


Abstract
Proximal promoter regions (PPR) are heavily transcribed yielding different types of small RNAs. The act of transcription within PPRs might regulate downstream gene expression via transcriptional interference (TI). For analysis, we investigated capped and polyadenylated small RNA transcripts within PPRs of human RefSeq genes in eight different cell lines. Transcripts of our datasets overlapped with experimentally determined transcription factor binding sites (TFBS). For TFBSs intersected by these small RNA transcripts, we established negative correlation of sRNA expression levels and transcription factor (TF) DNA binding affinities; suggesting that the transcripts acted via TI. Accordingly, datasets were designated as TFbiTrs (TF-binding interfering transcripts). Expression of most TFbiTrs was restricted to certain cell lines. This facilitated the analysis of effects related to TFbiTr expression for the same RefSeq genes across cell lines. We consistently uncovered higher relative TF/DNA binding affinities and concomitantly higher expression levels for RefSeq genes in the absence of TFbiTrs. Analysis of corresponding chromatin landscapes supported these results. ChIA-PET revealed the participation of distal enhancers in TFbiTr transcription. Enhancers regulating TFbiTrs, in effect, act as repressors for corresponding downstream RefSeq genes. We demonstrate the significant impact of TI on gene expression using selected small RNA datasets.
INTRODUCTION
Proximal promoter regions (PPRs, for a comprehensive list of abbreviations Supplementary File 1) of RNA polymerase II transcribed genes usually extend up to 1Kb upstream from transcription start sites (TSSs) (1). Transcription factor binding sites (TFBSs) within PPRs provide important cis-regulatory elements and, for example, integrate developmental programs or environmental stimuli in gene expression (2). In eukaryotes, PPRs are heavily interleaved with different classes of small transcripts (3–7). Promoter-associated RNAs (PARs) are transcripts located within several hundred nucleotides surrounding the TSS (3). The majority of these RNAs belong to three different classes (3). i) PASRs (promoter associated small RNAs) are transcribed bidirectionally; most of these small (<200 nt) transcripts map immediately downstream from the TSS, and sense transcription is generally prevalent (3,8,9). PASRs are capped and either represent small independently transcribed RNAs or, processing products of longer overlapping (heteronuclear or messenger) RNAs (3,9). ii) TSSa RNAs (TSS-associated RNAs) are often detected in association with strongly expressed genes (3,10). Most sense and antisense TSSa RNAs are located within 250 nt upstream and 50 nt downstream from the gene TSS (10). Northern blot hybridizations established RNA size ranges of 20 to 90 nt (10). Finally, iii) Transcription initiation RNAs (tiRNAs) are on average 18 nt long and map immediately downstream from the TSS of highly expressed genes (3). Even tiRNAs display bidirectional transcription, which coincides with regions of enriched RNAPII (RNA polymerase II) occupancy levels (11). The biogenesis of tiRNAs is not completely understood, but dependencies on the endonucleolytic cleavage activity of transcription elongation factor TFIIS are suggested (3,11). Therefore, these classes of PAR transcription are most often associated with promoters of highly expressed genes, and also occupy regions immediately downstream from the gene TSS (9). Actual functions of these transcripts are not completely resolved, but initial experiments implied an involvement in the regulation of gene expression (9,12). However, as the functional analysis has been limited to only a small number of transcripts, it cannot entirely be ruled out that many of these RNAs are mere by-products of gene expression (13,14).
Transcriptional interference (TI) and promoter occlusion describe cis-regulatory processes involving two adjacent promoters. Elongating RNA polymerases interfere with the transcription of downstream genes (15,16), and regulate gene expression throughout all major stages of the transcriptional cycle. Therefore, TI might inhibit the formation of preinitiation complexes or remove RNA polymerases that are slow to transit into active elongation. Clashing polymerases also regulate gene expression during the subsequent stages of transcriptional elongation. The efficacy of TI/promoter occlusion correlates with the relative promoter strength: stronger upstream promoters initiate transcription at higher frequencies, and cause greater inhibition at regulated downstream sites. Active RNA polymerases within PPRs also interfere with bound transcription factors (TFs) or inhibit effective binding in ways comparable to TI acting on core promoters (15). More broadly, ‘TI-related’ processes could be viewed as regulatory mechanisms controlling protein/DNA interactions via the act of transcription. We examined TI as a potential mode of action for subsets of small RNA transcription within PPRs of human RefSeq (hg19) protein coding genes. Here, candidate datasets were restricted to small RNAs (<200 nt) within 1Kb upstream regions overlapping experimentally identified TFBSs. Occlusion via TI or related mechanism was correlated with lower TF-binding affinities compared to sites for the same TF within PPRs devoid of candidate transcripts. The corresponding RefSeq genes that harbored TFBSs of reduced binding affinities in proximal promoters displayed only low mRNA expression levels. Hence, our datasets differed substantially from the previously reported PAR collections, which were associated with strongly expressed genes (see above) (3,9–11).
We refer to these transcripts as TFbiTrs (TF-binding interfering Transcripts) or TFbiTr candidates. Our analytical procedure (Supplementary File 6) employed ENCODE (ENCyclopedia Of DNA Elements) cDNA libraries from eight different human cell lines (K562, HeLa, A549, IMR90, MCF7, SKNSH, H1hESC and H1 neuron) as input data. The annotation of TFbiTrs uncovered minimal overlap between cell lines. This allowed the evaluation of TFbiTr-related effects across cell lines. Analysis of identical TFBSs via STAP/TRAP (sequence to affinity prediction/TF affinity prediction) revealed consistently higher relative binding affinities in cell lines and promoter regions devoid of candidate transcripts (17,18). Notably, the corresponding protein-coding RefSeq genes displayed significantly higher expression levels in cell lines and loci devoid of TFbiTrs. In summary, for our datasets TF-binding affinities and mRNA expression correlated negatively with TFbiTr transcription, suggestive of TI.
Detection of primary transcripts within 1Kb upstream regions of RefSeq mRNA encoding genes indirectly implied complex architectures of two promoters within comparatively close vicinity. To characterize these epigenetic landscapes underlying TFbiTr expression, we analyzed histone tail modifications indicative of active and poised transcription, respectively (19). TFbiTrs generally resided within regions enriched for histone tail modifications indicative of active transcription. Chromatin domains of associated downstream genes, however, displayed preferentially poised characteristics, and reconfirmed the RNA-seq analysis. Finally, the evaluation of promoter/enhancer interactomes via ChIA-PET (chromatin interaction analysis by paired-end tag sequencing) revealed that TFbiTr regions participate in cell line specific interactions (20,21). Accordingly, enhancers that trigger TFbiTr expression function indirectly as repressors of (RefSeq) gene expression via TI.
MATERIALS AND METHODS
Datasets: RNA-seq
All datasets included in this analysis are part of the ENCODE repository. References to the corresponding GEO (Gene Expression Omnibus) accession numbers for individual datasets are provided as URLs in Supplementary File 1 and Table S1.1–1.5.
Small RNA-seq for TFbiTr candidate isolation
The edgeR calcNormFactors function was used to obtain normalized expression values from read counts (BAM files) for biological sRNA-seq replicates (CPM) (22,23); featureCounts returned an R ‘List’ object, which includes raw cDNA read counts for each gene and library. Unannotated (i.e. according to GENCODE) cDNA contigs that represented small (<200) RNAs of eight different human cell lines (K562, HeLa, A549, IMR90, MCF7, SKNSH, H1hESC and H1 neuron) were the actual input data of our analysis (Supplementary File 1 and Table S1.1). The total RNA starting material was isolated from whole cell lysates and cDNA libraries were generated via CIP (calf intestinal alkaline phosphatase)/TAP (tobacco acid pyrophosphatase) treatment. All datasets were provided by the ENCODE repository (24). We refer these cDNA contigs as ‘novel’ or transcripts devoid of known function. Datasets were limited to 1Kb upstream regions from RefSeq annotated TSSs of mRNA encoding genes and intersected with experimentally identified TFBSs (see below).
Enrichments in CIP/TAP treated samples compared to untreated controls, allowed the detection of capped transcripts. Only cDNA contigs with ≥10-fold enrichment in CIP/TAP pre-treated starting material entered the analysis (Supplementary File 1 and Table S13). BCV (Biological coefficient of variation) values for sRNAs between biological replicates were calculated with edgeR’s estimateGLMCommonDisp function (Supplementary File 1 Table S14) (22).
Determination of genome-wide expression of messenger RNAs
BAM files for CSHL (Cold Spring Harbor Laboratory) long mRNA-seq (>200, polyA + RNA from whole cell lysates) data for K562, HeLa (main text) and the other six cell lines were accessed via GSE30567 (Supplementary File 1 and Table S1.2). Replicates were normalized using the calcNormFactors function (CPM); featureCounts returned an R ‘List’ object, which includes read counts for each gene and library (22,23). Messenger RNA expression for K562 and HeLa was analyzed via edgeR (rpkm) (22). BCV values between biological replicates within samples and across cell lines were calculated via edgeR’s estimateGLMCommonDisp function (Supplementary File 1 and Table S12) (23).
Calculation of correlation coefficient in expression data for TFbiTrs and corresponding downstream mRNA
We used RPKM calculated via edgeR to determine correlation coefficient between expression values obtained for TFbiTrs and mRNA datasets. rpkm gives expression values normalized by library size, TMM and gene length (23). The approach was used to consider the different feature lengths (TFbiTrs and corresponding downstream mRNAs) while comparing datasets within cell lines.
CAGE cluster analysis
CAGE (cap analysis of gene expression) was employed to restrict our datasets to products of primary transcription within 1Kb upstream regions of RefSeq mRNA encoding genes (25) (Supplementary File 1 and Table S1.3). CAGE clusters were selected via pre-calculated HMMs (hidden Markov models, scores-0.77/1.00) (26). The application of HMMs reduces the inclusion of false positives, which might be incorrectly interpreted as bona fide TSSs (27). The statistical significance of CAGE clusters intersecting with TFbiTr candidate regions was analyzed via two-tailed χ2 tests (Supplementary File 1 and Table S2.1 and 2.2).
RIP-seq (RNA Immunoprecipitation) with antibodies against polyA binding protein
PABP (polyA binding protein) binds to polyA tails of (m)RNAs (28). We analyzed BAM files for RNA RIP-seq libraries generated from K562 and HeLa cellular lysates and with antibodies raised against PABP with Piranha for confirmation of TFbiTr polyadenylation (29,30) (Supplementary File 1 and Table S1.4). The distribution of starting peaks at candidate 3′ termini was analyzed with default parameters (./Piranha rip.bam -o (output) -p (threshold = significant threshold for sites) -a (background threshold = 0.99) -b (bin size = 10) -l (log scale). The statistical significance of PABP clusters starting at TFbiTr candidate 3′ termini was evaluated via the GSC (genome structure correction, Supplementary File 1, Section A) (31,32).
PolyA-seq analysis
Pre-aligned polyA-seq (i.e. for cell lines other than K562 and HeLa) cDNA collections for single polyA sites representing five different human tissues (brain, kidney, liver, testis and muscle) were employed for analysis (33) (Supplementary File 1 and Table S1.5). The statistical significance of polyA-seq clusters intersecting with TFbiTr candidate regions was analyzed via two-tailed χ2 tests (Supplementary File 1 and Table S9).
CEAS analysis of candidate TFbiTr genomic regions
Genomic regions containing candidate transcripts were annotated with CEAS (cis-element annotation system), which computes the G/C content, identifies mapped genes, and reports enrichment of TFBSs (34). Regulatory regions reported by CEAS, which relies on RefSeq gene annotations (hg19), include: (i) PPRs (1Kb upstream from the TSS), (ii) enhancer domains (>1Kb upstream and downstream from the TSS and TTS [transcription termination site], respectively), (iii) exons, (iv) introns and (v) immediate downstream regions (extending up to 1Kb downstream from annotated 3′ ends of RefSeq genes). Only TFbiTrs, which are entirely located within PPRs (i.e. 1Kb upstream of the RefSeq-annotated TSS), were selected for further analyses. Therefore, our datasets do not contain small RNAs that intersect with the TSS of the corresponding downstream RefSeq genes.
Datasets: ChIP-seq (chromatin immunoprecipitation sequencing) and ChIA-PET (chromatin interaction analysis by paired-end tag sequencing)
All datasets for ChIP-seq, ChIA-PET and nucleosome positioning are part of the ENCODE repository (20,35). References to GEO accession numbers are provided as URLs in the Supplementary File 1 and Table S1.6–1.8. For analysis of ChIP-seq data we utilized the rmdup command from MACS2 (Model-based Analysis of ChIP-Seq v2) to filter duplicated reads starting within the analyzed regions (36).
Calculation of ChIP-seq peaks for transcription factor binding sites within proximal promoter regions
To quantify effects that are related to TI, we employed the ENCODE TFBS ChIP-seq data track for detectable binding sites that were intersected by TFbiTrs (Supplementary File 1 and Table S1.6) (37). Only TFs that directly interact with DNA and do not depend on chromatin-mediated interactions were considered for this analysis. Genome-wide peaks were identified with MACS2 and standard parameters (macs2 callpeak -t ChIP.bam -c Control.bam -f BAM -g hs -n output -B -q 0.01) (36). The application of the MACS2 bdgcmp command computes enrichment and removes background noise from BedGraph signal data for reported peaks. All subsequent analysis, i.e. (i) reading of peak datasets, (ii) occupancy analysis (for determining consensus peaksets) (iii) read counting (iv) differential binding affinity analysis and finally (v) plotting was conducted according to the DiffBind protocol (38). ChIP-seq peaks for TFBSs were intersected within H3K4me3 peaks to ensure the analysis of actual promoter regions (19). Datasets for investigated TF were obtained via the GEO; URLs are provided in Supplementary File 1 and Table S1.6. The impact of sRNA expression on TF-binding was further investigated via STAP/TRAP analysis tools (see below).
Histone modifications- ChIP-seq data and peak calling
Default parameters were applied for MACS2 analysis for calculation of genome-wide peaks of histone tail modifications within ENCODE ChIP-seq datasets as input (macs2 callpeak -t ChIP.bam -c Control.bam -f BAM -g hs -n output -B -q 0.01) (Supplementary File 1 and Table S1.7). The MACS2 bdgcmp command computes enrichment and removes background noise from BedGraph signal data for reported peaks. Biological replicates for each cell line were pooled separately. Peaks were selected via q-values (FDR [False Discovery Rate], 0.01 = default) and mfold = 10 (default = 5.5).
Active promoter states were defined by combined enrichments of H3K4me3, H3K4me2, H3K27ac, RNAPII (i.e. including RNAPII with phosphorylated C-terminal domains) and poised (promoter) states by H3K4me3 and H3K27me3, respectively (39). H3K4me3 BED regions (broad peaks) were the (input) reference for detection of the other overlapping peaks (i.e. H3K27ac, RNAPII, H3K4me2 for active and H3K27me3 for poised promoter states, respectively).
For analysis of transcription within candidate regions (i.e. +50 to −150 from the TSS of TFbiTrs) and RefSeq core promoters (i.e. +50 to −150 from the TSS of downstream RefSeq genes), H3K79me2, H3K36me3 and H4K20me1 histone tail modifications were evaluated (40,41). These datasets were provided by the Broad Institute Histone and accessed via the GEO. URLs are provided in Supplementary File 1 and Table S1.7. Local enrichments for individual histone tail modifications or combinations thereof were statistically analyzed via GSC (Supplementary File 1, Section A) for TFbiTr and core promoter regions, respectively. Calculations of consensus peaks between replicates were performed via DiffBind (consensusObj←dba.peakset (dbaObj, consensus = DBA_CONDITION, minOverlap = 1), data.peakset ← dba.peakset (consensusObj, bRetrieve = TRUE) (38).
Differential analysis of RNA-seq and ChIP-seq data across cell lines (identical loci for K562 and HeLa cells)
Generalized linear models as provided by edgeR and DiffBind were utilized to quantify RefSeq mRNA expression and TF-binding for identical loci across cell lines (22,38). The calcNormFactors (edgeR), glmTreat (edgeR with L2FC > 1.5) and dba_edgeR (DiffBind) functions were employed for normalization and further analysis. Data are represented as boxplots and scatterplots as provided by DiffBind (38).
Measurement of relative transcription factor/DNA binding affinities
In order to analyze relative TF/DNA binding affinities within TFbiTr regions, i.e. for TFBSs overlapped by TFbiTrs, and non-TFbiTr regions, i.e. for TFBSs within PPRs devoid of TFbiTrs, we adhered to the following workflow:
- Extraction of binding motifs and calculation of Position Weight Matrix from ChIP-seq peaks
- DNA sequences of ChIP-seq peak regions for JunD, c-Jun and c-Myc (for K562 and HeLa, for other TFs and cell lines Supplementary File 3 and Table S1) were input data for PhysBinder to extract corresponding binding motifs. The ‘Max. Precision value (PPV)’ option was used for the analysis (42).
- The resulting binding motifs were converted into PWMs (position weight matrices) via the make_pwm motility tool (https://github.com/ctb/motility/blob/master/doc/python-tutorial.html). The make_pwm tool generates PWMs based on log frequencies of each nucleotide and position. PWMs were converted to IUPAC symbols with the make_iupac_motif tool, which is part of the motility package, and employed for further analysis (https://github.com/ctb/motility/blob/master/doc/python-tutorial.html). The similarities between PWMs for case and control data were quantified via the KL divergence test in R (43,44) (Supplementary File 1 and Table S11).
- The PWMs for all the TFs from TFbiTr and non-TFbiTr regions were utilized individually for calculation of TF/DNA binding affinities with STAP version 2 (sequence to affinity prediction v2) (17).
-
Calculation and comparison of TF/DNA binding affinities in TFbiTr and non-TFbiTr regions
STAP (sequence to affinity prediction) requires as input: (a) sequences underlying the corresponding ChIP-seq peaks for TFs in TFbiTr regions (i.e. TFBSs overlapped by TFbiTrs), (b) control peaks in non-TFbiTr regions (i.e. TFBSs within PPRs devoid of TFbiTrs) for the same TF and (c) the TFbiTr associated PWMs (17).
For the calculation of relative TF/DNA binding affinities via STAP we adhered to the following workflow:-
The TF/DNA binding affinities predicted via STAP are based on biophysical models. As a relative measure of binding affinity, the program computes the expected number N of bound TF molecules for a given TF matrix of length W and a given DNA sequence of length ‘l’. This quantity is computed as the sum of individual contributions from all sites ‘l’ contained within the sequences of interest (17).
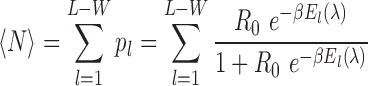 1/β = kBT denotes temperature times Boltzmann constant (Ro) E = energy and p l = probability of sites occurring in a sequence of length ‘l’.
1/β = kBT denotes temperature times Boltzmann constant (Ro) E = energy and p l = probability of sites occurring in a sequence of length ‘l’. - Predicted PWMs to chart TF/DNA affinities as consequence of TFbiTr transcription were derived from non-TFbiTr regions (i.e. TFBSs for the same TF within PPRs devoid of TFbiTrs) by the identical procedure as detailed above (17).
- STAP employs the predicted affinity (based on PWMs) to score DNA/TF-binding affinities in regions of interest (i.e. expected versus observed binding). By a single iteration STAP quantifies whether adding the motif to the model will significantly improve the internally computed Pearson's correlation coefficient. The significance of this improvement is assessed with randomized motifs as negative control. After these iterations STAP re-trains the model parameters (17).
- The STAP output consists of: (i) the binding parameter (i.e. a relative measure of how strongly the TF binds with its binding site): here values greater than 1 signify favorable interaction, and less than 1 unfavorable binding. (ii) The Pearson's correlation coefficients for predicted and observed binding scores (17).
-
-
Extraction of binding motifs computed across identical loci for K562 and HeLa cells
PscanChIP with pre-computed cell line specific background files representing JunD, c-Jun and c-Myc ChIP-seq binding signals in K562 and HeLa cell lines were used to calculate binding motifs in TFbiTr and non-TFbiTr loci (pscan_chip -r input.bed -g hg19 -M -bg BG/K562/HeLa.transfac.bg) (45). Resulting motifs were converted into PWMs as described above and utilized for further analysis via STAP tools.
-
TRAP graphical representations
TRAP v3.05 (transcription factor affinity prediction) generated the graphical output for STAP analysis and displays the predictions of relative binding affinities for TFbiTr and non-TFbiTr regions (18).
Analysis of TFbiTr expression thresholds for occlusion of TF-binding
ENCODE BAM files for sRNA-seq (<200) were used to calculate expression levels (i.e. CPM) for TFbiTrs that were associated with TFBSs of unfavorable TF-binding affinities. This procedure enabled to establish threshold expression levels for occluding transcripts: the ‘-dt flag’ in the STAP command line aided in the identification of TFbiTrs associated with sites of favorable or unfavorable TF-binding affinities.
TFbiTrs and the correlation with downstream RefSeq gene expression
For the selection of appropriate control datasets for RefSeq mRNA expression within cell lines, we categorized the corresponding RPKM values as low, medium or high. RPKM values were employed to account for length difference of RNAs and to allow internal ranking of RNA expression (46–48). RPKM values were calculated via edgeR (22). Using K means in R, expression levels of RefSeq protein coding genes within the K562 cell line were separated into three groups: high (RPKM ≥ 11 ≤ 18), medium (RPKM > 3 ≤ 10) and low (RPKM ≤ 3) (49) (Supplementary File 1 and Table S8 for HeLa and other cell lines).
ChIA-PET data analysis
For K562 and HeLa cell lines, genome-wide ChIA-PET (chromatin interaction analysis by paired-end tag sequencing) interaction clusters (i.e. enhancer/promoter) for RNAPII were accessed via GEO (Supplementary File 1 and Table S1.8). Interactomes for K562 and HeLa cells were intersected with TFbiTr candidate regions via BEDTools’ intersectBed command (intersectBed –a file.bed –b file.bed –f 0.8 –r; where f = Minimum overlap (80%) required as a fraction of a.bed and b.bed and -r = fraction of overlap reciprocal for a.bed and b.bed) (50). ChIA-PET interaction data was normalized for differential peak enrichment and identification of genomic proximity using Mango with default parameters (51).
Analysis of TFbiTr interactomes
TFbiTr interaction partners (i.e. as revealed by ChIA-PET) were scanned for enrichment of promoter, enhancer, and insulator elements as represented by ChIP-seq peaks for P300, CTCF and H3K4me1, respectively. Enhancers were further differentiated into active and poised domains according to the over-representation of H3K27ac and H3K27me3. More specifically, active enhancer states were identified by combined enrichments of H3K4me1, H3K27ac and H3K9ac histone tail modifications. Poised enhancer domains, however, displayed over-representation of H3K27me3 (i.e. in place of H3K27ac and H3K9ac) histone tails (19,52–54). All datasets were accessed via the GEO; URLs are provided within Supplementary File 1 and Table S1.8. Visualizations of the TFbiTr interactomes were generated with CIRCOS (55). The statistical analysis for enrichments of histone tail modifications within TFbiTr interacting arms was conducted via the GSC and two-tailed chi-square (χ2) tests, respectively (See Supplementary File 1 and Tables S5.1–6.2).
Nucleosome occupancy
Nucleosome occupancy levels were analyzed via MNase-seq (sequencing of micrococcal nuclease sensitive sites, Supplementary File 1 and Table S1.7) for TFbiTr containing PPRs. Individual nucleosomal peaks were calculated with DANPOS (dynamic analysis of nucleosome position and occupancy by sequencing) and standard parameters (danpos -b [background] -c count [specify count of reads per replicate] -o [out] -q [occupancy/height] -t [p = 1e-5] -n [data normalization] -F [fold normalization] -w [width] -d [distance between peaks, 100bps] -e [edge = 1] -z [smooth width]) (56). DANPOS analysis proceeds in five steps: (i) calculation of nucleosome occupancy based on mapped reads, (ii) quantile normalization, global scaling and bootstrap sampling to adjust occupancy levels, (iii) calculation of nucleosome signals at single-nucleotide resolution with control and treatment samples applying Poisson test, (iv) peak calling and finally, (v) classification of differential peaks into nucleosome position shifts, fuzziness and occupancy changes (56). Homer was used to compare nucleosome peaks for PPRs with TFbiTrs and promoters of RefSeq genes with similar expression levels to the TFbiTr containing counterparts (57).
Statistical data analysis
Statistical materials and methods are summarized in Supplementary File 1 Sections A and B.
Visualization of ChIP-seq signals for histone modifications and TFBSs
Logos of PWMs in TFbiTr and non-TFbiTr regions were generated using WebLogo (58) All boxplots were drawn with BoxPlotR and DiffBind (38,59). Boxplot notches indicate the 95% confidence interval for the median value, calculated as ± 1.58 × IQR/√n, where IQR is the interquartile range or distance between the first and third quartiles, and n is the number of cells (60). The lower and upper hinges of the boxplots correspond to the first and third quartiles i.e. (the 25th and 75th percentiles). The upper and lower whiskers extend from the hinge to ± 1.5 * IQR of the hinge.
RESULTS
Identification of capped TFbiTr candidates
To restrict datasets to small RNAs that represent products of primary transcription and to reduce the number of potential processing or degradation intermediates, we inspected RNA terminal modifications of candidate TFbiTrs (Supplementary File 6). For identification of capped TFbiTr candidates, we utilized ENCODE small RNA libraries generated with CIP/TAP pre-treated total RNA starting material. Only candidates that displayed at least 10-fold enrichment in CIP/TAP pre-treated starting material entered the analysis (Table 1) (61). Resulting cDNA contigs were intersected with CAGE clusters to reconfirm the identification of capped transcripts (Table 1).
Table 1. Numbers for cDNA contigs and TFbiTr candidates.
| Cell line | Total number of cDNA-contigs | Number of cDNA-contigs after CIP/TAP enrichment | Number of cDNA-contigs overlapping TFBSs of lower TF/DNA binding affinities within PPRs of RefSeq protein coding genes | TFbiTr candidates with capped and polyadenylated transcript termini that overlapped TFBSs of lower binding affinity and correlated with lower expression levels of corresponding downstream RefSeq genes | |
|---|---|---|---|---|---|
| 1 | A549 | 54 089 | 28 993 (53%) | 3282 (11%) | 997 |
| 2 | HeLa | 40 310 | 21 457 (53%) | 4269 (20%) | 1953 |
| 3 | K562 | 39 531 | 18 903 (47%) | 5213 (28%) | 1232 |
| 4 | IMR90 | 26 224 | 16 792 (64%) | 2167 (13%) | 566 |
| 5 | MCF7 | 110 576 | 23 215 (21%) | 5671 (25%) | 2345 |
| 6 | Sknsh | 115 581 | 22 256 (19%) | 6544 (30%) | 1993 |
PPRs are generally enriched in CAGE clusters (62–64). To statistically evaluate the contribution of TFbiTrs to CAGE clusters within PPRs, we analyzed two datasets. The first comprised of all 1Kb upstream regions from the RefSeq gene TSS containing TFbiTrs. The second dataset consisted of the same PPRs, from which we artificially excluded CAGE clusters associated with TFbiTr regions (Supplementary File 1 Section B, two-tailed chi square (χ2) test for details). This allowed the identification of specific associations between CAGE clusters and TFbiTrs compared to flanking regions. The analysis revealed significant enrichment of CAGE clusters within TFbiTr containing PPRs for HeLa and K562 cell lines (χ2, P < 0.01, Supplementary File 1 and Table S2.1 and 2.2), which implied the identification of capped transcripts (Table 1).
TFbiTrs are polyadenylated transcripts—PABP RIP in K562 and HeLa cell lines
We analyzed ENCODE RIP-seq data generated with antibodies against PABP (polyA binding protein) to investigate TFbiTr polyadenylation in K562 and HeLa cells (Supplementary File 6). Regions 30nt up- and downstream from TFbiTr 3′ ends served as control (Supplementary File 2 and Figure S8). The results were statistically evaluated with the GSC tools (GSC tools Supplementary File 1, Section A) (31,32). Here, the number of peaks starting at candidate 3′ ends (intersection) were compared to the number of peaks beginning within 30nt regions up- and downstream from TFbiTr 3′ ends (union) (Supplementary File 1 and Table S3.1a and 3.1b).
Jaccard indices for TFbiTr 3′ termini and flanking regions (0.87 and 0.81 for TFbiTr 3′ ends in K562 and HeLa cells compared with 0.15 and 0.16 for control regions, i.e. the 30nt flanks) revealed candidate polyadenylation for TFbiTrs in K562 and HeLa cells. The identification of TFbiTrs for K562 and HeLa cell lines is summarized in Table 1.
TFbiTrs overlap with polyA-seq clusters indicating candidate polyadenylation
To potentially extend this analysis to TFbiTrs detected within cell lines other than K562 and HeLa, for which no PABP RIP-seq data were available, candidate small RNA contigs were intersected with polyA-seq clusters collected from five different human tissues (‘Materials and Methods’ section) (33) (Supplementary File 6 and Figure S1). Similar to the CAGE cluster analysis, the numbers of polyA-seq clusters within TFbiTr containing PPRs were compared with those detected within the same regions in which TFbiTrs were artificially removed (Supplementary File 1 Section B, two-tailed chi square).
Resulting enrichments were analyzed via χ2-tests and suggestive of candidate polyadenylation (χ2-tests, P < 0.01 Supplementary File 1 and Table S9). Table 1 summarizes the results for TFbiTrs detection quantitatively per cell line.
Analysis of ChIP-seq signals for c-Myc, c-Jun and JunD binding within TFbiTr loci
Due to TI, significantly lower TF/DNA binding affinities in TFbiTr loci were anticipated (15). For this analysis, we computed the overlap of TFbiTrs with annotated TFBSs within PPRs via CEAS (34) (Supplementary File 6 and Figure S1). The analysis of TF-binding required ChIP-seq datasets. We identified that 95% of TFbiTrs in HeLa and 85% in K562 intersected with the ENCODE TFBS ChIP-seq data track (Jaccard indices for significant TFs 0.52 and 0.73). Only TFs present in both HeLa and K562 cell lines (analysis across cell lines, see below) were considered for further investigation. Also, we required that the analyzed TFs bind directly to DNA templates and do not depend on bridging chromatin interactions. Given that our cellular models represent immortalized cancer cell lines, which might be deregulated in various key cellular processes, we included an additional six cell lines with different TFs to establish that TFbiTr-related effects are connected to the general mechanism of TI and are not a consequence of regulation inherent to certain cell lines (Table 2). Quantitative differences for TF-occupancy levels for c-Myc, c-Jun and JunD in K562 and HeLa cell lines as revealed by ChIP-seq analysis for TFbiTr and non-TFbiTr regions (i.e. PPRs devoid of TFbiTrs) demonstrated the predicted effect (Figure 1).
Table 2. TFs included for analysis.
| Cell line | TFs |
|---|---|
| A549, MCF7, Sknsh | GABP, GATA3, JunD, Max, NRSF and TEAD4 |
| IMR90 | JunD |
| K562, HeLa | c-Myc, c-Jun and JunD |
| H1 hESC, H1 neuron | NRSF |
Figure 1.

ChIP-seq analysis of TFs c-Jun, c-Myc and JunD within PPRs (i.e. 1Kb upstream regions from the RefSeq TSS) of human protein coding RefSeq (hg19) genes; plots display ChIP-seq signals for TF-binding sites (A) with and (B) without TFbiTr expression in K562 cells. For the same analysis in HeLa cells see (C) and (D). STAP (sequence to affinity prediction) analysis for TFbiTr versus non-TFbiTr regions (i.e. PPRs devoid of TFbiTrs) revealed ‘unfavorable binding’ for TFs within candidate TFbiTr loci (see main text, Table 3 and Supplementary File 3 for details). Signals for TF-binding were calculated within the major H3K4me3 peak for PPRs. The results suggested the occlusion of TF-binding via TFbiTr expression.
STAP analysis for quantification of relative TF/DNA binding affinities
STAP analysis evaluates TF/DNA binding interactions with ChIP-seq data as input; the method builds on individual training datasets and PWMs for the analyzed TFs (Supplementary File 6). Binding affinities revealed by STAP analysis are therefore relative values and reflect differences in TF/DNA interactions for case and control datasets. PWMs are position-specific weight matrices and represent TF-motifs within input sequence data. To ensure that TFbiTr regions are not associated with particularly low affinity sites, which could explain the lower TF ChIP-seq signal intensities within candidate regions (compared to control regions), we generated PWMs for case (TFbiTr regions) and control (non-TFbiTr regions) datasets. The results clearly revealed that PWMs derived from TFbiTr and non-TFbiTr regions were essentially the same (see maxBindingWts in Tables 3-5Supplementary File 2 and Figure S13). These results were also confirmed via the KL test to analyze the actual similarity between sets of PWMs (Supplementary File 1 and Table S11). Therefore, the fortuitous association of TFbiTrs with low affinity binding sites was unlikely the reason for reduced ChIP-seq signal intensities in the case of TFBSs intersected by candidate sRNAs. Finally, the relative TF/DNA binding efficacies for TFBSs overlapped by TFbiTrs (case) were matched against those derived from non-TFbiTr regions (control) (‘Materials and Methods' section). Our results revealed ‘unfavorable’ binding for TFbiTr regions compared to non-TFbiTr regions (Table 3). STAP output data were also represented graphically via TRAP tools (Figure 2). Our results suggested that the effective reduction of TF/DNA binding in case of candidate datasets is correlated with regulatory TFbiTr expression.
Table 3. STAP (sequence to affinity prediction) results for TFs c-Myc, c-Jun and JunD in TFbiTr and non-TFbiTr regions (PPRs devoid of TFbiTrs) computed for K562 and HeLa cell lines, respectively.
| maxBindingWts | inFactorIntMat | expRatios | ||||||
|---|---|---|---|---|---|---|---|---|
| Cell line | TF | TFbiTr | non-TFbiTr | TFbiTr | non-TFbiTr | TFbiTr | non-TFbiTr | |
| K562 | 1. | c-Myc | 82.15 | 86.69 | 0.001 | 1.34 | 0.04 | 0.81 |
| 2. | c-Jun | 79.93 | 78.52 | 0.11 | 1.89 | 0.01 | 0.86 | |
| 3. | JunD | 87.18 | 93.29 | 0.02 | 1.28 | 0.014 | 0.69 | |
| HeLa | 1. | c-Myc | 79.02 | 82.14 | 0.03 | 1.38 | 0.01 | 0.72 |
| 2. | c-Jun | 69.12 | 68.20 | 0.12 | 1.28 | 0.115 | 0.85 | |
| 3. | JunD | 86.29 | 85.17 | 0.22 | 1.27 | 0.3 | 0.81 | |
Key parameters displayed are (i) maxBindingWts = PWM scores, (ii) inFactorIntMat = favorable (>1)/unfavorable (<1) TF-binding and (iii) expRatios = Pearson’s correlation (‘Materials and Methods’ section). The maxBindingWts scores were similar for TFbiTr and non-TFbiTr control regions. Hence, TFbiTrs are unlikely to be associated with TF-binding sites of lower relative affinity. The results revealed substantially reduced relative binding affinities within TFbiTr loci compared to control datasets (see the inFactorIntMat and expRatios for non-TFbiTr and TFbiTr loci) and suggested the occlusion of otherwise productive TF-binding.
Figure 2.

TRAP analysis for relative TF-binding affinities for loci with and without TFbiTr expression in (A) K562 and (B) HeLa cell lines. Graphs display TF-binding affinities (y-axis) against sequence positions (x-axis) for the investigated TFs. Resulting TF-binding affinities were consistently higher in non-TFbiTr (i.e. 1Kb upstream regions devoid of TFbiTrs) compared to TFbiTr loci (i.e. 1Kb upstream regions containing TFbiTrs)
To demonstrate that these effects are not restricted to specific TFs or cell lines, we investigated TF-binding of seven additional TFs within A549, MCF7, Sknsh and IMR90 cell lines, respectively (Supplementary File 3). The results were entirely consistent with those obtained for HeLa and K562, and revealed reduced TF-binding within regions intersected by TFbiTrs. Furthermore, the analysis of relative TF/DNA binding affinities across cell lines (‘Materials and Methods’ section) strongly implied TI for TFbiTr regions (Table 4) and hinted at a general mechanism of transcriptional regulation, independent of a specific TF or cell line.
Table 4. STAP (sequence to affinity prediction) results for TFs c-Myc, c-Jun and JunD in TFbiTr and non-TFbiTr regions (PPRs devoid of TFbiTrs) computed across identical loci for K562 and HeLa cell lines, respectively.
| maxBindingWts | inFactorIntMat | expRatios | ||||||
|---|---|---|---|---|---|---|---|---|
| Cell line | TF | TFbiTr | non-TFbiTr | TFbiTr | non-TFbiTr | TFbiTr | non-TFbiTr | |
| K562 | 1. | c-Myc | 83.69 | 87.75 | 0.001 | 1.03 | 0.004 | 0.64 |
| 2. | c-Jun | 76.12 | 78.50 | 0.13 | 1.18 | 0.01 | 0.62 | |
| 3. | JunD | 79.69 | 81.72 | 0.02 | 1.36 | 0.014 | 0.75 | |
| HeLa | 1. | c-Myc | 81.34 | 85.06 | 0.03 | 1.56 | 0.01 | 0.79 |
| 2. | c-Jun | 62.20 | 66.23 | 0.12 | 1.35 | 0.05 | 0.79 | |
| 3. | JunD | 82.17 | 80.12 | 0.21 | 1.39 | 0.03 | 0.62 | |
Key parameters displayed are (i) maxBindingWts = PWM scores, (ii) inFactorIntMat = favorable (>1)/unfavorable (<1) TF-binding and (iii) expRatios = Pearson’s correlation (‘Materials and Methods’ section). For each analysis, datasets were reduced to TFbiTrs that are expressed in one cell line (K562 or HeLa) only. The comparison across cell lines enables monitoring of identical TF-binding as a consequence of TFbiTr expression. The results revealed consistently reduced relative binding affinities for TFbiTr loci and agreed with TI as a functional mode of TFbiTr expression (compare inFactorIntMat and expRatios for non-TFbiTr and TFbiTr loci).
The identification of occluding and non-occluding transcripts via STAP, both within and across cell lines, allowed establishing expression thresholds (CPM) that are minimally required for effective TI (Supplementary File 1 Table S10, ‘Materials and Methods’ section).
Analysis of ChIP-seq c-Myc, c-Jun and JunD binding signals overlapping TFbiTr loci across cell lines
To directly monitor effects of TFbiTr expression on identical TF-binding sites, we investigated ChIP-seq signals across cell lines. Here, we restricted datasets to TFbiTrs, which are expressed in one cell line only (HeLa or K562). These transcript collections enabled the quantitative analysis for (the same) TF/DNA interaction with and without influences derived from the potentially regulatory act of TFbiTr expression. DNA binding affinities were investigated with STAP analytical tools. The corresponding relative affinities were on an average much higher in the absence of TFbiTr expression as revealed by ‘unfavorable binding’ in TFbiTr loci (Table 4 and Figure 3).
Figure 3.

Analysis across cell lines; scatterplots for TFs (A) c-Myc (green) (B) c-Jun (blue) and (C) JunD (red) in TFbiTr regions (TFbiTrs +) compared to the same loci that are devoid of TFbiTrs (TFbiTrs −). P-values corrected for multiple testing (q-value) K562 ≥ HeLa: c-Myc q = 1.3 × 10–9, c-Jun q = 7.7 × 10–5, JunD q = 8.8 × 10–1 and HeLa ≥ K562, c-Myc q = 1.5 × 10–7, c-Jun q = 5.6 × 10–4, JunD q = 9.7 × 10–1.
Thresholds of TFbiTr expression for occlusion of TF/DNA binding
Regulation via TI indirectly implies that there are thresholds of TFbiTr expression for effective (‘Introduction’ section) occlusion of otherwise productive TF/DNA binding (16,65). These thresholds likely depend on both the specific TF and analyzed cell line. STAP enables delineation of individual TFBSs, which are associated with favorable or unfavorable TF-binding. Finally, analysis of TFbiTr expression levels established these specific thresholds, which are minimally required to cause effective occlusion of TF-binding (Supplementary File 1 and Table S10). As anticipated, STAP analysis delivered no indications of ‘unfavorable’ TF/DNA binding for TFbiTr candidates that were represented by cDNA contigs with expression levels below these thresholds (Table 5).
Table 5. STAP results for c-Myc, c-Jun and JunD TFs in K562 and HeLa cells for TFbiTr and non-TFbiTr regions (PPRs devoid of TFbiTrs) with sRNA expression below thresholds.
| maxBindingWts | inFactorIntMat | expRatios | ||||||
|---|---|---|---|---|---|---|---|---|
| Cell line | TF | TFbiTr | non-TFbiTr | TFbiTr | non-TFbiTr | TFbiTr | non-TFbiTr | |
| K562 | 1. | c-Myc | 79.29 | 82.75 | 1.01 | 1.01 | 0.67 | 0.72 |
| 2. | c-Jun | 79.32 | 76.50 | 1.42 | 1.78 | 0.48 | 0.66 | |
| 3. | JunD | 82.29 | 87.02 | 1.03 | 1.16 | 0.74 | 0.68 | |
| HeLa | 1. | c-Myc | 73.44 | 78.15 | 1.24 | 1.64 | 0.73 | 0.67 |
| 2. | c-Jun | 81.20 | 79.23 | 1.62 | 1.42 | 0.77 | 0.79 | |
| 3. | JunD | 79.27 | 71.52 | 1.78 | 1.32 | 0.71 | 0.62 | |
Key parameters displayed are: (i) maxBindingWts = PWM scores, (ii) inFactorIntMat = favorable (>1)/unfavorable (<1) TF-binding and (iii) expRatios = Pearson’s correlation (‘Materials and Methods’ section). Note that the maxBindingWts scores were similar in TFbiTr and non-TFbiTr regions. Hence, TFbiTrs are unlikely to be associated with TF-binding sites of lower relative affinity. The analysis revealed no substantial difference for TFbiTrs below threshold and control datasets, which confirmed thresholds for effective TF occlusion (see the inFactorIntMat and expRatios for non-TFbiTr and TFbiTr loci).
TI as functional mode for TFbiTrs also implied that candidate sRNAs are on an average stronger expressed than the regulated downstream RefSeq genes (Figure 4A and B). Correlation tests for mRNA and TFbiTr expression confirmed the anticipated effect (Supplementary File 7). Notably, no such correlation could be established for TFbiTrs expressed below threshold levels for occlusion of TF-binding (Figure 5). Analysis across cell lines with TFbiTrs that were specific to K562 or HeLa cell lines demonstrated that RefSeq genes are significantly stronger expressed in the absence of candidate RNAs (Figure 4C and D, Table 6). We concluded that TF-binding and concomitant RefSeq gene expression are inhibited via TFbiTr expression.
Figure 4.

The panel displays the analysis of mRNA expression for RefSeq genes harboring TFbiTrs in PPRs in (A) K562 and (B) HeLa cell lines. The lower panel depicts mRNA expression for RefSeq genes harboring TFbiTrs in PPRs in (C) K562 (mRNA TFbiTrs +) cells compared to the same genes in HeLa cells devoid of TFbiT5rs (mRNA TFbiTrs −, after correction for multiple testing = 962 sites in analysis) and (D) vice versa (after correction for multiple testing = 1527 sites in analysis). P-values corrected for multiple testing (q-value) K562 ≥ HeLa, q = 1.0 × 10−2 and HeLa ≥ K562 q = 1.2 × 10−3. Black dots represent the non-differentially (non-DE) expressed dataset.
Figure 5.

Results for mRNA expression for RefSeq genes harboring TFbiTrs in PPRs in (A) K562 and (B) HeLa cell lines; the impact of TFbiTr candidates with expression levels above or below thresholds, which are minimally required for effective TF-occlusion, on the corresponding RefSeq gene expression is demonstrated (Supplementary File 1 and Table S10).
Table 6. Expression levels of protein-coding genes containing TFbiTrs in PPRs compared to identical genes devoid of TFbiTrs across cell lines (see main text).
| Cell line | Number of TFbiTrs detected in cell line | Identical TFbiTr loci analyzed in | Number of mRNAs with significantly higher expression values in loci devoid of TFbiTrs | |
|---|---|---|---|---|
| 1 | A549 | 997 | IMR90 | 861 (86%) |
| 2 | HeLa | 1953 | K562 | 1496 (76%) |
| 3 | K562 | 1232 | HeLa | 967 (78%) |
| 4 | IMR90 | 566 | A549 | 449 (88%) |
| 5 | MCF7 | 2345 | Sknsh | 2094(89%) |
| 6 | Sknsh | 1993 | MCF7 | 1872 (93%) |
Histone tail modifications support the RNA-seq results—analysis of TFbiTr and core promoters via GSC (genome structure correction)
Analysis of genome-wide epigenetic landscapes enables the identification of transcriptionally active and inactive genes (19,66) (Supplementary File 6). TI as the prevalent mode of TFbiTr action suggested the following: (i) TFbiTr regions are enriched for histone tail modifications indicative of active promoters; and reversely (ii) the associated downstream protein coding RefSeq genes display preferentially poised promoter characteristics. Specifically, combinations of H3K4me3 and H3K27ac define active promoters, whereas poised promoters display local enrichments of H3K4me3 and H3K27me3, respectively (19,67). We investigated H3K27ac and H3K27me3 occupancy levels along the 1Kb flanks (relative from the TSS) of RefSeq genes, harboring TFbiTrs to test our hypothesis (Figure 6, for results in HeLa cells Supplementary File 2 and Figure S2) (19,66).
Figure 6.

Average enrichments for histone tail modifications in (A) proximal and (B) core promoter regions for RefSeq genes with TFbiTrs in K562 cells; black bars denote the relative positions of TFbiTrs and core promoters. (C) For the same regions chromatin environments representing activation (H3K27ac) and (D) repression (H3K27me3) are also displayed as notched boxplots; chromatin landscapes of candidate loci were specific to PPRs harboring TFbiTrs.
TFbiTr expression within the 1Kb upstream regions also implied that these PPRs harbor cis-acting elements responsible for TFbiTr expression (i.e. apart from those driving the RefSeq gene). For analysis, we investigated chromatin landscapes associated with promoters transcribing TFbiTr and core promoters (i.e. regions −50 to +150 surrounding the TSS of candidate TFbiTrs or RefSeq genes, respectively) via the GSC tools to test the actual significance for local overlaps of histone tail modifications signifying active and poised states within critical regions (Supplementary File1 Section A and Supplementary File 2 and Figure S12).
Enrichments of histone tail modifications signifying transcriptional activity for TFbiTrs and associated core promoter regions
Our analysis indicated that TFbiTrs preferentially reside within active chromatin domains (Figure 6, for HeLa cells Supplementary File 2 and Figure S2, Jaccard indices 0.85 K562 and 0.90 HeLa in TFbiTr regions; Supplementary File 1 Tables S4.1 and S4.3 for K562 and HeLa cell lines). Conversely, connected downstream core promoters displayed predominantly poised characteristics (Jaccard indices 0.06 and 0.09 in K562 and HeLa cells; Supplementary File 1 Tables S4.2 and S4.4). This might imply that candidate regions host two promoters: one responsible for active TFbiTr expression upstream and a poised one, associated with the TSS of downstream mRNAs. Notably, even poised promoters harbor the initiation competent form of RNAPII (19,40,68,69). Therefore, the detection of RNAPII over-representation within TFbiTr containing 1Kb upstream regions and near the RefSeq TSS agreed with this interpretation (i.e. the bimodal distribution of RNAPII). Further confirmatory results were obtained via the analysis of nucleosome positioning based on MNase-treatment for active (i.e. TFbiTr containing 1Kb upstream regions) and poised promoter states (i.e. regions 1Kb downstream from the RefSeq gene TSS), respectively.
Nucleosome occupancies were unperturbed for 1Kb downstream regions from the TSS; displaced nucleosomes, which are strongly indicative of active transcription, were detectable only within the corresponding TFbiTr regions. To avoid wrong conclusion we also compared TFbiTr containing PPRs to those that are devoid of candidate sRNAs. Notably, mRNA expression levels for these control datasets were similar to their TFbiTr-containing counterparts (Figure 7). For PPRs within control data there were no signals of active transcription identifiable as revealed by regular nucleosome occupancies.
Figure 7.

Comparative analysis of nucleosome occupancy levels for genes (A) harboring TFbiTrs (+) and (B) RefSeq genes devoid of TFbiTrs (−) that are expressed at similar levels (‘Materials and Methods’ section) compared to TFbiTr-containing counterparts in K562 cells.
Histone tail modifications indicating transcriptional activity in PPRs with and without TFbiTrs
We compared histone tail modifications detected within PPRs containing TFbiTrs with epigenetic landscapes of PPRs for RefSeq genes devoid of candidate transcripts. In order to avoid bias, this analysis was restricted to RefSeq genes that displayed expression levels similar to those containing TFbiTrs within 1Kb upstream regions (RPKM 0.3–1.4 K562 and 0.2–1.9 HeLa). For this purpose, all datasets were categorized according to corresponding RefSeq gene expression levels (‘Materials and Methods’ section). The absence of H3K27ac enrichments within PPRs for genes devoid of TFbiTrs differentiated both data (Figure 8, for HeLa cell Supplementary File 2 and Figure S3). Even strongly expressed RefSeq genes devoid of TFbiTrs (RPKM 11.8–18.4 K562 and 13.7–18.6 HeLa) displayed specific enrichment of H3K27ac only in the vicinity of the RefSeq TSS, but were devoid of the activating mark within PPRs (Supplementary File 2 and Figure S4.1 and 4.2). In addition, we monitored H3K36me3 and H4K20me1 occupancy levels, which signify transcriptional activity, for highly expressed genes, and observed significant enrichments of either histone tail modification only within downstream core promoters. However, the corresponding PPRs did not display any detectable enrichment of either modification (Supplementary File 4 and Figures S1 and 2). Therefore, promoter landscapes of TFbiTr upstream regions are specific to candidate transcription and no feature of RNAPII promoters per se.
Figure 8.

Average enrichments for histone tail modifications (A) in proximal and (B) core promoter regions for RefSeq genes devoid of TFbiTrs in K562 cells. RefSeq genes were of similar expression levels as datasets containing TFbiTrs in PPRs (‘Materials and Methods’ section). The black bar denotes the core promoter region. (C) and (D) boxplots for the same regions, representing chromatin environments of activation (H3K27ac) and repression (H3K27me3) are displayed.
Analysis of phosphorylated RNAPII ChIP-seq footprints in TFbiTr loci
The largest subunit of RNAPII contains a repetitive carboxyl-terminal domain (CTD) that is phosphorylated during elongation (70). To gain further insight into the promoter structures of TFbiTr regulated genes and as additional control, we analyzed ENCODE ChIP-seq data for local enrichments of phosphorylated RNAPII within 1Kb flanks from the RefSeq gene TSS. Peaks of phosphorylated RNAPII were significantly over-represented in regions of active TFbiTrs compared to downstream mRNAs (Figure 9A and B) (Jaccard index 0.84 and 0.92 for K562 and HeLa cell lines, respectively for TFbiTr promoter regions and 0.012 and 0.005 for core promoters, Supplementary File 1 and Tables S4.5–4.6, χ2 tests P < 0.01, Supplementary File 1 and Tables S4.7 and 4.8). Analysis of analogous regions for non-TFbiTr regulated RefSeq genes with expression levels similar to those containing TFbiTr in PPRs served as control. PPRs of control datasets did not display enrichment for phosphorylated RNAPII. Confirmatory results were also obtained for the distribution of phosphorylated RNAPII in PPRs when non-TFbiTr regulated genes with high expression were analyzed (Figure 9C and D). Here RNA polymerase enrichment, as predicted, within core promoter regions surrounding the RefSeq TSS but was barely detectable within the corresponding 1Kb upstream regions. Hence, the distribution of specific histone tail modifications, occupancy levels of phosphorylated RNAPII and nucleosome positioning distinguish PPRs of TFbiTr-regulated from RefSeq genes devoid of candidate transcription. Figure 10 summarizes our findings for the KIF2A gene (for more examples Supplementary File 2 and Figures S9–11).
Figure 9.

ChIP-seq footprints for phosphorylated RNAPII (P+) in PPRs with TFbiTrs (RNAPII CTD P+ TFbiTr containing PPRs) in and corresponding 1Kb downstream regions in (A) K562 and (B) HeLa cells. ChIP-seq footprints of phosphorylated RNAPII (P+) for highly expressed non-TFbiTr genes (TFbiTr −) in (C) K562 and (D) HeLa cell lines are displayed as notched boxplots.
Figure 10.

TI via TFbiTr expression within the 1Kb upstream region of the KIF2A gene for K562 cells (left); histone tail modifications indicative of active and poised promoter states, respectively: H3K4me3 (mustard yellow), H3K4me2 (purple) and H3K27ac (green), H3K27me3 (aquamarine), H3K36me3 (dark green), H3K79me2 (brown). Peaks for RNAPII (RNA polymerase II) are displayed in gray and KIF2A mRNA expression in black. Lowered c-Myc binding and reduced mRNA expression as a consequence of TFbiTr expression is represented on the left. The right site displays results for the same region analyzed in HeLa cells (right); here in the absence of TFbiTr expression KIF2A gene expression was significantly higher than in K562 cells. Therefore, changes in c-Myc DNA-binding (light green) were associated with altered KIF2A expression. For further examples see Supplementary File 2 and Figures S9–11. The figure was drawn with the aid of the Human Epigenome Browser at http://epigenomegateway.wustl.edu/.
TFbiTr loci interact with enhancer regions
Enhancer and promoter regions are closely embedded in a complex mesh of cell-type specific interactions (71,72). ChIA-PET (chromatin interaction analysis by paired-end tag sequencing) integrates ChIP-based enrichment followed by chromatin proximity ligation (21). The technique utilizes paired-end tags and high-throughput sequencing for the determination of long-range chromatin interactions. The majority (>80%) of TFbiTr loci intersected with regions enriched in ChIA-PET signals in HeLa and K562 cell lines and the interacting regions revealed over-representation of enhancer marks (Supplementary File 1 Tables S5.1–5.6 for K562 and HeLa cell lines, Figure 11).
Figure 11.

Comparison of chromatin environments for distal enhancers associated with TFbiTrs across cell lines. Only enhancers connected with TFbiTrs that were actively expressed in (A) K562 but silent in (B) HeLa cells entered the analysis. ChIP-seq signals for histone tail modifications indicative of active enhancers within TFbiTr interacting arms (as identified via ChIA-PET) for K562 cells suggested that TFbiTr expression is dependent on the activity of these distal enhancers. Identical loci, representing active enhancers in K562,displayed only poised enhancer characteristics in HeLa cells. The results agreed with the predominantly cell line specific expression of TFbiTrs. Signals for the analyzed histone tail modifications were calculated within major H3K4me1 peaks. Boxplot notches indicate the 95% confidence interval for the estimated median value. Log base 2-fold change (L2FC) and P-values corrected for multiple testing (q-value) K562 ≥ HeLa: H3K27ac L2FC = 6.15, q = 1.0 × 10–2, H3K9ac L2FC= 8.17, q = 3.2 × 10–2, H3K27me3 L2FC = 7.11,q = 3.7 × 10–3.
ChIA-PET identified cell line specific interactions of TFbiTr loci with enhancer regions
Comparison of ChIA–PET interactomes between K562 and HeLa cell lines uncovered mostly cell line specific looping interactions. This finding might, at least in part, explain the overall limited intersection of TFbiTr datasets. Of all candidates, 932 were specific to K562 and 1509 to HeLa cells, respectively. To establish the hypothesis that TFbiTr expression is dependent on interacting enhancers of cell line specific activity, the following analysis was conducted: First, we selected PPRs of non-TFbiTr regulated genes, with expression values similar to those of TFbiTr-controlled protein coding genes (RPKM 0.1–1.6 and 0.5–1.8 in K562 and HeLa), and uncovered significantly less enhancer interactions compared to 1Kb upstream regions of mRNA genes regulated via TFbiTrs (χ2 test P < 0.01, Supplementary File 1 and Tables S6.1 and 6.2; Supplementary File 2 and Figure S5). Identical mRNA expression levels for case and control data suggested—albeit indirectly—that differences of enhancer/promoter looping interactions in case of TFbiTr controlled genes are relevant to candidate sRNA transcription. Second, the predominantly cell line-specific mode of TFbiTr transcription enabled the monitoring of enhancer/promoter interactions in relation to TFbiTr activity. We compared looping interactions within the 1Kb upstream regions of active TFbiTrs in K562 to identical sites in HeLa cells in the absence of candidate RNAs and uncovered significantly less enhancer interactions (Supplementary File 2 and Figure S6).
Based on this analysis, we suggest that enhancer loops detected within TFbiTr regions were linked to candidate transcripts and in turn responsible for their expression. ChIP-seq footprints of H3K27ac and H3K9ac enrichment for TFbiTr-linked enhancers revealed that the vast majority (1158/1232 in K562 and 1768/1953 in HeLa) of them were indeed active (73). Notably, when analyzed across cell lines, identical enhancer domains displayed poised characteristics (Figure 11, Supplementary File 2 and Figure S7 for HeLa). Our results therefore suggested functional switches of active enhancers into bona fide repressors via candidate transcription. The cartoon in Figure 12 serves as an illustration.
Figure 12.

(A) ChIA-PET loops representing enhancer–promoter interactions for a TFbiTr candidate, located within the PPR of the NCOA7 gene are displayed by black curves for K562 cells. Black arrows outside of the circle indicate the orientation for RefSeq gene and TFbiTr transcription. Messenger RNA expression levels are shown in blue and RefSeq annotations are displayed in black. Expression of NCOA7 was barely detectable in K562 cells. Orange, yellow and gray circles represent H3K4me3, H3K27ac histone tail modifications and RNAPII, respectively. This combination signifies active promoter states. Active enhancers are displayed in light green, light red and lightyellow for H3K4me1, H3K9ac and H3K27ac, respectively. This figure was drawn with the aid of the Human Epigenome Browser at http://epigenomegateway.wustl.edu/. (B) ChIA-PET loops representing enhancer–promoter interactions for the same region (as in A) in the absence of the TFbiTr candidate in HeLa cells are indicated by black curves. Note the absence of interacting loops for the 1Kb upstream region of the NCOA7 gene. Black arrows outside of the circle indicate the orientation of RefSeq gene and TFbiTr transcription. Messenger RNA expression levels are shown in blue and RefSeq annotations are displayed in black. In the absence of TFbiTr transcription NCOA7 mRNA expression was elevated. Orange, yellow and gray circles represent H3K4me3, H3K27ac histone tail modifications and RNAPII, respectively. This combination signifies active promoter states. Active enhancers are displayed in light green, light red and light yellow for H3K4me1, H3K9ac and H3K27ac, respectively. This figure was drawn with the aid of the Human Epigenome Browser at http://epigenomegateway.wustl.edu/. (C) The cartoon summarizes histone tail modifications and chromatin environments of enhancers interacting with TFbiTr promoters. Active enhancers display enrichments of H3K4me1, H3K9ac, H3K27ac and minimal H3K27me3 occupancies. (D) The cartoon depicts the analogous region as displayed in (C), in the absence of TFbiTr expression. Chromatin environments of poised enhancers display enrichments of H3K4me1 along with H3K27me3. H3K27ac and H3K9ac histone tail modifications, signifying active enhancer states, are barely detectable. Changing chromatin environments for the interacting promoters are indicated schematically.
DISCUSSION
Pervasive transcription generates myriads of non-protein coding transcripts (74,75). However, functions for most of these RNAs remain enigmatic (13). Promoter proximal regions are heavily interleaved with different types of potentially regulatory sRNAs (3,4,8,10,11,76). Overlapping modules of RNA transcription and TFBSs might establish regulatory networks where the act of transcription itself controls TF/DNA interactions (77). Here, as a proof of principle, we analyzed small RNAs (<200 nt) to evaluate the potential of TI to act within PPRs of human RefSeq (hg19) protein coding genes.
Our input datasets were specifically confined to small RNAs within the 1Kb upstream regions, and hence differ from the repertoire of promoter proximal RNAs which included transcripts from downstream regions of the gene TSS (8–11). We identified small transcripts that act via the occlusion of otherwise productive TF/DNA interactions. Only RNAs overlapping with TFBSs of significantly lower relative TF-binding affinities compared to controls were analyzed (24). We established TFbiTr expression thresholds that were minimally required to interfere with productive TF/DNA interactions. These thresholds do not represent absolute values and reflect also the experimental/computational design of our analysis. For control and to avoid false inferences, we also screened for sRNAs that overlapped TFBSs associated with productive TF-binding (i.e. ‘favorable’ binding), and found that these transcripts consistently did not meet the critical expression thresholds (Table 5). For 2% of our dataset we uncovered TFbiTr candidates that were expressed above the threshold and were associated with TFBSs of relatively higher TF-binding affinities. In future studies, these transcripts should be investigated, as they might function as transcriptional activators, maybe in ways analogous to eRNAs (enhancer RNAs) in eukaryotic enhancer domains (78,79). These transcripts might act in complex with proteins as RNPs, e.g. by increasing the local concentration of chromatin modifying activities within promoter proximal regions, which ultimately would lead to stronger TF/DNA interactions.
TI is commonly related to the relative promoter strength of the regulating and to be regulated promoters. For TFbiTrs, we uncovered that candidate transcripts were generally associated with mRNAs of comparatively lower expression. This finding suggested that the occlusion of otherwise productive TF/DNA interactions is the cause for diminished mRNA expression. Hence, TFbiTr expression levels are possibly the most important criterion to define functions associated with these transcripts. Consequently, we consider TFbiTrs to represent simple byproducts of TI, as the actual regulation is exerted via the act of transcription itself and is not connected to any RNA encoded function. The term ‘transcripts’ might be utilized to emphasize differences in TFbiTrs and other sRNAs, e.g. tRNAs or rRNAs that encode functions mediated via (higher order) RNA structures (14). However, our analysis does not necessarily rule out that TFbiTrs act also via additional, trans-regulatory mechanisms, e.g. based on local competition between TFbiTrs and TFBSs for effective TF-binding (12,80).
For the analysis of PPRs containing TFbiTrs, we reckon the precise identification of candidate 5′ termini is essential to unambiguously define TFbiTr in comparison to RefSeq core promoters. Notably, the intersection of candidate 5′ termini as identified with CIP/TAP pretreated total RNA starting material and CAGE clusters reduced the original datasets significantly. This procedure might correct for false positive detection of small RNAs, which potentially escaped sufficient CIP treatment. Furthermore, the confinement to sRNAs that harbor both terminal modifications, i.e. RNA 5′ caps and 3′ terminal polyA tails, helped to exclude transcripts or degradation products, which potentially are derived from hnRNAs with promoters located further upstream from the actual RefSeq TSS and to enrich for full-length transcripts.
Various transcript classes may have escaped identification due to the specific confinements of our cDNA datasets. For instance, CUTs (cryptic unstable transcripts), SUTs (stable uncharacterized transcripts) and PROMPTs (promoter upstream transcripts), which on average are much longer than 200 nt, potentially function via similar mechanisms (81,82,83,84). Also, non-polyadenylated transcripts or RNAs that do not possess 5′ terminal cap structures could act via the same mechanism. Indeed, analysis of these originally excluded transcripts revealed that they—depending on their actual expression levels are competent to occlude productive TF/DNA interactions (Supplementary File 1 and Table S7a–d). This implies that there are possibly (unreported) additional transcript classes within PPRs that regulate gene expression in ways analogous to TFbiTrs. On the other hand, it cannot be ruled that these transcripts represent processed or degraded TFbiTrs.
The interaction of TFbiTr promoters with human tissue-specific enhancer elements could explain the limited intersection of TFbiTrs between cell lines, and suggests that TFbiTrs may be involved in regulation of gene expression in response to developmental stimuli. Also, TI as a regulatory principle might be extended to the occlusion of transcriptional repressors (Supplementary File 5). The analysis of effects mediated by TFbiTr expression on NRSF (neuron-restrictive silencer factor) binding in human Embryonic Stem (ES) and neuronal progenitor cells agreed with both assumptions. Therefore, enhancers might, depending on the occlusion module, act as repressor or activator of corresponding downstream gene expression.
Given the ubiquitous nature of pervasive transcription and the number of regulatory sites encoded within eukaryotic genomes, we envision that numerous pervasive transcripts are best considered as byproducts of TI. Potentially, TI could contribute to the regulation of other functional modules that depend on protein/DNA interactions (Pande et al., in preparation).
Supplementary Material
ACKNOWLEDGEMENTS
We would like thank Philipp Bucher for comments on an earlier version of the manuscript, the reviewers for valuable suggestions and Stephanie Klco-Brosius for language consultation throughout the drafting of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Institute of Bioinformatics; Institute of Experimental Pathology; University of Muenster; Brandenburg Medical School (MHB). Funding for open access charge: University of Muenster, Internal Funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Maston G.A., Evans S.K., Green M.R.. Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet. 2006; 7:29–59. [DOI] [PubMed] [Google Scholar]
- 2. Warnatz H.J., Querfurth R., Guerasimova A., Cheng X., Haas S.A., Hufton A.L., Manke T., Vanhecke D., Nietfeld W., Vingron M. et al. Functional analysis and identification of cis-regulatory elements of human chromosome 21 gene promoters. Nucleic Acids Res. 2010; 38:6112–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taft R.J., Kaplan C.D., Simons C., Mattick J.S.. Evolution, biogenesis and function of promoter-associated RNAs. Cell Cycle. 2009; 8:2332–2338. [DOI] [PubMed] [Google Scholar]
- 4. Yan B.X., Ma J.X.. Promoter-associated RNAs and promoter-targeted RNAs. Cell. Mol. Life Sci. 2012; 69:2833–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jensen T.H., Jacquier A., Libri D.. Dealing with pervasive transcription. Mol. Cell. 2013; 52:473–484. [DOI] [PubMed] [Google Scholar]
- 6. Venkatesh S., Li H., Gogol M.M., Workman J.L.. Selective suppression of antisense transcription by Set2-mediated H3K36 methylation. Nat. Commun. 2016; 7:13610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mellor J., Woloszczuk R., Howe F.S.. The Interleaved Genome. Trends Genet. 2016; 32:57–71. [DOI] [PubMed] [Google Scholar]
- 8. Kapranov P., Cheng J., Dike S., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermuller J., Hofacker I.L. et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007; 316:1484–1488. [DOI] [PubMed] [Google Scholar]
- 9. Affymetrix E.T.P., Cold Spring Harbor Laboratory, E.T.P. Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature. 2009; 457:1028–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seila A.C., Calabrese J.M., Levine S.S., Yeo G.W., Rahl P.B., Flynn R.A., Young R.A., Sharp P.A.. Divergent transcription from active promoters. Science. 2008; 322:1849–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taft R.J., Glazov E.A., Cloonan N., Simons C., Stephen S., Faulkner G.J., Lassmann T., Forrest A.R., Grimmond S.M., Schroder K. et al. Tiny RNAs associated with transcription start sites in animals. Natu. Genet. 2009; 41:572–578. [DOI] [PubMed] [Google Scholar]
- 12. Song X., Wang X., Arai S., Kurokawa R.. Promoter-associated noncoding RNA from the CCND1 promoter. Methods Mol. Biol. 2012; 809:609–622. [DOI] [PubMed] [Google Scholar]
- 13. Brosius J. Waste not, want not–transcript excess in multicellular eukaryotes. Trends Genet. 2005; 21:287–288. [DOI] [PubMed] [Google Scholar]
- 14. Brosius J., Raabe C.A.. What is an RNA? A top layer for RNA classification. RNA Biol. 2016; 13:140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shearwin K.E., Callen B.P., Egan J.B.. Transcriptional interference–a crash course. Trends Genet. 2005; 21:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martens J.A., Laprade L., Winston F.. Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature. 2004; 429:571–574. [DOI] [PubMed] [Google Scholar]
- 17. He X., Chen C.C., Hong F., Fang F., Sinha S., Ng H.H., Zhong S.. A biophysical model for analysis of transcription factor interaction and binding site arrangement from genome-wide binding data. PLoS One. 2009; 4:e8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomas-Chollier M., Hufton A., Heinig M., O’Keeffe S., Masri N.E., Roider H.G., Manke T., Vingron M.. Transcription factor binding predictions using TRAP for the analysis of ChIP-seq data and regulatory SNPs. Nat. Protoc. 2011; 6:1860–1869. [DOI] [PubMed] [Google Scholar]
- 19. Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A. et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007; 39:311–318. [DOI] [PubMed] [Google Scholar]
- 20. Fullwood M.J., Han Y., Wei C.L., Ruan X., Ruan Y.. Chromatin interaction analysis using paired-end tag sequencing. Curr. Protoc. Mol. Biol. 2010; doi:10.1002/0471142727.mb2115s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li G., Fullwood M.J., Xu H., Mulawadi F.H., Velkov S., Vega V., Ariyaratne P.N., Mohamed Y.B., Ooi H.S., Tennakoon C. et al. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010; 11:R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCarthy D.J., Chen Y., Smyth G.K.. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012; 40:4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F. et al. Landscape of transcription in human cells. Nature. 2012; 489:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Valen E., Pascarella G., Chalk A., Maeda N., Kojima M., Kawazu C., Murata M., Nishiyori H., Lazarevic D., Motti D. et al. Genome-wide detection and analysis of hippocampus core promoters using DeepCAGE. Genome Res. 2009; 19:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kodzius R., Kojima M., Nishiyori H., Nakamura M., Fukuda S., Tagami M., Sasaki D., Imamura K., Kai C., Harbers M. et al. CAGE: cap analysis of gene expression. Nat. Methods. 2006; 3:211–222. [DOI] [PubMed] [Google Scholar]
- 27. Down T.A., Hubbard T.J.. Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res. 2002; 12:458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kahvejian A., Roy G., Sonenberg N.. The mRNA closed-loop model: the function of PABP and PABP-interacting proteins in mRNA translation. Cold Spring Harb. Symp. Quant. Biol. 2001; 66:293–300. [DOI] [PubMed] [Google Scholar]
- 29. Baroni T.E., Chittur S.V., George A.D., Tenenbaum S.A.. Advances in RIP-chip analysis: RNA-binding protein immunoprecipitation-microarray profiling. Methods Mol. Biol. 2008; 419:93–108. [DOI] [PubMed] [Google Scholar]
- 30. Uren P.J., Bahrami-Samani E., Burns S.C., Qiao M., Karginov F.V., Hodges E., Hannon G.J., Sanford J.R., Penalva L.O., Smith A.D.. Site identification in high-throughput RNA-protein interaction data. Bioinformatics. 2012; 28:3013–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bickel P.J., Boley N., Brown J.B., Huang H.Y., Zhang N.R.. Subsampling methods for genomic inference. Ann. Appl. Stat. 2010; 4:1660–1697. [Google Scholar]
- 32. Favorov A., Mularoni L., Cope L.M., Medvedeva Y., Mironov A.A., Makeev V.J., Wheelan S.J.. Exploring massive, genome scale datasets with the GenometriCorr package. PLoS Comput. Biol. 2012; 8:e1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Derti A., Garrett-Engele P., Macisaac K.D., Stevens R.C., Sriram S., Chen R., Rohl C.A., Johnson J.M., Babak T.. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012; 22:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shin H.J., Liu T., Manrai A.K., Liu X.S.. CEAS: cis-regulatory element annotation system. Bioinformatics. 2009; 25:2605–2606. [DOI] [PubMed] [Google Scholar]
- 35. Johnson D.S., Mortazavi A., Myers R.M., Wold B.. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007; 316:1497–1502. [DOI] [PubMed] [Google Scholar]
- 36. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nussbaum C., Myers R.M., Brown M., Li W. et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Euskirchen G.M., Rozowsky J.S., Wei C.L., Lee W.H., Zhang Z.D., Hartman S., Emanuelsson O., Stolc V., Weissman S., Gerstein M.B. et al. Mapping of transcription factor binding regions in mammalian cells by ChIP: comparison of array- and sequencing-based technologies. Genome Res. 2007; 17:898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ross-Innes C.S., Stark R., Teschendorff A.E., Holmes K.A., Ali H.R., Dunning M.J., Brown G.D., Gojis O., Ellis I.O., Green A.R. et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012; 481:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou V.W., Goren A., Bernstein B.E.. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011; 12:7–18. [DOI] [PubMed] [Google Scholar]
- 40. Barski A., Zhao K.. Genomic location analysis by ChIP-Seq. J. Cell. Biochem. 2009; 107:11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kolasinska-Zwierz P., Down T., Latorre I., Liu T., Liu X.S., Ahringer J.. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet. 2009; 41:376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Broos S., Soete A., Hooghe B., Moran R., van Roy F., De Bleser P.. PhysBinder: Improving the prediction of transcription factor binding sites by flexible inclusion of biophysical properties. Nucleic Acids Res. 2013; 41:W531–W534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hausser J., Strimmer K.. Entropy inference and the James-Stein Estimator, with application to nonlinear gene association networks. J. Mach. Learn. Res. 2009; 10:1469–1484. [Google Scholar]
- 44. Kullback S., Leibler R.A.. On information and sufficiency. Ann. Math. Statist. 1951; 22:79–86. [Google Scholar]
- 45. Zambelli F., Pesole G., Pavesi G.. PscanChIP: finding over-represented transcription factor-binding site motifs and their correlations in sequences from ChIP-Seq experiments. Nucleic Acids Res. 2013; 41:W535–W543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Conesa A., Madrigal P., Tarazona S., Gomez-Cabrero D., Cervera A., McPherson A., Szczesniak M.W., Gaffney D.J., Elo L.L., Zhang X.G. et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016; 17:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Phipson B., Zappia L., Oshlack A.. Gene length and detection bias in single cell RNA sequencing protocols [version 1; referees: 4 approved]. F1000Res. 2017; 6:595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Evans C., Hardin J., Stoebel D.M.. Selecting between-sample RNA-Seq normalization methods from the perspective of their assumptions. Brief. Bioinform. 2017; doi:10.1093/bib/bbx008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hartigan J.A., Wong M.A.. Algorithm AS 136: A k-means clustering algorithm. Appl. Stat. 1979; 28:100–108. [Google Scholar]
- 50. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Phanstiel D.H., Boyle A.P., Heidari N., Snyder M.P.. Mango: a bias-correcting ChIA-PET analysis pipeline. Bioinformatics. 2015; 31:3092–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karmodiya K., Krebs A.R., Oulad-Abdelghani M., Kimura H., Tora L.. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics. 2012; 13:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Heinz S., Romanoski C.E., Benner C., Glass C.K.. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015; 16:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A.. Circos: an information aesthetic for comparative genomics. Genome Res. 2009; 19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen K., Xi Y., Pan X., Li Z., Kaestner K., Tyler J., Dent S., He X., Li W.. DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013; 23:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spitzer M., Wildenhain J., Rappsilber J., Tyers M.. BoxPlotR: a web tool for generation of box plots. Nat. Methods. 2014; 11:121–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mcgill R., Tukey J.W., Larsen W.A.. Variations of Box Plots. Am. Stat. 1978; 32:12–16. [Google Scholar]
- 61. Gu W.F., Lee H.C., Chaves D., Youngman E.M., Pazour G.J., Conte D., Mello C.C.. CapSeq and CIP-TAP Identify Pol II Start Sites and Reveal Capped Small RNAs as C. elegans piRNA Precursors. Cell. 2012; 151:1488–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Davuluri R.V., Suzuki Y., Sugano S., Plass C., Huang T.H.. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet. 2008; 24:167–177. [DOI] [PubMed] [Google Scholar]
- 63. Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R. et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Abdelhamid R.F., Plessy C., Yamauchi Y., Taoka M., de Hoon M., Gingeras T.R., Isobe T., Carninci P.. Multiplicity of 5′ cap structures present on short RNAs. PLoS One. 2014; 9:e102895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Callen B.P., Shearwin K.E., Egan J.B.. Transcriptional interference between convergent promoters caused by elongation over the promoter. Mol. Cell. 2004; 14:647–656. [DOI] [PubMed] [Google Scholar]
- 66. Kimura H. Histone modifications for human epigenome analysis. J. Hum. Genet. 2013; 58:439–445. [DOI] [PubMed] [Google Scholar]
- 67. Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K.. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129:823–837. [DOI] [PubMed] [Google Scholar]
- 68. Hon G.C., Hawkins R.D., Ren B.. Predictive chromatin signatures in the mammalian genome. Hum. Mol. Genet. 2009; 18:R195–R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011; 473:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Komarnitsky P., Cho E.J., Buratowski S.. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 2000; 14:2452–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A.. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013; 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang Y., Wong C.H., Birnbaum R.Y., Li G., Favaro R., Ngan C.Y., Lim J., Tai E., Poh H.M., Wong E. et al. Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature. 2013; 504:306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Calo E., Wysocka J.. Modification of enhancer chromatin: what, how, and why. Mol. Cell. 2013; 49:825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Berretta J., Morillon A.. Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep. 2009; 10:973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Clark M.B., Amaral P.P., Schlesinger F.J., Dinger M.E., Taft R.J., Rinn J.L., Ponting C.P., Stadler P.F., Morris K.V., Morillon A. et al. The reality of pervasive transcription. PLoS Biol. 2011; 9:e1000625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. ENCODE Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature. 2009; 457:1028–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Greger I.H., Aranda A., Proudfoot N.. Balancing transcriptional interference and initiation on the GAL7 promoter of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:8415–8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kim T.K., Hemberg M., Gray J.M., Costa A.M., Bear D.M., Wu J., Harmin D.A., Laptewicz M., Barbara-Haley K., Kuersten S. et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010; 465:182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Orom U.A., Derrien T., Beringer M., Gumireddy K., Gardini A., Bussotti G., Lai F., Zytnicki M., Notredame C., Huang Q. et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010; 143:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Martianov I., Ramadass A., Serra Barros A., Chow N., Akoulitchev A.. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature. 2007; 445:666–670. [DOI] [PubMed] [Google Scholar]
- 81. Thompson D.M., Parker R.. Cytoplasmic decay of intergenic transcripts in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007; 27:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Buratowski S. TRANSCRIPTION gene expression-where to start. Science. 2008; 322:1804–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Preker P., Nielsen J., Kammler S., Lykke-Andersen S., Christensen M.S., Mapendano C.K., Schierup M.H., Jensen T.H.. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008; 322:1851–1854. [DOI] [PubMed] [Google Scholar]
- 84. Berretta J., Morillon A.. Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep. 2009; 10:973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.