

Abstract
Non‐natural oligonucleotides represent important (bio)chemical tools and potential therapeutic agents. Backbone modifications altering hybridization properties and biostability can provide useful analogues. Here, we employ an artificial nucleosyl amino acid (NAA) motif for the synthesis of oligonucleotides containing a backbone decorated with primary amines. An oligo‐T sequence of this cationic DNA analogue shows significantly increased affinity for complementary DNA. Notably, hybridization with DNA is still governed by Watson–Crick base pairing. However, single base pair mismatches are tolerated and some degree of sequence‐independent interactions between the cationic NAA backbone and fully mismatched DNA are observed. These findings demonstrate that a high density of positive charges directly connected to the oligonucleotide backbone can affect Watson–Crick base pairing. This provides a paradigm for the design of therapeutic oligonucleotides with altered backbone charge patterns.
Keywords: backbone modifications, DNA, oligonucleotides, peptides, stereoselective synthesis
Introduction
Oligonucleotides have unique binding properties, rendering them essential molecules in living organisms and providing a platform for the modulation of biological functions through antigene, antisense, or RNA interference approaches.1 In natural nucleic acids (DNA and RNA), nucleotides are linked by phosphate diester moieties, thus leading to a polyanionic backbone at physiological pH.2 The polyanionic character contributes to the low cellular uptake of such oligonucleotides, thereby compromising in vivo applications. In addition, DNA and RNA possess low stability toward nucleases, which are ubiquitous in biological systems. These limitations have led to the development of artificial, biostable nucleic acids (e.g., phosphorothioates and “locked” nucleic acids (LNA)).3 In many cases, these analogues exhibit altered binding properties, providing insights into the structural contributions to duplex formation. For instance, the charge pattern of the oligonucleotide backbone can influence both hybridization and pharmacokinetic properties. Therefore, artificial electroneutral internucleotide linkages, for example, amide,4 sulfone,5 or triazole6 moieties, have been developed. Triazole linkers have even been shown to be biocompatible, that is, triazole‐modified DNA can be recognized by polymerases in cells and can therefore be employed to construct genes.6b–6e Peptide nucleic acid (PNA) is an example of an electroneutral nucleic acid mimic with an amide‐based backbone exhibiting only limited resemblance to DNA and RNA.7 However, fully electroneutral nucleic acid analogues often suffer from low water solubility and a tendency to aggregate in aqueous solution. These limitations and the quest for oligonucleotides with fundamentally different properties have led to the development of positively charged nucleic acids. In most cases, positive charges were introduced through a modification of the 2′‐hydroxy groups (in RNA) or nucleobases leaving the phosphate diester backbone unchanged. These strategies furnished zwitterionic structures,8 but resulted in densely charged oligonucleotides.
An alternative approach involves the replacement of phosphate diester units by non‐natural positively charged linkers, thus providing an oligomer with a retained overall number of charges, but reversed polarity. This may be advantageous for biomedical applications, in particular with respect to cellular uptake, as indicated by the favorable properties of cationic cell‐penetrating peptides (CPPs).9 Only a few positively charged internucleotide linkages have been reported so far:10 1) Bruice's guanidine11 and S‐methylthiourea12 linkages; 2) Letsinger's phosphoramidate linkages, in which amines were connected to the backbone through alkyl linkers;13 and 3) our recently reported NAA‐modified oligonucleotides.14
Bruice's rather rigid guanidine linkage and Letsinger's flexible aminoalkyl moiety provide cationic oligonucleotides with high affinity for DNA, but conformational properties that deviate significantly from native nucleic acids. This raises the question of how moderately flexible internucleoside linkages would impact the hybridization properties of cationic oligonucleotide analogues. In principle, the NAA modification (with its rigid amide bond and the adjacent 5′‐C‐6′‐C single bond, Figure 1) could be used to assemble corresponding oligonucleotides. However, previously reported “dimeric” phosphoramidite building blocks only allow incorporation of the NAA modification adjacent to phosphate diester units, thus resulting in partially zwitterionic DNA analogues.14
Figure 1.
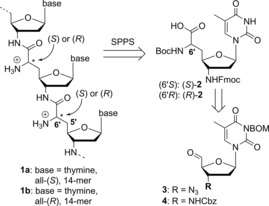
NAA‐derived fully cationic oligonucleotides 1 a and 1 b including their retrosynthesis. SPPS: solid‐phase peptide synthesis; BOM=benzyloxymethyl.
The favorable properties of these partially zwitterionic NAA‐modified DNAs and the general interest in fully cationic oligonucleotides have inspired us to design oligomers of type 1, which are completely assembled from NAA internucleoside linkages (Figure 1). The availability of such cationic oligonucleotide analogues provides the basis for answering some of the questions highlighted above. In particular, it would allow an assessment of the influence of the fully cationic backbone on duplex stability and sequence specificity of hybridization with native DNA.
Results and Discussion
For the synthesis of nucleoside‐derived δ‐peptide‐like oligomers 1, we envisioned connecting monomeric units of type 2 through amide formation in analogy to solid‐phase peptide synthesis (SPPS, Figure 1). Building block 2 should be obtained either from the 3′‐azido‐substituted nucleoside‐5′‐aldehyde 3, or via its protected 3′‐amino analogue 4. We decided to focus on cationic oligomers with thymine as nucleobase, aiming to prepare both the all‐(S)‐ and the all‐(R)‐configured oligomers (with respect to the stereochemical configuration at the 6′‐position). For subsequent hybridization experiments and biophysical characterizations, we designed 14‐mer oligomers 1 a and 1 b (Figure 1).
The synthesis of building blocks (S)‐2 and (R)‐2 started from 3‐N‐benzyloxymethyl‐(BOM)‐protected 3′‐azido‐3′‐deoxy‐thymidine 5 and its 3′‐N‐Cbz‐protected 3′‐amino congener 6, respectively, to compare the routes via azide 3 and protected amine 4 (Scheme 1; see Supporting Information for synthesis of 5 and 6). Aldehydes 3 and 4 were obtained by IBX oxidation of 5 and 6 in quantitative yields. On the basis of our previously reported syntheses of nucleosyl amino acids,15 we applied a sequence of Wittig–Horner olefination and asymmetric hydrogenation to introduce the amino acid motif. Wittig–Horner transformations of aldehydes 3 and 4 with phosphonate 7 (see Supporting Information) furnished didehydro amino acids 8 and 9 in yields of 73 % and 68 %, respectively, with high stereoselectivities toward the desired Z‐isomers (93:7 and 91:9, respectively). However, the concomitantly formed E‐isomers could not be fully removed, and thus, asymmetric hydrogenations were performed with the Z/E‐mixtures. Hydrogenation of 8 and 9 in the presence of chiral RhI catalysts (S,S)‐ and (R,R)‐Me‐DuPHOS‐Rh16 afforded the nucleosyl amino acid products 10 and 11 in yields of 54–92 %, with the major isomer (6′S or 6′R) depending on the employed catalyst [(S,S)‐catalyst for (6′S), (R,R)‐catalyst for (6′R); for stereochemical assignments see Experimental Section]. In contrast to our previous syntheses of nucleosyl amino acids,15 the hydrogenation products were not obtained in diastereomerically pure form, but with diastereomeric ratios ranging from 85:15 to 95:5. HPLC purification of (S)‐11 and (R)‐11 gave the pure 6′‐epimers (d.r.>99:1), to be followed by the efficient concomitant hydrogenolytic removal of Cbz, Bn, and BOM groups. As this hydrogenation step proceeded less satisfactorily for the 3′‐azido congeners, the synthesis of the target structures through the 3′‐N‐Cbz‐amino route (using aldehyde 4) was superior overall. Subsequent 3′‐N‐Fmoc protection furnished diastereomerically pure building blocks (S)‐2 and (R)‐2 in yields of 58 % and 48 %, respectively, over the last two steps (Scheme 1). Fmoc‐based SPPS employing either (S)‐4 or (R)‐4, followed by final acidic cleavage and deprotection (reactions not shown), gave the two 14‐mer diastereomers 1 a and 1 b, respectively.
Scheme 1.
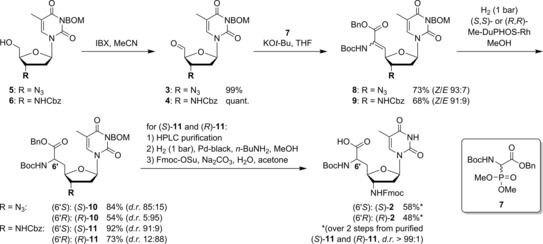
Synthesis of building blocks (S)‐2 and (R)‐2 for the preparation of cationic target oligomers 1 a and 1 b.
For investigation of the hybridization properties with DNA, fully cationic 6′‐all‐(S)‐14‐mer 1 a was incubated with complementary 14‐mer DNA (A14). The influence of ion strength on duplex formation was investigated by varying the concentration of NaCl (50–125 mm, all in phosphate buffer, pH 7.4). Under all conditions, duplex formation was observed, as indicated by hyperchromicity upon heating, leading to the typical sigmoidal (though slightly broadened) melting curves (Figure S2, Supporting Information) allowing the determination of melting temperatures (T m) (Table 1, entries 1 to 4, and Figure S2). As expected,13 we observed a decrease in T m with increasing NaCl concentration, which can be ascribed to salt‐mediated shielding of the backbone in 1 a (positive charges) and in DNA (negative charges). We then decided to use 100 mm NaCl, as this represents a commonly applied concentration. As a reference, the native DNA–DNA duplex (T14‐A14) was used. At 100 mm NaCl, the duplexes of both cationic analogues (1 a and 1 b) with fully complementary DNA (A14) were more stable than the corresponding DNA–DNA duplex, as indicated by the differences in melting temperatures (ΔT m 9 °C and 17 °C, respectively; Table 1, entry 3; melting curves shown in Figure 2, solid lines). The cationic 14‐mers 1 a and 1 b contained 13 non‐native internucleoside linkages (“modifications”, mod.), so these results were equivalent to ΔT m/mod. values of +0.7 °C (1 a) and +1.3 °C (1 b), respectively.
Table 1.
T m values (in °C±SD)[a] of cationic oligonucleotide analogues 1 a and 1 b (as well as the T14 DNA reference) with native DNA strands.
| NaCl | T14 ref. | 1 a (6′S) | 1 b (6′R) | ||||
|---|---|---|---|---|---|---|---|
| DNA[b] | [mm] | T m | T m | ΔT m [c] | T m | ΔT m [d] | |
| 1 | A14 | 50 | n.d.[e] | 53.6±0.9 | – | n.d. | – |
| 2 | A14 | 75 | n.d. | 51.1±0.6 | – | n.d. | – |
| 3 | A14 | 100 | 36.4±0.6 | 45.1±0.9 | +9 | 53.8±0.4 | +17 |
| 4 | A14 | 125 | n.d. | 43.1±0.4 | – | n.d. | – |
| 5 | A7 CA6 | 100 | 23.8±0.3 | 49.8±0.3 | +26 | 49.7±0.5 | +26 |
| 6 | A7 GA6 | 100 | 23.7±0.2 | 50.0±0.3 | +26 | 50.3±1.4 | +27 |
| 7 | A7 TA6 | 100 | 23.1±1.5 | 45.9±0.0 | +23 | 53.5±0.3 | +30 |
| 8 | G6TTG6 | 100 | –[f] | –[f] | –[f] | –[f] | –[f] |
[a] In aqueous 10 mm NaH2PO4 (pH 7.4) and NaCl. [b] Base mismatches underlined and in bold. [c] T m(1 a–DNA)‐T m(DNA–DNA); T m(DNA–DNA): T m value of the corresponding native DNA–DNA duplex. [d] T m(1 b–DNA)‐T m(DNA–DNA). [e] n.d.=not determined. [f] No sigmoidal melting curve was observed.
Figure 2.
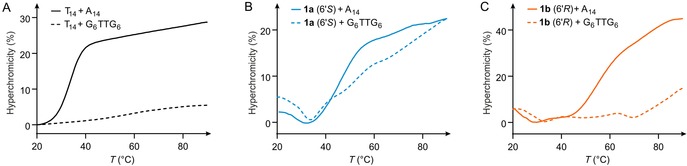
Melting curves (average of triplicates) for A) native DNA oligonucleotide T14, B) cationic oligonucleotide analogue 1 a (6′S), and C) cationic oligonucleotide analogue 1 b (6′R) with native complementary DNA (A14, solid lines), as well as with native fully mismatched DNA (G6TTG6, dashed lines).
Subsequently, we studied the base‐specificity of duplex formation of 1 a and 1 b with partially mismatched DNA strands. Upon introduction of a single base mismatch (C, G, or T instead of A in the middle of the DNA sequence), sigmoidal UV melting curves with 1 a and 1 b were observed, indicating duplex formation (Figures S3 and S4, Supporting Information). Remarkably, both cationic oligomers 1 a and 1 b showed nearly retained duplex stability upon incorporation of this single mismatch (Table 1, entries 5–7 vs. entry 3). In contrast, the corresponding DNA–DNA duplexes encountered the expected destabilization of approximately 13 °C. This led to ΔT m values (difference from the corresponding single mismatched native DNA duplex) of up to around 30 °C. In the case of T‐C and T‐G mismatches, isomer 1 a furnished slightly more stable duplexes with the single‐mismatched DNA than with the fully complementary A14 strand. For the same base mismatches, the 1 b–DNA duplexes were slightly destabilized relative to the fully complementary analogues (Table 1, entries 5 and 6 vs. entry 3). Overall, the (6′R)‐configured NAA linkage (1 b) furnished more stable duplexes than the (6′S)‐configured congener in the case of the fully matched sequence and of the T‐T mismatch (Table 1, entries 3 and 7, 1 b vs. 1 a). Remarkably, 1 b shows some preference for fully complementary DNA, resulting in T m values that are 0.3–4.1 °C lower with the single mismatched counterstrands.
These results indicate that oligonucleotide analogues 1 a and 1 b were relatively insensitive to single base mismatches in the DNA counterstrand. Hence, we aimed to probe whether Watson–Crick base pairing contributes to duplex formation, or if hybridization merely results from electrostatic attraction of the two backbones (oligocation 1 a,b with oligoanionic DNA). Therefore, melting curves were recorded for equimolar mixtures of thymidine‐derived oligomers 1 a or 1 b with a fully mismatched 14‐mer DNA (G6TTG6, Figure 2, Table 1, entry 8). The resultant curves indicate no specific melting process, that is, no defined transition between an aggregated and a non‐aggregated state was observed (Figure 2 B, C, dashed lines). However, 1 a in particular and also 1 b to some extent showed considerable hyperchromicity with G6TTG6 upon heating (up to ≈25 % for 1 a+G6TTG6 and ≈15 % for 1 b+G6TTG6), whereas the DNA–DNA reference T14+G6TTG6 only displays a moderate hyperchromicity of up to ≈5 %, steadily rising over the temperature range 20–90 °C (Figure 2 A, dashed line). In contrast, the hyperchromicity of the mixture 1 a+G6TTG6 starts to increase at around 35 °C, but this is less pronounced for 1 b+G6TTG6, setting in at approximately 70 °C. A possible explanation for this behavior is a charge‐mediated unspecific formation of aggregates at lower temperatures for both 1 a and 1 b with fully mismatched DNA. Elevated temperatures can then be expected to induce the disassembly of these structures. An additional indication of the presence of charge‐mediated unspecific interactions are the less defined transitions in the melting curves of 1 a and 1 b with the fully matched A14 DNA counterstrand (Figure 2 B, C, solid lines). These duplexes appear to undergo complex temperature‐induced transitions, with several changes in UV absorbance in addition to the main transition (that is, melting of the duplex). In comparison, the native DNA–DNA (T14‐A14) reference duplex (Figure 2 A, solid line) shows a sharp transition.
To obtain insights into the structural properties of cationic oligomers 1 a and 1 b and the resultant duplexes, we performed circular dichroism (CD) spectroscopy. Initially, the single‐stranded oligonucleotides were investigated (Figure 3 A), including the corresponding native DNA (T14, black) and complementary native DNA (A14, grey). A comparison of their CD spectra reveals differences between all oligomeric thymidine analogues (T14, 1 a and 1 b). Notably, the spectra of native T14 (black) and cationic 1 a (blue) are more similar in comparison to 1 b (orange), which differs particularly at lower wavelengths. This indicates substantial structural differences between both cationic oligomers in their single‐stranded form (note that 1 a and 1 b are diastereomers).
Figure 3.
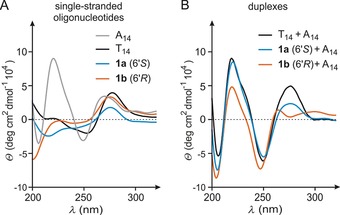
A) CD spectra of single‐stranded cationic oligonucleotides 1 a (6′S), 1 b (6′R), and of native single‐stranded DNA oligonucleotides T14 and A14. B) CD spectra of the aggregates 1 a (6′S)‐A14 and 1 b (6′R)‐A14 and of the native DNA–DNA reference duplex (T14‐A14).
Subsequently, duplexes of both cationic oligomers (1 a and 1 b) and of native DNA (T14) with complementary DNA (A14) were investigated (Figure 3 B). The CD spectrum of the 1 a‐A14 duplex (blue) shows resemblance to the CD signals of the native T14‐A14 DNA–DNA duplex17 (black, Figure 3 B). On the other hand, differences are observed for the 1 b‐A14 duplex (orange, Figure 3 B): although the pattern of signals for λ<260 nm is rather similar to that of the native duplex, it significantly differs for λ>260 nm. Instead of a single maximum at approximately 275 nm, 1 b‐A14 exhibits two maxima at around 260 nm and 300 nm, respectively. Overall, the similarities in the CD spectra suggest that binding of both isomers 1 a and 1 b to complementary DNA probably furnished duplexes with mainly DNA‐like helical topologies. However, 1 b (which, remarkably, formed the most stable duplex with A14) displayed the most pronounced deviations both in its single‐stranded form and in its complex with complementary native DNA A14.
We then studied the CD spectra of equimolar mixtures of oligomers 1 a or 1 b with fully mismatched 14‐mer DNA (G6TTG6, Figure 4). For the (6′S)‐configured oligomer 1 a, the CD spectrum of the mixture with mismatched DNA (Figure 4 A, solid line) does not show any signal pattern indicative of a helical duplex structure (as compared with that of the corresponding matched duplex, Figure 3 B). To assess the possibility of nonspecific interactions between both single strands in this mixture, we determined the CD spectrum of G6TTG6 alone (Figure S5, Supporting Information) and added it to the spectrum of single‐stranded 1 a. This combined spectrum (Figure 4 A, dashed line) differs significantly from the experimentally obtained spectrum of the mixture. This suggests Watson–Crick‐independent nonspecific interactions between 1 a and fully mismatched G6TTG6 at ambient temperature, and is in line with the aforementioned melting behavior (Figure 2). Similar results were obtained for the mixture of (6′R)‐configured oligomer 1 b with fully mismatched DNA (Figure 4 B).
Figure 4.
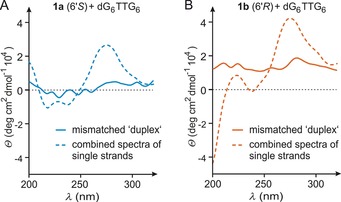
A) CD spectrum of the mixture of oligomer 1 a (6′S) with fully mismatched native DNA (G6TTG6, solid line) and calculated superposition of the CD spectra of both single strands (dashed line). B) CD spectrum of the mixture of oligomer 1 b (6′R) with fully mismatched native DNA (G6TTG6, solid line) and calculated superposition of the CD spectra of both single strands (dashed line).
Conclusion
We report the synthesis of a novel amide‐linked cationic oligonucleotide analogue based on NAA internucleoside linkages. Two cationic oligomers (1 a and 1 b, 6′‐epimers) were synthesized and investigated by UV melting studies and CD spectroscopy. Both oligomers form very stable, presumably helical, duplexes with native complementary DNA strands. The high duplex stability (compared with the native DNA–DNA duplex) resembles that of some previously reported cationic oligomers, in particular Bruice's guanidine‐linked oligonucleotides.11 Notably, hybridization of 1 a and 1 b with DNA was insensitive to single base mismatches, thus indicating robustness of hybridization toward limited local perturbations within the duplex. However, duplex formation was not detected in the case of a fully mismatched DNA counterstrand, demonstrating Watson–Crick base pairing to be a requirement for hybridization and the occurrence of a defined topology. The most likely explanation for our observations is that electrostatic attraction can compensate for single base mismatches, but is not sufficient to foster the formation of a defined structure in the absence of Watson–Crick base pairing. In the latter case, sequence‐unspecific charge‐mediated aggregation phenomena occur. This behavior is in sharp contrast to the pronounced mismatch sensitivity of partially zwitterionic oligonucleotides containing up to four NAA linkages.14a
Interestingly, both isomers differ moderately in their DNA‐binding affinity as well as selectivity, and on the basis of their CD spectra, also in their structural topology. Apparently, the (6′R)‐configuration in 1 b is slightly more beneficial for hybridization. Notably, an analogous behavior had also been observed for previously reported partially zwitterionic NAA‐modified oligonucleotides.14a Both isomers 1 a and 1 b appear to adopt different structures in their single‐stranded form. Furthermore, the CD spectrum of duplex 1 b‐A14 reveals a maximum at approximately 300 nm, which is unusual for fully helical oligonucleotide duplexes.
Overall, our findings will contribute to the future design of oligonucleotides for potential biomedical applications. The favorable properties of cationic cell‐penetrating peptides (CPPs)9 indicate that the introduction of positive charges into the oligonucleotide backbone might be beneficial for their therapeutic or diagnostic use, in particular with respect to cellular uptake. However, as demonstrated in this work, fully cationic (in contrast to partially zwitterionic) oligonucleotides can suffer from impaired base pairing fidelity and unspecific aggregation in the absence of Watson–Crick base pairing. In our future work, we will therefore study how the ratio of negatively and positively charged linkages impacts base‐pairing fidelity. In addition, more detailed structural studies will be performed, with the long‐term goal of obtaining a thorough understanding of the interplay of conformation, base pairing, and electrostatic attraction in duplexes of (partially) cationic and anionic oligonucleotide strands.
Experimental Section
General methods
The syntheses of starting materials 5 and 6 and of phosphonate 7 are described in the Supporting Information. All other chemicals were purchased from standard suppliers. Reactions involving oxygen‐ and/or moisture‐sensitive reagents were performed under an atmosphere of argon using anhydrous solvents. Anhydrous solvents were obtained in the following manner: THF was dried over sodium/benzophenone and distilled, CH2Cl2 was dried over CaH2 and distilled, MeOH was dried over activated molecular sieves (3 Å) and degassed, MeCN was dried over P2O5 and distilled, pyridine was dried over CaH2 and distilled, toluene was dried over sodium/benzophenone and distilled. The thus‐obtained solvents were stored over molecular sieves (4 Å; in case of MeOH and MeCN, 3 Å). All other solvents were of technical quality and distilled prior to use, and deionized water was used throughout. Column chromatography was performed on silica gel 60 (0.040–0.063 mm, 230–400 mesh ASTM, VWR) under flash conditions unless otherwise indicated. TLC was performed on aluminum plates precoated with silica gel 60 F254 (VWR). Visualization of the spots was achieved using UV light (254 nm) and/or staining under heating (H2SO4 staining solution: 4 g vanillin, 25 mL conc. H2SO4, 80 mL AcOH, and 680 mL MeOH; KMnO4 staining solution: 1 g KMnO4, 6 g K2CO3, and 1.5 mL NaOH (1.25 m) solution, all dissolved in 100 mL H2O; ninhydrin staining solution: 0.3 g ninhydrin, 3 mL AcOH, and 100 mL 1‐butanol). Analytical chiral HPLC was performed on a Jasco system equipped with a pu 2080 Plus pump, an AS 2055 Plus autosampler, an MD 2010 Plus multiwavelength detector, and an IB ChiralpakTM column (0.8×27.5 cm) purchased from Diacel. Method: isocratic eluent 70:30 n‐hexane‐EtOAc; flow 0.8 mL min−1; injection volume 10 μL (c≈4 mg mL−1 in EtOAc). Preparative chiral HPLC was performed on a Jasco system equipped with a pu 2080 Plus pump, an MD 2010 Plus multiwavelength detector, and an IB ChiralpakTM column (1.5×28 cm) purchased from Diacel. Method: isocratic eluent 73:27 n‐hexane‐EtOAc; flow 5 mL min−1; injection volume 100 μL (c≈100 mg mL−1 in EtOAc). 300 MHz‐ and 500 MHz‐1H, 75 MHz‐ and 126 MHz‐13C, and 121 MHz‐31P NMR spectra were recorded on Varian MERCURY 300, UNITY 300, INOVA 500, and INOVA 600 spectrometers. All 13C NMR spectra were H‐decoupled. All spectra were recorded at room temperature unless indicated otherwise, and were referenced internally to solvent reference frequencies. For calibration of 31P NMR signals, 85 % phosphoric acid was used as an external standard. Chemical shifts (δ) are quoted in ppm and coupling constants (J) are reported in Hz. Signals were assigned by using H,H‐COSY, HSQC, and HMBC spectra obtained on the spectrometers detailed above. Mass spectra of small molecules were measured on a Finnigan LCQ ion‐trap mass spectrometer or on a Bruker microTOF spectrometer. For ESI measurements in the negative mode, solutions of the compounds in pure MeOH were used, whereas for measurements in the positive mode, solutions in MeOH containing 0.1 % formic acid were employed. High‐resolution spectra were measured on a Bruker 7 Tesla Fourier transform ion cyclotron resonance (FTICR) mass spectrometer. Melting points (m.p.) were measured on a Büchi instrument and were not corrected. Optical rotations were recorded on a PerkinElmer polarimeter 241 with a Na source using a 10 cm cell. Solutions of the compounds (≈10 mg) in CHCl3 or pyridine (1 mL) were used, and concentrations are given in g/100 mL. Infrared (IR) spectroscopy was performed on a PerkinElmer Vektor 22 spectrometer with solids measured as KBr pills or on a Jasco FT/IR‐4100 spectrometer equipped with an integrated ATR unit (GladiATR™, PIKE Technologies). Wavenumbers (ν) are quoted in cm−1. UV spectroscopy of small molecules was performed on a PerkinElmer Lambda 2 spectrometer. Measurements were performed with solutions of approximately 0.1 mg of the compound in 10 mL MeCN and in the range 190–500 nm. Wavelengths of maximum absorption (λ max) are reported in nm with the corresponding logarithmic molar extinction coefficient (log ϵ) given in parentheses (ϵ in dm3 mol−1 cm−1).
Syntheses and characterization data
6′‐N‐Boc‐3′‐N‐Fmoc‐amino‐3′‐deoxy‐(S)‐thymidinyl amino acid (S)‐2: Pd‐black (1.28 g, 12.0 mmol) and n‐butylamine (1.80 g, 2.43 mL, 24.2 mmol) were added to a solution of diastereomerically pure NAA (S)‐11 (vide infra, 900 mg, 1.21 mmol) in MeOH (28 mL). The resultant suspension was stirred for 24 h under a hydrogen atmosphere (1 bar). It was then filtered and the filter cake was washed with MeOH (3×10 mL). The combined filtrates were evaporated and the residue was coevaporated with pyridine (3×4 mL). The solid thus obtained was used for the subsequent Fmoc protection without further purification.
Na2CO3 (115 mg, 1.09 mmol) and Fmoc‐OSu (368 mg, 1.09 mmol) were added to a solution of the obtained product (435 mg, 1.09 mmol) in a mixture of acetone and water (1:1, 3 mL). The reaction mixture was stirred for 1 h at RT. It was then acidified to pH 2 with 2 m HCl and partitioned between a mixture of CH2Cl2 (15 mL) and brine (15 mL). The aqueous layer was extracted with CH2Cl2 (3×10 mL). The combined organics were dried over Na2SO4, filtered, and evaporated. The resultant crude product was purified by column chromatography (9:1 CH2Cl2‐MeOH, 0.5 % AcOH). The obtained product was coevaporated with toluene (3×5 mL) to give (S)‐2 as a fine white powder (393 mg, 58 % over two steps from (S)‐11). M.p. decomposition >110 °C; TLC: R f=0.26 (9:1 CH2Cl2‐MeOH); =+39.7 (c=1.2, CHCl3); 1H NMR (500 MHz, CD3OD): δ=1.44 (s, 9 H, C(CH3)3), 1.93 (s, 3 H, 7‐H), 2.06–2.16 (m, 1 H, 5′‐Ha), 2.21–2.39 (m, 3 H, 2′‐Ha, 2′‐Hb, 5′‐Hb), 3.89–3.97 (m, 1 H, 4′‐H), 4.05–4.14 (m, 1 H, 3′‐H), 4.18–4.27 (m, 2 H, Fmoc‐CH2), 4.32–4.48 (m, 3 H, 9′′‐H, 6′‐H, 6′‐NH), 6.08–6.15 (m, 1 H, 1′‐H), 7.31 (dd, J=7.3, 7.3 Hz, 2 H, 2′′‐H, 7′′‐H), 7.34 (dd, J=7.3, 7.3 Hz, 2 H, 3′′‐H, 6′′‐H), 7.53 (brs, 1 H, 6‐H), 7.65 (d, J=7.3 Hz, 2 H, 4′′‐H, 5′′‐H), 7.79 ppm (d, J=7.3 Hz, 2 H, 1′′‐H, 8′′‐H); 13C NMR (126 MHz, CD3OD): δ=11.1 (C‐7), 27.3 (C(CH3)3), 35.3 (C‐5′), 36.8 (C‐2′), 46.8 (Fmoc‐CH2), 51.3 (C‐6′), 54.2 (C‐3′), 66.4 (C‐9′′), 79.2 (C(CH3)3), 81.0 (C‐4′), 84.4 (C‐1′), 110.5 (C‐5), 119.5 (C‐1′′, C‐8′′), 124.7, 124.8 (C‐4′′, C‐5′′), 126.8, 127.4 (C‐2′′, C‐7′′), 127.8, 128.5 (C‐3′′, C‐6′′), 136.4 (C‐6), 141.3 (C‐8′′a, C‐9′′a), 143.9 (C‐4′′a, C‐4′′b), 150.8 (C‐2), 156.3 (Boc‐ C=O), 156.3 (Fmoc‐C=O), 165.0 (C‐4), 174.4 ppm (COOH); IR (ATR): =1680, 1519, 1447, 1250, 1161, 1050, 1022, 759, 736 cm−1; UV (MeCN): λ max (log ϵ)=206 (4.95), 264 nm (4.65); HRMS (ESI) calcd for C32H36N4NaO9: 643.2374; found: 643.2368 [M+Na]+.
6′‐N‐Boc‐3′‐N‐Fmoc‐amino‐3′‐deoxy‐(R)‐thymidinyl amino acid (R)‐2: The synthesis of (6′R)‐configured NAA (R)‐2 was performed according to the procedure for the synthesis of (6′S)‐configured NAA (S)‐2 with diastereomerically pure NAA (R)‐11 (vide infra, 370 mg, 0.498 mmol), Pd‐black (528 mg, 4.98 mmol), n‐butylamine (731 mg, 1.00 mL, 10.0 mmol), MeOH (12 mL), Na2CO3 (53 mg, 0.50 mmol), Fmoc‐OSu (169 mg, 0.502 mmol), and acetone/water (1:1, 1.4 mL). The crude product was purified by column chromatography (9:1 CH2Cl2‐MeOH, 0.5 % AcOH). The obtained product was coevaporated with toluene (3×5 mL) to give (R)‐2 as a fine white powder (150 mg, 48 % over two steps from (R)‐11). M.p. decomposition >110 °C; TLC: R f=0.26 (9:1 CH2Cl2‐MeOH); = +23.5 (c 1.1, CHCl3); 1H NMR (500 MHz, CD3OD): δ=1.44 (s, 9 H, C(CH3)3), 1.93 (s, 3 H, 7‐H), 2.03–2.14 (m, 1 H, 5′‐Ha), 2.14–2.24 (m, 1 H, 5′‐Hb), 2.24–2.32 (m, 1 H, 2′‐Ha), 2.32–2.42 (m, 1 H, 2′‐Hb), 3.84–3.91 (m, 1 H, 4′‐H), 4.03–4.11 (m, 1 H, 3′‐H), 4.19–4.26 (m, 2 H, Fmoc‐CH2), 4.26–4.35 (m, 1 H, 6′‐H), 4.38–4.48 (m, 2 H, 9′′‐H, 6′‐NH), 6.14–6.21 (m, 1 H, 1′‐H), 7.33 (dd, J=7.3, 7.3 Hz, 2 H, 2′′‐H, 7′′‐H), 7.40 (dd, J=7.3, 7.3 Hz, 2 H, 3′′‐H, 6′′‐H), 7.50 (brs, 1 H, 6‐H), 7.66 (d, J=7.3 Hz, 2 H, 4′′‐H, 5′′‐H), 7.80 ppm (d, J=7.3 Hz, 2 H, 1′′‐H, 8′′‐H); 13C NMR (126 MHz, CD3OD): δ=11.0 (C‐7), 27.3 (C(CH3)3), 35.4 (C‐5′), 36.5 (C‐2′), 46.8 (Fmoc‐CH2), 51.3 (C‐6′), 54.4 (C‐3′), 66.3 (C‐9′′), 79.2 (C(CH3)3), 80.6 (C‐4′), 84.7 (C‐1′), 110.6 (C‐5), 119.5 (C‐1′′, C‐8′′), 124.7, 124.9 (C‐4′′, C‐5′′), 126.8, 127.4 (C‐2′′, C‐7′′), 127.8, 128.5 (C‐3′′, C‐6′′), 136.4 (C‐6), 141.3 (C‐8′′a, C‐9′′a), 143.9 (C‐4′′a, C‐4′′b), 150.8 (C‐2), 156.7 (Boc‐C=O), 157.0 (Fmoc‐C=O), 164.9 (C‐4), 174.8 ppm (COOH); IR (ATR): =1685, 1519, 1447, 1255, 1161, 1070, 1050, 1022, 759, 736 cm−1; UV (MeCN): λ max (log ϵ)=206 (4.58), 264 nm (4.25); HRMS (ESI) calcd for C32H36N4NaO9: 643.2374; found: 643.2373 [M+Na]+.
3‐N‐BOM‐3′‐azido‐3′‐deoxythymidine‐5′‐aldehyde 3: IBX (3.15 g, 11.2 mmol) was added to a solution of 3‐N‐BOM‐3′‐azido‐3′‐deoxythymidine 5 (1.74 g, 4.49 mmol) in MeCN (43 mL), and the reaction mixture was stirred for 45 min under reflux. It was then cooled to RT and filtered. The filter cake was washed with EtOAc (3×20 mL), and the combined filtrates were evaporated under reduced pressure. The resultant residue was kept under high vacuum to remove remaining volatiles to give 3 as a colorless foam (1.72 g, 99 %). With respect to its limited stability, 3 was only characterized by NMR spectroscopy and then used directly in the next reaction. 1H NMR (500 MHz, CDCl3): δ=1.94 (d, J=1.2 Hz, 3 H, 7‐H), 2.35–2.45 (m, 1 H, 2′‐Ha), 2.43 (ddd, J=14.1, 6.7, 4.0 Hz, 1 H, 2′‐Hb), 4.42 (d, J=3.4 Hz, 1 H, 4′‐H), 4.60 (ddd, J=7.0, 3.5, 3.4 Hz, 1 H, 3′‐H), 4.68 (s, 2 H, 2′′‐H), 5.46 (s, 2 H, 1′′‐H), 6.00 (dd, J=6.7, 6.6 Hz, 1 H, 1′‐H), 7.23–7.35 ppm (m, 6 H, 6‐H, aryl‐H); 13C NMR (126 MHz, CDCl3): δ=13.1 (C‐7), 36.6 (C‐2′), 61.7 (C‐3′), 70.6 (C‐2′′), 72.4 (C‐1′′), 88.6 (C‐1′), 90.4 (C‐4′), 110.7 (C‐5), 127.6 (C‐6′′), 127.7 (C‐4′′, C‐8′′), 128.3 (C‐5′′, C‐7′′), 135.3 (C‐6), 137.9 (C‐3′′), 150.7 (C‐2), 163.2 (C‐4), 198.3 ppm (C‐5′).
3‐N‐BOM‐3′‐N‐Cbz‐amino‐3′‐deoxythymidine‐5′‐aldehyde 4: IBX (2.07 g, 7.38 mmol) was added to a solution of 3‐N‐BOM‐3′‐N‐Cbz‐amino‐3′‐deoxythymidine 6 (1.46 g, 2.95 mmol) in MeCN (28 mL), and the reaction mixture was stirred for 45 min under reflux. It was then cooled to RT and filtered. The filter cake was washed with EtOAc (3×15 mL), and the combined filtrates were evaporated under reduced pressure. The resultant residue was kept under high vacuum to remove remaining volatiles to give 4 as a colorless foam (1.45 g, quant.). With respect to its limited stability, 4 was only characterized by NMR spectroscopy and then used directly in the next reaction. 1H NMR (500 MHz, CDCl3): δ=1.96 (s, 3 H, H‐7), 2.06–2.16 (m, 1 H, 2′‐Ha), 2.38–2.47 (m, 1 H, 2′‐Hb), 4.40 (m, 1 H, 3′‐H), 4.53–4.59 (m, 1 H, 4′‐H), 4.65 (s, 2 H, 1′′‐H), 5.04–5.14 (m, 2 H, 1′′′‐H), 5.45 (s, 2 H, 2′′‐H), 5.81–5.89 (m, 1 H, 3′‐NH), 6.25–6.35 (m, 1 H, 1′‐H), 7.20–7.39 (m, 10 H, aryl‐H), 7.64 (s, 1 H, 6‐H), 9.71 ppm (s, 1 H, 5′‐H); 13C NMR (126 MHz, CDCl3): δ=13.3 (C‐7), 36.5 (C‐2′), 52.0 (C‐3′), 67.3 (C‐1′′′), 70.6 (C‐1′′), 72.3 (C‐2′′), 87.7 (C‐1′), 88.5 (C‐4′), 110.9 (C‐5), 127.7, 127.7, 128.2, 128.3, 128.3, 128.7 (aryl‐C), 134.7 (C‐6), 135.7, 135.8 (C‐3′′, C‐2′′′), 137.9 (Cbz‐C=O), 151.0 (C‐2), 163.3 (C‐4), 198.0 ppm (C‐5′); HRMS (ESI) calcd for C26H26N3O7: 492.1776; found: 492.1780 [M−H]−.
(Z)‐6′‐N‐Boc‐3‐N‐BOM‐3′‐azido‐5′,6′‐didehydro‐3′‐deoxythymidinyl amino acid benzyl ester 8: A solution of phosphonate 7 (1.67 g, 4.49 mmol) in THF (34 mL) was added to a precooled (−78 °C) solution of KOtBu (504 mg, 4.49 mmol) in THF (43 mL) at −78 °C. The resultant solution was stirred for 5 min at −78 °C. Subsequently, a solution of aldehyde 3 (1.45 g, 2.95 mmol) in THF (18 mL) was added. The reaction mixture was stirred for 16 h and was allowed to warm slowly to RT during this time period. The resultant suspension was cooled to 0 °C and MeOH (5 mL) was added, after which the solution was diluted with EtOAc (200 mL). It was then washed with water (1×100 mL) and brine (1×100 mL), dried over Na2SO4, filtered, and evaporated under reduced pressure. The resultant crude product was purified by column chromatography (3:2 iso‐hexanes‐EtOAc) to give 8 as a colorless foam (2.06 g, 73 %, diastereomeric mixture Z/E 93:7). As described before,14, 15a,15b, 16c the stereochemical assignment (Z/E) was based on the empirical rules for NMR data established by Mazurkiewicz et al.18 Z‐8: TLC: R f=0.62 (2:3 iso‐hexanes‐EtOAc); 1H NMR (500 MHz, CDCl3): δ=1.48 (s, 9 H, C(CH3)3), 1.83 (d, J=1.2 Hz, 3 H, 7‐H), 2.24 (ddd, J=13.9, 7.4, 6.6 Hz, 1 H, 2′‐Ha), 2.49 (ddd, J=13.9, 5.9, 2.9 Hz, 1 H, 2′‐Hb), 4.35 (ddd, J=6.0, 3.0, 3.0 Hz, 1 H, 3′‐H), 4.69 (s, 2 H, 2′′‐H), 4.86 (dd, J=8.4, 2.8 Hz, 1 H, 6′‐H), 5.22 (d, J=12.2 Hz, 2 H, 1′′′‐Ha), 5.27 (d, J=12.2 Hz, 2 H, 1′′′‐Hb), 5.46 (d, J=9.7 Hz, 1 H, 1′′‐Ha), 5.48 (d, J=9.7 Hz, 1 H, 1′′‐Hb), 6.07 (dd, J=7.4, 6.0 Hz, 1 H, 1′‐H), 6.31 (d, J=8.4 Hz, 1 H, 5′‐H), 6.60 (s, 1 H, 6′‐NH), 7.09 (q, J=1.2 Hz, 1 H, 6‐H), 7.22–7.26 (m, 1 H, aryl‐H), 7.28–7.33 (m, 2 H, aryl‐H), 7.34–7.39 ppm (m, 7 H, aryl‐H); 13C NMR (126 MHz, CDCl3): δ=13.2 (C‐7), 28.1 (C(CH3)3), 37.4 (C‐2′), 65.0 (C‐3′), 68.1 (C‐1′′′), 70.6 (C‐1′′), 72.3 (C‐2′′), 81.9 (C‐6′), 87.7 (C‐1′), 110.4 (C‐5), 125.3 (C‐5′), 127.6, 128.3, 128.5, 128.7, 128.7, 134.0 (aryl‐C), 134.9 (C‐6), 138.4 (aryl‐C), 150.7 (C‐2), 153.1 (Boc‐C=O), 163.2 (C‐4), 163.9 ppm (ester‐C=O); IR (KBr): =2101, 1708, 1652, 1242, 1151, 1065, 1027, 769, 736, 693 cm−1; UV (MeCN): λ max (log ϵ)=206 (4.56), 261 nm (4.17); HRMS (ESI) calcd for C32H36N6NaO8: 655.2492; found: 655.2487 [M+Na]+.
(Z)‐6′‐N‐Boc‐3‐N‐BOM‐3′‐N‐Cbz‐amino‐5′,6′‐didehydro‐3′‐deoxythymidinyl amino acid benzyl ester 9: A solution of phosphonate 7 (1.21 g, 3.25 mmol) in THF (25 mL) was added to a precooled (−78 °C) solution of KOtBu (330 mg, 2.95 mmol) in THF (28 mL) at −78 °C. The resultant solution was stirred for 5 min at −78 °C. Subsequently, a solution of aldehyde 4 (1.45 g, 2.95 mmol) in THF (12 mL) was added. The reaction mixture was stirred for 16 h and was allowed to warm slowly to RT during this time period. The resultant suspension was cooled to 0 °C and MeOH (3 mL) was added, after which the solution was diluted with EtOAc (150 mL). It was then washed with water (1×80 mL) and brine (1×80 mL), dried over Na2SO4, filtered, and evaporated under reduced pressure. The resultant crude product was purified by column chromatography (3:2 iso‐hexanes‐EtOAc) to give 9 as a colorless foam (1.48 g, 68 %, diastereomeric mixture Z/E 91:9). As described before,14, 15a,15b, 16c the stereochemical assignment (Z/E) was based on the empirical rules for NMR data established by Mazurkiewicz et al.18 Z‐9: TLC:=R f 0.31 (1:1 iso‐hexanes‐EtOAc); 1H NMR (500 MHz, CDCl3, 50 °C): δ=1.46 (s, 9 H, C(CH3)3), 1.88 (d, J=1.1 Hz, 3 H, 7‐H), 2.40 (ddd, J=13.9, 7.0, 7.0 Hz, 1 H, 2′‐Ha), 2.44 (ddd, J=13.9, 8.0, 6.2 Hz, 1 H, 2′‐Hb), 4.06–4.13 (m, 1 H, 3′‐H), 4.69 (s, 2 H, 1′′‐H), 4.79 (dd, J=8.4, 6.4 Hz, 1 H, 4′‐H), 5.11 (d, J=12.7 Hz, 1 H, 1′′′‐Ha), 5.13 (d, J=12.7 Hz, 1 H, 1′′′‐Hb), 5.23 (d, J=12.6 Hz, 1 H, 1iv‐Ha), 5.26 (d, J=12.6 Hz, 1 H, 1iv‐Hb), 5.47 (d, J=9.7 Hz, 1 H, 2′′‐Ha), 5.49 (d, J=9.7 Hz, 1 H, 2′′‐Hb), 5.95–6.01 (m, 1 H, 3′‐NH), 6.20 (dd, J=6.2, 6.2 Hz, 1 H, 1′‐H), 6.31 (d, J=8.4 Hz, 1 H, 5′‐H), 6.85 (s, 1 H, 6′‐NH), 7.09 (s, 1 H, 6‐H), 7.21–7.40 ppm (m, 15 H, aryl‐H); 13C NMR (126 MHz, CDCl3, 50 °C): δ=13.1 (C‐7), 28.1 (C(CH3)), 38.7 (C‐2′), 55.9 (C‐3′), 67.0 (C‐1′′′), 68.0 (C‐1iv), 70.7 (C‐1′′), 72.3 (C‐2′′), 79.4 (C‐4′), 81.8 (C(CH3)3), 85.8 (C‐1′), 125.5 (C‐5′), 127.5, 127.6, 128.0, 128.1, 128.2, 128.5, 128.5, 128.6 (aryl‐C), 128.6 (C‐6′), 133.5 (C‐6), 135.1, 136.3, 138.1 (C‐3′′, C‐2′′′, C‐2iv), 150.9 (C‐2), 153.4 (Boc‐C=O), 156.3 (Cbz‐C=O), 163.1 (C‐4), 164.0 ppm (ester‐C=O); IR (ATR): =1704, 1648, 1452, 1250, 1151, 1065, 1027, 774, 736, 698 cm−1; UV (MeCN): λ max (log ϵ)=206 (4.53), 260 nm (4.03); HRMS (ESI) calcd for C40H44N4NaO10: 763.2955; found: 763.2953 [M+Na]+.
6′‐N‐Boc‐3‐N‐BOM‐3′‐azido‐3′‐deoxy‐(S)‐thymidinyl amino acid benzyl ester (S)‐10: The reaction was performed under strict exclusion of oxygen. Nitrogen was bubbled through a solution of olefin 8 (Z/E 93:7, 950 mg, 1.50 mmol) in MeOH (50 mL) for 15 min. Subsequently, the catalyst (S,S)‐Me‐DuPHOS‐Rh (19 mg, 32 μmol) was added and the reaction was stirred for three days at RT under a hydrogen atmosphere (1 bar). A further portion of the catalyst (19 mg, 32 μmol) was added and the reaction was stirred further under a hydrogen atmosphere (1 bar) at RT for four days. Silica (approx. 1/3 of the solvent volume) was added to the solution and the solvent was removed under reduced pressure. The resultant crude product was purified by column chromatography (3:2 iso‐hexanes‐EtOAc) to give (S)‐10 as a colorless foam (800 mg, 84 %, diastereomeric mixture 6′S/6′R 85:15). As described before,14, 15a,15b the stereochemical assignment (6′S/6′R) was based on the catalyst‐controlled nature of the reaction and on an X‐ray crystal structure of a nucleosyl amino acid derivative.15b (S)‐10: TLC R f=0.62 (1:1 iso‐hexanes‐EtOAc); 1H NMR (300 MHz, C6D6, 70 °C): δ=1.39 (s, 9 H, C(CH3)3), 1.68 (ddd, J=13.8, 8.0, 6.0 Hz, 1 H, 2′‐Ha), 1.76–1.84 (m, 2 H, 2′‐Hb, 5′‐Ha), 1.88 (d, J=1.1 Hz, 3 H, 7‐H), 2.01 (ddd, J=14.4, 6.6, 4.0 Hz, 1 H, 5′‐Hb), 3.28 (ddd, J=7.4, 6.0, 6.0 Hz, 1 H, 3′‐H), 3.64 (ddd, J=8.7, 6.2, 4.0 Hz, 1 H, 4′‐H), 4.50–4.58 (m, 1 H, 6′‐H), 4.70 (s, 2 H, 2′′‐H), 4.90 (d, J=12.3 Hz, 1 H, 1′′′‐Ha), 4.93 (d, J=12.3 Hz, 1 H, 1′′′‐Hb), 5.11 (d, J=7.4 Hz, 1 H, 6′‐NH), 5.51 (s, 2 H, 1′′‐H), 5.68 (dd, J=6.0, 6.0 Hz, 1 H, 1′‐H), 6.75 (brs, 1 H, 6‐H), 7.00–7.13 (m, 8 H, aryl‐H), 7.31–7.34 ppm (m, 2 H, aryl‐H); 13C NMR (126 MHz, C6D6, 70 °C): δ=12.6 (C‐7), 28.0 (C(CH3)3), 35.6 (C‐5′), 36.4 (C‐2′), 51.1 (C‐6′), 62.8 (C‐3′), 66.8 (C‐1′′′), 70.6 (C‐1′′), 72.2 (C‐2′′), 79.6 (C(CH3)3), 80.1 (C‐4′), 85.9 (C‐1′), 110.1 (C‐5), 133.7 (C‐6), 127.1, 127.9, 127.9, 128.0, 128.2, 128.3 (aryl‐C), 135.4 (C‐3′′), 138.7 (C‐2′′′), 150.4 (Boc‐C=O), 154.9 (C‐2), 162.5 (C‐4), 171.1 ppm (ester‐C=O); IR (KBr): =2101, 1704, 1652, 1452, 1250, 1156, 1070, 774, 736, 698 cm−1; UV (MeCN): λ max (log ϵ)=206 (4.65), 266 nm (4.19); HRMS (ESI) calcd for C32H37N6O8: 633.2678; found: 633.2675 [M−H]−.
6′‐N‐Boc‐3‐N‐BOM‐3′‐azido‐3′‐deoxy‐(R)‐thymidinyl amino acid benzyl ester (R)‐10: The synthesis of (R)‐10 was performed according to the procedure for the synthesis of (S)‐10 with olefin 8 (Z/E 93:7, 950 mg, 1.50 mmol), (R,R)‐Me‐DuPHOS‐Rh (38 mg, 64 μmol), MeOH (50 mL), and a reaction time of 14 days to give (R)‐10 as a colorless foam (541 mg, 54 %, diastereomeric mixture 6′R/6′S 95:5). As described before,14, 15a,15b the stereochemical assignment (6′S/6′R) was based on the catalyst‐controlled nature of the reaction and on an X‐ray crystal structure of a nucleosyl amino acid derivative.15b (R)‐10: TLC: R f=0.62 (1:1 iso‐hexanes‐EtOAc); 1H NMR (300 MHz, C6D6, 70 °C): δ=1.39 (s, 9 H, C(CH3)3), 1.72–1.85 (m, 3 H, 2′‐Ha, 2′‐Hb, 5′‐Ha), 1.78 (d, J=1.2 Hz, 3 H, 7‐H), 1.86–1.95 (m, 1 H, 5′‐Hb), 3.27–3.34 (m, 1 H, 3′‐H), 3.61 (ddd, J=9.4, 6.4, 3.2 Hz, 1 H, 4′‐H), 4.50–4.58 (m, 1 H, 6′‐H), 4.70 (s, 2 H, 2′′‐H), 4.89 (d, J=12.3 Hz, 1 H, 1′′′‐Ha), 5.00 (d, J=12.3 Hz, 1 H, 1′′′‐Hb), 5.01–5.06 (m, 1 H, 6′‐NH), 5.44 (dd, J=6.8, 5.4 Hz, 1 H, 1′‐H), 5.45 (d, J=9.3 Hz, 2 H, 1′′‐Ha), 5.50 (d, J=9.3 Hz, 2 H, 1′′‐Hb), 6.44–6.47 (m, 1 H, 6‐H), 7.02–7.16 (m, 8 H, aryl‐H), 7.31–7.34 ppm (m, 2 H, aryl‐H); 13C NMR (126 MHz, C6D6, 70 °C): δ=12.7 (C‐7), 28.0 (C(CH3)3), 35.5 (C‐5′), 36.4 (C‐2′), 51.8 (C‐6′), 63.1 (C‐3′), 66.8 (C‐1′′′), 70.6 (C‐1′′), 72.2 (C‐2′′), 79.5 (C(CH3)3), 80.5 (C‐4′), 86.8 (C‐1′), 109.8 (C‐5), 127.2, 127.3, 127.7, 128.0, 128.1, 128.3 (aryl‐C), 133.9 (C‐6), 135.6 (C‐3′′), 138.7 (C‐2′′′), 150.3 (Boc‐ C=O), 155.1 (C‐2), 162.4 (C‐4), 171.3 ppm (ester‐C=O); IR (ATR): =2101, 1704, 1652, 1455, 1270, 1250, 1156, 1075, 736, 698 cm−1; UV (MeCN): λ max (log ϵ)=267 nm (3.99); HRMS (ESI) calcd for C32H37N6O8: 633.2678; found: 633.2679 [M−H]−.
6′‐N‐Boc‐3‐N‐BOM‐3′‐N‐Cbz‐amino‐3′‐deoxy‐(S)‐thymidinyl amino acid benzyl ester (S)‐11: The reaction was performed under strict exclusion of oxygen. Nitrogen was bubbled through a solution of olefin 9 (Z/E 91:9, 1.00 g, 1.35 mmol) in MeOH (49 mL) for 15 min. Subsequently, the catalyst (S,S)‐Me‐DuPHOS‐Rh (16 mg, 27 μmol) was added and the reaction was stirred for four days at RT under a hydrogen atmosphere (1 bar). Silica (approx. 1/3 of the solvent volume) was added to the solution and the solvent was removed under reduced pressure. The resultant crude product was purified by column chromatography (3:2 iso‐hexanes‐EtOAc) to give (S)‐11 as a colorless foam (918 mg, 92 %, diastereomeric mixture 6′S/6′R 91:9). The diastereomers were separated by preparative chiral HPLC to give the pure (6′S)‐diastereomer (S)‐11 (900 mg from 1.00 g of diastereomeric mixture, obtained from several reactions). As described before,14, 15a,15b the stereochemical assignment (6′S/6′R) was based on the catalyst‐controlled nature of the reaction and on an X‐ray crystal structure of a nucleosyl amino acid derivative.15b (S)‐11: M.p. 69 °C; TLC: R f=0.31 (3:2 iso‐hexanes‐EtOAc); HPLC (analytical): t R=30.5 min; HPLC (preparative): t R=32.0 min; = +34.4 (c 1.1, CHCl3); 1H NMR (500 MHz, C6D6, 50 °C): δ=1.39 (s, 9 H, C(CH3)3), 1.68 (ddd, J=13.9, 6.9, 6.9 Hz, 1 H, 2′‐Ha), 1.74 (ddd, J=13.9, 8.4, 5.9 Hz, 1 H, 2′‐Hb), 1.88 (s, 3 H, 7‐H), 1.95 (ddd, J=14.4, 7.3, 7.2 Hz, 1 H, 5′‐Ha), 2.17–2.26 (m, 1 H, 5′‐Hb), 3.59 (ddd, J=7.3, 4.2, 4.2 Hz, 1 H, 4′‐H), 3.83–3.94 (m, 1 H, 3′‐H), 4.63–4.75 (m, 1 H, 6′‐H), 4.71 (s, 2 H, 1′′‐H), 4.91 (d, J=12.5 Hz, 1 H, 1′′′‐Ha), 4.95 (d, J=12.5 Hz, 1 H, 1′′′‐Hb), 5.01 (d, J=12.3 Hz, 1 H, 1iv‐H), 5.06 (d, J=12.3 Hz, 1 H, Bn‐CH2), 5.41–5.49 (m, 1 H, 6′‐NH), 5.51 (s, 2 H, 2′′‐H), 5.97 (dd, J=5.9, 5.9 Hz, 1 H, 1′‐H), 6.93 (brs, 1 H, 6‐H), 6.99–7.19 (m, 11 H, aryl‐H), 7.23–7.27 (m, 2 H, aryl‐H), 7.33–7.37 ppm (m, 2 H, aryl‐H); 13C NMR (126 MHz, C6D6, 50 °C): δ=12.8 (C‐7), 28.0 (C(CH3)3), 35.6 (C‐5′), 37.3 (C‐2′), 51.2 (C‐6′), 54.1 (C‐3′), 66.8 (C‐1′′′), 70.6 (C‐1′′), 72.1 (C‐2′′), 79.5 (C(CH3)3), 80.8 (C‐4′), 84.7 (C‐1′), 110.3 (C‐5), 127.2, 127.5, 127.7, 127.9, 128.0, 128.1, 128.1, 128.3, 128.4 (aryl‐C), 133.4 (C‐6), 135.6, 136.7, 138.7 (C‐3′′, C‐2′′′, C‐2iv), 150.7 (C‐2), 155.2 (Boc‐C=O), 155.7 (Cbz‐C=O), 162.7 (C‐4), 171.5 ppm (ester‐C=O); IR (ATR): =1709, 1647, 1528, 1270, 1237, 1212, 1161, 1022, 736, 693 cm−1; UV (MeCN): λ max (log ϵ)=206 (4.66), 261 nm (4.27); HRMS (ESI) calcd for C40H45N4O10: 741.3141; found: 741.3144 [M−H]−.
6′‐N‐Boc‐3‐N‐BOM‐3′‐N‐Cbz‐amino‐3′‐deoxy‐(R)‐thymidinyl amino acid benzyl ester (R)‐11: The synthesis of (R)‐11 was performed according to the procedure for the synthesis of (S)‐11 with olefin 9 (Z/E 91:9, 300 mg, 0.405 mmol), (R,R)‐Me‐DuPHOS‐Rh (10 mg, 16 μmol), MeOH (15 mL), and a reaction time of 14 days to give (R)‐11 as a colorless foam (220 mg, 73 %, diastereomeric mixture 6′R/6′S 88:12). The diastereomers were separated by preparative chiral HPLC to give the pure (6′R)‐diastereomer (R)‐11 (370 mg from 430 mg of diastereomeric mixture, obtained from several reactions). As described before,14, 15a,15b the stereochemical assignment (6′S/6′R) was based on the catalyst‐controlled nature of the reaction and on an X‐ray crystal structure of a nucleosyl amino acid derivative.15b (R)‐11: M.p. 64 °C; TLC: R f=0.31 (3:2 iso‐hexanes‐EtOAc); HPLC (analytical): t R=37.5 min; HPLC (preparative): t R=39.0 min; =+39.1 (c 0.87, CHCl3); 1H NMR (500 MHz, C6D6, 50 °C): δ=1.38 (s, 9 H, C(CH3)3), 1.60–1.69 (m, 1 H, 2′‐Ha), 1.70–1.80 (m, 1 H, 2′‐Hb), 1.76 (s, 3 H, 7‐H), 1.88–2.00 (m, 1 H, 5′‐Ha), 2.00–2.09 (m, 1 H, 5′‐Hb), 3.52–3.59 (m, 1 H, 4′‐H), 3.77–3.87 (m, 1 H, 3′‐H), 4.60–4.70 (m, 1 H, 6′‐H), 4.71 (s, 2 H, 1′′‐H), 4.89 (d, J=12.2 Hz, 1 H, 1′′′‐Ha), 4.97–5.04 (m, 1 H, 1′′′‐Hb), 5.01 (d, J=12.5 Hz, 1 H, 1iv‐Ha), 5.06 (d, J=12.5 Hz, 1 H, 1iv‐Hb), 5.19–5.29 (m, 1 H, 6′‐NH), 5.48 (d, J=9.5 Hz, 1 H, 2′′‐Ha), 5.51 (d, J=9.5 Hz, 1 H, 2′′‐Hb), 5.75–5.85 (m, 1 H, 1′‐H), 6.93 (brs, 1 H, 6‐H), 7.00–7.19 (m, 11 H, aryl‐H), 7.23–7.27 (m, 2 H, aryl‐H), 7.33–7.37 ppm (m, 2 H, aryl‐H); 13C NMR (126 MHz, C6D6, 50 °C): δ=12.9 (C‐7), 28.0 (C(CH3)3), 35.7 (C‐5′), 37.3 (C‐2′), 52.0 (C‐6′), 54.5 (C‐3′), 66.7 (C‐1′′′), 70.6 (C‐1′′), 72.1 (C‐2′′), 79.5 (C(CH3)3), 80.9 (C‐4′), 85.3 (C‐1′), 110.1 (C‐5), 127.3, 127.5, 127.7, 127.9, 128.1, 128.1, 128.2, 128.4, 128.4 (aryl‐C), 133.2 (C‐6), 135.8, 136.6, 138.7 (C‐3′′, C‐2′′′, C‐2iv), 150.6 (C‐2), 155.4 (Boc‐C=O), 155.6 (Cbz‐C=O), 162.6 (C‐4), 171.8 ppm (ester‐C=O); IR (ATR): =1700, 1642, 1452, 1255, 1156, 1075, 1022, 736, 698 cm−1; UV (MeCN): λ max (log ϵ)=204 (4.60), 266 nm (3.94); HRMS (ESI) calcd for C40H45N4O10: 741.3141; found: 741.3139 [M−H]−.
Synthesis of oligonucleotide analogues 1 a and 1 b
The synthesis of fully cationic oligonucleotide analogues was performed manually on NovaSyn®TGR resin (Merck KGaA) according to standard Fmoc‐based solid‐phase peptide synthesis (SPPS).19 The building blocks were coupled using three equivalents of Fmoc‐protected building blocks relative to the initial Fmoc loading of the resin. The building blocks were mixed with three equivalents of benzotriazol‐1‐yl‐oxy‐tris‐pyrrolidino‐phosphonium hexafluoro‐phosphate (PyBOP) and six equivalents of N,N‐diisopropylethylamine (DIPEA) in N‐methyl‐2‐pyrrolidone (NMP), and twice incubated with the resin for 1 h. Fmoc deprotection was performed with 25 % piperidine in NMP for 15 min. After each double‐coupling step, unreacted amines were blocked with NMP/DIPEA/acetic anhydride (10:1:1; capping solution) for 10 min. For the final N‐terminal modification, the fully cationic oligonucleotide analogues were deprotected as mentioned above, and reacted twice for 10 min with capping solution for acetylation. The fully cationic oligonucleotide analogues were finally cleaved from the resin applying TFA/water/1,2‐ethanedithiol/triisopropylsilane (94:2.5:2.5:1) for 4 h, and precipitated with Et2O at −20 °C. After this final cleavage, crude products were dissolved in water/MeCN (7:3) and purified by RP‐HPLC using a Nucleodur C18 reverse‐phase column (10×125 mm, 110 Å, particle size 5 μm, Macherey–Nagel; solvent A: water+0.1 % TFA, solvent B: MeCN+0.1 % TFA; flow rate: 6 mL min−1). The thus‐obtained pure product fractions were combined, frozen in liquid nitrogen, and lyophilized with a Heto PowerDryTM LL1500 freeze‐drying system (Thermo Scientific). Analytical data of oligonucleotide analogues 1 a and 1 b are given in Table S1 and Figure S1 (Supporting Information).
Melting temperature experiments
Melting temperatures (T m values) were determined in phosphate buffer at pH 7.4 (10 mm NaH2PO4) with varying NaCl concentrations (50 mm, 75 mm, 100 mm, 125 mm). The final oligonucleotide duplex concentration was 1 μm. Prior to the measurement, the samples were heated to 90 °C for 2 min and subsequently cooled to 20 °C. Afterwards, the changes in absorption at λ=260 nm were detected with a CARY‐100 Bio UV/Vis Spectrophotometer (Varian), applying a temperature increase from 20 °C to 90 °C and a temperature decrease from 90 °C to 20 °C four times each with a heating rate of 1.0 °C min−1, respectively, and a data interval of 0.5 °C (bandwidth: 1.0 nm). The T m values correspond to the maximum of the first derivation of the melting curves and are the average of at least three measurements.
CD spectroscopy
CD spectra were recorded with a J‐715 CD spectrometer (Jasco) and a quartz cuvette (path length: 0.1 cm; Hellma). The samples were dissolved in a buffer composed of 10 mm NaH2PO4 and 100 mm NaCl (pH 7.4) to a final concentration of 20 μm. Five CD spectra between wavelengths of λ=190 and 320 nm were recorded in continuous scanning mode at 20 °C and averaged (sensitivity: 10 mdeg, resolution: 1.0 nm, response: 1.0 s, bandwidth: 1.0 nm, scanning speed: 50 nm min−1). Background correction was performed prior to data evaluation and the CD spectra were smoothed by applying an FFT filter. CD data are presented as the mean residual ellipticity (Θ) in degrees cm2 dmol−1.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the group of Prof. Roland Winter (TU Dortmund) for access to their CD spectrometer and the Deutsche Forschungsgemeinschaft (DFG, grant DU 1095/2‐1, Emmy Noether program GR 3592/2‐1) as well as the Fonds der Chemischen Industrie (FCI, Sachkostenzuschuss) for financial support. B.S. is grateful for a doctoral fellowship of the Studienstiftung des deutschen Volkes. This work was supported by AstraZeneca, Bayer CropScience, Bayer HealthCare, Boehringer Ingelheim, Merck KGaA, and the Max Planck Society.
B. Schmidtgall, A. Kuepper, M. Meng, T. N. Grossmann, C. Ducho, Chem. Eur. J. 2018, 24, 1544.
Contributor Information
Prof. Dr. Tom N. Grossmann, Email: t.n.grossmann@vu.nl.
Prof. Dr. Christian Ducho, Email: christian.ducho@uni-saarland.de.
References
- 1. Sharma V. K., Rungta P., Prasad A. K., RSC Adv. 2014, 4, 16618–16631. [Google Scholar]
- 2. Westheimer F. H., Science 1987, 235, 1173–1178. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Cobb A. J. A., Org. Biomol. Chem. 2007, 5, 3260–3275; [DOI] [PubMed] [Google Scholar]
- 3b. Deleavey G. F., Damha M. J., Chem. Biol. 2012, 19, 937–954. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Lebreton J., De Mesmaeker A., Waldner A., Fritsch V., Wolf R. M., Freier S. M., Tetrahedron Lett. 1993, 34, 6383–6386; [Google Scholar]
- 4b. De Mesmaeker A., Waldner A., Lebreton J., Hoffmann P., Fritsch V., Wolf R. M., Freier S. M., Synlett 1993, 733–736; [Google Scholar]
- 4c. De Mesmaeker A., Waldner A., Lebreton J., Hoffmann P., Fritsch V., Wolf R. M., Freier S. M., Angew. Chem. Int. Ed. Engl. 1994, 33, 226–229; [Google Scholar]; Angew. Chem. 1994, 106, 237–240; [Google Scholar]
- 4d. Selvam C., Thomas S., Abbott J., Kennedy S. D., Rozners E., Angew. Chem. Int. Ed. 2011, 50, 2068–2070; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 2116–2118; [Google Scholar]
- 4e. Tanui P., Kennedy S. D., Lunstad B. D., Haas A., Leake D., Rozners E., Org. Biomol. Chem. 2014, 12, 1207–1210; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4f. Mutisya D., Selvam C., Lunstad B. D., Pallan P. S., Haas A., Leake D., Egli M., Rozners E., Nucleic Acids Res. 2014, 42, 6542–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang Z., Benner S. A., J. Org. Chem. 2002, 67, 3996–4013. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. El-Sagheer A. H., Brown T., J. Am. Chem. Soc. 2009, 131, 3958–3964; [DOI] [PubMed] [Google Scholar]
- 6b. El-Sagheer A. H., Sanzone A. P., Gao R., Tavassoli A., Brown T., Proc. Natl. Acad. Sci. USA 2011, 108, 11338–11343; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Sanzone A. P., El-Sagheer A. H., Brown T., Tavassoli A., Nucleic Acids Res. 2012, 40, 10567–10575; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Birts C. N., Sanzone A. P., El-Sagheer A. H., Blaydes J. P., Brown T., Tavassoli A., Angew. Chem. Int. Ed. 2014, 53, 2362–2365; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2394–2397; [Google Scholar]
- 6e. Kukwikila M., Gale N., El-Sagheer A. H., Brown T., Tavassoli A., Nat. Chem. 2017, 9, 1089–1098. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Nielsen P. E., Egholm M., Berg R. H., Buchardt O., Science 1991, 254, 1497–1500; [DOI] [PubMed] [Google Scholar]
- 7b. Zhilina Z. V., Ziemba A. J., Ebbinghaus S. W., Curr. Top. Med. Chem. 2005, 5, 1119–1131. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Strauss J. K., Roberts C., Nelson M. G., Switzer C., L. J. Maher III , Proc. Natl. Acad. Sci. USA 1996, 93, 9515–9520; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Prakash T. P., Kawasaki A. M., Lesnik E. A., Sioufi N., Manoharan M., Tetrahedron 2003, 59, 7413–7422; [Google Scholar]
- 8c. Prhavc M., Prakash T. P., Minasov G., Cook P. D., Egli M., Manoharan M., Org. Lett. 2003, 5, 2017–2020; [DOI] [PubMed] [Google Scholar]
- 8d. Milton S., Honcharenko D., Rocha C. S. J., Moreno P. M. D., Smith C. I. E., Strömberg R., Chem. Commun. 2015, 51, 4044–4047. [DOI] [PubMed] [Google Scholar]
- 9. Brock R., Bioconjugate Chem. 2014, 25, 863–868. [DOI] [PubMed] [Google Scholar]
- 10. Jain M. L., Bruice P. Y., Szabo I. E., Bruice T. C., Chem. Rev. 2012, 112, 1284–1309. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Blaskó A., Dempcy R. O., Minyat E. E., Bruice T. C., J. Am. Chem. Soc. 1996, 118, 7892–7899; [Google Scholar]
- 11b. Barawkar D. A., Bruice T. C., Proc. Natl. Acad. Sci. USA 1998, 95, 11047–11052; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Linkletter B. A., Szabo I. E., Bruice T. C., J. Am. Chem. Soc. 1999, 121, 3888–3896; [Google Scholar]
- 11d. Challa H., Bruice T. C., Bioorg. Med. Chem. Lett. 2001, 11, 2423–2427. [DOI] [PubMed] [Google Scholar]
- 12. Arya D. P., Bruice T. C., J. Am. Chem. Soc. 1998, 120, 6619–6620. [Google Scholar]
- 13. Letsinger R. L., Singman C. N., Histand G., Salunkhe M., J. Am. Chem. Soc. 1988, 110, 4470–4471. [Google Scholar]
- 14.
- 14a. Schmidtgall B., Spork A. P., Wachowius F., Höbartner C., Ducho C., Chem. Commun. 2014, 50, 13742–13745; [DOI] [PubMed] [Google Scholar]
- 14b. Schmidtgall B., Höbartner C., Ducho C., Beilstein J. Org. Chem. 2015, 11, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Spork A. P., Ducho C., Org. Biomol. Chem. 2010, 8, 2323–2326; [DOI] [PubMed] [Google Scholar]
- 15b. Spork A. P., Wiegmann D., Granitzka M., Stalke D., Ducho C., J. Org. Chem. 2011, 76, 10083–10098; [DOI] [PubMed] [Google Scholar]
- 15c. Spork A. P., Büschleb M., Ries O., Wiegmann D., Boettcher S., Mihalyi A., Bugg T. D. H., Ducho C., Chem. Eur. J. 2014, 20, 15292–15297; [DOI] [PubMed] [Google Scholar]
- 15d. Wiegmann D., Koppermann S., Wirth M., Niro G., Leyerer K., Ducho C., Beilstein J. Org. Chem. 2016, 12, 769–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Burk M. J., J. Am. Chem. Soc. 1991, 113, 8518–8519; [Google Scholar]
- 16b. Masquelin T., Broger E., Müller K., Schmid R., Obrecht D., Helv. Chim. Acta 1994, 77, 1395–1411; [Google Scholar]
- 16c. Ducho C., Hamed R. B., Batchelar E. T., Sorensen J. L., Odell B., Schofield C. J., Org. Biomol. Chem. 2009, 7, 2770–2779. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Mitsuoka Y., Kodama T., Ohnishi R., Hari Y., Imanishi T., Obika S., Nucleic Acids Res. 2009, 37, 1225–1238; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Holm A. I. S., Munksgaard Nielsen L., Vrønning Hoffmann S., Brøndsted Nielsen S., Phys. Chem. Chem. Phys. 2010, 12, 9581–9596. [DOI] [PubMed] [Google Scholar]
- 18. Mazurkiewicz R., Kuźnik A., Grymel M., Kuźnik N. H., Magn. Reson. Chem. 2005, 43, 36–40. [DOI] [PubMed] [Google Scholar]
- 19. Kim Y. W., Grossmann T. N., Verdine G. L., Nat. Protoc. 2011, 6, 761–771. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary