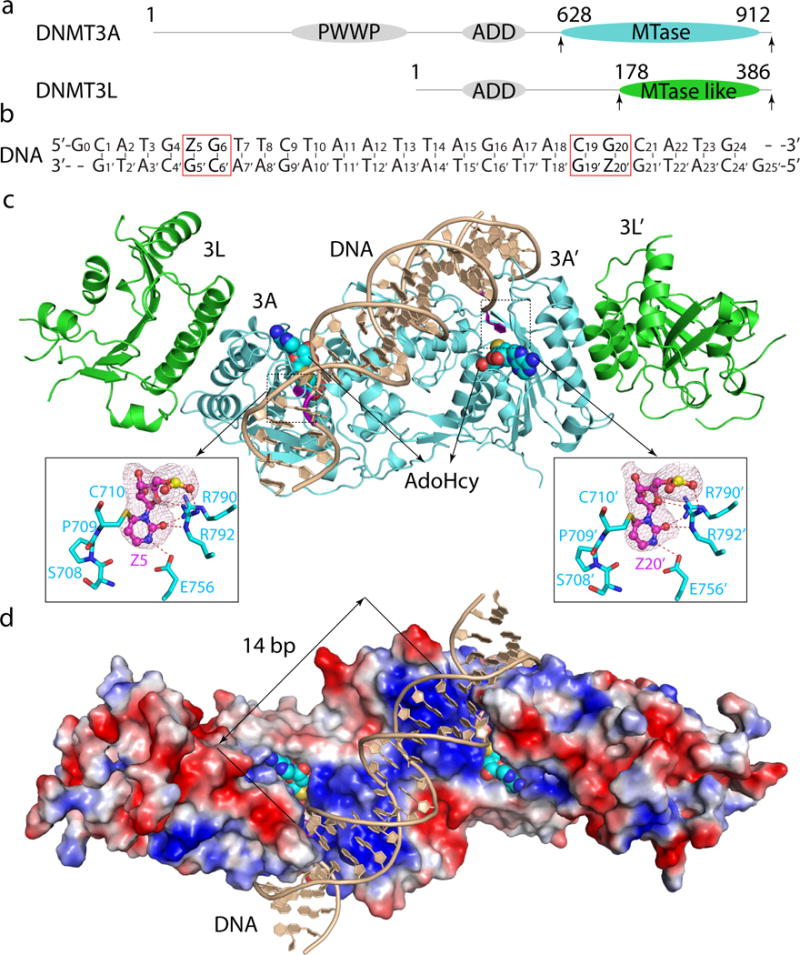
a, Domain architectures of DNMT3A and DNMT3L, with the C-terminal domains marked with arrowheads. b, DNA sequence used for structural study. Z, Zebularine. c, d, Ribbon (c) and surface (d) representations of DNMT3A–DNMT3L bound to DNA and AdoHcy. The Zebularines anchored at the two active sites are 14-bp away and shown in expanded views, with hydrogen-bonding interactions depicted as dashed lines and the Fo–Fc omit map (pink) of Z5 or Z20′ contoured at 3σ level.
The structure of this DNMT3A–DNMT3L–DNA complex reveals a tetrameric fold arranged in the order of DNMT3L–DNMT3A–DNMT3A–DNMT3L, reminiscent of its DNA-free form11, 12 (Extended Data Fig. 1d, 2a). Notably, two DNA duplexes, each bound to one DNMT3A monomer, are separated by ~15 Å, implying a total of 14-base pair (bp) spacing between the two active sites of DNMT3A (Extended Data Fig. 1d). This finding prompted us to design a longer DNA substrate involving a self-complementary 25-mer Zebularine (Z)-containing DNA, with two (CpG) • (ZpG) sites across 14 bp (Fig. 1b). The structure of the DNMT3A–DNMT3L–25-mer DNA complex was determined at 2.65 Å resolution (Extended Data Fig. 1c), revealing only one DNA duplex bending towards the DNMT3A-DNMT3L tetramer, with the two CpG sites simultaneously anchored by two DNMT3A monomers (Fig. 1c,d and Extended Data Fig. 2b). Our data thus support a notion that the two DNMT3A monomers can co-methylate two adjacent CpG dinucleotides in one DNA-binding event12, 13. Despite being crystalized under different conditions, both DNMT3A–DNMT3L–DNA complexes are well aligned with their DNA-free state, with a root-mean-square deviation of 0.87 Å and 1.12 Å over 790 and 826 Cα atoms, respectively (Extended Data Fig. 2a). Notably, in DNMT3A–DNMT3L–DNA complexes, Zebularines are flipped out of the DNA helix and inserted deep into DNMT3A catalytic pockets, where they are covalently anchored by the catalytic cysteine C710 and hydrogen bonded to E756, R790 and R792 (Fig. 1c and Extended Data Fig. 1d). Since both structures reveal productive reaction states with consistent protein–DNA interactions, we focus on the structure of DNMT3A–DNMT3L–25-mer DNA for further analysis.
Figure 2. Structure comparison of free and DNA-bound DNMT3A–DNMT3L tetramer.
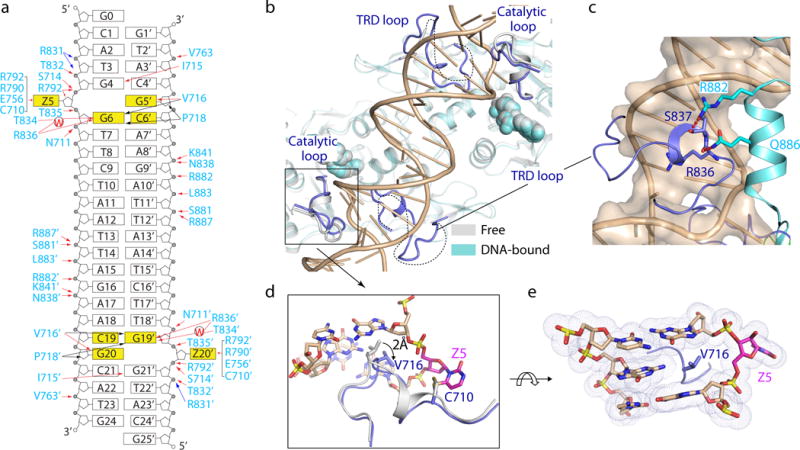
a, Schematic view of the intermolecular interactions between DNMT3A and DNA. The hydrogen-bonding, electrostatic and van der Waals contacts are represented by red, blue and black arrows, respectively. Water-mediated hydrogen bonds are labeled with letter ‘W’. b, Structural overlay of free (grey) and DNA-bound (cyan) DNMT3A–DNMT3L. The disordered TRD loops in free DNMT3A-DNMT3L are depicted as dotted lines. c, The TRD loop (blue) in DNA-bound DNMT3A is stabilized by hydrogen-bonding interactions (dashed lines) with R882 and Q886. d, Structural overlay of the catalytic loop between free (grey) and DNA-bound (blue) DNMT3A. e, DNA intercalation by DNMT3A V716.
DNMT3A binding to DNA is mainly mediated by a loop from the target recognition domain (TRD) (residues R831-F848), the catalytic loop (residues G707-K721) and the homodimeric interface of DNMT3A, which together create a continuous DNA-binding surface (Fig. 1d, 2a). Accordingly, these segments exhibit the most prominent structural changes upon DNA binding – the TRD loop lacked electron density in the DNA-free structure of DNMT3A–DNMT3L11, 12, but became well defined upon DNA binding and penetrated into the DNA major groove for intermolecular contacts (Fig. 2a–c); additionally, the TRD loop is stabilized through hydrogen-bonding interactions with R882, the DNMT3A mutational hotspot among leukemias9, 10, and Q886 from an adjacent helix (Fig. 2c). Meanwhile, the catalytic loop residue V716 moves towards the DNA minor groove by ~2 Å, intercalating into the DNA cavity vacated due to Zebularine base flipping (Fig. 2d,e). Although no protein-DNA contact was observed for DNMT3L, two DNMT3L-contacting helices of DNMT3A are preceded by DNA-binding loops (Extended Data Fig. 2c), reinforcing a notion that DNMT3L enhances DNMT3A functionality through stabilizing its DNA-binding sites12.
Recognition of CpG dinucleotides by DNMT3A is mediated by both catalytic and TRD loops. In particular, guanine of the target strand, G6 (G19′), is specified by a hydrogen bond between its O6 atom and the Nε atom of R836 from the TRD loop, as well as water-mediated hydrogen bonds between its N7 atom and the Nε and Oγ atoms of R836 and T834, respectively (Fig. 3a). Meanwhile, the catalytic loop approaches to the minor groove where the backbone carbonyl oxygen of V716 forms a hydrogen bond with the N2 atom of the unpaired guanine G5′ (G20) (Fig. 3b). Penetration of the catalytic loop also permits V716 and P718 to engage van de Waals contacts with the base of G6 (G19′), providing additional base-specific recognition (Fig. 3b). No protein interaction was associated with C6′ (C19) of the non-target strand, lending explanation for the observation that DNMT3A does not discriminate hemimethylated over unmethylated DNA2. Formation of the DNMT3A–DNA complex is also supported by various protein–DNA interactions flanking CpG, which involve electrostatic and/or hydrogen-bonding interactions of the TRD residues (R831, T832, T835, N838 and K841), catalytic loop residues (N711, S714 and I715) and DNMT3A–DNMT3A homodimeric interface residues (S881, R882, L883 and R887) with various DNA backbone or base sites (Fig. 2a and Extended Data Fig. 3a–f). These DNA-binding residues are highly conserved in DNMT3B (Extended Data Fig. 3g), suggesting a similar substrate engagement mechanism used by the DNMT3 family.
Figure 3. DNMT3A–CpG interactions.
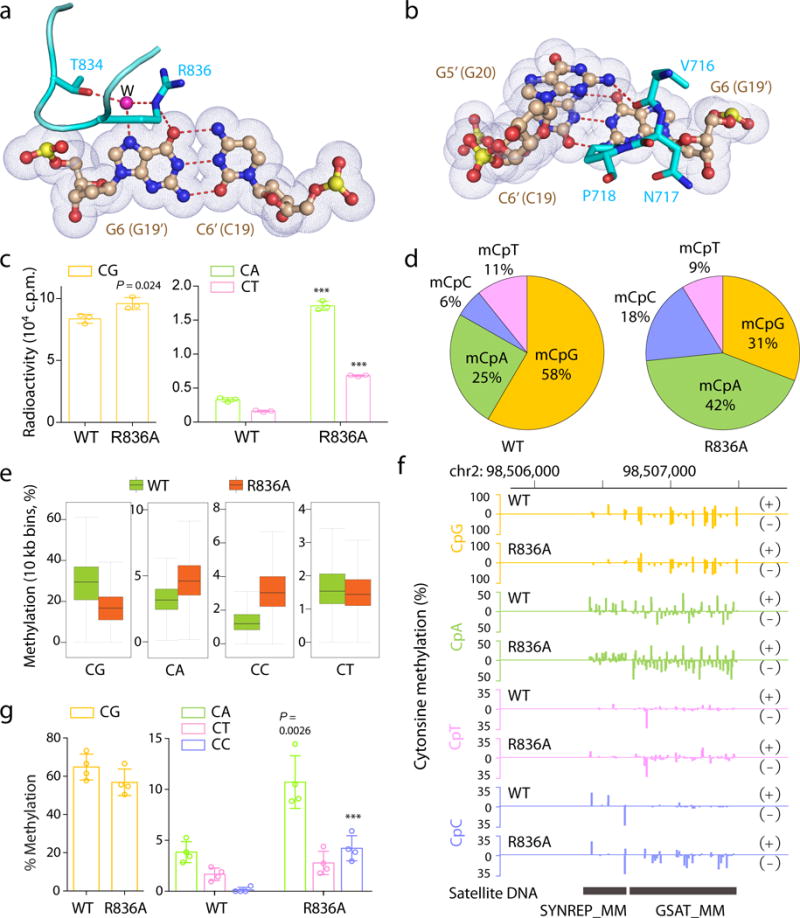
a, b, Interactions of R836 (a) and V716-P718 (b) with one CpG site. The hydrogen bonds and water molecule are shown as dashed lines and purple sphere, respectively. Nucleotides in the second CpG site are shown in parenthesis. c, 40-min in vitro methylation using DNMT3AWT–DNMT3L or DNMT3AR836A–DNMT3L complex (n = 3 biological replicates). d, Composition of all called methylated cytosines (mC) in TKO ESCs reconstituted with DNMT3AWT or DNMT3AR836A. An mC is defined using a binomial distribution-based filter with FDR less than 1% (n = 21,678,839 for WT and n = 25,272,362 for R836A). e, f, eRRBS revealing averaged CpG and non-CpG methylations, either at global levels (e) or at a representative major satellite DNA region (f), that are induced by DNMT3AWT versus DNMT3AR836A among 3 independent lines of TKO ESCs. Shown in e are boxplots of 10 kb-bin-averaged methylation levels for each sequence context. +/−, forward/reverse strand. Box plots and whiskers depict the interquartile and 1.5× interquartile ranges, respectively. g, Individual bisulfate sequencing detecting the methylation level of CG, CA, CT or CC sites within a major satellite DNA site at chromosome 2 in TKO cells expressing DNMT3AWT or DNMT3AR836A (n = 4). Data are mean ± s.d. Statistical analysis used two-tailed Student’s t-test. ***, p < 0.001.
To determine the roles for CpG-engaging residues R836 and V716 in regulation of DNMT3A activity, we performed mutagenesis followed by enzymatic studies using CpG-, CpA- or CpT-containing substrates (Fig. 3c and Extended Data Fig. 4a–b). First, wild-type (WT) DNMT3A showed methylation efficiency for CpG-containing DNA >20-fold higher than for CpA- or CpT-containing DNA, confirming its well-known CpG specificity14. In contrast, mutation of R836 to alanine (R836A) enhanced methylation of CpA- and CpT-containing DNA by 5.2- and 4.2-fold, respectively, but, as previously reported15, only led to slight change in CpG methylation. As a result, the relative CpG/CpA and CpG/CpT preference of the DNMT3AR836A enzyme was reduced by 4.5- and 3.7-fold, respectively, supporting a role for R836 in substrate specificity determination. In line with these observations, we solved the structure of DNMT3AR836A–DNMT3L–DNA complex, which lacks R836-mediated hydrogen bonds to CpG without causing overall structural alterations (Extended Data Fig. 4c). Meanwhile, mutation of V716 to glycine (V716G) abolished methylation of all tested substrates (Extended Data Fig. 4d). These observations support that R836-mediated CpG engagement contributes to substrate specificity whereas V716-mediated intercalation is essential for DNMT3A-mediated catalysis. The increased in vitro activity of DNMT3AR836A on CpA and CpT suggests that R836 might energetically influence enzymology of DNMT3A, in addition to target recognition. In the case of CpG DNA, such influence might be partly compensated by the R836-mediated hydrogen bond, thereby ensuring the CpG specificity of DNMT3A.
Next, we introduced comparable levels of DNMT3A, either WT or the above CpG-engagement-defective mutants, into ESCs with compound knockouts of DNMT1, DNMT3A and DNMT3B (TKO)16, and detected global increase in cytosine methylation after rescue with DNMT3AWT or DNMT3AR836A, but not DNMT3AV716G (Extended Data Fig. 4e,f). Furthermore, genome-wide methylation profiling with enhanced reduced representation bisulfite sequencing (eRRBS), followed by calling of methylation using the previously described binomial model and false discovery rate (FDR)-based threshold17, 18, revealed that, in TKO ESCs reconstituted with DNMT3AWT, 58% and 42% of methylated cytosines were presented at CpG and non-CpG sites, respectively (Fig. 3d and Extended Data Fig. 5a–c); in contrast, such distribution was reversed in cells expressing DNMT3AR836A, with 31% and 69% of methylated cytosines found at CpG and non-CpG contexts (Fig. 3d and Extended Data Fig. 5b,c). Consistently, relative to WT controls, the absolute methylation levels were found decreased at CpG but increased at CpA and CpC sites among cells with DNMT3AR836A, especially at sites showing intermediate to high levels of methylation (Fig. 3e and Extended Data Fig. 5d,6a). These changes were persistent among all chromosomes, at both DNA strands and over all annotated genes (Extended Data Fig. 6b–d), as exemplified by those detected at the major satellite DNA repeats (Fig. 3f) and gene-coding regions of Foxp1 and Dock1 (Extended Data Fig. 6e). Sanger bisulfite sequencing further validated eRRBS results at major satellite repeats in ESCs (Fig. 3g and Extended Data Fig. 7)19. Meanwhile, DNMT3AV716G abolished both CpG and non-CpG methylations at major satellite DNA (Extended Data Fig. 7b–d). The above observation that DNMT3AR836A decreases overall CpG methylations in TKO cells might be due to competition of non-CpG as potential substrate for this mutant enzyme. Collectively, we demonstrate that engaging CpG by the R836 side chain ensures DNMT3A substrate specificity.
Notably, heterozygous mutation of DNMT3A at its DNA-binding residues, such as S714, V716, P718, R792, T835, R836, N838, K841 and R882 (Fig. 2a and 4a,b), occurs recurrently in hematological cancer9, 10, 20 and overgrowth syndrome21. While recent studies support a dominant-negative effect of the hotspot R882H mutation on DNMT3A-mediated methylation possibly through affecting DNMT3A tetramerization22–25, our structural observation raises a possibility that interfering with the DNA binding via residue substitution also results in functional impairment of DNMT3A during pathogenesis. Indeed, in vitro enzymatic assays showed the significantly reduced activity for all tested DNA-binding mutants, with most pronounced effect observed for V716D, R792H and K841E (Fig. 4c and Extended Data Fig. 8a–d). Consistently, expression of these three mutants in TKO ESCs failed to restore global DNA methylation (Extended Data Fig. 8e–f). It is worth noting that, while DNMT3AR836W exhibited modestly reduced overall activities (Extended Data Fig. 8c, f), its activities for non-CpG methylations were found significantly increased at the major satellite DNA in TKO cells and in in vitro enzymatic assays (Extended Data Fig. 8g,h), suggesting a potential role of R836W in redistribution of CpG versus non-CpG methylations in diseased cells. Given a largely heterozygous feature of DNMT3A mutations in leukemia, we also queried whether the DNA-binding-defective mutants of DNMT3A inhibit functionality of DNMT3AWT. To test this, we turned to a co-expression system used previously for studying the domain-negative DNMT3AR882H mutant23, and reconstituted WT and mutant DNMT3A in equal amounts into TKO ESCs (Fig. 4d). Relative to expression of WT alone, co-expression of DNMT3AV716D, DNMT3AR792H or DNMT3AK841E with DNMT3AWT significantly decreased overall cytosine methylation (Fig. 4d). Together, we show that the DNMT3A mutants defective in substrate binding not only have decreased activity but also interfere with that of DNMT3AWT.
Figure 4. Hematological cancer-associated mutations of the DNMT3A–DNA interaction residues.
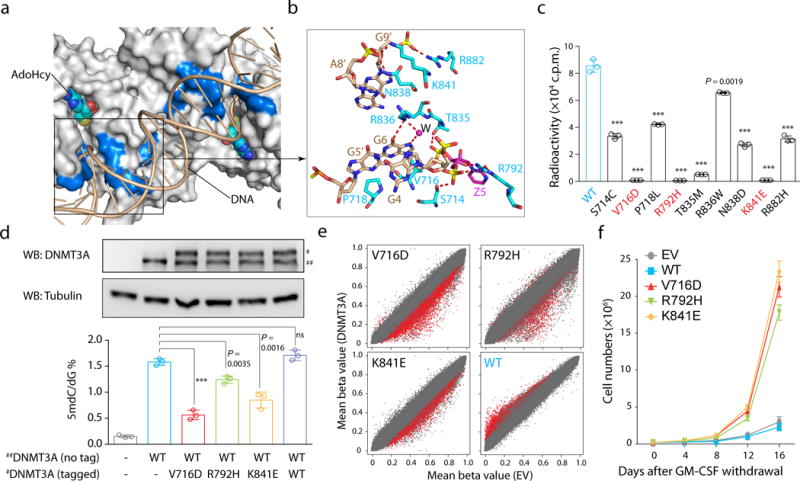
a, b, Surface (a) and stick (b) views of DNMT3A’s DNA-contacting residues found mutated in hematological cancer. Mutation sites are colored blue in a. The hydrogen-bonding interactions and water molecule are shown as dashed lines and purple sphere, respectively. c, In vitro methylation of CpG DNA using DNMT3AWT or its hematological cancer-associated mutants (n = 3). d, DNMT3A immunoblots (top) and LC-MS-based quantification of mC (bottom, n = 3 biological replicates) among TKO ESCs re-expressing DNMT3AWT alone (##, non-tagged) or together with the equal amount of the indicated DNMT3A mutant (#, tagged). Shown is a representative blot of two independent experiments. For gel source data, see Supplementary Figure 1. e, Scatter plots showing mean methylation beta values of each individual CpG among TF1 cells with stable expression of the indicated DNMT3A mutant (y-axis; n = 3-8 biological replicates) in comparison to empty vector controls (EV, x-axis), with significantly differentially methylated CpGs depicted in red. f, Proliferation of the indicated DNMT3A-expressing TF-1 cells under cytokine-poor conditions (n = 3). Data are mean ± s.d. Statistical analysis used two-tailed Student’s t-test. ***, p < 0.001.
We further ectopically expressed the above DNMT3A mutants in TF-1 cells, a model used for studying leukemia-associated gene mutation26. Through array profiling and bisulfite sequencing validation, we observed significant reduction of overall CpG methylations in TF1 cells stably expressing either DNMT3AV716D, DNMT3AR792H or DNMT3AK841E, relative to control; in contrast, ectopic expression of DNMT3AWT induced hyper-methylation (Fig. 4e and Extended Data Fig. 9). There is significant overlap among CpG sites showing hypo-methylation due to expression of DNMT3AV716D, DNMT3AR792H or DNMT3AK841E (Extended Data Fig. 10a), indicating their common effect on epigenomic deregulation. Reduced methylation of these commonly affected sites was also detected post-transduction of other leukemia-associated substrate-binding mutations of DNMT3A (P718L, T835M, R836W and N838D), although the latter did not induce hypo-methylation globally (Extended Data Fig 10b–c). Binding of WT or mutant DNMT3A was comparable at tested loci showing methylation changes (Extended Data Fig 10d–e). Given that epigenetic deregulation promotes TF-1 cell transformation characterized by cytokine-independent growth26, we queried whether the DNA-binding-defective mutation of DNMT3A causes similar transformation of this model. We found that, under cytokine-supporting conditions, TF-1 cells expressing WT or mutant DNMT3A exhibited comparable proliferation (Extended Data Fig. 10f). In contrast, those expressing a DNA-binding-defective mutant, but not DNMT3AWT, had significant cytokine-independent growth capability (Fig. 4f and Extended Data Fig. 10g). Collectively, we demonstrate that the DNA-binding residues of DNMT3A are vital for establishment of appropriate CpG methylation in hematological cells and that their somatic mutations detected in leukemia patients promote transformation.
Methods
Protein expression and purification
The gene fragments encoding residues 628-912 of human DNMT3A (NCBI accession NM_022552) and residues 178-386 of human DNMT3L were inserted in tandem into a modified pRSFDuet-1 vector (Novagen). The DNMT3A sequence was separated from the preceding His6-SUMO tag by a ubiquitin-like protease (ULP1) cleavage site. Expression and purification of the DNMT3A–DNMT3L complex followed a previously described protocol27. In short, the His6-SUMO-DNMT3A fusion protein and DNMT3L was co-expressed in E. coli BL21 DE3 (RIL) cell strains and purified using a Ni2+-NTA column. Subsequently, the His6-SUMO tag was removed through ULP1-mediated cleavage, followed by ion exchange chromatography on a Heparin column. For enzymatic assay, the DNMT3A–DNMT3L complex was further purified through size exclusion chromatography on a Superdex 200 16/60 column (GE Healthcare), and concentrated to 0.1-0.3 mM in a buffer containing 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 0.1% β-mercaptoethanol and 5% glycerol. To generate the covalent DNMT3A–DNMT3L–DNA complex, a 11-mer single-stranded DNA that was in-house synthesized to contain 2′-deoxy-Zebularine28 (5′- CATGdZGCTCTC -3′, dZ = 2′-deoxy-Zebularine) was annealed with a 10-mer single-stranded DNA (5′- AGAGCGCATG -3′) before reaction with the DNMT3A–DNMT3L complex in the presence of 20 mM Tris-HCl (pH 7.5), 50 mM NaCl, 20% Glycerol and 40 mM DTT at room temperature. In addition, a 25-mer Zebularine-containing DNA (5′- GCATGZGTTCTAATTAGAACGCATG -3′, Z = Zebularine) was self-annealed and used to form a second DNMT3A–DNMT3L–DNA complex or the DNMT3A (R836A)–DNMT3L–DNA complex. The reaction products were further purified through a HiTrap Q XL column (GE Healthcare), followed by size exclusion chromatography on a Superdex 200 16/60 column. The final samples for crystallization of the productive DNMT3A–DNMT3L–DNA complexes contain about 0.1-0.2 mM covalent DNMT3A–DNMT3L–DNA complexes, 0.3 mM AdoHCy, 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 0.1% β-mercaptoethanol and 5% glycerol.
Crystallization conditions and structure determination
The crystals for the covalent complex of DNMT3A–DNMT3L with the 10/11-mer DNA were generated by hanging-drop vapor-diffusion method at 23 °C, from drops mixed from 0.5 μl of DNMT3A–DNMT3L–DNA solution and 0.5 μl of precipitant solution [7% PEG4000, 0.1 M Tris-HCl (pH 8.5), 100 mM MgCl2, 166 mM imidazole (pH 7.0)]. The reproducibility and quality of crystals were further improved by the micro-seeding method. The crystals were soaked in cryoprotectant made of mother liquor supplemented with 30% PEG400, before flash frozen in liquid nitrogen. For the complex of DNMT3A (either wild-type or R836A mutant) with DNMT3L and the 25-mer DNA, crystals were generated by hanging-drop vapor diffusion method at 4 °C, from drops mixed from 1.5 μl of the protein solution and 1.5 μl of precipitation solution [0.1 M Tris-HCl (pH 7.0), 200 mM NaH2PO4 and 5% PEG4000]. The crystals were treated with cryoprotectant containing the precipitation solution and 30% glycerol before harvesting.
X-ray diffraction data sets for the covalent DNMT3A–DNMT3L–DNA complexes were collected at selenium peak wavelength on the BL501 or BL502 beamlines at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory, and the data set for the covalent DNMT3A (R836A)–DNMT3L–DNA complex was collected on the 24-ID-E NE-CAT beamline at the Advanced Photon Source (APS), Argonne National Laboratory. The diffraction data were indexed, integrated and scaled using the HKL 2000 program29 or the XDS program30. The structures of the productive covalent complexes of DNMT3A–DNMT3L–DNA were solved using the molecular replacement method in PHASER31, with the DNA-free structure of DNMT3A–DNMT3L (PDB 2QRV) serving as a search model. Further modeling of the covalent DNMT3A–DNMT3L–DNA complexes was carried out using COOT32 and then subject to refinement using the PHENIX software package33. The same R-free test set was used throughout the refinement. The final models for DNMT3A–DNMT3L complexed with the 25-mer and 10/11-mer DNAs were refined to 2.65 Å and 3.1 Å resolution, respectively. The final model for DNMT3A (R836A)–DNMT3L complexed with the 25-mer DNA was refined to 3.0 Å resolution.
The statistics for data collection and structural refinement of the productive covalent DNMT3A–DNMT3L–DNA complexes is summarized in Extended Data Fig. 1c.
In vitro DNA methylation assay
Synthesized (GAC)12, (AAC)12 and (TAC)12 DNA duplexes were used as CG-, CA- and CT-containing substrates, respectively. The DNA methylation assays were carried out in triplicate at 37 °C for 1 hr., unless otherwise indicated. Briefly, a 20-μL reaction mixture contained 2.5 μM S-adenosyl-L-[methyl-3H]methionine (AdoMet) (specific activity 18 Ci/mmol, PerkinElmer), 0.3 μM DNMT3A-DNMT3L, 0.75 μM DNA in 59 mM Tris-HCl, pH 8.0, 0.05% β-mercaptoethanol, 5% glycerol and 200 μg/mL BSA. The methylation reactions were stopped by flash freezing in liquid nitrogen, followed by precipitation and incubation on ice for 1 hr. in 1 ml of 15% trichloroacetic acid (TCA) solution plus 40 μg/ml BSA. The TCA-precipitated samples were then passed through a GF/C filter (GE Healthcare) using a vacuum-filtration apparatus. After sequential washing with 2 × 5 ml of cold 10% TCA and 5 ml of ethanol, the filters were dried and transferred to scintillation vials filled with 5 ml of ScintiVerse (Fisher), followed by measurement of tritium scintillation with a Beckman LS6500 counter.
Plasmid construction
Full-length human DNMT3A isoform 1 was cloned into EcoRI site of the pPyCAGIZ vector (a kind gift of Dr. Jianlong Wang, Ican School of Medicine at Mount Sinai). DNMT3A mutation was generated by QuikChange II XL Site-Directed Mutagenesis Kit (Agilent). To achieve co-expression of the wildtype (WT) and mutant DNMT3A at equal levels in cells, we engineered a T2A-based fusion construct consisting of the mutant cDNA, which was added with an N-terminal 3×Flag-(GGGGS)3-Myc tag to differentiate its protein size from non-tagged WT DNMT3A, followed by a T2A peptide sequence at its C-terminus and the cDNA of non-tagged WT DNMT3A. Myc-tagged full-length human DNMT3A isoform 1 were cloned into MSCV Pac retroviral vector as previously described24. All plasmid sequences were verified by sequencing.
Cell lines and cell culture
Dnmt3a, Dnmt3b and Dnmt1 triple knockout (TKO) mouse ES cells (a kind gift from Dr. Masaki Okano, RIKEN Center for Developmental Biology)16 were cultivated on gelatin-coated dishes in the high-glucose DMEM base medium (Invitrogen) supplemented with 15% of fetal bovine serum (FBS, Invitrogen), 1 × nonessential amino acids (Invitrogen), 0.1 mM β-mercaptoethanol, and 1000 U/ml leukemia inhibitory factor (ESGRO). The TF-1 human erythroleukemic cell line was obtained from ATCC and cultivated in the RPMI 1640 base medium (Invitrogen) supplemented with 10% of FBS and 2 ng/ml of recombinant human GM-CSF (R&D Systems). Acquisition of the cytokine-independent growth of TF1 cells due to introduction of cancer-associated gene mutation was examined and quantified upon GM-CSF removal as previously described26.
Authentication of cell line identities, including those of parental and derived lines, was ensured by the Tissue Culture Facility (TCF) affiliated to UNC-Chapel Hill Lineberger Comprehensive Cancer Center using the genetic signature profiling and fingerprinting analysis as previously described34. Every 1-2 month, a routine examination of cell lines in culture for any possible mycoplasma contamination was carried out using the commercially available detection kits (Lonza Walkersville Inc).
Generation of stable cell lines
TKO ES cells were transfected by Lipofectamine 2000 (Invitrogen) with the pPyCAGIZ empty vector (EV) or that carrying WT or mutant DNMT3A. 48 hours post transfection, the transduced ES cells were selected out in 50 μg/ml Zeocin (Invitrogen) for 10 days. The pooled stable-expression cell lines and independent single cell-derived clonal lines were continuously maintained in the medium with 25 μg/ml Zeocin. To generate TF-1 leukemia cell lines with stable expression of WT or mutant DNMT3A, the MSCV-based retrovirus was packaged in HEK293 and used for infection as previously described35. 48 hrs post infection, TF-1 cells were selected by 2 μg/ml puromycin for 4 days and maintained in medium with 1μg/ml puromycin.
Western blotting
Antibodies used for western blotting were α-MYC (Sigma, 9E10), α-DNMT3A (Santa Cruz, H-295), α-beta-Actin (Santa Cruz, sc-47778), and α-Tubulin (Sigma). Total protein samples were prepared by cell lysis with SDS-containing Laemmli sample buffer followed by brief sonication. Extracted samples equivalent to 100,000 cells were loaded to the SDS-PAGE gels for western blot analysis.
Quantification of 5-methyl-2´-deoxycytidine (5-mdC) and 2´-deoxyguanosine (dG) in genomic DNA
The measurement procedures for 5-mdC and dG in genomic DNA were described previously36, 37. Briefly, 1 μg of genomic DNA prepared from cells was enzymatically digested into nucleoside mixtures. Enzymes in the digestion mixture were removed by chloroform extraction, and the resulting aqueous layer was concentrated to 10 μL and subjected directly to LC-MS/MS and LC-MS/MS/MS analysis for quantification of 5-mdC and dG, respectively. The amounts of 5-mdC and dG (in moles) in the nucleoside mixtures were calculated from area ratios of peaks found in selected-ion chromatograms (SICs) for the analytes over their corresponding isotope-labeled standards, the amounts of the labeled standards added (in moles) and the calibration curves. The final levels of 5-mdC, in terms of percentages of dG, were calculated by comparing the moles of 5-mdC relative to those of dG.
Enhanced Reduced Representation Bisulfite Sequencing (eRRBS) and data analysis
Genomic DNA of each sample was added with 0.5% of unmethylated lambda DNA (Promega) as spike-in control and subjected to eRRBS using MethylMidi-seq (Zymo Research) as described before24. In brief, approximately 300 ng of DNA were digested with three restriction enzymes (80 units of MspI, 40 units of BfaI and 40 units of MseI) to improve genomic DNA fragmentation and coverage. The generated DNA fragments were ligated to the pre-annealed 5′-methyl-cytosine-containing adapters, followed by filling in overhangs and the A extension at 3′-terminus. The DNA fragments were then purified and subject to bisulfite treatment using the EZ DNA Methylation–Lightning kit (Zymo Research). After amplification, the quality of eRRBS libraries was checked with Agilent 2200 TapeStation, followed by deep sequencing using the Illumina HiSeq-2000 genome analyzer (50-bp and paired end as parameters). Obtained reads were aligned to in silico bisulfite-converted mouse reference genome mm9 and lambda DNA sequence (GenBank: J02459.1) using Bismark package in a strand-specific manner38. For identification of methylated cytosines, all mapped cytosines were subjected to a binomial distribution model-based methylation calling as described in the below section. To determine distribution of methylation levels, only those high-quality reads with at least 15 times of coverage were used. For convenience of data analysis and to increase data complexity, data from all three biological replicates were merged and cytosine sites covered with at least 15 reads in the merged dataset were used for downstream analysis such as analysis of averaged methylation levels in 10-kb window sliding and aggregated methylation levels across genes. Data representation and plots were generated with the ggplot2 package in R software using custom scripts.
Identification of methylated cytosines
We used a previously described binomial model to identify methylated cytosines17, 18. Specifically, with the unmethylated spike-in lambda DNA, we first determined the bisulfite non-conversion rate (probability, P) for each cytosine sequence context independently (i.e., CpG, CpA, CpC and CpT). For each mapped cytosine in our eRRBS data, we calculated the binomial p-value that methylated reads occur out of the total read number based on the binomial test, with bisulfite non-conversion rate as the success probability (P). If a p-value is under a threshold, we defined the cytosine as truly methylated. To determine the false discovery rate (FDR) for each different threshold, we created a control methylome for each eRRBS sample. In the control methylome, read depth at each cytosine was equal to the real data, and the methylated events were simulated by binomial distribution using previously defined non-conversion rate (P). The FDR was determined by the ratio between the number of identified methylated cytosine sites from the control methylome and that from the real data. For each eRRBS sample, we have chosen to use a p-value under which the FDR is less than 1% or 0.1%, as specified in figure legends.
DNA methylation array and data analysis
Genomic DNA was extracted and bisulfite-converted as described above. DNA methylation profiling using the lllumina Infinium HumanMethylation450 BeadChip array was performed by the UNC Genomics Core according to the manufacturer’s instructions. Methylation data were then subject to background subtraction and control normalization by executing preprocessIllumina in the R ‘minfi’ package39. Differentially methylated CpGs were identified using dmpFinder in a categorical mode. Methylation changes were considered significant at a q-value of less than 0.05 and a beta value difference of more than 0.1. Hierarchical clustering analysis, scatter plots and density plots were generated in R using ‘pheatmap’, and ‘ggplot2’ packages.
Sanger bisulfite sequencing
Sanger bisulfite sequencing was carried out as previously described24. Briefly, genomic DNA was prepared using the DNeasy Blood & Tissue Kit (Qiagen) and 1 μg genomic DNA subject to bisulfite conversion using the EZ DNA methylation gold kit according to manufacturer’s instructions (Zymo Research). Bisulfite-treated DNA was then used as template in PCR to amplify the target DNA region, followed by cloning of PCR products into pCR2.1-TOPO vector (Invitrogen) for direct sequencing of individual clones. Four biological replicates per cell line were tested, with at least 10 clonal sequences per replicate generated. The primers used for amplifying a major satellite DNA sequence located at chromosome 2 are 5′- GGG AAT TTT GGT GGT AGG GT -3′ and 5′- AAA AAA CAT CCA CTT AAC TAC TTA AAA A -3′. The primers used for validating 450k array data are listed as follows: for EIF4G1, 5′- AGG AGA TTG AGG TTT TAG TGA ATA TGT-3′ and 5′- CCC TAT ATC AAA TTC TTC CTA CCA TAA -3′; for HDLBP, 5′- GGA GGT GAA GTT ATG GAG ATA TTT TT -3′ and 5′- ATC CCA TAC CAA CAA AAA CTA ACA A-3′; for FOXK2, 5′- TAT GTT TGT ATT TGG GGT GTT TTT T -3′ and 5′- CTA AAA AAT CAA AAA CAT TTC CTA CC -3′.
Chromatin immunoprecipitation (ChIP)
Chromatin samples used for ChIP were prepared as previously described8. Briefly, chromatin samples extracted from cells expressing Myc-tagged DNMT3A were used for ChIP with the 9E10 anti-Myc antibody (Sigma), with cells expressing empty vector used as negative control. Real-time PCR was carried out for detecting DNMT3A binding at sites listed below. The primers for ChIP-PCR at each tested site are: for cg23189692, 5′- TTG GCA TGC TCA CAG AGA GG -3′ and 5′- GTC TTC CCA GGC TCA TTG CT -3′; for cg00704780. 5′- AGC AAA ACG GTC AGT AGC CA -3′ and 5′- TAC CAG CAA AAG CTG GCA GG -3′; for cg10460657, 5′- GCC TCT GAC CTG CTG TCT AC -3′ and 5′- AGG AAA TGC CCC AGA CGT G -3′; for cg07564962, 5′- GGC CGG CAC TAA TGT CTT TC -3′ and 5′- TTC CCT GCT CTG TGG GAA GG -3′; for cg13393476, 5′- CCT TGC GAG TGA GTC ACG G -3′ and 5′- GAG ATT CTG CCA GGC TCC AC -3′; for cg20509869, 5′- GTG GGA CGC TAA CCC TCT TC -3′ and 5′- GGC GGC TGA TTT ATC TGG GT -3′; and for GAPDH transcription start site (TSS), 5′- TCT CCC CAC ACA CAT GCA CTT -3′ and 5′- CCT AGT CCC AGG GCT TTG ATT -3′’.
Statistics
Data are presented as the mean ± SD of at least three independent experiments. Statistical analysis was performed with Student’s t test for comparing two sets of data with assumed normal distribution. A p value of less than 0.05 was considered to be significant.
Data availability
Coordinates and structure factors for the DNMT3A–DNMT3L–25-mer DNA, DNMT3A–DNMT3L–10/11-mer DNA, and DNMT3A (R836A)–DNMT3L–25-mer DNA complexes have been deposited in the Protein Data Bank with accession codes 5YX2, 6F57 and 6BRR, respectively. The eRRBS and Illumina Human Methylation 450K array data have been deposited in NCBI Gene Expression Omnibus (GEO) under accession code GSE99391.
Code availability
The scripts for genomic data analyses and all other data are available from the corresponding authors upon reasonable request.
Extended Data
Extended Data Figure 1. Structures of the DNMT3A–DNMT3L tetramer in complex with the 10/11-mer DNA.
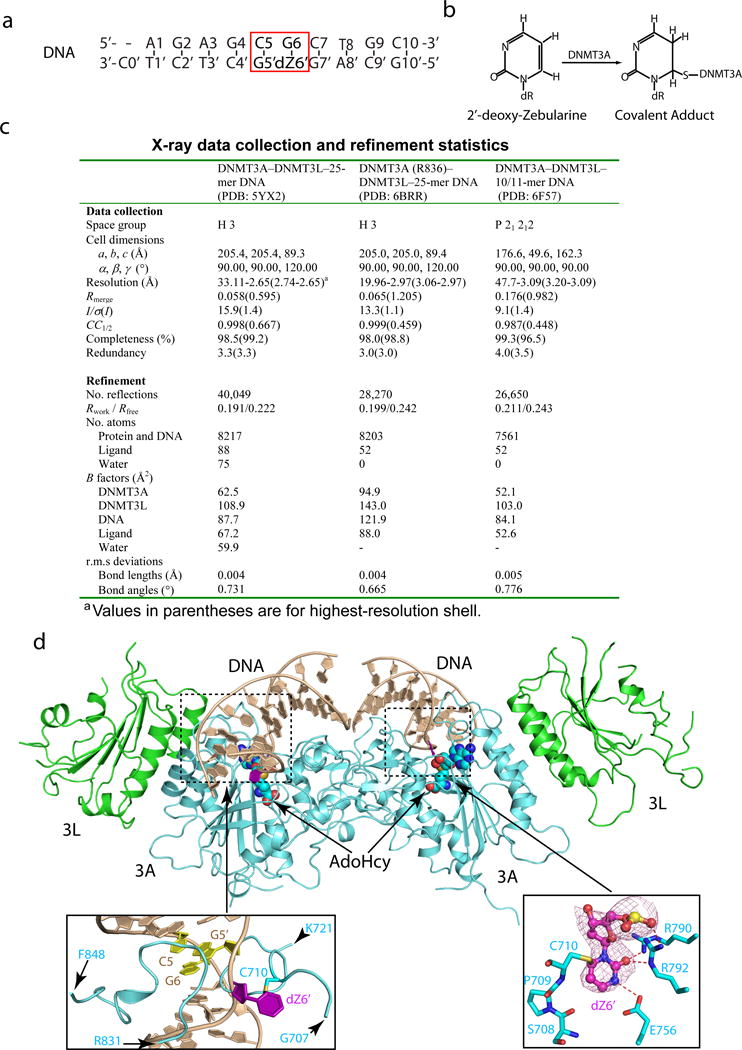
a, The sequence of the 10/11-mer DNA duplex used for structural study. b, Chemical formula of the covalent adduct of DNMT3A and 2′-deoxy-Zebularine. c, Data collection and refinement statistics. Each dataset was collected from a single crystal. d, Ribbon representations of the DNMT3A–DNMT3L tetramer in complex with the 10/11-mer DNA duplex and AdoHcy. DNMT3A, DNMT3L and DNA are colored in light blue, green and wheat, respectively, and AdoHcy shown in sphere representation. The boxed areas show expanded views for the CpG sites (purple and yellow), the DNA-binding TRD and catalytic loops (left box) and the flipped out 2′-deoxy-Zebularine (dZ6′) surrounded by conserved catalytic residues (right box). The Fo–Fc omit map of dZ6′ (pink) is contoured at 3σ level. The hydrogen-bonding interactions are depicted as dashed lines.
Extended Data Figure 2. Intermolecular interactions between the DNMT3A–DNMT3L tetramer and DNA.
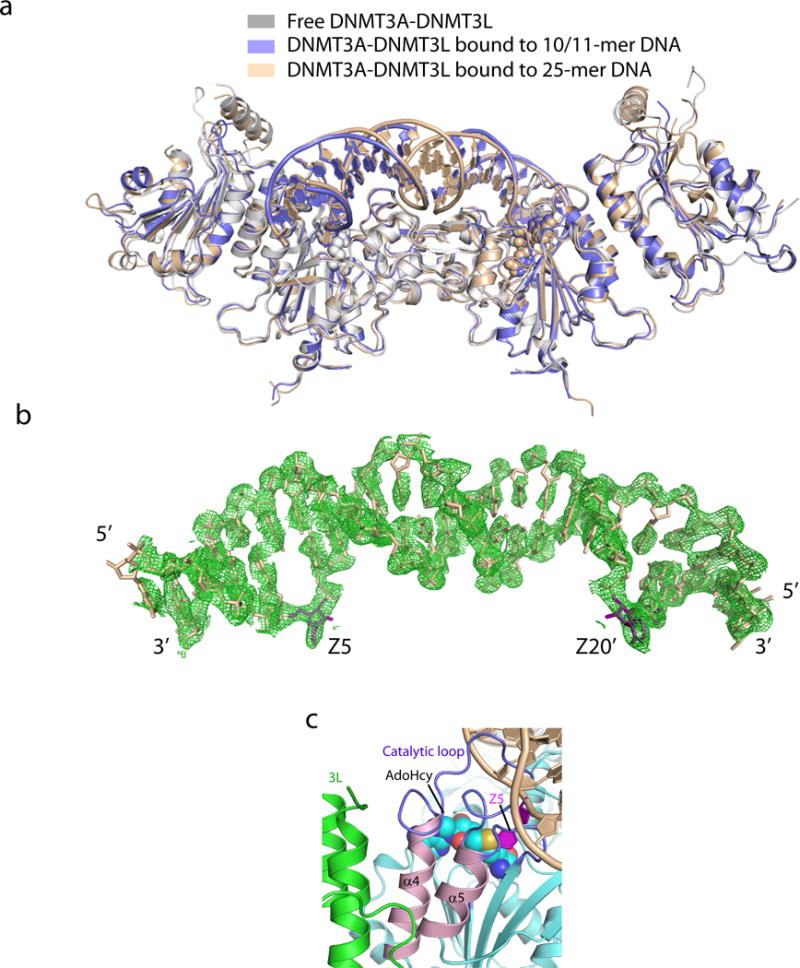
a, Structural overlay of free DNMT3A-DNMT3L (PDB 2QRV) with the 10/11-mer DNA and 25-mer DNA-bound states. b, Stick representation of the 25-mer DNA duplex bound to the DNMT3A-DNMT3L tetramer, with the 2Fo-Fc omit map contoured at 1σ level. c, The two helices of DNMT3A that interact with DNMT3L (shown in green) are colored in pink (α4 and α5 in accordance with the numeration in Extended Data Figure 3g) and preceded by two DNA contact loops, colored in blue. The flipped out Zebularine (Z5) is colored in purple. The bound AdoHcy molecule is shown in sphere representation.
Extended Data Figure 3. Various intermolecular interactions between the two DNMT3A monomers and DNA.
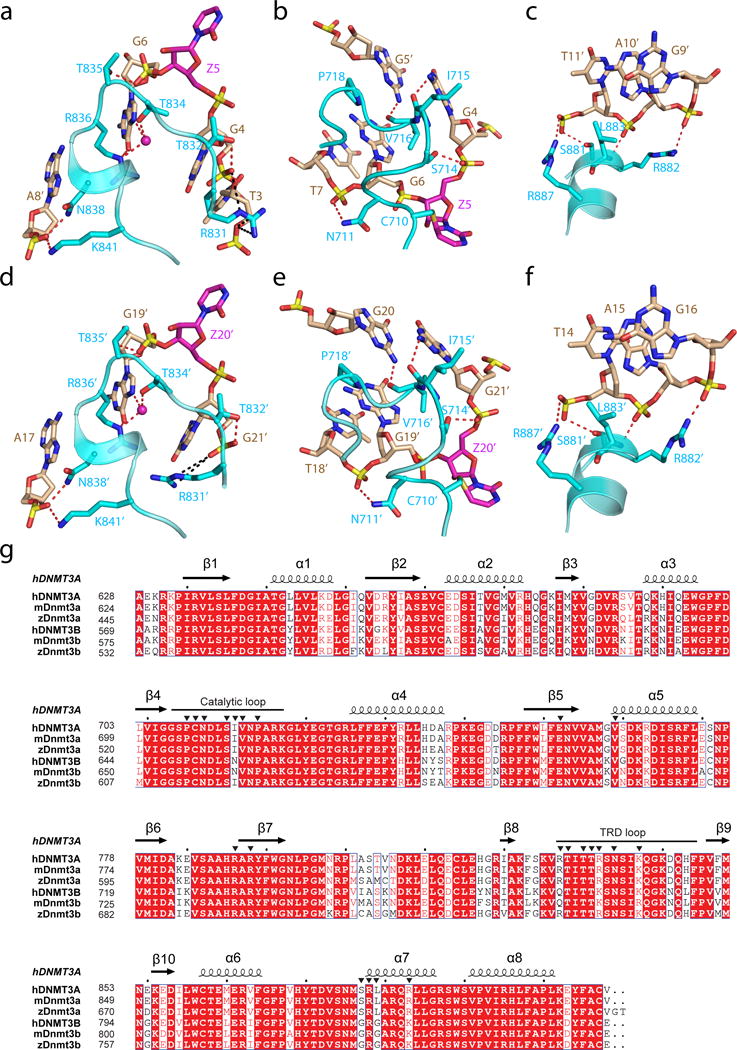
a–f, DNA binding by the first and second DNMT3A monomer (defined as 3A and 3A′, respectively, in Fig. 1c) includes the intermolecular interactions between DNMT3A’s TRD loop and DNA major groove (a,d), between DNMT3A’s catalytic loop and DNA minor groove (b,e), and between DNMT3A’s homodimeric interface and DNA backbone (c,f). The hydrogen-bonding interactions are shown as dashed lines. The water molecules are shown as purple spheres. g, Structure-based sequence alignment of DNMT3 proteins from human (hDNMT3A and hDNMT3B), mouse (mDnmt3a and mDnmt3b) and zebrafish (zDnmt3a and zDnmt3b). Completely conserved residues are colored in white and highlighted in red. Partially conserved residues are colored in red. Secondary structures are shown above the aligned sequences. The DNA binding residues as revealed by this study are marked with black triangles.
Extended Data Figure 4. The essential roles for the CpG-engaging residues of DNMT3A, R836 and V716, in DNMT3A-mediated CpG versus non-CpG methylations.
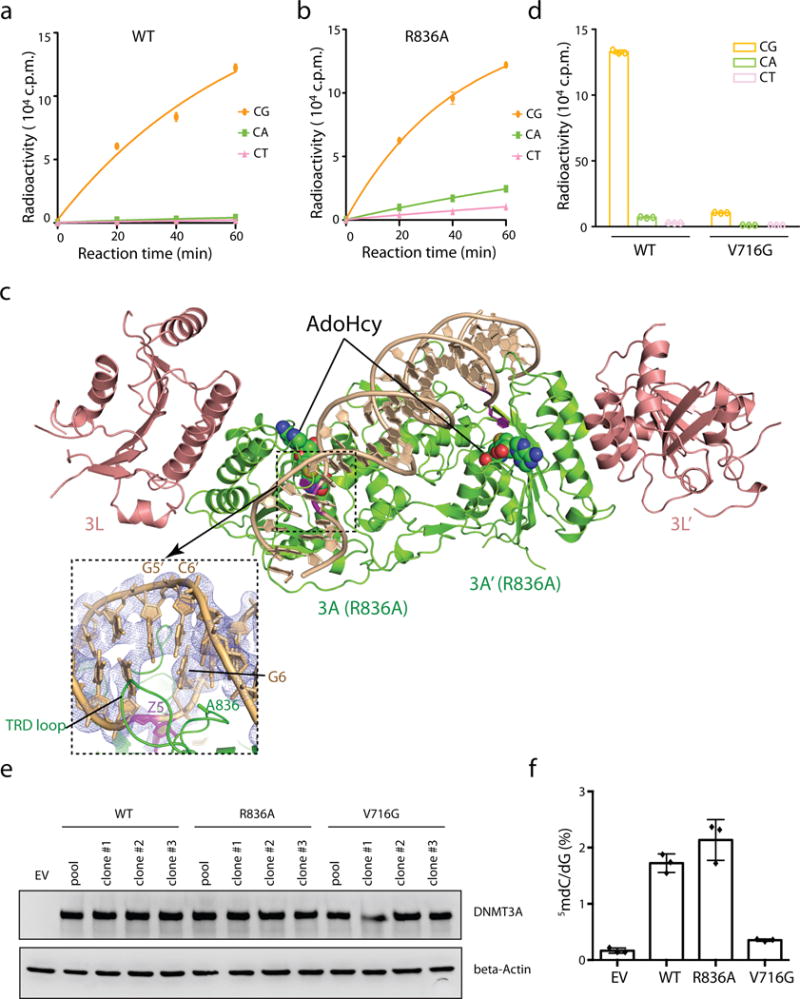
a, b, DNA methylation kinetics analysis of wild-type (DNMT3AWT, panel a) and R836A-mutated (DNMT3AR836A, panel b) DNMT3A using the CpG-, CpA- or CpT-containing DNA substrates (n = 3 biological replicates). Purified DNMT3A–DNMT3L tetramer complex was used for measurements, followed by fitting with first-order exponential equation. c, Ribbon representation of the crystal structure of DNMT3AR836A–DNMT3L tetramer in complex with the 25-mer DNA, with the CpG recognition by one of the DNMT3A monomer shown in expanded view. The 2Fo-Fc omit map of DNA was contoured at 0.8σ level, and colored in light blue. d, Methylation assay using either DNMT3AWT or DNMT3AV716G on CG-, CA- and CT-containing DNA (n = 3 biological replicates). e, Immunoblots detect reconstituted expression of the indicated DNMT3A among TKO mouse ES cells, either the pooled stable-expression cell population or single cell-derived clonal lines. Shown is a representative blot of two independent experiments. For gel source data, see Supplementary Figure 1. f, Liquid chromatography–mass spectrometry (LC-MS) analysis reveals the global 5-mC levels (as calculated by 5-mdC/dG in y-axis) in the TKO ES cells after stable transduction of EV or the indicated DNMT3A (n = 3 biological replicates). Data are mean ± s.d. EV, empty vector.
Extended Data Figure 5. eRRBS reveals distribution of cytosine methylations in each sequence context among TKO ES cells with reconstituted expression of either DNMT3AWT or DNMT3AR836A.
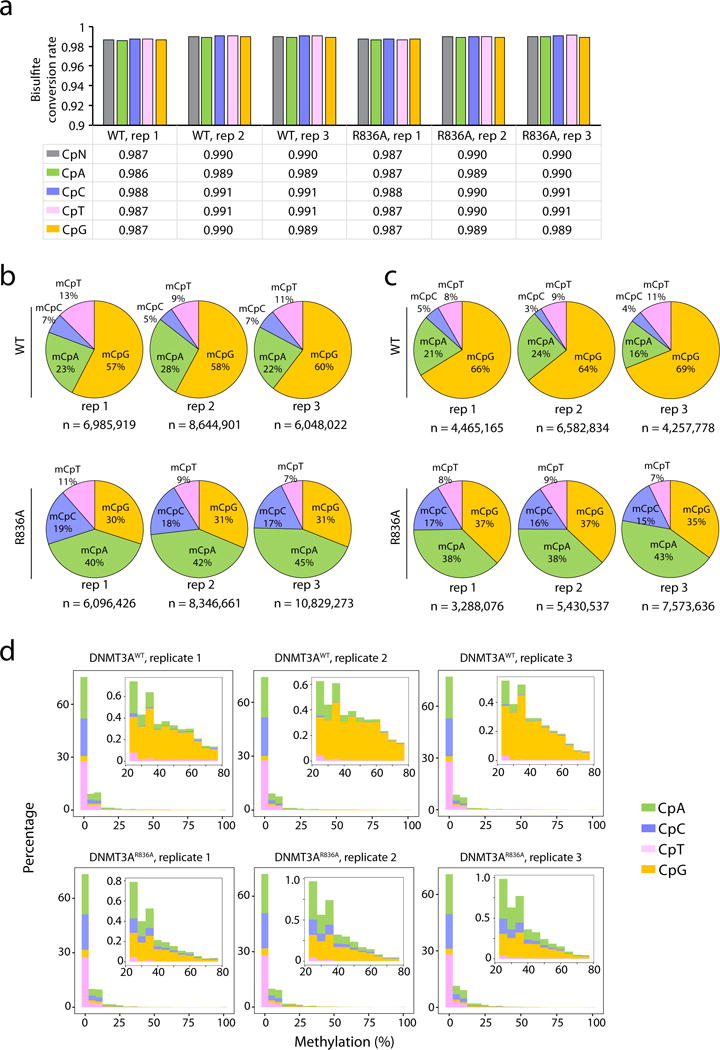
a, The rates of bisulfite conversion for the indicated sequence context in each sample as determined by the unmethylated lambda DNA spike-in control. CpN, all cytosines. b, c, Piecharts showing the percentage of methylated cytosines (total number n shown at the bottom of each plot) identified among the DNMT3AWT or DNMT3AR836A-expressing TKO ES cells in each sequence context. The methylated cytosines were called using a stringent binomial distribution–based filter to eliminate false positives from incomplete bisulfite conversion, with a false discovery rate (FDR) of 1% and 0.1% set for panel b and c, respectively. d, Distribution of methylation levels (% in x-axis) for the indicated sequence context. Insert panels show a closed view of the distribution at sites with intermediate to high levels of cytosine methylation.
Extended Data Figure 6. CpG and non-CpG methylations induced by DNMT3AWT versus DNMT3AR836A in the TKO ES cells.
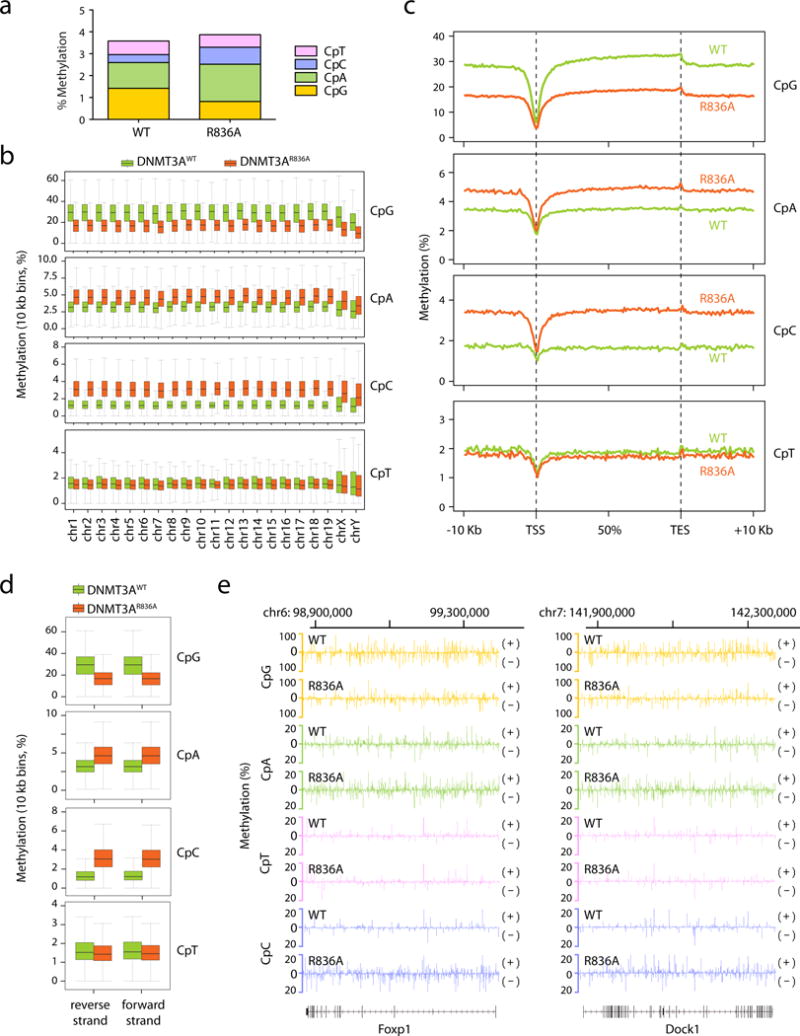
a, Overall methylation levels of cytosines at four different sequence contexts as detected by eRRBS. Shown on y-axis is the averaged methylation level of all cytosines within the mouse genome as calculated by normalization of the detected methylation cytosines over total cytosine numbers in the TKO ES cells reconstituted with either DNMT3AWT (left) or DNMT3AR836A (right). b, Global levels of CpG and CpH (H= A, C or T) methylation induced by DNMT3AWT (green) or DNMT3AR836A (red) across the mouse chromosomes of the TKO ES cells. Shown are boxplots of 10 kb-bin-averaged methylation levels of each mouse chromosome. c, Global levels of CpG and CpH methylation induced by DNMT3AWT (green) versus DNMT3AR836 (red) across all annotated genes. Each gene was divided into 100 equally sized bins and the 10 Kb flanking region was divided into 50 equally sized bins. Averaged methylation levels were plotted for each bin. TSS, transcription start site; TES, transcription end site. d, Global levels of CpG and CpH methylation induced by DNMT3AWT (green) versus DNMT3AR836A (red) on the two opposite DNA strands. Shown are boxplots of 10 kb-bin-averaged CpG, CpA, CpC and CpT methylation levels of each strand. e, Representative gene-wide views of CpG and CpH methylations at Foxp1 and Dock1, which are grouped into either forward (+) or reverse (-) DNA strand. Shown are cytosines covered by at least 15 reads from eRRBS data, with each site designated by a vertical line. Panels a-e use the combined dataset of three biological replicates per group. Box plots depict the interquartile range, and whiskers depict 1.5× interquartile range.
Extended Data Figure 7. Sanger bisulfite sequencing to validate the cytosine methylation levels mediated by DNMT3A, either WT or defective in recognizing the CpG substrate, among the TKO mouse ES cells.
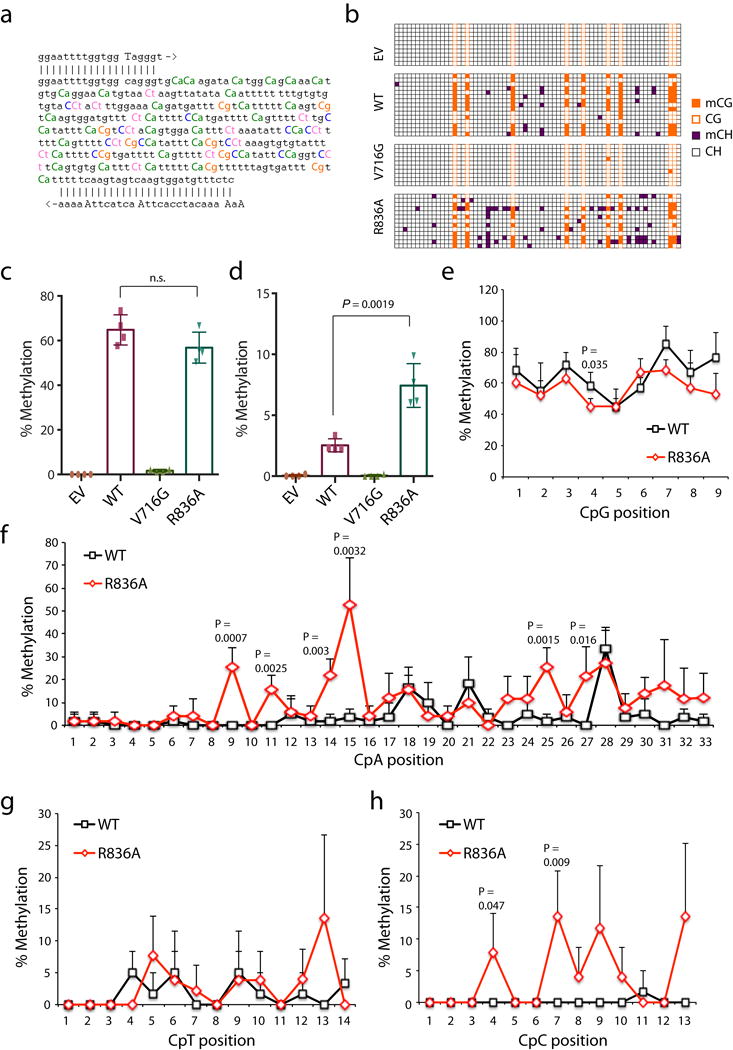
a, Sequence of the examined major satellite DNA region. Primers used for bisulfite PCR are denoted with 5′ and 3′ primer paring. The counts for cytosines, highlighted in color, are 9 for the CG dinucleotide, 33 for CA, 14 for CT, and 13 for CC. b, A representative result for bisulfite sequencing analysis of the major satellite repeat region described above in the TKO cells expressing EV, WT DNMT3A or the indicated mutant. Each row represents one DNA clone and each column represents one site of cytosine, either methylated (filled) or unmethylated (open). c, d, Percentage of methylation mediated by DNMT3A or the indicated mutant at CpG (c) and non-CG (d) sites within the examined major satellite DNA region in the TKO ES cells. Data are mean ± s.d.; n= 4 independent bisulfite sequencing experiments as shown in b. e-h, Average cytosine methylation levels at each individual site grouped by the CpG (e), CpA (f), CpT (g) or CpC (h) context in the examined major satellite DNA among the TKO ES cells reconstituted with DNMT3AWT versus DNMT3AR836A (n = 4 biological replicates; mean ± s.d., with the labeled p values). EV, empty vector. Statistical analysis used two-tailed Student’s t-test: n.s., not significant.
Extended Data Figure 8. Effect of hematological cancer-associated mutations of DNMT3A on DNA methylation in vitro and in mouse TKO ES cells.
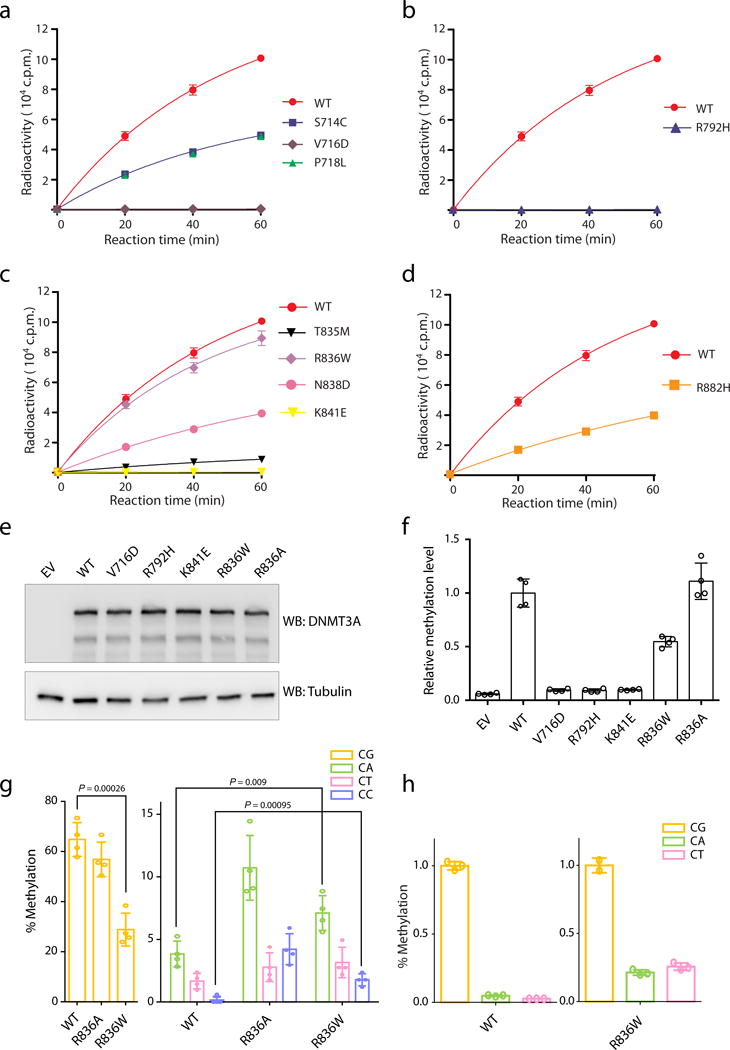
a, Methylation kinetics of DNMT3A with mutations located at the catalytic loop, in comparison to DNMT3AWT. b, Methylation kinetics of DNMT3A with an active site mutation, R792H. c, Methylation kinetics of DNMT3A with mutations located at the TRD loop. d, Methylation kinetics of DNMT3A with the hotspot mutation R882H. For panels a-d, DNMT3A–DNMT3L complex was used for the measurements (n = 3 biological replicates), followed by fitting with first-order exponential equation. These data were measured independently from data shown in Fig. 4c. e, Immunoblot detects stable reconstitution of DNMT3AWT or the indicated DNMT3A mutant in the TKO ES cells. Shown is a representative blot of two independent experiments. For gel source data, see Supplementary Figure 1. f, LC-MS analysis reveals the global 5-mC levels (as calculated by 5-mdC/dG) in the TKO ES cells after stable transduction of empty vector (EV) or the indicated DNMT3A. Plotted are the methylation levels relative to TKO cells expressing DNMT3AWT. Data are mean ± s.d.; n = 4 biological replicates. g, Individual bisulfate sequencing detects the methylation level of cytosines in each sequence context within a major satellite DNA site at chromosome 2 in the TKO cells reconstituted with DNMT3AR836W (right), in comparison to DNMT3AWT (left) or DNMT3AR836A (middle; as determined in Fig. 3g) (n = 4; mean ± s.d.). Statistical analysis used two-tailed Student’s t-test. h, In vitro methylation of CG, CA or CT-containing DNA using DNMT3AWT (left) or DNMT3AR836W (right) in complex with DNMT3L, reacted for 40 min (n = 3 biological replicates). Plotted are the methylation levels relative to CG-containing DNA substrates. Data are mean ± s.d.
Extended Data Figure 9. Effect of hematological cancer-associated mutations of DNMT3A on genomic DNA methylation in the TF-1 leukemia cells.
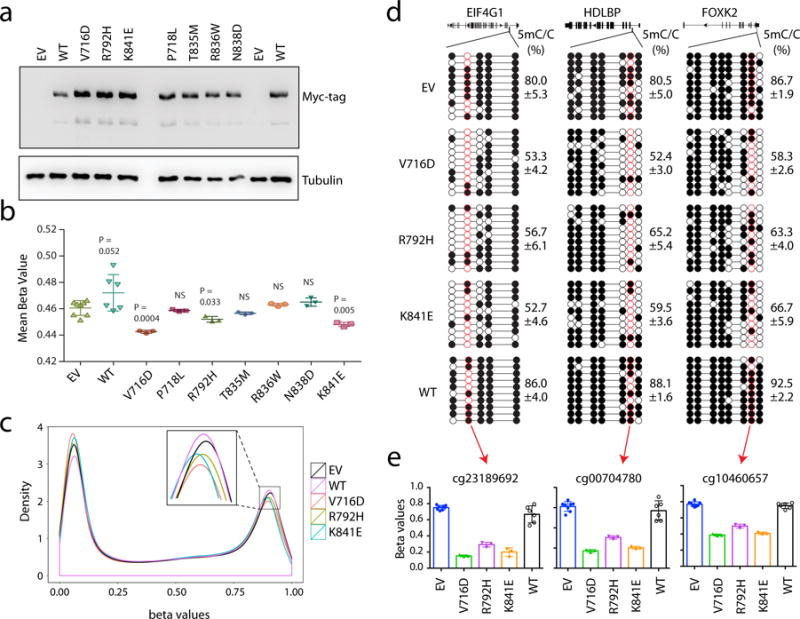
a, Immunoblot of the TF-1 cells stably transduced with Myc-tagged DNMT3A, either WT or the indicated cancer-associated mutants. EV, empty MSCV vector. Shown is a representative blot of two independent experiments. For gel source data, see Supplementary Figure 1. b, Profiling of the indicated DNMT3A-expressing TF-1 cell lines with the HumanMethylation_450K BeadChip array reveals the mean methylation beta values for all examined CpGs. Each dot represents a biological replicate, i.e., an independently derived stable-expression cell line (n = 3-8 biological replicates per group; mean ± s.d.). Statistical analysis used two-tailed Student’s t-test. NS, not significant. c, Density plot of methylation beta values for all examined CpGs in the indicated DNMT3A-expressing TF-1 cell lines. The inserted box shows a zoom-in view for densities for highly methylated DNA sites among the indicated samples. Data are mean ± s.d., with the labeled p values. Statistical analysis used two-tailed Student’s t-test: NS, not significant. d, Sanger bisulfite sequencing of the indicated regions from TF-1 cell lines stably transduced with EV, DNMT3AWT or the indicated cancer-associated mutant. Individual CpG sites (circles) are filled with black (methylated) or white (unmethylated). Red circles denote the CpG sites covered by the Illumina Infinium 450K DNA methylation array. Data are mean ± s.d.; n = 3 biological replicates. e, Methylation values of the indicated CpG sites (labeled by red circles in d) based on the measurements with the Infinium 450K DNA methylation arrays (n = 3-8 biological replicates; mean ± s.d.).
Extended Data Figure 10. Effect of hematological cancer-associated DNMT3A mutations on DNA hypo-methylation and cytokine-independent growth of the TF-1 leukemia cells.
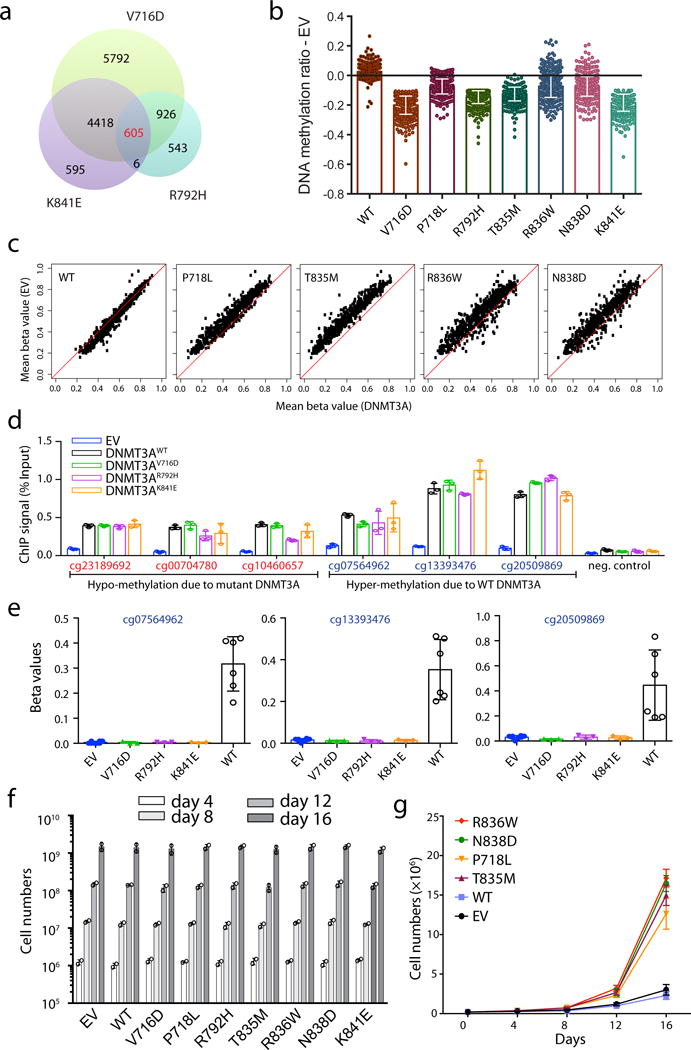
a, Venn diagram of CpG sites with hypo-methylation induced by either one of the three indicated strong DNA-binding-defective mutants of DNMT3A, V716D, K841E and R792H. b, c, Bar plots (b) and scatter plots (c) showing methylation difference at the 605 commonly hypo-methylated CpG sites identified in panel a among the TF-1 cells with stable transduction of either DNMT3AWT or the indicated mutant, in comparison to empty MSCV vector (EV). A black line in b indicates EV control (n = 3-8 biological replicates per group; mean ± s.d.). Plotted in c are mean methylation beta values of each individual CpG in the indicated DNMT3A experimental group (x-axis) and control EV group (y-axis; n = 3-8 biological replicates per group). d, e, Comparable occupancy of DNMT3A and its mutant forms at the indicated genomic loci with affected DNA methylation, as measured by ChIP analysis of the Myc-tagged DNMT3AWT or mutants in TF-1 stable-expression cell lines. Tested sites by ChIP-qPCR in panel d (n = 3 biological replicates; mean ± s.d.) included three CpG sites showing hypo-methylation due to expression of mutant DNMT3A (left; also see Extended Data Fig. 9e), three sites showing hyper-methylation due to expression of DNMT3AWT (middle; also see panel e, which shows mean methylation beta values from measurements with the Infinium 450K DNA methylation arrays, n = 3-8 biological replicates; mean ± s.d.), and a negative control locus (right; the GAPDH transcription start site). The anti-Myc antibody was used for ChIP and the EV-expressing TF-1 cells used as cell control for unspecific binding. f, Proliferation of the indicated DNMT3A stable-expression TF-1 cells in the presence of a supporting cytokine, GM-CSF (n = 2 biological replicates; mean ± s.d.). g, Proliferation of the indicated DNMT3A-expressing TF-1 cells after GM-CSF withdrawal (n = 3 biological replicates; mean ± s.d.).
Supplementary Material
Acknowledgments
We would like to thank Dr. Xiaodong Cheng for valuable comments of the manuscript, Drs. Masaki Okano, Jianlong Wang and Julie-Aurore Losman for providing reagents used in the study, and staff members at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory and at the Advanced Photo Source (APS), Argonne National Laboratory for access to X-ray beamlines. We are also grateful for professional support of UNC facilities including Genomics Core, which are partly supported by the UNC Cancer Center Core Support Grant P30-CA016086. This work was supported by Kimmel Scholar Awards (to J.S. and G.G.W.), March of Dimes Foundation (1-FY15-345 to J.S.), DoD Peer-reviewed Cancer Research Program (W81XWH-14-1-0232 to G.G.W.), Gabrielle’s Angel Foundation for Cancer Research (to G.G.W.) and NIH (1R35GM119721 to J.S, 5R21ES025392 to Y.W., and 1R01CA215284, 1R01CA218600 and 1R01CA211336 to G.G.W.). G.G.W. is an American Cancer Society (ACS) Research Scholar. R.L. was supported by a Lymphoma Research Foundation postdoctoral fellowship.
Footnotes
Author Contributions
Z-M.Z., R.L., P.W., Y.Y., D.C., L.G., S.L., D.J. and J.S. performed experiments. S.B.R. provided technical support. Y.W., G.G.W. and J.S. conceived and organized the study. Z-M.Z., R.L., G.G.W. and J.S. prepared the manuscript.
Author Information
The authors declare no competing financial interests
Online Content
Methods, Extended Data display items and Source Data are available in the online version of the paper; references unique to these sections appear only in the online paper.
References
- 1.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 2.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 3.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 4.Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 5.Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 6.Chedin F, Lieber MR, Hsieh CL. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Natl Acad Sci U S A. 2002;99:16916–16921. doi: 10.1073/pnas.262443999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hata K, Okano M, Lei H, Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129:1983–1993. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- 8.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 9.Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer. 2015;15:152–165. doi: 10.1038/nrc3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ley TJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo X, et al. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature. 2014 doi: 10.1038/nature13899. [DOI] [PubMed] [Google Scholar]
- 12.Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007;449:248–251. doi: 10.1038/nature06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jurkowska RZ, et al. Formation of nucleoprotein filaments by mammalian DNA methyltransferase Dnmt3a in complex with regulator Dnmt3L. Nucleic Acids Res. 2008;36:6656–6663. doi: 10.1093/nar/gkn747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gowher H, Jeltsch A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J Mol Biol. 2001;309:1201–1208. doi: 10.1006/jmbi.2001.4710. [DOI] [PubMed] [Google Scholar]
- 15.Gowher H, et al. Mutational analysis of the catalytic domain of the murine Dnmt3a DNA-(cytosine C5)-methyltransferase. J Mol Biol. 2006;357:928–941. doi: 10.1016/j.jmb.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 16.Tsumura A, et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes to cells : devoted to molecular & cellular mechanisms. 2006;11:805–814. doi: 10.1111/j.1365-2443.2006.00984.x. [DOI] [PubMed] [Google Scholar]
- 17.Guo JU, et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nature neuroscience. 2014;17:215–222. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen T, Tsujimoto N, Li E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol Cell Biol. 2004;24:9048–9058. doi: 10.1128/MCB.24.20.9048-9058.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forbes SA, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tatton-Brown K, et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. 2014;46:385–388. doi: 10.1038/ng.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holz-Schietinger C, Matje DM, Reich NO. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J Biol Chem. 2012;287:30941–30951. doi: 10.1074/jbc.M112.366625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim SJ, et al. A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood. 2013;122:4086–4089. doi: 10.1182/blood-2013-02-483487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu R, et al. Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer Cell. 2016;30:92–107. doi: 10.1016/j.ccell.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russler-Germain DA, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell. 2014;25:442–454. doi: 10.1016/j.ccr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Losman JA, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song J, Rechkoblit O, Bestor TH, Patel DJ. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science. 2011;331:1036–1040. doi: 10.1126/science.1195380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou L, et al. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol. 2002;321:591–599. doi: 10.1016/S0022-2836(02)00676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Adams PD, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 34.Yu M, et al. A resource for cell line authentication, annotation and quality control. Nature. 2015;520:307–311. doi: 10.1038/nature14397. [DOI] [PubMed] [Google Scholar]
- 35.Wang GG, et al. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nature methods. 2006;3:287–293. doi: 10.1038/nmeth865. [DOI] [PubMed] [Google Scholar]
- 36.Volz DC, et al. Tris(1,3-dichloro-2-propyl)phosphate Induces Genome-Wide Hypomethylation within Early Zebrafish Embryos. Environmental science & technology. 2016;50:10255–10263. doi: 10.1021/acs.est.6b03656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu Y, et al. Comprehensive Assessment of Oxidatively Induced Modifications of DNA in a Rat Model of Human Wilson’s Disease. Mol Cell Proteomics. 2016;15:810–817. doi: 10.1074/mcp.M115.052696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aryee MJ, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors for the DNMT3A–DNMT3L–25-mer DNA, DNMT3A–DNMT3L–10/11-mer DNA, and DNMT3A (R836A)–DNMT3L–25-mer DNA complexes have been deposited in the Protein Data Bank with accession codes 5YX2, 6F57 and 6BRR, respectively. The eRRBS and Illumina Human Methylation 450K array data have been deposited in NCBI Gene Expression Omnibus (GEO) under accession code GSE99391.