

ABSTRACT
The damage response of DNA single-stranded breaks(SSBs) and double-stranded breaks(DSBs) are two relatively independent processes involving different signaling pathways and protein factors, but there are still many overlapping parts. All of them can activate p53 protein, then the activated p53 regulates the damage response of single-stranded breaks or double-stranded breaks in transcriptional regulation and non-transcriptional regulation. Especially, the two types of damage would compete for RPA and ATR resources in damage repair process. The research has been focused on damage response of DNA single-stranded breaks or DNA double-stranded breaks. However, when single-stranded breaks and double-stranded breaks exist simultaneously, the DNA damage response remains to be elucidated. Here, we present a hybrid numerical model of p53 response and a hybrid numerical model of DNA damage repair exploring DNA damage repair and apoptosis mechanisms when DNA single-stranded breaks and DNA double-stranded breaks exist simultaneously. Firstly, when two kinds of damage are present at the same time, the response of p53 is graded, it means that p53 responds to single-stranded breaks preferentially; Secondly, DNA single-stranded breaks are repaired preferentially, and single-stranded breaks and double-stranded breaks can be repaired simultaneously after most of single-stranded breaks having been repaired; Moreover, single-stranded breaks are more likely to cause apoptosis, because the accumulation of p53 in DNA single-stranded breaks is faster than it in DNA double-stranded breaks and single-stranded breaks has lower threshold of apoptosis.
KEYWORDS: SSB, DSB, DNA damage response, p53 response, damage repair, apoptosis
Introduction
In response to DNA single-stranded breaks, the complex of ATR and its functional partner, ATR-Interacting protein(ATRIP), is recruited to sites of DNA damage by replication proteinA(RPA)-coated single-stranded DNA(RPA-ssDNA) [1–3], and then activated ATR phosphorylate downstream signals CHK1 and p53. Activated p53 regulates the process of DNA damage repair, the repair mechanism for single-stranded breaks is NER or BER, and they require the participation of various regulatory factors such as RPA, PCNA, p48 XP, poly ADP-ribose polymerase (PARP), X-ray repair cross-complementary protein1 (XRCC1) and DNA ligase. P53 is involved in the process of DNA damage repair mainly by interacting with p48, PCNA, RPA and PARP. DNA double-stranded breaks rapidly activate ATM kinase, then ATM phosphorylate downstream reaction elements CHK2 and p53, activated p53 will mediate cell cycle arrest to re pair the lesions. Typically, cells employ two main mechanisms to repair DSBs: homologous recombination(HR) and nonhomologous end joining(NHEJ). NHEJ is typically error-prone process, and it can occur throughout the cell cycle but is dominant in G0/G1 and G2 [4,5]. In this mechanism, the DSB is repaired by blunt end ligation independently of sequence homology, and requiring many factors such as Ku70/80, DNA-PKcs, PARP, MRN complex, BRCA1,BRCA2,XRCC4 and DNA ligase IV [6,7]. Alternatively, the resected DSB can be repaired by HR. Because HR uses a sister or homologous chromatid for repair, it requires strand invasion mediated by the recombinase RAD51 and the process is typically error-free [41], in this mechanism, many RPA molecules bind to the exposed single-stranded DNA to stabilize and protect it, preventing the formation of secondary structures. Though DNA single-stranded breaks and double-stranded breaks initiate different signaling pathways, the p53 activated by ATR/ATM is their common node in the different signaling pathways, the activated p53 regulates the two damage responses in form of transcription-dependent and transcriptional-independent. Moreover, they compete for RPA and ATR resources for their repair. So single-stranded breaks response and double-stranded breaks response are two independent and mutual restraint processes.
The response of p53 to single-stranded damage is graded, it is not excitability and is input-dependent [8]. While p53 responds to double-stranded damage in pulse, the response is excitability, the amplitudes and intervals between pulses seem to be independent of the extent of damage [9–11]. Activated p53 activates cell cycle arrest for repairing lesions, DNA damages are repaired by each pulse, and the lesions are exponentially attenuated with p53 pulses [12–14]; If the lesions are too serious to be repaired timely, the accumulation of p53 in large will activate the apoptotic proteins to induce apoptosis [15]. The thresholds of apoptosis increases with the time, the cells with slower accumulation of p53 have higher apoptosis thresholds [16]. Present research has been focused on DNA single-stranded breaks or DNA double-stranded breaks response. When the two lesions exist simultaneously, the damage responses are unclear.
Here, we present a numerical model of p53 response and a numerical model of DNA damage repair when DNA single-stranded breaks and DNA double-stranded breaks exist simultaneously. Based on the two hybrid numerical models, Firstly, when two kinds of damage are present at the same time, the response of p53 is graded; Secondly, DNA single-stranded breaks are repaired preferentially, and single-stranded breaks and double-stranded breaks can be repaired simultaneously after most of single-stranded breaks having been repaired; Moreover, single-stranded breaks are more likely to induce apoptosis, because of the faster accumulation of p53 in DNA single-stranded breaks.
Modeling methods and assumptions
The model is constructed at the protein levels; Values for the rate constants in the model were chosen by a trail-and-error method based on the results of some relative literature to give simulations that are consistent with known properties of p53 responds to SSB or DSB [8,11].
To explore the relationship between single-stranded damage response and double-stranded damage response, giving the model based on established biological fact supplemented by some assumptions and simplifications:
-
1)The protein synthesis: transcription regulation is replaced by regulation of corresponding p53 protein synthesis, and is incorporated using Hill function,
To characterize the effect of a transcription factor([TF] = x)on the rate of synthesis of the regulated protein [10,15]. While the transcription regulation of the activated p53 to MDM2/Wip1 is replaced by a stiff delay term, which make the production rate of Mdm2 depend directly on the concentration of p53 at an earlier time [10]. The transcriptional activation of p53 to Mdm2 is faster than Wip1, so the delay term about activation of Mdm2 has a smaller value.
-
2)
The protein degradation: the protein degradation is a ubiquitin-dependent process, composed by two parts, one is that the protein is ubiquitinated by others protein, another is the protein auto-ubiquitination [17]. We use the degradation rates to replace the degradation regulation, dependent on the protein levels.
-
3)
The DNA damage repair: the process of DNA damage repair depends on the p53, and the p53 mediates almost all the DNA damage repair pathways [12]. We simply assume that the extent of DNA damage decreases proportionally to p53 levels [13]. In addition, the competition for the resources is replaced by that the sum of the SSB repair rate k22 and the DSB repair rate k12 is a constant.
Results
Model
When DNA single-stranded breaks and double-stranded breaks exist simultaneously, we propose a mechanism of p53 response in Figure 1A and a mechanism of DNA damage repair response in Figure 1B, and then based on these assumptions, the mechanisms are translated into equations respectively (Table 1A and Table 1B).
Figure 1.
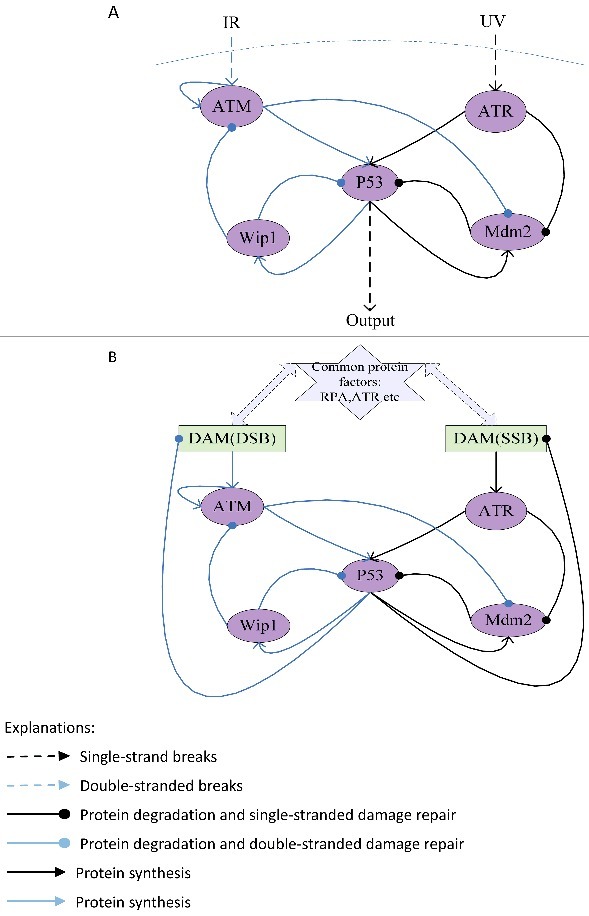
The network structure of p53 response (A) and the network structure of DNA damaged repaired (B) when SSB and DSB exist simultaneously.
Table 1.
Hybrid Model I: the numerical model of p53 response when single-stranded breaks and double-stranded breaks exist simultaneously; Hybrid Model II: the numerical model of DNA damage repair when single-stranded breaks and double-stranded breaks exist simultaneously.
| A |
| B |
In the models, DNA single-stranded breaks and double-stranded breaks can rapidly activate ATR/ATM and its downstream substrates Chk2/Chk1, synergistically catalyzing the phosphorylation of p53 at sites of Ser15 and Ser20; at the same time kinase of ATR/ATM would phosphorylate MDM2 and other E3 ligase, inhibiting p53 ubiquitination degradation.P53 regulates Mdm2 as a transcription factor, whereas Mdm2 regulates p53 by promoting it ubiquitination [18–21]; A negative feedback loop is that p53 can activate Wip1 expression and Wip1 dephosphorylates p53 to reduce it stability [22]. However, the activation process of ATM and the activation process of ATR are different [23–29]. ATM is activated by DSBs with blunt ends or short single-stranded overhangs(SSOs), the conformation of the chromosome changes caused by the DNA double-stranded breaks can induce ATM to be rapidly autophosphorylated and activated. But ATR does not require phosphorylation for its activity [40], ATR and its interacting proteins ATRIP need to be recruited on sites of DNA damage to be activated. So the ATM but not the ATR can be inactivated through dephosphorylation by wip1. Moreover, the two types of damage would compete for RPA and ATR resources in damage repair process.
The order response of DNA damage
P53 responds to single-stranded breaks firstly
Based on the hybrid model of p53 response (Figure 1A and Table 1A), the parameter values given in the Table 2. The output of p53 (Figure 2A) is graded response by setting input1 UV = 1 and input2 IR = 0, the width and amplitude of output increases with the stimulus. The output of p53 (Figure 2B) is digital impulse response by setting input1 UV = 0 and input2 IR = 1, the width and amplitude of the output seem to be independent of stimulus and the number of pulses increases with the stimulus, as experimentally observed by Bachelor [8,11]. While the single-stranded breaks and double-stranded breaks exist at the same time by setting input1 UV = 1 and input2 IR = 1, the output of p53 is graded response firstly, and then impulse response (Figure 2C).
Table 2.
Parameters and Values for the models.
| β1 = 0.8; β11 = 0.4; α15 = 2.7;β2 = 3; α2 = 2.5;β3 = 0.9 |
| β31 = 1.5(A),1.2(B);α34 = 1.4;α35 = 0.14;β4 = 0.9;α41 = 0.5;α42 = 0.5 |
| α4 = 1;β5 = 0.8(A),0.5(B);α5 = 0.7;τ1 = 0.7;τ2 = 1.25;T1 = 0.5 |
| T2 = 1;T3 = 1;n1 = 4;n2 = 4;n3 = 2;C = 0.1 |
| ATM0 = 0; ATR0 = 0;P530 = 0; Mdm20 = 0;Wip10 = 0.9(A),0.1(B) |
Figure 2.
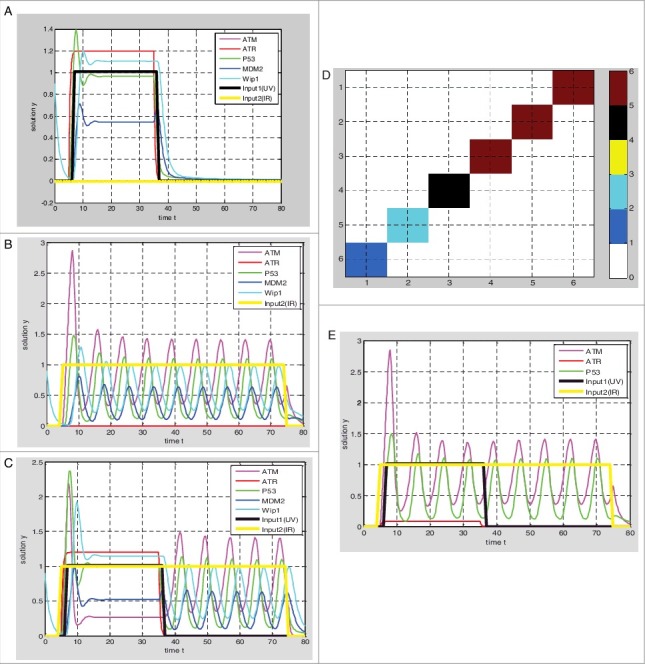
Simulations of p53 response based on hybrid Model I. (A) UV = 1, IR = 0, graded response of p53; (B) UV = 0, IR = 1, the digital impulse response of p53; (C) UV = 1, IR = 1, p53 preferentially responds to DNA single-stranded breaks; (D) the dark red block represents the graded response of the p53, the blue block represents the digital impulse response of p53, the green block and the black block represent the transition state, the ATR protein production rate decreasing from 3 to 0, when the ATR production rate is reduced to 0–0.5, the p53 responds to double-stranded breaks firstly, and in the range of 1.5–3 p53 responds to single-stranded breaks firstly; (E) when ATR production rates are 0.2, p53 responds to DNA double-stranded breaks firstly.
In addition, the output response will vary with the parameter values based on the hybrid model I, meanwhile we set input1 UV = 1 and input2 IR = 1. During the ATR protein production rate β2 decreases from 3 to 0 and other parameters remained, the output response is gradually changing from graded response to digital impulse response. The output response is not digital pulse until the β2 is less than 0.5 (Figure 2D, Figure 2E).
The Single-stranded breaks are repaired firstly
Based on the hybrid model II of DNA damage repair (Figure 1B and Table 1B), the parameter values given in the Table 2. Setting the initial value of SSB is 1 and the initial value of DSB is 0.5; 1) At the beginning, single-stranded breaks are rapidly repaired, while double-stranded breaks can hardly be repaired; 2) The SSB and the DSB can be repaired simultaneously until the amount of SSB is less than DSB, but the repair rate of SSB is still faster than DSB at this time; 3) With the reducing of SSB, the repair rate of SSB decreases and the repair rate of DSB increases; 4) The DNA double-stranded breaks can also be repaired by the graded response of p53, at this time double-stranded breaks may be repaired by NHEJ; 5) With the completion of SSB repair, the p53 responds to DSB in pulse; 6) There may be two reasons for repairing SSB preferentially, one is that the repair rate of single-stranded breaks is faster than the double-stranded breaks; Another is the amount of single-stranded breaks is much more than double-stranded breaks. Therefore, when a large of single-stranded breaks generated, it can quickly takes up the resources to repair and suppress DSB to be repaired. The repair rate of SSB decreases to give up resources to be used by DSB until the amount of SSB is less than the amount of DSB (Figure 3).
Figure 3.
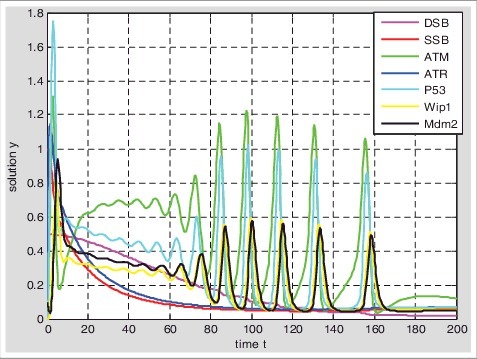
Simulations of DNA damage repaired response based on hybrid Model II. Setting the initial value of single-stranded damage is 1 and double-stranded damaged is 0.5.With the activation of p53(blue) the single strand breaks(red) and double-strand breaks(pink) were repaired competitively.
The single-stranded breaks are more likely to induce apoptosis
P53 responds to single-stranded breaks in graded and responds to double-stranded breaks in digital pulse (Figure 4); Calculating the cumulative value of p53 about t, we set: , then is the p53 cumulative value of single-stranded breaks about t, and is the p53 cumulative value of double-stranded breaks about t. We define that the changing rate of the cumulative value about the different time is the rate of p53 accumulation, then p53 accumulation rate of single-stranded breaks was faster than double-stranded breaks (Figure 4).
Figure 4.
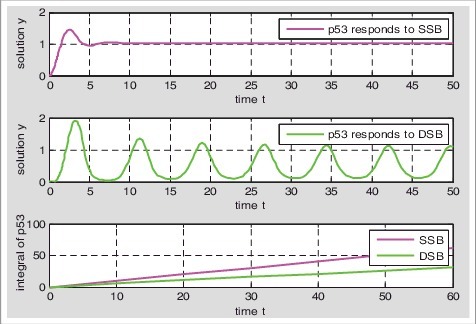
Accumulation rate ofp53 about single-stranded breaks and double-stranded breaks respectively: We calculate the D1, D2 values when t is 0,10,20,30,40,50,60, respectively. The changing rate of D1(D2) is the rate of p53 accumulation.
The low levels of p53 triggers cell-cycle arrest and high levels of p53 leads to apoptosis [31–33]. When the damage is serious, the p53 can accumulate to a certain value to reach the threshold of apoptosis and induce apoptosis. P53 accumulation rate determines the threshold of apoptosis, the slower cumulative rate, the higher threshold of apoptosis and the less likely to apoptosis [16,34]. Because the p53 accumulation rate of single-stranded breaks is faster than that of double-stranded breaks, double-stranded breaks have higher threshold of apoptosis, and single-stranded breaks are more likely to induce apoptosis than double-stranded breaks.
Discussion
In summary, when the two kinds of damage exist simultaneously, they compete to be responded due to the existence of collaborative signal pathways and common damage repair protein factors. Based on the hybrid numerical model of p53 response and the hybrid numerical model of DNA damage repair, we explore the response mechanism when the single-stranded breaks and double-stranded breaks exist simultaneously; P53 response is graded and then digital pulse when the two damages exist at the same time; And DNA single-stranded breaks can be repaired at first, they can be repaired simultaneously once the amount of SSB is less than DSB; Moreover, single- stranded breaks are more likely to induce apoptosis, because it has lower threshold of apoptosis than double-strand breaks.
Ultimately, we wish to understand why the p53 response is graded and why the single-stranded breaks can be repaired preferentially when the two damages coexist. One possibility is that the amount of single-stranded breaks is thousands more than the double-stranded breaks in cells, the ATR can be activated in a high level while at the same time suppressing the expression of ATM by Wip1. A second possibility is that if the single-stranded breaks can not be repaired preferentially, the system will crumble. We supposed that the single-stranded breaks are not repaired preferentially, then the single-stranded breaks that can not be repaired timely will be converted to DSB [34], these DNA double-stranded breaks in the cell cycle phase of S caused by replication fork must be repaired by HR, the single-stranded breaks can not be repaired effectively because the limited resources of RPA and ATR are used by HR, then there will be more single-stranded breaks converting into double-stranded breaks, this positive feedback process is fatal for cells, so the single-stranded breaks must be repaired preferentially. In addition, the risk of cancer increasing with age may attribute to this kind of repair mechanism, the DNA damage increased with age, then there will be relatively more double-stranded breaks that can not be repaired timely because of the competitive repair mechanism, and the double-stranded breaks have a higher apoptotic threshold, the cells with the accumulation of lesions that can not apoptosis timely will be cancerous.
With regard to our models, many improvements could be considered, for example, we only discussed the effects of ATR protein production rate to the output response in the hybrid model I, and further discussions of other parameters in all spaces is necessary; And for the hybrid model II, the introduction of specific molecular mechanisms is necessary, such as, considering the complexity of DNA double-stranded breaks, involving the fast repair process and slow repair process; Considering the relationship between the cell cycle and DNA damage repair; Considering the choosing of repair mechanism and the relationship between damaged repair and cell fate decision [35–40]; This work provides clues to design more effective and less toxic treatments for cancer.
Acknowledgments
Aiqing Ma and X. H. Dai designed and implemented the numerical models, Aiqing Ma accomplished the simulations, all of them analyzed the simulation results and drafted the manuscript.
Disclosure of potential conflicts of interest
NO potential conflict of interest was reported by the authors.
References
- [1].Byun TS, Pacek M, Yee MC, et al.. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. PMID:15833913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Costanzo V, Shechter D, Lupardus PJ, et al.. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell. 2003;11:203–213. doi: 10.1016/S1097-2765(02)00799-2. PMID:12535533 [DOI] [PubMed] [Google Scholar]
- [3].Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. PMID:12791985 [DOI] [PubMed] [Google Scholar]
- [4].Chiruvella KK, Liang Z, Wilson TE. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol. 2013;5:a012757. doi: 10.1101/cshperspect.a012757. PMID:23637284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Karanam K, Kafri R, Loewer A, et al.. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell. 2012;47:320–329. doi: 10.1016/j.molcel.2012.05.052. PMID:22841003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Takata M, Sasaki MS, Sonoda E, et al.. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17(18):5497–5508. doi: 10.1093/emboj/17.18.5497. PMID:9736627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. PMID:20690856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Batchelor E, Loewer A, Mock C, et al.. Stimulus-dependent dynamics of p53 in single cells. Mol Syst Biol. 2011;7(1):488. doi: 10.1038/msb.2011.20. PMID:21556066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lahav G, Rosenfels N, Sigal A, et al.. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36(2):147–150. doi: 10.1038/ng1293. PMID:14730303 [DOI] [PubMed] [Google Scholar]
- [10].Gevazatorsky N, Rosenfeld N, Itzkovitz S, et al.. Oscillations and variability in the p53 system. Mol Syst Biol. 2006;2(1):2006.0033. doi: 10.1038/msb4100068. PMID:16773083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Batchelor E, Mock C, Bhan I, et al.. Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol Cell. 2008;30(3):277–289. doi: 10.1016/j.molcel.2008.03.016. PMID:18471974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sengupta S, Harris CC. p53:traffic cop at crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol. 2005;6:44–55. doi: 10.1038/nrm1546. PMID:15688066 [DOI] [PubMed] [Google Scholar]
- [13].Lobrich M, Rydberg B, Cooper PK. Repair of x-ray-induced DNA double-strand breaks in specific not I restriction fragments in human fibroblasts: joining of correct and incorrect ends. Proc Natl Acad Sci USA. 1995;92:12050–12054. doi: 10.1073/pnas.92.26.12050. PMID:8618842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ciliberto A, Novak B, Tyson JJ. Steady states and oscillations in the p53/mdm2 network. Cell Cycle. 2005;4(3):488–493. doi: 10.4161/cc.4.3.1548. PMID:15725723 [DOI] [PubMed] [Google Scholar]
- [15].Zhang T, Brazhnik P, Tyson JJ. Exploring mechanisms of the dna-damage response: p53 pulses and their possible relevance to apoptosis. Cell Cycle. 2007;6(1):85–94. doi: 10.4161/cc.6.1.3705. PMID:17245126 [DOI] [PubMed] [Google Scholar]
- [16].Paek AL, Liu JC, Loewer A, et al.. Cell-to-cell variation in p53 dynamics leads to fractional killing. Cell. 2016;165(3):631. doi: 10.1016/j.cell.2016.03.025. PMID:27062928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Haupt Y, Maya R, Kazaz A, et al.. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296–299. doi: 10.1038/387296a0. PMID:9153395 [DOI] [PubMed] [Google Scholar]
- [18].Barak Y, Juven T, Haffner R, et al.. mdm2 expression isinduced by wild type p53 activity. EMBO J. 1993;12:461–468. PMID:8440237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Haupt Y, Maya R, Kazaz A, et al.. Mdm2 promotes the rapiddegradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. PMID:9153395 [DOI] [PubMed] [Google Scholar]
- [20].Kubbutat MH, Jones SN, Vousden KH. Regulation of p53stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. PMID:9153396 [DOI] [PubMed] [Google Scholar]
- [21].Wu X, Bayle JH, Olson D, et al.. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. PMID:8319905 [DOI] [PubMed] [Google Scholar]
- [22].Shreeram S, Demidov ON, Hee WK, et al.. Wip1 phosphatase modulates ATM dependent signaling pathways. Mol Cell. 2006;23:757–764. doi: 10.1016/j.molcel.2006.07.010. PMID:16949371 [DOI] [PubMed] [Google Scholar]
- [23].Kozlov SV, Graham ME, Peng C, et al.. Involvement of novel autophosphorylation sites in atm activation. Embo J. 2006;25(15):3504. doi: 10.1038/sj.emboj.7601231. PMID:16858402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506. doi: 10.1038/nature01368. PMID:12556884 [DOI] [PubMed] [Google Scholar]
- [25].Shiotani B, Zou L. Single-stranded dna orchestrates an atm-to-atr switch at dna breaks. Mol Cell. 2009;33(5):547. doi: 10.1016/j.molcel.2009.01.024. PMID:19285939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Barlow JH, Faryabi RB, Callén E, et al.. Identification of early replicating fragile sites that contribute to genome instability. Cell. 2013;152:620–632. doi: 10.1016/j.cell.2013.01.006. PMID:23352430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. PMID:10691732 [PMC free article] [PubMed] [Google Scholar]
- [28].Murga M, Bunting S, Montaña MF, et al.. A mouse model of atr-seckel shows embryonic replicative stress and accelerated aging. Nature Genetics. 2009;41(8):891. doi: 10.1038/ng.420. PMID:19620979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Toledo LI, Murga M, Zur R, et al.. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18:721–727. doi: 10.1038/nsmb.2076. PMID:21552262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cortez KACD. Atr: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9(8):616. doi: 10.1038/nrm2450. PMID:18594563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Das S, Raj L, Zhao B, et al.. Hzf Determines cell survival upon genotoxic stress by modulating p53 transactivation. Cell. 2007;130:624–637. doi: 10.1016/j.cell.2007.06.013. PMID:17719541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Samuels-Lev Y, O'Connor DJ, Bergamaschi D, et al.. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell. 2001;8:781–794. doi: 10.1016/S1097-2765(01)00367-7. PMID:11684014 [DOI] [PubMed] [Google Scholar]
- [33].Tang Y, Luo J, Zhang W, et al.. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. PMID:17189186 [DOI] [PubMed] [Google Scholar]
- [34].Zhang XP, Liu F, Cheng Z, et al.. Cell fate decision mediated by p53 pulses. Proc Nat Acad Sci USA. 2009;106(30):12245–12250. doi: 10.1073/pnas.0813088106. PMID:19617533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Keith W. Single-strand break repair and genetic disease. Nat Rev. 2008;1(1):1–8. [DOI] [PubMed] [Google Scholar]
- [36].Stewart RD. Two-lesion kinetic model of dsb rejoining and cell killing. Radiat Res. 2001;156(4):365–378. doi: 10.1667/0033-7587(2001)156%5b0365:TLKMOD%5d2.0.CO;2. PMID:11554848 [DOI] [PubMed] [Google Scholar]
- [37].Ma L, Wagner J, Rice JJ, et al.. A plausible model for the digital response of p53 to DNA damage[J]. Proc Natl Acad. Sci U S A. 2005;102(40):14266–14271. doi: 10.1073/pnas.0501352102. PMID:16186499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li Y, Reynolds P, O'Neill P, et al.. Modeling damage complexity-dependent non-homologous end-joining repair pathway. Plos One. 2014;9(2):e85816. doi: 10.1371/journal.pone.0085816. PMID:24520318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ceccaldi R, Rondinelli B, D'Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26(1):52–64. doi: 10.1016/j.tcb.2015.07.009. PMID:26437586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Abbotts R. Coordination of DNA single strand break repair[J]. Free Radical Biol Med. 2017;107:228–224. doi: 10.1016/j.freeradbiomed.2016.11.039. PMID:27890643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ceccaldi R, Rondinelli B, D'Andrea AD. Repair Pathway Choices and Consequences at the Double-Strand Break[J]. Trends in Cell Biology, 2016;26(1):52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]