

Abstract
A composite adhesive capable of inducing cellular infiltration was prepared by incorporating control clustered silica microparticle (MP) derived from the aggregation of silica nanoparticle (NP) into a catechol-terminated poly (ethylene glycol) bioadhesive (PEG-DA). Incorporation of MP into PEG-DA significantly improved the mechanical and adhesive properties of the bioadhesive. There was no statistical difference between the measured values for NP- and MP-incorporated adhesives, indicating that MP was equally as effective in enhancing the material properties of PEG-DA as NP. Most importantly, MP was significantly less cytotoxic when compared to NP when these particles were directly exposed to L929 fibroblast. When the adhesives were implanted subcutaneously in rats, MP-containing PEG-DA also exhibited reduced inflammatory responses, attracted elevated levels of regenerative M2 macrophage to its interface, and promoted cellular infiltration due to increased porosity within the adhesive network. Control clustered silica microparticle can be used to improve the performance and biocompatibility of PEG-based adhesive while minimizing undesirable cytotoxicity of silica nanoparticle.
Keywords: silica particle, nanoparticle, clustered particle, microparticle, bioadhesive, biological response, catechol
1. Introduction
The incorporation of inorganic nanoparticles (NP) into polymeric matrices provides a simple approach to create nanocomposites with improved mechanical properties and tailored biological responses (1, 2). This reinforcement strategy of using NP to form physical and chemical crosslinks within a polymer network has been rigorously studied in the past decades. Specifically, nanoparticles used in biomedical applications include silica (Si) particle, bioactive glass, and hydroxyapatite (1, 3–5). Among these particles, Si particles are one of the most commonly used, with key examples such as magneto-luminescence composites (6), drug releasing composite (7), and composites with elevated mechanical property (2, 8). Additionally, Si NP degrades into silicic acid, which has been found to be beneficial for bone and connective tissue formation and remodeling (9, 10).
Despite the advantages associated with Si NP, some reported the toxicity of these particles due to its reduced size (9, 11, 12). The cytotoxicity of NP is associated with the direct physical interaction between the particle and cell membrane, resulting in the cellular internalization of NP (13). Particles with a larger size exhibited reduced cytotoxicity, as the internalization process is hindered when the particle size approaches micron scale (9, 11, 12, 14). However, the performance of the micron-sized particle is inferior to that of NP in improving the mechanical properties of polymer matrices, due to a significant reduction in the surface area as well as the limitation in particle dispersion (15, 16). To overcome the cytotoxicity associated with NP and the reduced performance of micron-sized particles as a filler, we prepared clustered microparticles (MP) that were formed through the clustering of NP. We hypothesized that the clustered MP will have reduced cytotoxicity due to its increased size while retaining nano-scaled surface features with an enhanced surface area for interacting with the polymer matrix.
To evaluate the biocompatibility and effectiveness of MP in functioning as a filler, these particles were incorporated into a biodegradable poly(ethylene glycol) (PEG)-based bioadhesive. A branched PEG was end-functionalized with dopamine (PEG-DA) (Scheme 1). Dopamine contains a catechol adhesive moiety that mimics the curing and interfacial chemistries found in mussel adhesive proteins (17–19). Catechol forms strong interfacial bonds (e.g., hydrogen bonds, electrostatic interaction) with inorganic surfaces such as SiO2 and SiOH on the surface of Si particle (Scheme 1A) (17, 19–21). We previously demonstrated that the incorporation of nanosilicate particles (e.g., Laponite®) into catechol containing hydrogel, significantly improved the mechanical property of the hydrogel (22–25). When catechol is oxidized, it forms the highly reactive quinone which can result in catechol polymerization and curing of the catechol-containing adhesive (26, 27). Quinone can also react with a nucleophilic group (e.g., -NH2 of lysine), resulting in interfacial covalent bond formation with the tissue surface (28, 29).
Scheme 1.
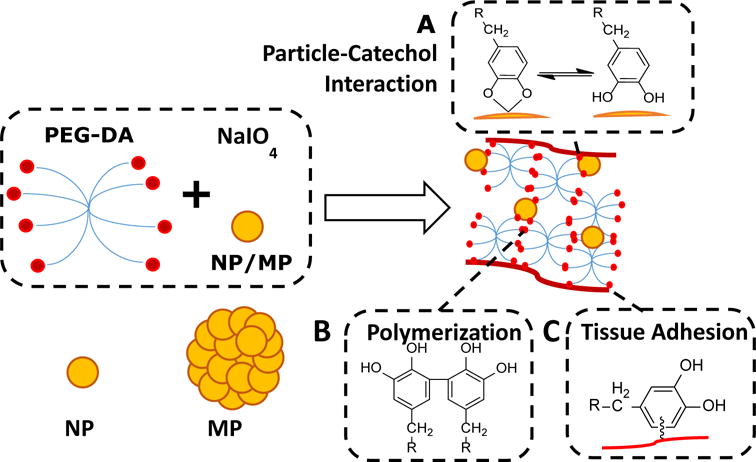
Schematic representation of the chemical interactions involved in the curing and adhesion processes of the composite adhesive. PEG-DA contains catechol end-groups that can interact with silica nano- and microparticles (NP and MP, respectively) through strong reversible interactions (A). Oxidation of catechol in the presense of NaIO4 forms highly reactive quinone, which results in catechol polymerization and curing of the adhesive (B), as well as formation of interfacial covalent bond with nucleophilic function groups (i.e., -NH2 of lysine) and adhesion to the surface of soft tissue (C).
In this paper, Si NP was first prepared by sol-gel method and further clustered to form MP. The effect of incorporating the clustered particle on the mechanical property, adhesive property, and degradation rate of PEG-DA was examined. Additionally, the biocompatibility of the composite adhesive was assessed both in culture and through subcutaneous implantation in rats.
2. Material and Method
Sodium periodate (NaIO4, > 99.8%) was obtained from Acros Organics (Fair Lawn, NJ). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide 98% (MTT) was purchased from Alfa Aesar (Ward Hill, MA). Phosphate buffer saline (PBS), tetraethyl orthosilicate (TEOS, 99.8%), ethanol (200 proof), sodium hydroxide, and acetic acid (Glacier) were purchased from Fisher Scientific Co. (Pittsburgh, PA). Histology mounting medium Polyfreeze, Trichrome Stain (Masson) Kit, Bouin’s solution, and Weigert’s iron hematoxylin solution were purchased from Sigma-Aldrich (St. Louis, MO). Anti-CD163 antibody (ab87099), goat anti-rabbit IgG H&L (Alexa Fluor 488) (ab150077), anti-CD68 antibody (ab125212), and goat anti-rabbit IgG H&L (Alexa Fluor 647) were purchased from Abcam (Cambridge, MA). 4,6-Diamidino-2-phenylindole (DAPI) was obtained from Invitrogen (Grand Island, NY). PEG-DA (Figure S1) was prepared using an 8-armed PEG (MW = 20 kDa, JenKem USA, TX) while following previously published protocols (22, 30).
2.1. Preparation of silica nano- and microparticle (NP and MP)
NP was prepared using a modified Stöber method (31). Silica precursor solution was prepared by mixing 4 g of TOES with 50 mL of ethanol and sonicated for 10 minutes. The reactor solution was prepared with 25 mL deionized water mixed with 30 mL ethanol and vigorously stirred (15 minutes at 1000 rpm) using a Real Torque Digital overhead mechanical stirrer with an HR 18 Half-Moon Blade (Fisher Scientific Co., Pittsburgh, PA) at 55°C. The pH of the reactor solution was maintained at pH 11 by adding 0.1 M NaOH. The silica precursor solution was added dropwise into the reactor solution at a rate of approximately 300 μl/second while stirring at 1000 rpm. After stirring for 1 hour at 55 °C, the pH of the reaction mixture was neutralized by adding 0.1 M acetic acid. The reaction mixture was further stirred for an additional 12 hr at room temperature. The reaction mixture was filtered using a filter paper with a mesh size of 20 μm to remove large contaminants. Ultra high-speed centrifugation (10000 Gs) was used to separate the particles from the solution. The particles were washed rigorously for 3 times with ethanol to ensure complete dehydration and removal of sodium acetate. After drying under a vacuum of 24 hrs, the yield of NP was 1.5 g.
MP was prepared similarly as NP. Silica precursor solution was added to the reactor solution and vigorously stirred (1000 rpm) at 55°C for 1 hr as described above to form NP. To form clustered MP, the pH of the reaction mixture was reduced to pH 3 with the addition of 0.2 M acetic acid. The reaction mixture was further stirred for an additional 12 hr at room temperature. Then, the solution was neutralized by adding 0.1 M NaOH. Ultra high-speed centrifugation (5 minutes at 10000 G) was used to separate the particles from the solution. The particle was washed 3 times with ethanol and dried under vacuum for 24 hrs. MP was further sifted through ASTM 200 (75 μm) and ASTM 450 (32 μm) sifters. The final yield of MP was 1.2 g.
2.2. Preparation of composite adhesive
The adhesive hydrogels were formed by mixing an equal volume of the PEG-DA precursor solution (300 mg/mL in 20 mM PBS at pH 7.4 with or without 4–20 wt% NP or MP) and NaIO4 solution (54.5 mM in deionized H2O). The final concentration of PEG-DA was 150 mg/mL, and the final concentration of NP or MP in the composite adhesive were 0–10 wt%. The final NaIO4 to dopamine molar ratio was 0.5. Cure time was determined when the mixture ceased to flow in a tilted vial (Figure S2) (30), as measured using a digital timer. Unless specified otherwise, the adhesive was allowed to further crosslinked after curing for 24 hours and equilibrated in PBS (pH = 7.4) for further characterization. The composite adhesives containing NP and MP were referred to as PEGx%N and PEGx%M, respectively, where x is the weight percentage of the particle in the composite adhesive.
2.3. Characterization of the adhesive
Samples were freeze-dried for at least 48 hours and characterized using Fourier transform infrared (FTIR) spectroscopy (Perkin Elmer Spectrum One). The morphologies of the particles, cured PEG-DA, and its composites were observed using field emission scanning electron microscopy (FE-SEM, Hitachi S-4700). The pore size of composite adhesive was analyzed using ImageJ (National Institutes of Health, Bethesda, MD). The calculation of the equilibrium water content (EWC) of the gels was performed as previously described (22).
2.4. In vitro degradation
Composite adhesive discs (diameter = 10 mm, thickness = 1.5 mm, n = 3) were incubated in 5 ml of PBS (pH = 7.4) at 37 °C. The PBS solution was removed and replaced with fresh PBS every 7 days. Samples were dried to determine their relative remaining mass (Mr) at 2, 4, 6, and 8 weeks after incubation. The calculation of the residual dry mass of the adhesive was performed according to the following equation:
where M0 and Mt are the initial mass of the adhesive and its mass after degradation, respectively (22).
2.5. Rheological testing
Rheological properties of the composite adhesives were characterized using a Discovery Hybrid Rheometer HR2 (TA instrument, Newcastle DE). Both strain sweep (0.01–100% strain, at 0.1 Hz) and frequency sweep (0.01–50 Hz at 10% strain) experiments were performed to determine the storage (G′) and loss (G″) moduli. Composite adhesive discs (diameter = 10 mm, thickness = 1.5 mm, n = 3) were tested using a parallel plate set up with a gap distance that is set at 87.5% of the individual hydrogel thickness, as measured by a digital caliper. PBS was introduced around the composite adhesive to prevent dehydration. To monitor the changes in the moduli during the curing process, precursor solutions (100 μL of PEG precursor solution with and without NP and MP) were directly added to the rheometer and mixed in situ with a NaIO4 solution (100 μL) at a gap distance of 0.5 mm with a cone and plate set up. The cone dimension was 20 mm diameter with the 2° inclination angle.
2.6. Lap shear adhesion testing
Adhesive properties of the composite adhesives were determined using lap shear adhesion test according to American Society for Testing and Materials (ASTM) standard F2255-05 (32). Briefly, the bovine pericardium was cut into 2.5 cm × 2.5 cm strips and hydrated in PBS. PEG-DA composite adhesives were cured between two partially overlapping bovine pericardial with an overlapping area of 2.5 cm × 1 cm. The adhesive joint was compressed with a 100-g weight for 15 min and further conditioned in PBS (pH=7.4) at 37 °C for overnight before testing. The dimensions of the contact area of each adhesive joint were measured using a digital caliper immediately before testing. The adhesive joints were pulled to failure at a rate of 0.1 mm/sec until the tissues separate using an Electroforce® machine (Bose Electroforce Group, MN). The adhesive strength was determined by the max load divided by the initial contact area of the adhesive joint (22).
2.7. In vitro cytotoxicity study
A quantitative MTT assay was used to determine the viability of cells exposed to NP and MP, extracts of these particles, and the extracts of the composite adhesives containing these particles while following ISO 10993-5 guideline (33). The composite adhesives were sterilized using ethanol following a published protocol (34). Briefly, disc-shaped samples (5-mm diameter, 2-mm thick) were submerged in 70 % (v/v) ethanol for 45 min followed by washing three times with 20 ml of sterile PBS for 90 min. The adhesives were further incubated in cell culture medium (10 mg/mL) for 24 hours at 37 °C to obtain the adhesive extracts. NP and MP were treated with UV radiation for 30 minutes before incubation with cell culture medium. The treated cell culture medium was used as is or filtered with syringe filter with a mesh size of 0.22 μm for direct and indirect cytotoxicity testing, respectively. L929 cells were suspended in cell culture medium and seeded into 96-well microculture plates at a density of 104 cells/100 μL/well. The cells were incubated in a humidified incubator (37 °C, 5% CO2) for 24 hours to obtain a confluent monolayer of cells. Then, the medium was replaced with 100 μL of adhesive extract, particle extract, or particles suspended in cell culture medium. After incubation for 24 hours, the medium was removed and replaced with 50 μL of MTT solution (1 mg/ml in PBS) and further incubated for another 2 hours. Finally, all solution was removed, and 100 μL of DMSO was added to dissolve MTT. The absorbance of each well was measured at 570 nm (reference 650 nm) using a Synergy HT Multi-Mode Microplate Reader (BioTek, USA). The relative cell viability was calculated as the ratio between the mean absorbance value of the sample and that of cells cultured in the culture medium (22).
2.8. Subcutaneous implantation
Healthy, weight matched Sprague-Dawley rats were obtained from Michigan Technological University (MTU) animal facility. The subcutaneous implantation was performed following the approved protocol by the Institutional Animal Care and Use Committee (IACUC). PEG-DA, PEG-DA with 10wt% NP (PEG10%N), and PEG-DA with 10wt% MP (PEG10%M) (diameter = 10 mm, thickness = 1.5 mm) were bilaterally implanted subcutaneously in the backs of Sprague-Dawley rats. Adhesive samples were sterilized using the same procedure as in the in vitro cytotoxicity study (34). Rats were anesthetized using an isofluorane-oxygen gas mixture and fur around the implantation site were removed. A pouch was formed using a pair of fine scissors, and a composite adhesive disc was placed in this pouch. 4 weeks post-surgery, the animals were euthanized and implanted composite adhesive along with surrounding tissues was collected, embedded in Polyfreeze, and then flash frozen in liquid nitrogen. The frozen samples were stored in −80 °C freezer before sectioning. All tissues were cryosectioned into 10 μm thick sections and stained with Masson’s trichrome staining for morphology and collagen production evaluation. For immunofluorescence staining, CD68 and CD163 were used to identify total macrophage population and macrophage type 2 (M2), respectively. All histological imaging analyses were performed on an Olympus microscope. Cells infiltration and local collagen content were quantified by ImageJ in a 50 μm × 50 μm area at the tissue-composite adhesive interface.
2.9. Statistical analysis
Statistical analysis was performed using Origin software. One-way analysis of variance (ANOVA) with Tukey’s HSD analysis and Student’s t-test were performed for comparing means of multiple groups and two groups, respectively, using a p-value of 0.05.
3. Results and discussion
3.1. Preparation of PEG-DA composite adhesives
NP and MP were successfully synthesized via the modified Stöber method. The average diameters of the particles were 360 ± 60 nm and 42 ± 25 μm, respectively (Figure 1). MP was prepared by controlled clustering of NP resulting in spherical clusters (Figure 1C) with nanosized surface features (Figure 1D). MP was forced to cluster via the reduction in pH of the reactor solution during the early portion of the condensation process. This allowed Si-O-Si bond and polysilicic chains to form at the boundaries of the particles, causing the clustering of MP similar to aggregation model reported by Kobayashi et al. (35).
Figure 1.

FE-SEM images of NP (A, B), MP (C) and the enlarged surface of MP (D).
Up to 10 wt% of NP and MP were incorporated into PEG-DA adhesives. The adhesive precursor solutions solidified to create opaque gels (Figure S2). PEG-DA adhesive cured through the polymerization of catechol with the introduction of the oxidizing agent, sodium periodate (NaIO4) (30, 36). The cure time of the adhesive significantly decreased from 35.3 ± 0.7 seconds down to less than 8 seconds with the incorporation of either 10 wt% NP or MP (Table 1). The presence of these Si particles promoted cohesive crosslinking and reduced the number of catechol-catechol crosslinking required to form a 3-dimensional polymer network, resulting in a reduction in the cure time (22, 37). There was no difference in the cure time for adhesive incorporated with NP and MP, suggesting that there was no significant alteration in the catechol crosslinking behavior as a result of the size differences of the particles.
Table 1.
Composite adhesive formulations, cure time, pore size, and equilibrium water content
| Formulation | Silica particle (wt%) | Cure time (second) | Pore size (μm) | Equilibrium water content (%) | |
|---|---|---|---|---|---|
| Nano | Micro | ||||
| PEG-DA | 0 | 0 | 35.3±0.7 | 1.54±0.0800 | 94.0±0.680 |
| PEG2%N | 2 | 0 | 14.0±1.0* | – | 90.0±0.250 |
| PEG10%N | 10 | 0 | 7.3±0.7* | 14.5±2.13* | 82.4±0.920* |
| PEG2%M | 0 | 2 | 16.7±1.3* | – | 92.9±1.65 |
| PEG10%M | 0 | 10 | 8.3±0.7* | 15.1±3.40* | 86.8±1.33* |
p < 0.05 when compare to PEG-DA;
Both PEG10%N and PEG10%M also exhibited pores that were an order of magnitude larger (~15 μm) when compared to that of PEG-DA (Figure 2 and Table 1). The large pore size can potentially facilitate cellular infiltration by providing unhindered openings for cell migration and proliferation (38, 39). NP appeared clustered in the adhesive network (Figure 2B) as untreated silica NP are known to cluster or aggregate in a neutral pH solution (40–42). In contrast to the FE-SEM image of the individual MP, particles found in PEG10%M appeared smaller in size (10 to 40 μm, Figure 2C). We speculated that freeze-drying and sample sectioning for FE-SEM imaging likely damaged MP in the adhesive network to form smaller particulates. Network-encapsulated NP and MP are distinguishable from one another as individual NP appeared to be more clearly defined due to much weaker particle-particle interaction. On the other hand, the encapsulated MP appeared to be less well defined. There was no significant difference in pore sizes between PEG10%N and PEG10%M, suggesting a similar interaction between the polymer matrix and these particles. Without surface modification, NP aggregated at a neutral pH (21, 35) and behaved similarly as MP in a polymer network.
Figure 2.

FE-SEM images of the cross-section of PEG-DA (A), PEG10%N (B), and PEG10%M (C). Scale bar is 10 μm.
The equilibrium water content (EWC) of PEG-DA averaged around 94 wt% (Table 1). Incorporation of NP and MP into the composite adhesive resulted in a significant reduction in the EWC value. EWC values are inversely proportional to both the crosslinking density and the stiffness of crosslinked network, indicating that incorporating silica particles into PEG-DA significantly increase the crosslinking density of the PEG network (43, 44).
The FTIR spectrum of PEG-DA showed characteristic peaks for ether bonds (1000–1150 cm−1, -C-O-C-), alkyl groups (2880 cm−1, -CH2-), carbonyl (1729 cm−1, ester bonds), and aromatic ring (1400–1500 cm−1) (Figure 3). The spectra for PEG10%N and PEG10%M exhibited an increase in the peak intensity at 1100 cm−1 corresponding to the known characteristic peak of Si-O-Si, indicating that the particles were successful incorporation into the adhesive. There was no noticeable difference between the spectra of PEG10%N and PEG10%M, indicating that both composite adhesives have the same chemical composition. Both NP and MP have the same building block (i.e., Si NP), which enabled us to examine the effect of particle size on the performance of the composite adhesive while keeping the chemical composition constant.
Figure 3.
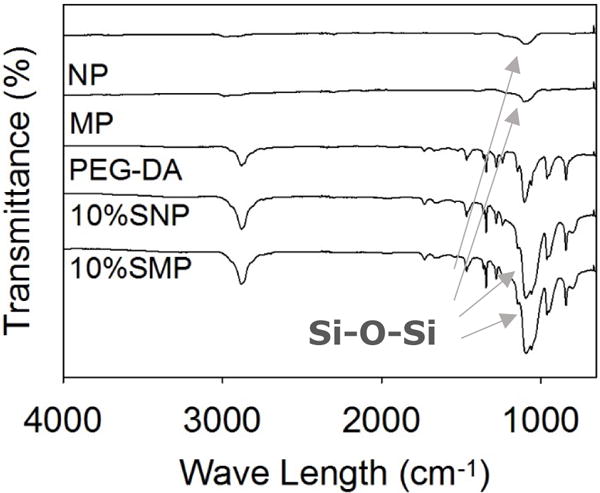
FTIR spectra of the silica nano and micro particles, PEG-DA, PEG10%N, and PEG10%M. The arrows point to peaks corresponding to Si-O-Si structure (1100 cm−1).
3.2. Rheological testing
Oscillatory rheometry was used to track the curing process of the adhesive (Figure S3) as well as determining the viscoelastic properties of the cured composite adhesives (Figures 4 and S4). For all the formulations tested, the storage modulus (G′) values were greater than those of the loss moduli (G″) for both PEG10%N and PEG10%M even at the very first measurable time point, indicating that the composite adhesive had already cured during the process of sample loading (Figure 3S). As such, we were unable to determine the time point when the crossover of the G′ and G″ values occurred. However, G′ values for both PEG10%N and PEG10%M rose sharply at an earlier time point (10 s) when compared to that of PEG-DA (30 s), confirming that the encapsulation of either NP or MP drastically reduced cure time (Table 1). The amplitude sweep experiment was used to determine the linear viscoelastic range of the adhesive (strain = 0.01–100%, Figure S4). Based on the frequency sweep experiment, G′ values of all the adhesive formulations were independent of the frequency (< 20 Hz, Figure 4A), further confirming that these polymer networks were covalently crosslinked (22, 45). G′ values increased with further increase in the rate of deformation as the polymer networks stiffened when polymer chains were not given sufficient time to relax (22).
Figure 4.
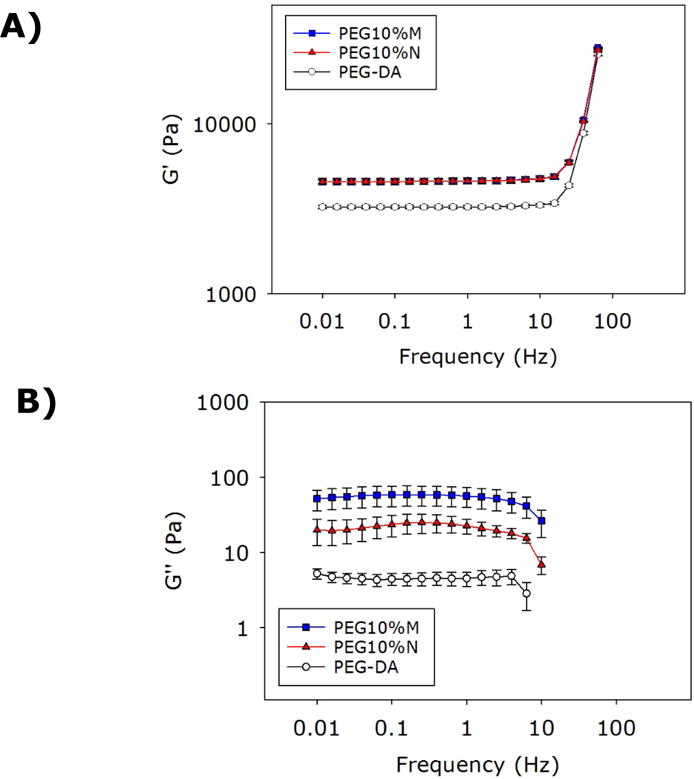
Storage (A) and loss (B) moduli of adhesvei during oscillatory frequency sweep (0.01–50 Hz, strain of 0.1%) experiment. The G′ values for PEG10%M and PEG10%N appeared to overlap in the frequency range tested.
Incorporating either NP or MP into PEG-DA increased both G′ and G″ values (approximately 1.5- and 10-fold increases, respectively; Figure 4). The increase in the G′ values indicated an increase in the stiffness and the crosslinking density of the composite adhesives. This increase coincided in the reduced EWC for the composite adhesives (Table 1). Elevated G″ values indicated that both PEG10%N and PEG10%M demonstrated an increased viscous dissipation properties due to the presence of reversible bonds between dopamine and the silica particles in the composite adhesive network (46–48). Catechol forms hydrogen bonds or electrostatic interaction with the SiO2 surface on the particles, which contribute to the increase in the measured viscoelastic properties (20). However, there was no difference in the values measured between PEG10%M and PEG10%N, indicating that MP was equally as effective in enhancing the mechanical properties of the PEG-DA matrix as that of NP.
3.3. Lap shear adhesion testing
Incorporation of the Si particles significantly enhanced (~1.5 fold increase) the adhesive properties of PEG-DA (Figure 5). Surprisingly, only PEG10%M showed statistically significant improvement in the measured adhesion strength when compared to PEG-DA. The adhesive properties of PEG10%N may have been affected by the poor particle dispersion of NP, which hindered particle-adhesive interaction (20). Nevertheless, there were no statistical differences between the measured values for PEG10%N and PEG10%M. Additionally, the both composite adhesives failed at a higher displacement when compared to PEG-DA (Figure S5). Improvement of adhesive properties is attributed to the strong catechol-silica interaction, which resulted in an increased fracture energy to separate the adhesive joint. When compared to mechanical testing results, incorporation of the silica particles significantly increased the bulk mechanical properties of the adhesive, which resulted in improved adhesive performance. This observation is consistent with previously published reports that bulk cohesive properties of an adhesive contribute to its adhesive properties (49, 50). Additionally, Rose et al. (51) also reported that incorporation of nanosilicates increases the interfacial adhesion force between soft tissue interfaces which may have partially contributed to the increase in adhesion strength. The lap shear strength values reported here are lower when compared to previously published results for catechol-modified PEG systems with similar architectures (10–40 kPa) potentially due to differences in the testing protocols (e.g., preparation of adhesive joint, strain rate, etc.) and the test substrates used (52, 53).
Figure 5.
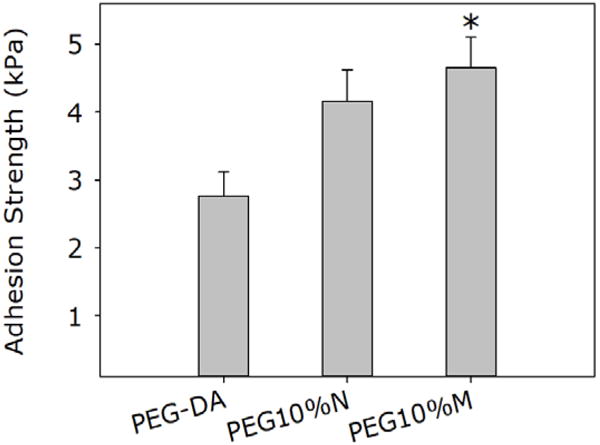
Lap shear adhesion test results for PEG-DA, PEG10%N, and PEG10%M. * p < 0.05 when compared to PEG-DA.
3.4. In vitro degradation
The degradation rate of the PEG-DA adhesive was determined by tracking the changes in the dry mass of the samples over time (Figure 6). PEG-DA degraded slowly over the first 4 weeks (~5 wt% loss) but degraded much faster over the later weeks (~95 wt% loss between weeks 4 and 8) before completely degrading in week 8. PEG-DA degraded through the hydrolysis of the ester bond between PEG and the glutaric acid (Figure S1) (54). As degradation progressed, the reduction in the crosslinking density and the increase in the water content of the adhesive matrix likely hastened the rate of degradation PEG-DA. On the other hand, both PEG10%N and PEG10%M degraded steadily over the same period, retaining 20 and 40 wt% of its dry mass, respectively, by week 8. When exposed to water, silica particles form a hydrolyzed surface layer that is hydrophilic (9). This increase in the local hydrophilicity may have increased water uptake and accelerated the degradation of these composite adhesives when compared to PEG-DA. The porosity of the adhesive matrix may have also affected the degradation rate as it governs the liquid diffusion within the matrix (55–57). Specifically, both PEG10%M and PEG10%N exhibited pores that were an order of magnitude larger than those of PEG-DA (Figure 2 and Table 1), which resulted in a faster degradation rate during the earlier time points. The degradation product of the composite may contain shorter PEG chains, glutaric-catechol groups, and silicic acid (58, 59). The release of silicic acid may have mildly reduced the local pH within the adhesive network, resulting in a delayed rate of hydrolysis of composite adhesive over time (54). These combined effects resulted in a more constant rate of degradation for PEG-DA containing either NP or MP. Interestingly, PEG10%M degraded slower than PEG10%N starting from week 4. Given that NP is much smaller compared to the pore size of PEG10%N, this accelerated mass loss mass may be due to the loss of the NP into the surrounding fluid.
Figure 6.
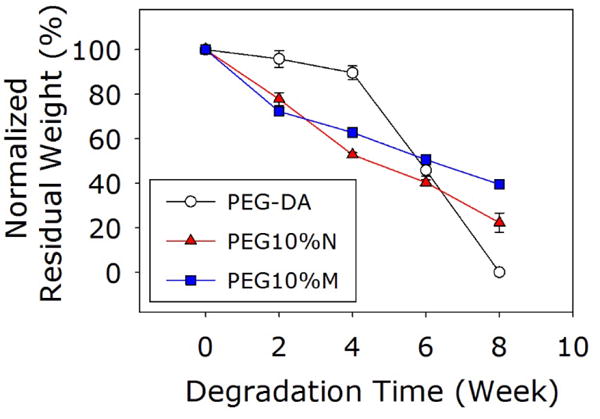
Residue dry mass of the adhesives incubated in PBS.
3.5. In vitro cytotoxicity
When L929 cells were exposed directly to NP and MP, relative cell viability decreased with increasing particle concentration (Figure 7). Specifically, 10 wt% of NP was highly cytotoxic with a cell viability of only 20%. However, at the same concentration, MP was significantly less cytotoxic when compared to NP with a cell viability of 60%. When the particles were removed by filtration (0.22 μm filter), the filtrate solutions became non-cytotoxic for both NP and MP. Our results confirmed published findings that Si NP is cytotoxic through direct contact with cells. Silica particle is known to bypass and disrupt membrane function and increase reactive oxygen species generation when it comes in contact or internalized by cells in a dose and size dependent manner (11, 12, 60). The clustered MP with a particle size in the micrometer range was significantly less cytotoxic as its larger size hindered cellular internalization. The filtered extracts were non-cytotoxic for both NP and MP. The main degradation product of these Si particles is silicic acid, which is found in numerous human tissues and organs (e.g., bone, tendon, liver, and kidney tissues). It had been demonstrated to promote synthesis of type I collagen and osteoblast differentiation in vitro (61, 62). Similarly, the extracts of the different adhesive formulations were found to be non-cytotoxic (Figure 8). Other catechol-modified polymeric adhesives have been demonstrated to be biocompatible both in vitro and in vivo (22, 37, 63, 64). Given that directly exposing L929 cells to elevated concentrations of the silica particles reduced cell viability, these results suggested that both NP and MP were securely sequestered within the adhesive matrix.
Figure 7.
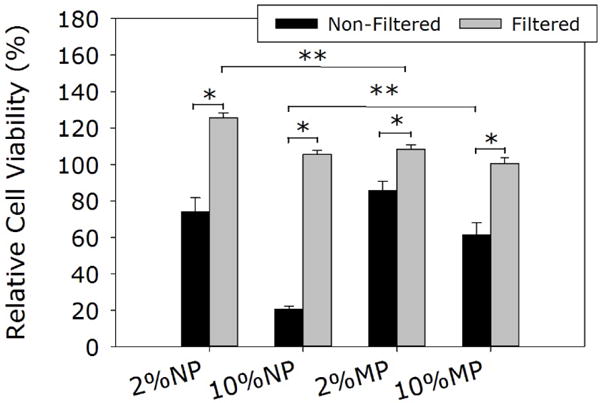
Relative cell viability of L929 cells after being treated with silica particles (Non-Filtered) and their extracts after filtration (0.22 μm, Filtered). * p < 0.05 when compared with the filtered samples. ** p < 0.05 when compared with different particle types at the same wt%.
Figure 8.
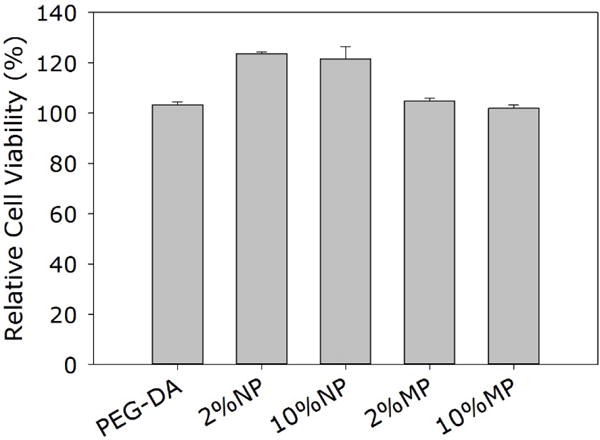
Relative cell viability of L929 cells treated with the extracts of PEG-DA adhesive and its composites.
3.6 Subcutaneous implantation
To further evaluate the effect of the particles on the biocompatibility of the composite adhesives and their ability to support cellular infiltration in vivo, PEG-DA adhesive with either 10 wt% NP or MP were implanted subcutaneously in rat for 4 weeks. Masson’s trichrome staining revealed no definite formation of a fibrous capsule at the interface between the implant and subcutaneous tissue regardless of the formulation (Figure 9). Both PEG10%N and PEG10%M demonstrated a cellular infiltration layer rich with cells near the surface of the adhesive (between the red arrows in Figure 9B and C). From SEM images (Figure 2), these composite adhesives contained pores (~15 μm) that are larger than the typical size of the cells, which enabled cellular infiltration before the complete degradation of the adhesive. On the other hand, PEG-DA contained pores that were an order of magnitude smaller (~1.5 μm), which physically hindered cellular ingrowth. The depth of cellular infiltration for PEG10%M was 2.5 fold higher when compared to that of PEG10%N (Table 2), even though both adhesive formulations exhibited similar pore sizes. This difference may be attributed to the cytotoxicity of NP. The increase in cellular infiltration depth may accelerate wound healing associating with the rate of migration of cells into the material allowing faster new tissue formation (22, 65). MP was noticeably present even after 4 weeks of implantation (black arrows in Figure 9F) and in some instances surrounded by the infiltrating cells (66). This observation further confirms that the larger MP was not cytotoxic.
Figure 9.
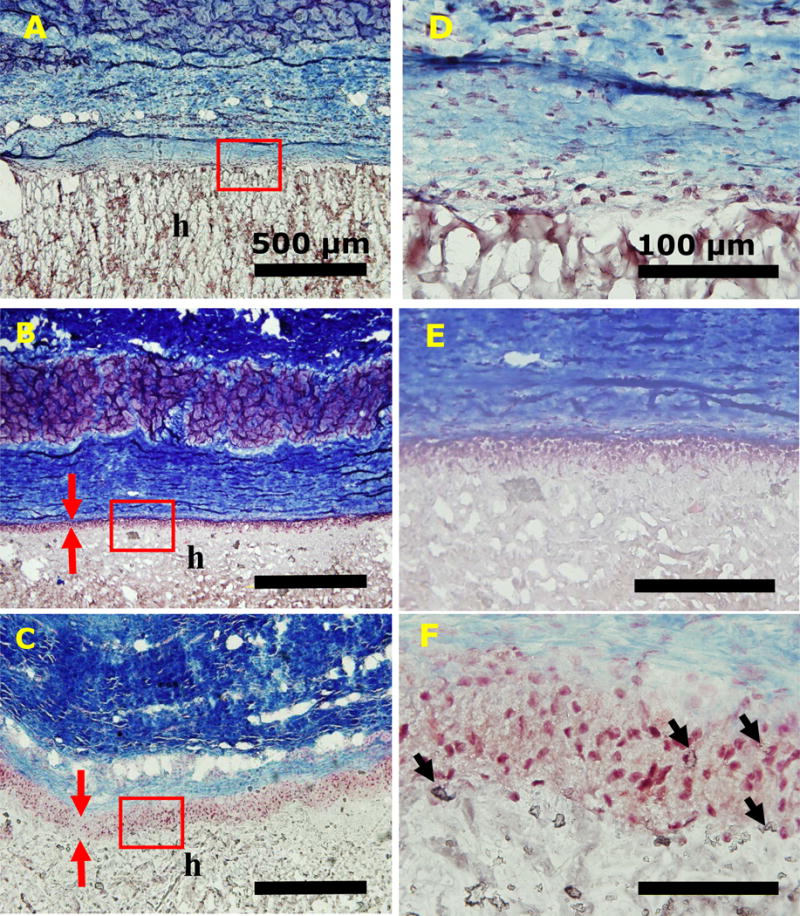
Trichrome staining of PEG-DA (A, D), PEG10%N (B, E), and PEG10%M (C, F) and their surrounding tissue after 4-week subcutaneous implantation. Letters h indicate the location of the adhesive. Red arrows indicate the thickness of cellular infiltration layer. Black arrow indicates silica particle clusters. Scale bars are 500 μm (A, B, and C) and 100 μm (D, E, and F).
Table 2.
Depth of cellular infiltration and macrophage count surrounding the implanted adhesive after 4 weeks of subcutaneous implantation
| Formulation | Cell infiltration (μM) | Average macrophage count at the interface (#/2,500μm2) |
|---|---|---|
| PEG-DA | 0 | 18.7 ± 4.25 |
| PEG10%N | 44.8 ± 7.67* | 15.5 ± 5.75 |
| PEG10%M | 112 ± 15.5*,& | 5.25 ± 2.35*,& |
p < 0.05 when compared to PEG-DA;
p < 0.05 and to PEG10%N;
Based on immunofluorescence staining, PEG10%M attracted 3 times lower the amount of macrophage to its surrounding tissue when compared to that of PEG-DA and PEG10%N (Figure 10 and Table 2). This result indicated that the incorporation of MP elicited a reduced level of immune response. Most noticeably, M2 macrophages were found near PEG10%M but not near the other two adhesive formulations (Figures 10 and S6). Unlike the pro-inflammatory response that M1 macrophages exhibit, M2 macrophages are responsible for anti-inflammatory responses and matrix remodeling, which are critical in promoting tissue regeneration (67, 68).
Figure 10.
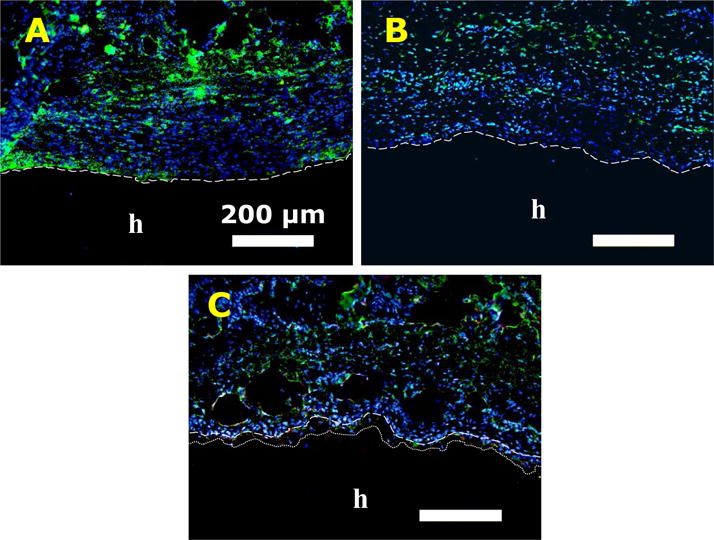
Immunofluorescence staining of PEG-DA (A), PEG10%N (B), and PEG10%M (C) after 4 weeks of subcutaneous implantation. Letters h indicate the location of the adhesive. Dash lines indicate the tissue-adhesive interface. Dotted line in (C) indicates the depth of cellular infiltration. Blue (DAPI): cell nuclei; Green (CD68): macrophage; Red (CD163): M2 macrophage. Scale bars are 200 μm.
PEG-DA formulated with either NP or MP were compositionally identical. As such, the observed differences in the mechanical and adhesive performances and the biological responses of the composite adhesives can be fully attributed to the difference in the particle size of the incorporated Si particles. Unlike other reported micron-sized particles that demonstrated reduced effectiveness as a matrix reinforcement filler (15, 16), adhesive formulated MP demonstrated equivalent or better mechanical and adhesive properties when compared with those formulated with NP. Most importantly, MP exhibited reduced cytotoxicity and promoted cellular infiltration and M2 macrophage polarization in vivo which will be critical for promoting rapid wound healing.
There are numerous commercially available PEG-based sealants and bioadhesives (e.g., Coseal, Baxter Healthcare Corporation and DuraSeal, Integra LifeSciences). However, due to the bioinert nature of PEG and small pore size typically associated with covalently crosslinked networks, PEG-based biomaterials lack the ability to promote cellular attachment and infiltration needed for rapid tissue repair and regeneration (69). PEG-based hydrogels have been chemically modified with various peptide sequences or bioligands (e.g., Arg-Gly-Asp (70), Arg-Glu-Asp-Val (71), and cysteine-containing peptides (72)) to improve their bioactivity. However, these approaches require a multi-step chemical synthesis, which is associated with low yield and high cost. Most importantly, these peptide-functionalization approaches do not increase the mechanical properties of these PEG-based hydrogels. The incorporation of MP provided a facile method for simultaneously increasing the adhesive property of a PEG-based bioadhesive and its ability to promote cellular infiltration. Additionally, Si particles have been previously reported to provide binding sites for cell attachment and proliferation (40, 63) and their soluble degradation products have been demonstrated to increase fibroblast proliferation and collagen type I synthesis (10). Compositing strategy reported here can potentially be used for designing functional bioadhesive and sealant that can promote rapid wound healing.
4. Conclusions
Micron-sized particles were formed by clustering silica nanoparticle and incorporated into PEG-DA adhesive. PEG-DA incorporated with MP exhibited equivalent or better mechanical and adhesive properties as adhesive formulated with NP, potentially due to nano-scaled surface features derived from the clustering of NP. Cytotoxicity tests confirmed that MP was significantly less cytotoxic than NP when these particles were brought into direct contact with L929 cells. Subcutaneous implantation in rats revealed that PEG-DA incorporated with MP demonstrated enhanced cellular infiltration and reduced inflammation when compared to adhesives formulated with NP. Most noticeably, MP containing adhesive attracted M2 macrophages to the implantation site. Incorporation of clustered silica microparticle is potentially a simple and cost-effective method to improve bioactivity and adhesive performance of a bioinert PEG-based adhesive.
Supplementary Material
Acknowledgments
This project was supported by National Institutes of Health under the award numbers R15GM104846 (BPL) and R15GM112082 (RMR) as well as Portage Health Foundation (BPL). RP was supported by the Royal Thai Government Scholarship.
Footnotes
Supporting Information
Supporting Information is available at the Wiley Online Library or from the author including chemical structure of the adhesive polymer, photographs of the adhesive, additional rheology data, lap shear testing results, and Immunofluorescence staining results.
References
- 1.Gibson RF. A review of recent research on mechanics of multifunctional composite materials and structures. Composite structures. 2010;92(12):2793–810. [Google Scholar]
- 2.Hussain F, Hojjati M, Okamoto M, Gorga RE. Review article: polymer-matrix nanocomposites, processing, manufacturing, and application: an overview. Journal of composite materials. 2006;40(17):1511–75. [Google Scholar]
- 3.Rezwan K, Chen Q, Blaker J, Boccaccini AR. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials. 2006;27(18):3413–31. doi: 10.1016/j.biomaterials.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 4.Jones JR. Reprint of: Review of bioactive glass: From Hench to hybrids. Acta biomaterialia. 2015;23:S53–S82. doi: 10.1016/j.actbio.2015.07.019. [DOI] [PubMed] [Google Scholar]
- 5.Zhou H, Lee J. Nanoscale hydroxyapatite particles for bone tissue engineering. Acta biomaterialia. 2011;7(7):2769–81. doi: 10.1016/j.actbio.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 6.Althues H, Henle J, Kaskel S. Functional inorganic nanofillers for transparent polymers. Chemical Society Reviews. 2007;36(9):1454–65. doi: 10.1039/b608177k. [DOI] [PubMed] [Google Scholar]
- 7.Slowing II, Trewyn BG, Giri S, Lin VY. Mesoporous silica nanoparticles for drug delivery and biosensing applications. Advanced Functional Materials. 2007;17(8):1225–36. [Google Scholar]
- 8.Fu S-Y, Feng X-Q, Lauke B, Mai Y-W. Effects of particle size, particle/matrix interface adhesion and particle loading on mechanical properties of particulate–polymer composites. Composites Part B: Engineering. 2008;39(6):933–61. [Google Scholar]
- 9.Jurkić LM, Cepanec I, Pavelić SK, Pavelić K. Biological and therapeutic effects of ortho-silicic acid and some orthosilicic acid-releasing compounds: new perspectives for therapy. Nutrition & metabolism. 2013;10(1):1. doi: 10.1186/1743-7075-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reffitt D, Ogston N, Jugdaohsingh R, Cheung H, Evans BAJ, Thompson R, et al. Orthosilicic acid stimulates collagen type 1 synthesis and osteoblastic differentiation in human osteoblast-like cells in vitro. Bone. 2003;32(2):127–35. doi: 10.1016/s8756-3282(02)00950-x. [DOI] [PubMed] [Google Scholar]
- 11.Napierska D, Thomassen LC, Rabolli V, Lison D, Gonzalez L, Kirsch‐Volders M, et al. Size ‐ Dependent Cytotoxicity of Monodisperse Silica Nanoparticles in Human Endothelial Cells. Small. 2009;5(7):846–53. doi: 10.1002/smll.200800461. [DOI] [PubMed] [Google Scholar]
- 12.Brunner TJ, Wick P, Manser P, Spohn P, Grass RN, Limbach LK, et al. In vitro cytotoxicity of oxide nanoparticles: comparison to asbestos, silica, and the effect of particle solubility. Environmental science & technology. 2006;40(14):4374–81. doi: 10.1021/es052069i. [DOI] [PubMed] [Google Scholar]
- 13.Monopoli MP, Åberg C, Salvati A, Dawson KA. Biomolecular coronas provide the biological identity of nanosized materials. Nature nanotechnology. 2012;7(12):779. doi: 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- 14.Eglin D, Maalheem S, Livage J, Coradin T. In vitro apatite forming ability of type I collagen hydrogels containing bioactive glass and silica sol-gel particles. Journal of Materials Science: Materials in Medicine. 2006;17(2):161–7. doi: 10.1007/s10856-006-6820-6. [DOI] [PubMed] [Google Scholar]
- 15.Cho J, Joshi M, Sun C. Effect of inclusion size on mechanical properties of polymeric composites with micro and nano particles. Composites Science and Technology. 2006;66(13):1941–52. [Google Scholar]
- 16.Zhou S, Wu L, Sun J, Shen W. The change of the properties of acrylic-based polyurethane via addition of nano-silica. Progress in Organic Coatings. 2002;45(1):33–42. [Google Scholar]
- 17.Kord Forooshani P, Lee BP. Recent approaches in designing bioadhesive materials inspired by mussel adhesive protein. Journal of Polymer Science Part A: Polymer Chemistry. 2016 doi: 10.1002/pola.28368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee BP, Messersmith PB, Israelachvili JN, Waite JH. Mussel-inspired adhesives and coatings. Annual review of materials research. 2011;41:99–132. doi: 10.1146/annurev-matsci-062910-100429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cha HJ, Hwang DS, Lim S. Development of bioadhesives from marine mussels. Biotechnology journal. 2008;3(5):631–8. doi: 10.1002/biot.200700258. [DOI] [PubMed] [Google Scholar]
- 20.Narkar AR, Barker B, Clisch M, Jiang J, Lee BP. pH Responsive and Oxidation Resistant Wet Adhesive based on Reversible Catechol–Boronate Complexation. Chemistry of Materials. 2016;28(15):5432–9. doi: 10.1021/acs.chemmater.6b01851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bagwe RP, Hilliard LR, Tan W. Surface modification of silica nanoparticles to reduce aggregation and nonspecific binding. Langmuir. 2006;22(9):4357–62. doi: 10.1021/la052797j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Meng H, Konst S, Sarmiento R, Rajachar R, Lee BP. Injectable dopamine-modified poly (ethylene glycol) nanocomposite hydrogel with enhanced adhesive property and bioactivity. ACS applied materials & interfaces. 2014;6(19):16982–92. doi: 10.1021/am504566v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skelton S, Bostwick M, O’Connor K, Konst S, Casey S, Lee BP. Biomimetic adhesive containing nanocomposite hydrogel with enhanced materials properties. Soft Matter. 2013;9(14):3825–33. [Google Scholar]
- 24.Ding X, Vegesna GK, Meng H, Winter A, Lee BP. Nitro‐Group Functionalization of Dopamine and its Contribution to the Viscoelastic Properties of Catechol‐Containing Nanocomposite Hydrogels. Macromolecular chemistry and physics. 2015;216(10):1109–19. doi: 10.1002/macp.201500010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Lee BP. Recovery property of double-network hydrogel containing a mussel-inspired adhesive moiety and nano-silicate. Journal of Materials Chemistry B. 2016;4(40):6534–40. doi: 10.1039/C6TB01828A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waite JH. Nature’s underwater adhesive specialist. International Journal of Adhesion and Adhesives. 1987;7(1):9–14. [Google Scholar]
- 27.Lee BP, Dalsin JL, Messersmith PB. Synthesis and Gelation of DOPA-Modified Poly(ethylene glycol) Hydrogels. Biomacromolecules. 2002;3(5):1038–47. doi: 10.1021/bm025546n. [DOI] [PubMed] [Google Scholar]
- 28.Lee H, Scherer NF, Messersmith PB. Single Molecule Mechanics of Mussel Adhesion. Proceedings of the National Academy of Sciences. 2006;103:12999–3003. doi: 10.1073/pnas.0605552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugumaran M, Dali H, Semensi V. Chemical- and cuticular phenoloxidase-mediated synthesis of cysteinyl-catechol adducts. Arch Insect Biochem Physiol. 1989;11(2):127–37. [Google Scholar]
- 30.Lee BP, Dalsin JL, Messersmith PB. Synthesis and gelation of DOPA-modified poly (ethylene glycol) hydrogels. Biomacromolecules. 2002;3(5):1038–47. doi: 10.1021/bm025546n. [DOI] [PubMed] [Google Scholar]
- 31.Stöber W, Fink A, Bohn E. Controlled growth of monodisperse silica spheres in the micron size range. Journal of colloid and interface science. 1968;26(1):62–9. [Google Scholar]
- 32.ASTM F2255-05. STMfSPoTAiL-SbTL. ASTM International; West Conshohocken, PA: 2010. 2010. www.astm.org. [DOI] [Google Scholar]
- 33.ISO B. 10993-5. Biological evaluation of medical devices. Tests for in vitro cytotoxicity. 1999 [Google Scholar]
- 34.Huebsch N, Gilbert M, Healy KE. Analysis of sterilization protocols for peptide ‐ modified hydrogels. Journal of Biomedical Materials Research Part B: Applied Biomaterials. 2005;74(1):440–7. doi: 10.1002/jbm.b.30155. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi M, Juillerat F, Galletto P, Bowen P, Borkovec M. Aggregation and charging of colloidal silica particles: effect of particle size. Langmuir. 2005;21(13):5761–9. doi: 10.1021/la046829z. [DOI] [PubMed] [Google Scholar]
- 36.Silverman HG, Roberto FF. Understanding marine mussel adhesion. Marine Biotechnology. 2007;9(6):661–81. doi: 10.1007/s10126-007-9053-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Meng H, Liu Y, Narkar A, Lee BP. Gelatin Microgel Incorporated Poly(ethylene glycol)-Based Bioadhesive with Enhanced Adhesive Property and Bioactivity. ACS Applied Materials & Interfaces. 2016;8(19):11980–9. doi: 10.1021/acsami.6b01364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy CM, Haugh MG, O’Brien FJ. The effect of mean pore size on cell attachment, proliferation and migration in collagen–glycosaminoglycan scaffolds for bone tissue engineering. Biomaterials. 2010;31(3):461–6. doi: 10.1016/j.biomaterials.2009.09.063. [DOI] [PubMed] [Google Scholar]
- 39.Zeltinger J, Sherwood JK, Graham DA, Müeller R, Griffith LG. Effect of pore size and void fraction on cellular adhesion, proliferation, and matrix deposition. Tissue engineering. 2001;7(5):557–72. doi: 10.1089/107632701753213183. [DOI] [PubMed] [Google Scholar]
- 40.Gaharwar AK, Rivera CP, Wu CJ, Schmidt G. Transparent, elastomeric and tough hydrogels from poly(ethylene glycol) and silicate nanoparticles. Acta biomaterialia. 2011;7(12):4139–48. doi: 10.1016/j.actbio.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 41.Wu S-H, Mou C-Y, Lin H-P. Synthesis of mesoporous silica nanoparticles. Chemical Society Reviews. 2013;42(9):3862–75. doi: 10.1039/c3cs35405a. [DOI] [PubMed] [Google Scholar]
- 42.Schexnailder P, Schmidt G. Nanocomposite polymer hydrogels. Colloid and Polymer Science. 2009;287(1):1–11. [Google Scholar]
- 43.Anseth KS, Bowman CN, Brannon-Peppas L. Mechanical properties of hydrogels and their experimental determination. Biomaterials. 1996;17(17):1647–57. doi: 10.1016/0142-9612(96)87644-7. [DOI] [PubMed] [Google Scholar]
- 44.Peppas NA, Bures P, Leobandung W, Ichikawa H. Hydrogels in pharmaceutical formulations. European Journal of Pharmaceutics & Biopharmaceutics. 2000;50(1):27–46. doi: 10.1016/s0939-6411(00)00090-4. [DOI] [PubMed] [Google Scholar]
- 45.Cencer M, Murley M, Liu Y, Lee BP. Effect of nitro-functionalization on the cross-linking and bioadhesion of biomimetic adhesive moiety. Biomacromolecules. 2014;16(1):404–10. doi: 10.1021/bm5016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Narita T, Mayumi K, Ducouret G, Hébraud P. Viscoelastic Properties of Poly (vinyl alcohol) Hydrogels Having Permanent and Transient Cross-Links Studied by Microrheology, Classical Rheometry, and Dynamic Light Scattering. Macromolecules. 2013;46(10):4174–83. [Google Scholar]
- 47.Rose SV, Marcellan A, Hourdet D, Creton C, Narita T. Dynamics of Hybrid Polyacrylamide Hydrogels Containing Silica Nanoparticles Studied by Dynamic Light Scattering. Macromolecules. 2013;46(11):4567–74. [Google Scholar]
- 48.Yang J, Han C-R, Zhang X-M, Xu F, Sun RC. Cellulose Nanocrystals Mechanical Reinforcement in Composite Hydrogels with Multiple Cross-Links: Correlations between Dissipation Properties and Deformation Mechanisms. Macromolecules. 2014 [Google Scholar]
- 49.da Silva LF, Rodrigues T, Figueiredo M, De Moura M, Chousal J. Effect of adhesive type and thickness on the lap shear strength. The journal of adhesion. 2006;82(11):1091–115. [Google Scholar]
- 50.Murphy JL, Vollenweider L, Xu F, Lee BP. Adhesive performance of biomimetic adhesive-coated biologic scaffolds. Biomacromolecules. 2010;11(11):2976. doi: 10.1021/bm1007794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rose S, Prevoteau A, Elziere P, Hourdet D, Marcellan A, Leibler L. Nanoparticle solutions as adhesives for gels and biological tissues. Nature. 2014;505(7483):382. doi: 10.1038/nature12806. [DOI] [PubMed] [Google Scholar]
- 52.Brubaker CE, Messersmith PB. Enzymatically degradable mussel-inspired adhesive hydrogel. Biomacromolecules. 2011;12(12):4326–34. doi: 10.1021/bm201261d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burke SA, Ritter-Jones M, Lee BP, Messersmith PB. Thermal gelation and tissue adhesion of biomimetic hydrogels. Biomedical Materials. 2007;2:203–10. doi: 10.1088/1748-6041/2/4/001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin M-H, Anderson J, Pinnaratip R, Meng H, Konst S, DeRouin AJ, et al. Monitoring the long-term degradation behavior of biomimetic bioadhesive using wireless magnetoelastic sensor. IEEE Transactions on Biomedical Engineering. 2015;62(7):1838–42. doi: 10.1109/TBME.2015.2405251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahmed EM. Hydrogel: Preparation, characterization, and applications: A review. Journal of Advanced Research. 2015;6(2):105–21. doi: 10.1016/j.jare.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bose S, Roy M, Bandyopadhyay A. Recent advances in bone tissue engineering scaffolds. Trends in biotechnology. 2012;30(10):546–54. doi: 10.1016/j.tibtech.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu L, Ding J. Effects of porosity and pore size on in vitro degradation of three ‐ dimensional porous poly (D, L‐lactide‐co‐glycolide) scaffolds for tissue engineering. Journal of Biomedical Materials Research Part A. 2005;75(4):767–77. doi: 10.1002/jbm.a.30487. [DOI] [PubMed] [Google Scholar]
- 58.Alexander GB, Heston W, Iler RK. The solubility of amorphous silica in water. The Journal of Physical Chemistry. 1954;58(6):453–5. [Google Scholar]
- 59.Chen S-L, Dong P, Yang G-H, Yang J-J. Kinetics of formation of monodisperse colloidal silica particles through the hydrolysis and condensation of tetraethylorthosilicate. Industrial & engineering chemistry research. 1996;35(12):4487–93. [Google Scholar]
- 60.Kim I-Y, Joachim E, Choi H, Kim K. Toxicity of silica nanoparticles depends on size, dose, and cell type. Nanomedicine: Nanotechnology, Biology and Medicine. 2015;11(6):1407–16. doi: 10.1016/j.nano.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 61.Carlisle EM. Silicon: A Possible Factor in Bone Calcification. Science. 1970;167(3916):279–80. doi: 10.1126/science.167.3916.279. [DOI] [PubMed] [Google Scholar]
- 62.Reffitt DM, Ogston N, Jugdaohsingh R, Cheung HFJ, Evans BAJ, Thompson RPH, et al. Orthosilicic acid stimulates collagen type 1 synthesis and osteoblastic differentiation in human osteoblast-like cells in vitro. Bone. 2003;32(2):127–35. doi: 10.1016/s8756-3282(02)00950-x. [DOI] [PubMed] [Google Scholar]
- 63.Liu Y, Meng H, Qian ZC, Fan N, Choi WY, Zhao F, et al. A Moldable Nanocomposite Hydrogel Composed of a Mussel-Inspired Polymer and a Nanosilicate as a Fit-to-Shape Tissue Sealant. Angew Chem Int Edit. 2017;56(15):4224–8. doi: 10.1002/anie.201700628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meng H, Liu Y, Lee BP. Model polymer system for investigating the generation of hydrogen peroxide and its biological responses during the crosslinking of mussel adhesive moiety. Acta Biomaterialia. 2017;48:144–56. doi: 10.1016/j.actbio.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Griffin DR, Weaver WM, Scumpia P, Di Carlo D, Segura T. Accelerated wound healing by injectable microporous gel scaffolds assembled from annealed building blocks. Nature materials. 2015;14(7):737. doi: 10.1038/nmat4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garcia-Santamaria F, Miguez H, Ibisate M, Meseguer F, Lopez C. Refractive index properties of calcined silica submicrometer spheres. Langmuir. 2002;18(5):1942–4. [Google Scholar]
- 67.Spiller KL, Anfang RR, Spiller KJ, Ng J, Nakazawa KR, Daulton JW, et al. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials. 2014;35(15):4477–88. doi: 10.1016/j.biomaterials.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marchetti V, Yanes O, Aguilar E, Wang M, Friedlander D, Moreno S, et al. Differential Macrophage Polarization Promotes Tissue Remodeling and Repair in a Model of Ischemic Retinopathy. Sci Rep. 2011;1 doi: 10.1038/srep00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shin H, Ruhe PQ, Mikos AG, Jansen JA. In vivo bone and soft tissue response to injectable, biodegradable oligo (poly (ethylene glycol) fumarate) hydrogels. Biomaterials. 2003;24(19):3201–11. doi: 10.1016/s0142-9612(03)00168-6. [DOI] [PubMed] [Google Scholar]
- 70.Shin H, Jo S, Mikos AG. Modulation of marrow stromal osteoblast adhesion on biomimetic oligo [poly (ethylene glycol) fumarate] hydrogels modified with Arg‐Gly‐ Asp peptides and a poly (ethylene glycol) spacer. Journal of biomedical materials research. 2002;61(2):169–79. doi: 10.1002/jbm.10193. [DOI] [PubMed] [Google Scholar]
- 71.Hubbell JA, Massia SP, Desai NP, Drumheller PD. Endothelial cell-selective materials for tissue engineering in the vascular graft via a new receptor. Nature Biotechnology. 1991;9(6):568–72. doi: 10.1038/nbt0691-568. [DOI] [PubMed] [Google Scholar]
- 72.Chung I-M, Enemchukwu NO, Khaja SD, Murthy N, Mantalaris A, García AJ. Bioadhesive hydrogel microenvironments to modulate epithelial morphogenesis. Biomaterials. 2008;29(17):2637–45. doi: 10.1016/j.biomaterials.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.