

Abstract
Nrf2, a redox regulated transcription factor, has recently been shown to play a role in cartilage integrity but the mechanism remains largely unknown. Osteoarthritis (OA) is a multifactorial disease in which focal degradation of cartilage occurs. Here, we studied whether Nrf2 exerts chondroprotective effects by suppressing the oxidative stress and apoptosis in IL-1β stimulated human OA chondrocytes. Expression of Nrf2 and its target genes HO-1, NQO1 and SOD2 was significantly high in OA cartilage compared to normal cartilage and was also higher in damaged area compared to smooth area of OA cartilage of the same patient. Human chondrocytes treated with IL-1β resulted in robust Nrf2/ARE reporter activity, which was inhibited by pretreatment with antioxidants indicating that Nrf2 activity was due to IL-1β-induced ROS generation. Ectopic expression of Nrf2 significantly suppressed the IL-1β-induced generation of ROS while Nrf2 knockdown significantly increased the basal as well as IL-1β-induced ROS levels in OA chondrocytes. Further, Nrf2 activation significantly inhibited the IL-1β-induced activation of extrinsic and intrinsic apoptotic pathways as determined by inhibition of DNA fragmentation, activation of Caspase-3,-8,-9, cleavage of PARP, release of cytochrome-c, suppression of mitochondrial dysfunction and mitochondrial ROS production in OA chondrocytes. Nrf2 over-expression in OA chondrocytes increased the expression of anti-apoptotic proteins while pro-apoptotic proteins were suppressed. Importantly, Nrf2 over-expression activated ERK1/2 and its downstream targets-ELK1, P70S6K and P90RSK and suppressed the IL-1β-induced apoptosis whereas inhibition of ERK1/2 activation abrogated the protective effects of Nrf2 in OA chondrocytes. Taken together, our data demonstrate that Nrf2 is a stress response protein in OA chondrocytes with anti-oxidative and anti-apoptotic function and acts via activation of ERK1/2/ELK1-P70S6K-P90RSK signaling axis. These activities of Nrf2 make it a promising candidate for the development of novel therapies for the management of OA.
Keywords: Osteoarthritis, Nrf2, ERK1/2, Redox, Apoptosis
Graphical Abstract
Introduction
Osteoarthritis (OA), is a leading cause of joint dysfunction in the affected population and the disease is characterized by progressive destruction of the articular cartilage [1]. At molecular level, loss of cartilage is attributed to multifactorial events including oxidative stress, mitochondrial dysfunction, apoptosis and associated changes in inflammatory and catabolic gene expression in chondrocytes-the only cell type present in the cartilage [1, 2]. Pro-inflammatory cytokine interleukin-1β (IL-1β) is a critical player in OA pathogenesis and has been shown to induce oxidative stress, apoptosis and catabolic gene expression in OA [3]. Oxidative stress in chondrocytes occurs due to excess generation of reactive oxygen species (ROS) by exogenous factors such as overload, trauma, local intra-articular lesions and synovial inflammation [4, 5]. Recent studies have demonstrated that elevated levels of ROS modulates multiple signaling pathways initiated by cytokines and further amplifies inflammatory, catabolic and apoptotic response in chondrocytes leading to the destruction of cartilage matrix in OA [6]. Additionally, the magnitude of cartilage matrix damage positively correlates with the extent of chondrocyte apoptosis induced by pro-inflammatory cytokines in OA [7–9]. Since oxidative stress and apoptotic signaling pathways are integral component of OA pathogenesis, therefore it is reasonable to hypothesize that pharmacological attenuation of these pathways may be a promising approach for the management of OA.
The nuclear factor-erythroid 2-related factor 2 (Nrf2) is a redox-regulated transcription factor that orchestrates the expression of a battery of approximately 250 genes involved in a wide variety of cellular functions and contributes to cytoprotection against environmental electrophiles and oxidative stresses [10–12]. Nrf2 plays a crucial role in protection against the oxidative damage, hyperoxia, nitrosative stress and ER stress by inducing the expression of cytoprotective and antioxidant genes including Heme-Oxygenase-1 (HO-1), Peroxiredoxin 1 (PRDX1), Superoxide Dismutase-2 (SOD2) and elevating the intracellular glutathione levels [13]. Additionally, Nrf2 has been identified as an inhibitor of apoptosis in a number of studies including Fas-induced apoptosis [14], nitric oxide induced apoptosis [15] and cell death in an in vitro model of ischemia/reperfusion injury [16]. These studies thus point to a beneficial role of Nrf2 activation in a wide variety of diseases generally associated with oxidative stress. The Nrf2-activating drug Dimethyl Fumarate (Tecfidera) has recently been approved by the US Food and Drug Administration (FDA) for the treatment of multiple sclerosis [17], in part based on its anti-oxidative and anti-apoptotic effects. These observations indicate that Nrf2/ARE pathway is of great interest as an attractive drug target for the pharmacological control of oxidative and apoptotic response in different cell types and organ-specific diseases. However, despite these findings, little attention has been given to explore its role in OA.
Recent studies have shown that activation of Nrf2 with phytochemicals suppressed the catabolic and inflammatory mediator’s production by chondrocytes and inhibited the cartilage damage in an experimental model of OA [18–24]. Other studies have shown that Nrf2 deficient mice had severe cartilage damage compared to wild type mice in a model of post-traumatic OA thereby highlighting the crucial role of Nrf2 in maintaining cartilage homeostasis [25]. However, the mechanism of Nrf2 mediated inhibition of cartilage damage was not reported and the data underlying the role of Nrf2 in maintaining cartilage homeostasis is completely lacking. Here, we studied how Nrf2 suppresses oxidative stress and inhibit apoptosis in OA chondrocytes under pathological conditions. Our results demonstrate that Nrf2/ARE signaling exerts chondroprotection by diminishing the oxidative stress and inhibiting the apoptosis via activation of ERK1/2/ELK1-P70S6K-P90RSK signaling axis in OA chondrocytes.
Material and Methods
Reagents
Pronase and Collagenase were from Roche Diagnostics (Indianapolis, IN). Ham’s F12 Medium was from Lonza (Walkersville, MD). Dulbecco’s modified Eagle’s Medium (DMEM) and other reagents for cell culture were from HyClone Laboratories (Logan, UT). Recombinant human IL-1β and TdT In Situ Apoptosis Detection Kit-Fluorescein were from R&D Systems (St Paul, MN). Dichlorodihydrofluorescein Diacetate (H2DCF-DA), Dihydro-Rhodamine (DHR123), PD98059 and MTT (#M2128) were from Sigma Aldrich Chemicals Inc (St. Louis, MO). Amplex™ Red Hydrogen Peroxide/Peroxidase Assay Kit was purchased from Thermo Fischer Scientific. Nrf2/ARE-luciferase reporter plasmid was from Signosis (Santa Clara, CA). Dual-Glo® Luciferase Assay System was purchased from Promega (Madison, WI). Antibodies specific for p-ERK1/2, total ERK1/2, p-JNK, total JNK, p-P38-MAPK, total P38-MAPK, Nrf2, p-Elk1 (Ser383), p-P90RSK (Ser380), p-MEK1/2 (Ser217/221), MEK1/2, Mcl-1, cleaved Caspase-3, cleaved PARP, Caspase-9, p-Iκβα (Ser 32) and Iκβα were from Cell Signaling Technology (Beverly, MA). Antibodies specific for β-Actin, Bcl-2, Bcl-xl, Bax, Bad, Bid, Cytochrome c, Caspase-8, p-P70S6K (Ser411), were from Santa Cruz Biotechnology (Santa Cruz, CA). Appropriate horseradish peroxidase (HRP) conjugated secondary antibodies were from Cell Signaling Technology.
Human cartilage samples and isolation of chondrocytes
The study protocol was reviewed and approved by the Institutional Review Board (IRB) of Northeast Ohio Medical University, Rootstown, Ohio and SUMMA Health System, Akron, OH as a “non-human subject study under 45 CFR”. All the methods used in this study were carried out in accordance with the approved protocol and guidelines. Discarded and de-identified knee cartilage samples were from donors who underwent total knee arthroplasty due to OA at SUMMA Health System Hospitals, Akron, OH. Normal/unaffected articular cartilage samples (no recorded history of arthritis) were from post mortem donors and obtained from the National Disease Research Interchange (NDRI), Philadelphia, PA.
Cartilage specimen were washed with sterile phosphate buffered saline (PBS) and stained with India ink. Areas of cartilage with a smooth articular surface (no staining with India ink) and area with a damaged articular surface (intense staining with India ink) were used for the histopathologic grading according to the Mankin scoring system [26] and the regions of the cartilage samples with Mankin score of 0–1 were considered unaffected and regions with a score of 2–3 were graded as damaged. Chondrocytes were prepared by enzymatic digestion of the cartilage as described previously [22, 27, 28].
Treatment of human chondrocytes with IL-1β
Primary human OA or normal chondrocytes (1×106/well of 6-well plate) were seeded in DMEM/F-12 mixture supplemented with 10% fetal calf serum (FCS), 100 units/ml Penicillin, and 100 mg/ml Streptomycin for 2–3 days after plating. At about 80% confluence, chondrocytes were serum starved overnight and then stimulated with IL-1β (1 or 10 ng/ml) for indicated time points.
Over-expression and knockdown of Nrf2 in OA chondrocytes
Nrf2 over-expression plasmid (pcDNA3-EGFP-C4-Nrf2) was a gift from Yue Xiong (Addgene plasmid # 21549) [29]. Primary human OA chondrocytes were transfected with pcDNA3-EGFP-C4-Nrf2 plasmid or pcDNA3.1 empty plasmid as controls (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit and the Amaxa Nucleofection System (Lonza, Walkersville, MD) as described earlier [22]. For knockdown experiments, chondrocytes were transfected with 100 nM Nrf2 siRNA (SMARTpool: ON-TARGETplus NFE2L2 siRNA Dharmacon, Lafayette, CO) or MISSION® siRNA Universal Negative Controls (Sigma Aldrich, St. Louis, MO) using X-tremeGENE™ siRNA transfection reagent (Sigma Aldrich, St. Louis, MO) according to the instructions provided. Forty-eight hours later, chondrocytes were serum starved overnight and stimulated with IL-1β (1 or 10 ng/ml) in serum free medium for indicated time. Total RNA and lysates were prepared for the determination of gene and protein expression levels by quantitative RT-PCR and Western blot analysis respectively as described [22, 27, 28, 30].
Total RNA isolation and quantitative RT-PCR
Chondrocytes (1×106/well of 6-well plate) were serum starved overnight and stimulated with IL-1β (1 ng/ml) for 24h. Total RNA from isolated chondrocytes was prepared essentially as previously described [27, 30, 31] and the levels of Nrf2, HO-1, NQO1 and SOD2 mRNA was quantified using SYBR® green assays as previously described [27].
Western Immunoblotting
Chondrocytes were harvested, washed with cold PBS and lysed in ice-cold RIPA buffer and equivalent amounts of lysate protein (20–35 μg) were resolved by 10% or 12% SDS-PAGE and transferred to a PVDF membrane (Bio-Rad, USA). Western blots were incubated with primary antibodies overnight at 4°C and were then incubated with an appropriate HRP-conjugated secondary antibody, followed by washing thrice with TBST [27]. Blots were developed using Luminata Western HRP substrate (EMS Millipore) and the immunoreactive proteins were visualized by chemiluminescence and imaged using the Pxigel imaging system (Syngene, Frederick, MD).
Luciferase reporter assay
Chondrocytes were transfected with pNrf2/ARE-Luc reporter vector (LR-2016, Signosis, Santa Clara, CA, USA) and pRL-TK (10 nM) as internal control by Nucleofection as described above. Forty-eight hours later, chondrocytes were serum starved overnight and then stimulated with IL-1β (1 ng/ml) in serum free medium for indicated time and luciferase activity was measured using a Dual-Glo® Luciferase Assay System (Promega, Madison, WI, USA) according to the instructions provided. The luciferase reporter activity was normalized to the Renilla luciferase activity of the internal control.
Cell viability assay
Forty-eight hours after transfection with pcDNA3-EGFP-C4-Nrf2 vector or siRNA against Nrf2, human chondrocytes (40,000 cells/well of 96-well plate) were serum starved overnight, and then stimulated with IL-1β (10 ng/ml) for 48 h in serum free medium and viability was determined using the MTT assay as described earlier [27].
Estimation of apoptosis by TUNEL assay
Human OA chondrocytes transfected with the pcDNA3-EGFP-C4-Nrf2 vector or the control plasmid were stimulated with IL-1β (10 ng/ml) for 48 h in serum free medium and the extent of apoptosis was quantified using TUNEL assay as described previously [32, 33].
Measurement of Reactive Oxygen Species (ROS) levels
Basal and induced ROS levels were measured by using cell-permeable fluorogenic probes DHR123 and H2DCF-DA as described previously [34]. Briefly, OA chondrocytes transfected as above were stained with oxidation-sensitive DCFH2-DA (20 μM) or DHR123 (5 μM) probes for 0.5 h at 37 °C and were stimulated with IL-1β (1 ng/ml) for 5 minutes and ROS levels were estimated by measuring the fluorescence emission due to oxidation of DHR123 to Rhodamine123 at 500 nm excitation and 536 nm emission or oxidation of H2DCF to DCF at an excitation and emission wavelengths of 485 and 525 nm respectively using the Synergy H1 multi-mode plate reader.
Estimation of H2O2 in the culture supernatant using Amplex Red® assay
We measured H2O2 generation in the supernatants of Nrf2 knockdown or over-expressing human chondrocytes stimulated with IL1β according to the manufacturer’s protocol. Amplex® Red reagent reacts with H2O2 in the presence of peroxidase (HRP) and produces the red-fluorescent oxidation product Resorufin which was estimated by measuring the fluorescence at the excitation and emission wavelengths of 571 and 585 nm respectively using the Synergy H1 multi-mode plate reader as previously described [22].
Estimation of mitochondrial O2− using MitoSOX
For the measurement of mitochondrial O2−, chondrocytes treated as above were stained with MitoSOX ™ Red (5 μM) for 10 minutes at 37°C and O2− generation was estimated by measuring fluorescence using 510 nm excitation and 580 nm emission as previously described [22].
Statistical Analyses
The values are presented as the Mean±SD and the difference in the values between the experimental groups and controls was analyzed using one-way ANOVA followed by post hoc analyses by the Tukey’s test. Unless otherwise stated, each experiment was repeated three times using chondrocytes prepared from three independent cartilage samples from different donors. P<0.05 was considered statistically significant.
Results
Activated Nrf2/ARE signaling was detected in human OA cartilage
To examine the potential role of Nrf2 in OA, articular cartilage from patients with OA and of non-OA donors was examined for the expression of Nrf2. The protein and mRNA expression of Nrf2 was significantly high in the cartilage of patients with OA compared to the levels detected in cartilage samples from non-OA donors (Figure 1A, B). Since temporal expression pattern of target genes is a key to determine the activity and function of a transcription factor, therefore, we first determined the expression of Nrf2 dependent genes HO-1 and SOD2. The protein and mRNA expression of HO-1 and SOD2 was considerably high in the articular cartilage of OA patients compared with cartilage from non-OA donors (P<0.05, Figure 1A, B). To further confirm these findings, we next examined the smooth and damaged cartilage samples from the same OA patient for the expression of Nrf2 and its signaling targets. The expression levels of Nrf2 and its target genes HO-1 and SOD2 were also significantly higher in the damaged OA cartilage compared with the levels detected in the smooth cartilage area from the same patients (P<0.05, Figure 1C–E). These data suggest that Nrf2 signaling was activated and its target genes were highly expressed in OA cartilage.
Figure 1. Nrf2/ARE signaling was activated in articular cartilage of patients with OA.

Cartilage from OA patients and healthy donors was processed for lysate preparation or RNA isolation using method described in method section. (A) Protein expression of Nrf2 and its signaling target HO-1 and SOD2 was investigated by immunoblotting using antibodies against indicated protein. β-actin was used as a control for equal loading. Fold change relative to β-actin for the expression these proteins were quantified by measuring specific signal intensities using ImageJ software. Immunoblot results are representatives of three blots performed on samples obtained from three individuals. *p≤0.05, as compared to normal cartilage. (B) Expression of Nrf2, HO-1 and SOD2 was measured by quantitative PCR using SYBR® green assay (Life Technologies). β-actin was used as endogenous expression control. (C–E) RNA was isolated from damaged or smooth cartilage for same patients and mRNA levels of (C) Nrf2, (D) HO-1 and (E) SOD2 were determined by SYBR® green assay (Life Technologies). Each symbol represents a single patient and horizontal lines shows the mean value of expression levels in the indicated number of samples. Bar graph represents mean±SD from three subjects. *p≤0.05, as compared to normal cartilage or smooth cartilage.
Nrf2/ARE signaling was activated in human OA chondrocytes under pathological conditions
To maintain the chondrogenic phenotype, chondrocytes were cultured in monolayer at high density (1×106/well of 6-well plate) and phenotype was determined by mRNA expression of chondrocyte marker genes. As shown in Supplementary Figure 1, human chondrocytes cultured in monolayer expressed COL2A1 and ACAN genes robustly with little or no expression of COL10A1 and COL1A1 at the time of analysis (Supplementary Figure 1). These conditions were thus maintained throughout the study.
Since activation of Nrf2/ARE signaling was found in OA cartilage, we determined whether Nrf2/ARE signaling was activated in human OA chondrocytes under pathological conditions (stimulation with IL-1β). In OA chondrocytes transfected with the Nrf2/ARE-reporter vector and stimulated with IL-1β, a time-dependent increase in the activity of Nrf2 was observed and a significant activation was seen after 3 h of stimulation with IL-1β (P<0.05, Figure 2A).
Figure 2. Nrf2/ARE signaling was activated in human OA chondrocytes under pathological conditions.

Primary human chondrocytes were isolated from cartilage of OA patients. (A) Nrf2/ARE luciferase reporter vector was transfected in OA chondrocytes and 48 hours later, chondrocytes were stimulated with IL-1β (1 ng/ml) for indicated time and luciferase activity was measured by Dual-Glo® reporter assay. Renilla luciferase was cotransfected for normalization purposes. Values are the mean±SD of 2 experiments each performed in triplicate. (B–C) Human OA chondrocytes were stimulated with IL-1β (1 ng/ml) for indicated time. At the end of treatment, chondrocytes were harvested and cell lysates were prepared using RIPA buffer for immunoblot analysis. (B) Protein expression of Nrf2, HO-1, NQO1 and SOD2 was investigated by immunoblotting using antibodies against indicated protein. β-actin was used as a control for equal loading. Immunoblot results are representatives of three blots performed on samples obtained from three individuals. (C) Fold change relative to β-actin for the expression these proteins were quantified by ImageJ software. *p≤0.05, as compared to control. (D) Human OA chondrocytes were pre-treated with anti-oxidants-NAC(10 mM) or GSH (10 mM) or DPI (5μM) for 2 h followed by treatment with IL-1β (1 ng/ml) for 16 h and RNA were isolated from harvested chondrocytes and expression of Nrf2 and HO-1 was measured by quantitative PCR using the SYBR® green assay (Life Technologies). β-actin was used as endogenous expression control. Bar graph represents mean±SD from three subjects. *p≤0.05, as compared to control, # ≤0.05, as compared to IL-1β.
To explore Nrf2/ARE signaling in depth in IL-1β stimulated OA chondrocytes, we performed time and dose kinetics studies and results showed that in comparison to untreated control, treatment of OA chondrocytes with IL-1β significantly (P<0.05) increased the mRNA (Supplementary Figure 2A) and protein expression of Nrf2 and its signaling targets HO-1, NQO1 and SOD2 in a time (Figure 2B, C) and dose dependent manner (Supplementary Figure 2B, C). Since Nrf2 is an oxidative stress responsive transcription factor, therefore, we hypothesized that increase in the level of Nrf2 activity may be a response to oxidative stress present in OA cartilage and chondrocytes under pathological conditions. To test this hypothesis, we first treated the OA chondrocytes with different antioxidants and then determined the expression of Nrf2 and its target gene HO-1 after stimulation with IL-1β. The results showed that treatment with antioxidants NAC, GSH and DPI significantly inhibited the IL-1β-induced expression of Nrf2 and HO-1 in OA chondrocytes (Figure 2D). Taken together these results suggest that activation of Nrf2/ARE signaling in OA chondrocytes was in response to the generation of prooxidants under pathological conditions.
Human OA chondrocytes under pathological conditions had enhanced levels of ROS
To determine whether enhanced Nrf2 activation was due to oxidative stress in damaged OA cartilage and IL-1β stimulated OA chondrocytes, we monitored the extent of oxidative stress in chondrocytes isolated from normal cartilage and the damaged cartilage from patients with OA. We monitored both the basal and induced oxidative stress by measuring the ROS levels using oxidation-sensitive dye DHR123 and our results showed that basal level of ROS was significantly higher in OA chondrocytes as compared to normal chondrocytes (P<0.05, Figure 3A). Of importance is our finding that stimulation with IL-1β resulted in little to no induction of ROS in normal chondrocytes, whereas the generation of ROS was significantly higher in OA chondrocytes (P<0.05, Figure 3A), which probably reflects the preactivated state of OA chondrocytes due to the disease.
Figure 3. Oxidative stress was enhanced in human OA chondrocytes under pathological conditions.

(A) Chondrocytes isolated from cartilage obtained from OA patients and healthy individuals were stained with DHR123 (5 μM) for 0.5 h at 37°C, and stimulated with IL-1β (1 ng/ml) for indicated time and ROS generation was estimated by measuring fluorescence emission at 535 nm. Bar graph showing relative fluorescence levels was calculated with respect to untreated control. Data points represent mean±SD from four replicates. *p≤0.05, as compared to untreated control OA chondrocytes, # ≤0.05, as compared to OA chondrocytes in each respective groups. (B) OA chondrocytes were incubated with PEG-catalase (1000 unit/ml) or PEG-SOD (200 unit/ml) for 1 h, stained with H2DCF-DA (20 μM) for 0.5 h at 37°C and then stimulated with IL-1β (1 ng/ml) for 30 minutes at 37°C. Fluorescence emission was measured at 535 nm using Synergy H1 multi-mode plate reader. Bar graph shows arbitrary fluorescence units indicating ROS levels. (C) Chondrocytes were incubated with DPI (5 μM) or Rotenone (10 μM) for 2 h, stained with H2DCF-DA (20 μM) for 0.5 h at 37°C and then stimulated with IL-1β (1 ng/ml) for 30 minutes at 37°C and ROS levels were estimated by measuring the fluorescence emission at 535 nm using Synergy H1 multi-mode plate reader. Data points represent mean±SD of four replicates from a representative experiment and three such independent experiments were carried out. *p≤0.05, as compared to control, # ≤0.05, as compared to IL-1β.
Since ROS include superoxide (O2−) and hydroxyl radical (HO·), along with non-radical species such as hydrogen peroxide (H2O2), therefore, we determined the nature of ROS generated by IL-1β in OA chondrocytes. OA chondrocytes were pretreated with antioxidant enzymes including PEG-Catalase or PEG-SOD, prior to the stimulation with IL-1β and it was observed that IL-1β-induced increase in H2DCF-DA fluorescence was significantly reduced in the presence of both Catalase and SOD suggesting that IL-1β stimulation induces the generation of both H2O2 and O2− in human OA chondrocytes (Figure 3B). Further, to identify the intracellular source of IL-1β stimulated generation of ROS, human OA chondrocytes were incubated with inhibitors of putative sources of ROS such as NADPH oxidase (DPI, 5μM), or mitochondrial complex I inhibitor (Rotenone, 10 μM). While Rotenone did not have any effect, the treatment with DPI completely inhibited the IL-1β-induced generation of ROS suggesting that NADPH oxidase (NOX) was the major source of ROS in IL-1β stimulated human OA chondrocytes (Figure 3C). Taken together with previous reports [35], our data demonstrate that enhanced NOX activity is a critical player in IL-1β-induced generation of high levels of ROS to induce oxidative stress in OA chondrocytes.
Nrf2 deficiency intensified the oxidative stress while its over-expression inhibited the oxidative stress in OA chondrocytes under pathological conditions
Our data shown in Figure 2D indicated that increase in Nrf2 levels in OA chondrocytes upon stimulation with IL-1β reflects cellular response to the induced oxidative stress. However, this increase in Nrf2 levels is not sufficient to fully counteract the pathogenic events induced by the oxidative stress in IL-1β-stimulated OA chondrocytes. We hypothesized that a deficiency in Nrf2-mediated antioxidant defenses plays a pivotal role in OA pathogenesis and therefore, over-expression of Nrf2 may alleviate the catabolic events through attenuation of oxidative stress in OA chondrocytes. To test this hypothesis, we employed both gain- and loss-of function studies of Nrf2 expression and monitored the oxidant levels in OA chondrocytes upon stimulation with IL-1β. Genetic ablation of Nrf2, using specific siRNA against Nrf2 (siNrf2) resulted in >90% depletion of mRNA and protein levels of Nrf2 and its signaling targets HO-1, NQO1 and SOD2 (Supplementary Figure 3A, B). Similarly, the ectopic expression of Nrf2 showed a significantly enhanced expression of Nrf2 and its downstream targets at mRNA and protein levels in OA chondrocytes (P<0.05, Supplementary Figure 3C, D).
We next determined whether over-expression or knockdown of Nrf2 had any effect on IL-1β-induced ROS levels in OA chondrocytes. Our results showed that IL-1β treatment significantly increased the ROS production in OA chondrocytes whereas over-expression of Nrf2 in OA chondrocytes suppressed the IL-1β-mediated generation of ROS (P<0.05, Figure 4A). Interestingly, knockdown of Nrf2 enhanced the basal ROS levels and augmented the ROS levels induced by IL-1β in OA chondrocytes (P<0.05, Figure 4B). Since H2O2 is one of the major ROS produced by IL1β stimulation, we specifically measured H2O2 levels using Amplex® Red assay in the supernatant of OA chondrocytes with either over-expression or depleted expression of Nrf2. In agreement with the above data, it was observed that OA chondrocytes with Nrf2 over-expression had significantly less basal levels of H2O2 and produced less H2O2 upon stimulation with IL-1β. On the other hand, OA chondrocytes with depleted expression of Nrf2 had considerably enhanced basal and induced levels of H2O2 (P<0.05, Figure 4C, D). These results indicated that increased expression and activity of Nrf2 in OA chondrocytes can effectively suppress IL-1β-induced ROS generation and may protect against oxidative stress under pathological conditions.
Figure 4. Nrf2 over-expression in OA chondrocytes inhibited oxidative stress while its deficiency intensified the oxidative stress under pathological conditions.

Human OA chondrocytes were transfected with siRNA specific for Nrf2 (siNrf2, 100 nM) or negative controls siRNA (siCNT, 100 nM) using X-tremeGENE™ siRNA transfection reagent or transfected with Nrf2 expression plasmid (pNrf2, 1μg) or pcDNA3.1 as control plasmid (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit on 4D on Amaxa Nucleofection System. (A, B) Forty-eight hours after the transfection with (A) siRNA or (B) expression vector, OA chondrocytes were stained with DHR123 (5 μM) for 0.5 h at 37°C, and stimulated with IL-1β (1 ng/ml) for 30 minutes at 37°C and ROS generation was estimated by measuring fluorescence emission at 535 nm. Bar graph shows arbitrary fluorescence units indicating ROS levels. (C–D) Forty-eight hours after the transfection with (C) siRNA or (D) expression vector, OA chondrocytes were stained with stimulated with IL-1β (1 ng/ml) for 30 minutes at 37°C, supernatants were collected and generation of H2O2 in the supernatant was estimated using Amplex red assay as described in method section. Data points represent mean±SD of three replicates from a representative experiment and three such independent experiments were carried out.*p≤0.05, as compared to control, # ≤0.05, as compared to IL-1β.
Enhanced expression of Nrf2 suppressed both extrinsic and intrinsic apoptotic pathways and increased the survival of OA chondrocytes under pathological conditions
Since degeneration of articular cartilage, a hallmark of OA pathogenesis, is primarily attributed to compromised chondrocyte function and survival, hence chondrocyte death due to induction of apoptosis is considered as a central feature in OA progression [9]. We therefore, investigated the effect of Nrf2 overexpression on apoptosis induction in OA chondrocytes under pathological conditions. As shown previously [27], treatment with IL-1β for 48 h considerably reduced the viability of OA chondrocytes (Figure 5A). Interestingly, Nrf2 over-expression in OA chondrocytes significantly inhibited the IL-1β-mediated loss of viability (P<0.05, Figure 5A). In contrast, OA chondrocytes with depleted expression of Nrf2 showed significantly decreased viability after stimulation with IL-1β (Supplementary Figure 4A). The cytoprotective effect of Nrf2 was further confirmed by quantitative evaluation of apoptosis using TUNEL and DNA fragmentation assays and in corroboration with MTT data, results showed that Nrf2 over-expression significantly inhibited IL-1β-mediated apoptosis in OA chondrocytes (P<0.05, Figures 5B, C and Supplementary Figure 4B). Apoptosis is the culmination of sequential events executed through the formation of apoptosome complex, cleavage of pro-Caspase-3 into the active form, and finally the fragmentation of PARP. We observed that upregulation of Nrf2 expression significantly inhibited the formation of active Caspase-3 and fragmentation of PARP in IL-1β-stimulated OA chondrocytes (Figure 6A, B). These results suggest that increased expression of Nrf2 inhibit apoptosis by suppressing the activation of Caspase-3 in OA chondrocytes under pathological conditions.
Figure 5. Nrf2 over-expression inhibited apoptosis in OA chondrocytes under pathological conditions.

Human OA chondrocytes were transfected with Nrf2 expression plasmid (pNrf2, 1μg) or pcDNA3.1 as control plasmid (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit on 4D on Amaxa Nucleofection System. Forty-eight hours after the transfection, OA chondrocytes were serum starved and then stimulated with or without IL-1β (10 ng/ml) for 48 h. (A) Cell viability was measured by MTT assays using method described in method sections. Unstimulated chondrocytes were served as control and viability was expressed relative to control cells. Data point was expressed as a mean ± SD of two replicates from a representative experiment and three such independent experiments were carried out. *p≤0.05, as compared to pcDNA (control), # ≤0.05, as compared to pcDNA+IL-1β group. (B) Apoptosis was estimated by TUNEL assay as described in method section. Flow cytometric histograms showing percent TUNEL positive chondrocytes were shown. Twenty thousand cells in each group were acquired using a flowcytometer and percent apoptosis was calculated using flow Jo software and expressed as a mean±SD of three replicates from a representative experiment and three such independent experiments were carried out. (C) Apoptosis was visualized by DNA fragmentation assay as described in method section and DNA samples were electrophoresed in 1.8% agarose gel and DNA fragmentation pattern in control and simulated groups were visualized by EtBr staining. Results are representatives of three experiments performed on samples obtained from three individuals.
Figure 6. Nrf2 over-expression inhibited both extrinsic and intrinsic apoptotic pathways in OA chondrocytes under pathological conditions.

Human OA chondrocytes were transfected with Nrf2 expression plasmid (pNrf2, 1μg) or pcDNA3.1 as control plasmid (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit on 4D on Amaxa Nucleofection System. Forty-eight hours after the transfection, OA chondrocytes were serum starved and then stimulated with or without IL-1β (10 ng/ml) for 48 h. (A). At end of treatment, cell lysates were prepared using RIPA buffer and protein levels of cleaved caspase-3, cleaved caspase-8, pro-caspase-9, cleaved PARP and Bid were investigated by immunoblotting using antibodies against indicated protein. β-actin was used as a control for equal loading. Immunoblot results are representatives of three blots performed on samples obtained from three individuals. (B) Fold change relative to β-actin for the expression these proteins were quantified by measuring specific signal intensities using ImageJ software. *p≤0.05, as compared to pcDNA, # ≤0.05, as compared to pcDNA+IL-1β. (C) After transfection, OA chondrocyte were stimulated IL-1β (1 ng/ml) for 30 minutes at 37°C and then stained with JC-1 (2.5 μM) and mitochondrial membrane potential loss was measured by ratio of red to green fluorescence. Values were expressed relative to unstimulated control and graph shows mean ± SD of four replicates from a representative experiment and three such independent experiments were carried out. *p≤0.05, as compared to control, # ≤0.05, as compared to IL-1β. (D) Cytosolic lysates were prepared after 48 h of IL-1β (10 ng/ml) stimulation in transfected chondrocytes and levels of cytosolic cytochrome-c was estimated by immunoblotting using validated antibody. β-actin was used as a control for equal loading. Immunoblot results are representatives of three blots performed on samples obtained from three individuals.
We next determined the molecular events of apoptosis suppression for the observed protective effects of Nrf2 in OA chondrocytes. Several reports on chondrocytes apoptosis in OA suggest the activation of signaling effectors involved in both extrinsic and intrinsic pathways [7–9, 36]. Extrinsic apoptotic pathways are mediated by activation of Caspase-8, whereas Caspase-9 occupies a central role in mediating intrinsic apoptosis [37]. Therefore, we explored whether Nrf2 had suppressive effects on the extrinsic or intrinsic pathways in IL-1β stimulated OA chondrocytes. OA chondrocytes stimulated with IL-1β showed cleavage of Caspase-8 and Caspase-9 suggesting the activation of both extrinsic and intrinsic apoptotic pathways (Figure 6A, B). Further, the over-expression of Nrf2 in OA chondrocytes inhibited the IL-1β induced cleavage of both Caspase-8 and Caspase-9 indicating that Nrf2 suppresses the activation of both the extrinsic and intrinsic apoptotic pathways in OA chondrocytes (Figure 6A, B). We next asked how Nrf2 suppressed both the pathways of apoptosis in chondrocytes. Molecular crosstalk between the extrinsic and intrinsic apoptotic pathways require Caspase-8 mediated cleavage and activation of the Bid (BH3-interacting domain death agonist), the product of which tBid (truncated BID) protein is required for intrinsic apoptosis in some cell types [38]. Therefore, we determined whether Nrf2 had any effect on the cleavage of Bid in IL-1β-stimulated OA chondrocytes and our results showed that Nrf2 over-expression inhibited the IL-1β induced cleavage of Bid (Figure 6A, B). Taken together, these findings indicate that the suppression of Bid cleavage was probably due to the inhibition of Caspase-8 activation and thus may be a common denominator of Nrf2 mediated suppression of both extrinsic and intrinsic apoptotic pathways in OA chondrocytes under pathological conditions.
Mitochondrial depolarization has been proposed as the early event during intrinsic apoptotic pathway and previous studies demonstrated the impairment of mitochondrial membrane potential and loss of mitochondrial ATP production in IL-1β-stimulated chondrocytes [39]. To investigate whether Nrf2 expression had any effects on mitochondrial depolarization, we examined the mitochondrial dysfunction by measuring changes in mitochondrial membrane potential using the mitochondria specific cationic dye JC-1. Our data showed that IL-1β induced a mitochondrial permeability transition (loss of Δ ψm) suggesting that mitochondrial damage and dysfunction are involved in IL-1β-induced apoptosis in OA chondrocytes (Figure 6C). Further over-expression of Nrf2 completely inhibited the IL-1β mediated loss of mitochondrial membrane potential in OA chondrocytes (Figure 6C). Since high levels of mitochondrial ROS (mtROS) initiate the molecular events leading to mitochondrial membrane permeabilization [40], we measured mtROS levels using a mitochondrial superoxide-sensitive fluorescent dye, MitoSOX™ red. While IL-1β stimulation resulted in enhanced generation of mtROS, over-expression of Nrf2 significantly inhibited the basal and induced levels of mtROS (Supplementary Figure 5A). Mitochondrial apoptotic pathway is further exemplified by Bax dependent release of Cytochrome c, which is considered as point of no return in the apoptotic death pathway [37]. We observed that Nrf2 over-expression significantly inhibited the IL-1β mediated release of Cytochrome-c in OA chondrocytes (Figure 6D and Supplementary Figure 5B). Taken together these results suggest that Nrf2 is a critical regulator of mitochondrial health in human OA chondrocytes and plays a vital role in inhibiting mitochondrial dysfunction and apoptosis implicated in OA pathogenesis.
Mitochondrial apoptosis is mediated through increased expression of pro-apoptotic proteins such as Bax and Bad which promote the formation of apoptosome consisting of Cytochrome-c, Caspase-9 and Apaf-1 [40]. IL-1β treatment resulted in a profound increase in the expression of Bax and Bad and inhibited the expression of anti-apoptotic proteins-Bcl-2, Bcl-xl and Mcl-1 in OA chondrocytes (Supplementary Figure 5C). Importantly, over-expression of Nrf2 reversed these effects of IL-1β in human OA chondrocytes (Supplementary Figure 5C). Thus, in combination with the data presented above, we demonstrate that Nrf2 plays a crucial role in chondroprotection by maintaining the mitochondria integrity and suppressing both the extrinsic and intrinsic apoptosis pathways in human OA chondrocytes under pathological conditions.
Nrf2 over-expression did not inhibit the activation of NF-κB in human OA chondrocytes under pathological conditions
Recent studies suggest that Nrf2 inhibits the transcriptional activation of NF-κB by preventing the degradation of IκB-α [41–43]. Therefore, we hypothesized that Nrf2 may exerts its protective effects through the suppression of NF-κB activation in OA chondrocytes under pathological conditions. Stimulation of OA chondrocytes with IL-1β has been shown to induce the phosphorylation and degradation of the NF-κB inhibitor IκBα [28, 44, 45]. Therefore, we evaluated the effect of Nrf2 over-expression on the phosphorylation and degradation of IκBα. Stimulation of OA chondrocytes with IL-1β caused the phosphorylation and degradation of IκBα within 15 minutes of treatment followed by reappearance of IκBα at 30 minutes (Supplementary Figure 6A). However, contrary to other reports [41–43], we observed that Nrf2 over-expression in OA chondrocytes results in phosphorylation and degradation of IκBα leading to activation of NF-κB and also prolonged the IL-1β-mediated activation of NF-κB upto 30 minutes (Supplementary Figure 6A). Taken together these results indicate the absence of cross talk between Nrf2 and NF-κB and further suggest that suppressive and chondroprotective effects of Nrf2 were independent of NF-κB signaling pathways in OA chondrocytes under pathological conditions.
Nrf2 over-expression did not inhibit the activation of p38MAPK and JNK in human OA chondrocytes under pathological conditions
We next examined the involvement of MAPKs in the protective effects of Nrf2 in OA chondrocytes as IL-1β induced activation of p38MAPK and JNK is involved in OA pathogenesis [46, 47]. Expression of Nrf2 has been shown to inhibit TNF-α induced activation of p38 MAP kinase in endothelial cells [48]. Therefore, we determined whether Nrf2 mediates its chondroprotective effects through the suppression of p38MAPK activation by measuring the phosphorylation in IL-1β stimulated OA chondrocytes. Stimulation of OA chondrocytes with IL-1β resulted in the phosphorylation of p38MAPK within 15 minutes of treatment. However, Nrf2 over-expression did not inhibit the phosphorylation of p38MAPK, but instead stimulated its activation in OA chondrocytes (Supplementary Figure 6B). This was surprising observation as activation of p38 MAPK is recognized as primary signal for apoptosis in chondrocytes [49–51]. We further examined the activation of JNK and results showed that expression of Nrf2 did not have any effects on IL-1β mediated activation of JNK1/2 in IL-1β stimulated OA chondrocytes (Supplementary Figure 6B). These results indicated that Nrf2 exerts chondroprotective effect without involving p38MAPK and JNK signaling pathways in OA chondrocytes.
Nrf2 over-expression inhibited apoptosis through activation of ERK1/2 signaling in human OA chondrocytes under pathological conditions
It has been shown that ERK1/2 signaling is recognized as a primary survival signaling in chondrocytes and function as anti-apoptotic since inhibition of ERK causes cellular apoptosis [50, 52, 53]. Therefore, we determined the possible involvement of ERK1/2 signaling in the observed protective effects of Nrf2 in OA chondrocytes under pathological conditions. The activation of ERK1/2 was examined by measuring the phosphorylation and results showed that IL-1β stimulation resulted in the phosphorylation of ERK1/2 within 15 minutes followed by a slow decline in phosphorylation levels at 30 minutes in OA chondrocytes (Figure 7A and Supplementary Figure 6C). Interestingly, OA chondrocytes with over-expression of Nrf2 showed robust phosphorylation of ERK1/2 and also enhanced the IL-1β-mediated activation of ERK in OA chondrocytes (Figure 7A and Supplementary Figure 6C) with the stimulatory effect on the phosphorylation of ERK1/2 being prominent after 30 minutes of IL-1β stimulation (Figure 7A and Supplementary Figure 6C). To further elucidate the signaling pathway leading to Nrf2 mediated activation of ERK1/2, we examined the phosphorylation of MEK1/2 in OA chondrocytes as Ras/Raf/MEK signaling axis is the canonical signaling pathway for the activation of ERK [54]. However, Nrf2 expression did not resulted in the phosphorylation of MEK1/2 in OA chondrocytes indicating that Nrf2 did not activate the ERK1/2 signaling through MEK1/2 and most likely involve an alternate mechanism of ERK activation (Figure 7A).
Figure 7. Nrf2 over-expression activated ERK1/2 signaling in human OA chondrocytes under pathological conditions.

Human OA chondrocytes were transfected with Nrf2 expression plasmid (pNrf2, 1μg) or pcDNA3.1 as control plasmid (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit on 4D on Amaxa Nucleofection System. Forty-eight hours after the transfection, OA chondrocytes were serum starved and then stimulated with or without IL-1β (10 ng/ml) for 15 and 30 minutes at 37°C (A–C). At end of treatment, cell lysates were prepared using RIPA buffer and immunoblotting was performed to detect the protein levels of (A) p-ERK1/2 (Thr180/Tyr182), total ERK1/2, p-MEK1/2 (Ser217/221), total MEK1/2, p-ELK-1 (Ser383), p-P70S6K (Ser411), p-P90RSK (Ser380). β-actin was used as a control for equal loading. Immunoblot results are representatives of three blots performed on samples obtained from three individuals. (B–C) At end of transfection, chondrocytes were treated with ERK inhibitor PD98059 (30 μM) for 2 h and then stimulated with IL-1β (10 ng/ml) for (B) 15 minutes and (C) 16 h at 37°C and processed for (B) immunoblotting with p-ERK1/2 and total ERK1/2 and (C) TUNEL assay as described in method section. Bar graph shows mean±SD of three replicates from a representative experiment and three such independent experiments were carried out. *p≤0.05, as compared to pcDNA, # ≤0.05, as compared to pcDNA+IL-1β, $≤0.05, as compared to pNrf2+IL-1β.
To further dissect the role of ERK1/2 signaling in the protective effect of Nrf2, we determined the activity of ERK in Nrf2 over-expressed and IL-1β stimulated OA chondrocytes by evaluating the activation of downstream targets. More than 150 targets of ERK are known in mammalian cells, we focused on the effector molecules having anti-apoptotic roles [54]. The ERK downstream target p70S6 kinase and p90 ribosomal S6 kinase (RSK) are involved in the inhibition and down regulation of pro-apoptotic protein BAD and Bim [54–56]. Corresponding with temporal activation of ERK, our results showed that over-expression of Nrf2 increased the ERK activity as judged from the levels of p-ELK1, p-P70S6K and p-P90RSK in OA chondrocytes (Figure 7A). To further substantiate the role of ERK1/2 signaling in chondroprotective effects of Nrf2, a pharmacological inhibitor of ERK (PD98059) was used in Nrf2 over-expressed chondrocytes which effectively inhibited the phosphorylation of ERK1/2 (Figure 7B). Results of TUNEL assay showed that inhibition of ERK1/2 in IL-1β stimulated OA chondrocytes significantly abrogated the protective effects of Nrf2 (Figure 7C). All together, these results suggest that activated Nrf2 is an effective inhibitor of IL-1β mediated apoptosis through the activation of ERK1/2 signaling in OA chondrocytes.
Discussion
Oxidative stress due to perturbations in cellular redox status is an established risk factor in the pathogenesis of OA and likely mediates the effects of emerging, less well-defined variables that contribute to residual risk not explained by traditional factors [57]. Inside the cell, high levels of ROS are derived from many sources including mitochondria, xanthine oxidase, uncoupled nitric oxide synthases and NADPH oxidase [58]. In addition to generalized oxidation resulting in cell dysfunction, necrosis or apoptosis, ROS also induce specific post-translational modifications that alter the function of important cellular proteins and signaling pathways. The Nrf2/ARE signaling pathway is mainly responsible for cellular defense against oxidative stress and maintaining the cellular redox balance at physiological levels. The relation between Nrf2/ARE signaling and regulation of apoptosis and oxidative stress in osteoarthritis is not understood. Towards this goal, in the present study we analyzed the mechanisms underlying Nrf2 mediated chondroprotection under pathological and oxidative stress microenvironment. We employed an in vitro cellular model of chondrocyte dysfunction and death by treating human chondrocytes, the only resident cell type present in the cartilage, with IL-1β, a critical player in the induction and the pathogenesis of OA. Our results provide considerable evidence that the chondroprotective effects of Nrf2 were mediated through the suppression of molecular events involved in oxidative stress and apoptosis in OA chondrocytes. In the present study, we show for the first time that Nrf2 exerts potent anti-apoptotic effects through the activation of ERK1/2/ELK-1-P70S6K-P90RSK signaling axis in human OA chondrocytes under pathological conditions. These results also identify the Nrf2/ARE signaling as an endogenous chondroprotective system for protection against the prooxidants mediated cellular dysfunction and suppression of apoptotic events suggesting that targeting the Nrf2/ARE pathway may represent a novel therapeutic approach for the maintenance of cartilage integrity and suppressing disease progression in OA.
Increasing evidence supports a key role that oxidative stress play in OA pathogenesis, as increased levels of oxidized molecule such as TBARS (thiobarbituric acid reactive substances), oxidized nucleotide 8-oxoguanine and decreased levels of antioxidants including ascorbic acid, GSH, catalase and GPx were reported in OA patients [4, 59]. Oxidative stress disturbs intracellular signaling involved in the regulation of multiple events associated with OA pathogenesis including chondrocyte senescence and apoptosis, ECM synthesis and degradation, synovial inflammation and dysfunction of the subchondral bone [6]. We have shown that the expression and activity of the oxidative stress responsive transcription factor Nrf2 was increased in the damaged cartilage and chondrocytes isolated from the damaged area of the OA cartilage compared to chondrocytes prepared from the normal cartilage or the unaffected cartilage of the same patients (Figure 1A–E, 2A–C). These findings were in agreement with earlier studies where increased expression of Nrf2 was observed in the affected joints of arthritic mice and patients with rheumatoid arthritis [60]. The increased Nrf2 expression and activity may represent a self-adaptive mechanism of chondrocytes to cope with and survive in the disease microenvironment characterized by increased levels of inflammatory molecules and oxidative stress. We have provided experimental evidence to support this hypothesis and our data showed that increased Nrf2 activity in IL-1β stimulated chondrocytes was significantly suppressed by treatment with antioxidants suggesting that Nrf2 activation in OA chondrocytes and cartilage was response to oxidative stress (Figure 2D). Further, Nrf2 activation upon stimulation with IL-1β might also be the result of activation of upstream MAPK as increased ERK1/2 activity were reported in IL-1β-stimulated OA chondrocytes (Figure 7A) [45]. However, the intrinsic activity of Nrf2 in OA cartilage and IL-1β-stimulated OA chondrocytes is not sufficient to fully encounter the catabolic events associated with OA pathogenesis, our data suggest that a deficiency imposed by the suppression of Nrf2 expression and activity in chondrocytes under pathological conditions plays a significant role in OA pathogenesis and therefore pharmacological interventions to enhance expression and activity of Nrf2 would be an efficient and effective therapeutic strategy for the management of OA.
Targeting oxidative stress by stimulation of endogenous antioxidants through activation of Nrf2/ARE signaling to make their own natural antioxidants seems to be more promising as these become available locally inside the cells to reduce the level of ROS. Activation of Nrf2 signaling leads to induction of several antioxidants effectors and cytoprotective genes such as SOD2, Catalase, and HO-1 that have been shown to inhibit OA pathogenesis. A recent study showed that HO-1 induction reduced the severity of OA like changes in two distinct mouse models of OA [61]. Additional studies using phytochemicals and polyphenols including Wogonin, Plumbagin, Sesamin, Protandim and 6-Gingerol also reported similar findings where induction of endogenous antioxidants defenses through the activation of Nrf2 signaling exerted chondroprotective and disease suppressing effects in experimental models of OA [18, 20–23]. Collectively, these data support the notion that activation of Nrf2/ARE signaling is a powerful tool for the restoration of the redox status of OA chondrocytes that is protective under pathological conditions.
Compromising the chondrocyte function and survival would lead to the failure of articular cartilage leading to OA, therefore loss of chondrocytes survival due to induction of apoptosis is considered a central feature in OA progression [7]. However, signaling pathways involved in chondrocytes apoptosis are not characterized in detail and further it is not known whether chondrocyte apoptosis is the inducer of cartilage degeneration or a byproduct of cartilage destruction. Therefore, characterization of novel pathways involved in chondrocytes apoptosis leading to cartilage disintegration is of importance for the effective management of OA. Present study was undertaken to characterize the molecular pathways involved in chondrocytes apoptosis using IL-1β stimulated OA chondrocytes as a cellular model because this model closely resembles chondrocytes damage in OA [1, 27]. We found that treatment of OA chondrocytes with IL-1β activated both the extrinsic and intrinsic apoptotic pathways as evidenced by activation Caspase-8, -9 and -3 and cleavage of PARP (Figure 6A). Our data supports the earlier findings which demonstrated that treatment of bovine chondrocytes with IL-1 results in induction of apoptosis [8]. Our findings support the recent studies where treatment with IL-1β activated Caspase-3 and cleavage of PARP was observed in chondrocytes [62, 63]. In addition, our data extend the knowledge by demonstrating that IL-1β-induced apoptosis was associated with loss of mitochondrial membrane potential and induction of mtROS generation (Supplementary Figure 5A) and provide further experimental support to an earlier finding where mitochondrial dysfunction was reported in OA [64]. In our studies, release of cytochrome-c further substantiated the role of mitochondrial dysfunction in IL-1β-mediated intrinsic apoptosis in OA chondrocytes (Figure 6D and Supplementary Figure 5B). Similar to previous reports in rat chondrocytes [59], we also observed that IL-1β treatment resulted in increased expression of Bax and decreased levels of Bcl2 in human OA chondrocytes (Supplementary Figure 5C) [62]. However, in addition to these effector molecules, our study also demonstrated for the first time the involvement of Bad and Bid proteins as pro-apoptotic and Bcl-xl and Mcl-1 as anti-apoptotic proteins in IL-1β-stimulated human chondrocytes (Supplementary Figure 5C). Our data thus identify the molecular effectors involved in chondrocytes apoptosis, which is controlled by multiple signaling players and involves cross talk between these regulatory molecules.
Recent highlights on the role of Nrf2 activation in the suppression of apoptosis have been described in periodontal ligament stem cells (PDLSCs), THP-1-derived macrophage and ethanol induced apoptosis in H9C2 cardiomyocytes and PC12 cells [65–68]. Here, we undertook a detailed investigation of the role of Nrf2 in the suppression of apoptosis in OA chondrocytes using an Nrf2 expression vector, as genetic over-expression is more specific than pharmacological activation. Our results showed that Nrf2 over-expression effectively diminished TUNEL staining, reduced PARP cleavage and decreased the activation of Caspase-3, Caspase-8, and Caspase-9 in IL-1β-stimulated OA chondrocytes (Figure 5B, C and 6A). A recent report demonstrated that Nrf2 activation exerts anti-apoptotic effects by upregulating the levels of Bcl-2, in addition to downregulating Bax and c-fos expression in a different cell type [67]. In agreement with this study, we found that Nrf2 over-expression inhibited the expression of pro-apoptotic proteins Bax, Bad and Bid and upregulated the expression of anti-apoptotic proteins Bcl2, Bcl-xl and Mcl-1 in IL-1β-stimulated human OA chondrocytes (Supplementary Figure 5C). These data provide experimental support to the previously reported findings by demonstrating that the activation of Nrf2 inhibits stress induced apoptosis and increase cell survival by enhancing the expression of Bcl-2 [69]. We next examined the role of Nrf2 in maintaining the mitochondrial homeostasis which is known be disrupted in OA. Our data extend the previous findings on mitochondrial function in OA [70] by demonstrating that expression of Nrf2 improved the mitochondrial health and suppressed apoptosis by downregulating the mtROS production, preventing mitochondrial dysfunction and inhibited the release of cytochrome-c from mitochondria to cytosol (Figure 6C, D and Supplementary Figure 5A, B).
Nrf2 expression has been shown to inhibit NF-κB and p38 MAP kinase, the known therapeutic targets of OA [48]. Our data rule out the possible involvement of NF-κB and p38 MAPK signaling in the observed protective effect of Nrf2 and goes a step further and suggests that the anti-apoptotic effect of Nrf2 was exerted independent of NF-κB and p38 MAP kinase signaling pathways in OA chondrocytes (Supplementary Figure 6A, B). Earlier reports suggested that ERK1/2 functions as an inhibitory signal for apoptosis and further promote the maintenance of chondrocyte phenotype [50]. Pharmacological activation of ERK1/2 using Curcumin was shown to be protective by suppressing the IL-1β-mediated apoptosis in primary chondrocytes [52]. Our data demonstrated that increased Nrf2 expression resulted in increased phosphorylation of ERK1/2 and its downstream targets ELK1, P70S6K and P90RSK (Figure 7A). Our data further revealed that increased kinase activity of ERK1/2 was not mediated through the activation of the upstream kinase MEK1/2 in OA chondrocytes (Figure 7A). Our findings of ERK1/2 activation and inhibition of apoptosis in chondrocytes with over expression of Nrf2 are similar to previous reports in other cell types where ERK1/2 mediated activation of Nrf2 prevented apoptosis induced by 6-Hydroxydopamine in neuroblastoma cells [71]. Thus, this study for the first time demonstrated that increased expression of Nrf2 in human OA chondrocytes resulted in coordinated and temporal activation of ERK1/2 and inhibition of apoptosis under pathological conditions.
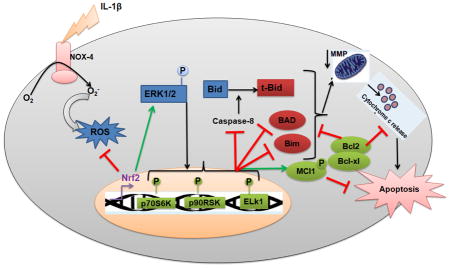
In conclusion, our study shows for the first time that Nrf2 plays a pivotal role in protecting OA chondrocytes against IL-1β-induced redox imbalance and apoptotic death and subsequently improves the chondrocytes survival by preventing the mitochondrial damage and dysfunction under pathological conditions. Furthermore, we characterized the role of extrinsic and intrinsic signaling events involved in chondrocytes apoptosis in vitro under pathological conditions. Our data provide strong evidence that Nrf2 exerts chondroprotective effect by repressing oxidative stress and apoptosis through inhibition of extrinsic and intrinsic pathways, suppression of pro-apoptotic and expression of anti-apoptotic proteins via activation of ERK1/2/ELK1-P70S6K-P90RSK signaling axis in human OA chondrocytes (Fig. 8). These findings suggest that approaches to induce Nrf2/ARE signaling have the potential to be developed as a novel therapeutic approach for the management of OA.
Figure 8. Schematic representation of proposed mechanism of Nrf2/ARE mediated protective effects in human OA chondrocytes.

Nrf2 plays a pivotal role in protecting osteoarthritis chondrocytes against IL-1β-induced oxidative stress and apoptotic events through inhibition of extrinsic apoptosis (caspase-8) and intrinsic apoptotic pathways (caspase-9 and 3), mitochondria dysfunction, release of cytochrome-c in cytosol, expression of pro-apoptotic proteins-Bax and Bad and stimulation of anti-apoptotic proteins-Bcl-2, Bcl-xl and Mcl-1 via activation of ERK1/2/ELK1-P70S6K-P90RSK signaling axis in human OA chondrocytes.
Supplementary Material
Supplementary Figure 1. Chondrocytes phenotype was maintained in monolayer culture: Human OA chondrocytes were cultured in monolayer and total RNA was isolated and mRNA expression of COL2A1, ACAN, COL10A1 and COL1A1 were quantified using TaqMan assay. Gene expression was determined by the ΔCt method and results were normalized to β-actin expression.
Supplementary Figure 2. Treatment of OA chondrocytes with IL-1β activated Nrf2/ARE signaling: Human OA chondrocytes were stimulated with (A) IL-1β (1 ng/ml) for indicated time or (B–C) IL-1β (0–10 ng/ml) for 16 hours. At the end of treatment, chondrocytes were harvested and RNA was isolated for real time PCR analysis or cell lysates were prepared using RIPA buffer for immunoblot analysis. (A, B) Expression of Nrf2 and its downstream signaling targets HO-1, NQO1 and SOD2 was measured by quantitative PCR using the SYBR® green assay (Life Technologies). β-actin was used as endogenous expression control. Bar graph represents mean±SD from two subjects. *p≤0.05, as compared to control, # ≤0.05, as compared to IL-1β. (C) Protein expression of Nrf2, HO-1, NQO1 and SOD2 was investigated by immunoblotting using antibodies against indicated protein. β-actin was used as a control for equal loading. Immunoblot results are representatives of two blots performed on samples obtained from two individuals.
Supplementary Figure 3. Transfection efficacy of Nrf2 knockdown and over-expression in human OA chondrocytes: Human OA chondrocytes were transfected with siRNA specific for Nrf2 (siNrf2, 100 nM) or negative controls siRNA (siCNT, 100 nM) using (A, B) X-tremeGENE™ siRNA transfection reagent or (C, D) transfected with Nrf2 expression plasmid (pNrf2, 1μg) or pcDNA3.1 as control plasmid (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit on 4D on Amaxa Nucleofection System. Forty-eight hours after the transfection, RNA was isolated for real time PCR analysis or cell lysates were prepared using RIPA buffer for immunoblot analysis. Expression of Nrf2 and its downstream signaling targets HO-1, NQO1 and SOD2 was measured by (A, C) quantitative PCR using SYBR® green assay (Life Technologies) and (B, D) immunoblotting using antibodies against indicated protein. β-actin was used as endogenous expression control.
Supplementary Figure 4. Nrf2 over-expression inhibited apoptosis in OA chondrocytes under pathological conditions: Human OA chondrocytes were transfected with (A) siRNA specific for Nrf2 (siNrf2, 100 nM) or negative controls siRNA (siCNT, 100 nM) or (B) Nrf2 expression plasmid (pNrf2, 1μg) or pcDNA3.1 as control plasmid (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit on 4D on Amaxa Nucleofection System. Forty-eight hours after the transfection, OA chondrocytes were serum starved and then stimulated with or without IL-1β (10 ng/ml) for 48 h. (A) Cell viability was measured by MTT assays using method described in method sections. Unstimulated chondrocytes were served as control and viability was expressed relative to control cells. Data point was expressed as a mean ± SD of two replicates from a representative experiment and three such independent experiments were carried out. *p≤0.05, as compared to siCNT, # ≤0.05, as compared to siCNT+IL-1β. (B) Apoptosis was estimated by TUNEL assay as described in method section. Bar chart corresponding to Flow cytometric histograms showed percent TUNEL positive chondrocytes. Percent apoptosis was calculated using flow Jo software and expressed as a mean±SD of three replicates from a representative experiment and three such independent experiments were carried out. *p≤0.05, as compared to pcDNA, # ≤0.05, as compared to pcDNA+IL-1β.
Supplementary Figure 5. Nrf2 over-expression inhibited intrinsic apoptotic pathways in OA chondrocytes under pathological conditions: Human OA chondrocytes were transfected with Nrf2 expression plasmid (pNrf2, 1μg) or pcDNA3.1 as control plasmid (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit on 4D on Amaxa Nucleofection System. (A) Forty-eight hours after the transfection, OA chondrocytes were stained with MistoSOX red (5 μM) and then stimulated with IL-1β (1 ng/ml, 30 minutes) and the production of mitochondrial O2− was estimated by measuring the fluorescence emission at 580 nm following the excitation at 510 nm. Bar graph shows mean±SD of four replicates from a representative experiment and three such independent experiments were carried out. (B) Fold change relative to β-actin for the expression cytosolic cytochrome-c was quantified by measuring specific signal intensities using ImageJ software. *p≤0.05, as compared to pcDNA, # ≤0.05, as compared to pcDNA+IL-1β. (C) Forty-eight hours after the transfection, OA chondrocytes were serum starved and then stimulated with or without IL-1β (10 ng/ml) for 48 h and protein expression in the cell lysates prepared above was investigated by immunoblotting using antibodies against Bax, Bad, Bcl2, Bcl-xl and Mcl-1. β-actin was used as a control for equal loading. The β-actin in Figure 6A and supplementary Figure 5C are from the same experiment. Immunoblot results are representatives of three blots performed on samples obtained from three individuals with or without for 48 h.
Supplementary Figure 6. Nrf2 over-expression did not inhibit the activation of NF-κB, p38 MAPK and JNK in human OA chondrocytes under pathological conditions: Human OA chondrocytes were transfected with Nrf2 expression plasmid (pNrf2, 1μg) or pcDNA3.1 as control plasmid (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit on 4D on Amaxa Nucleofection System. Forty-eight hours after the transfection, OA chondrocytes were serum starved and then stimulated with or without IL-1β (10 ng/ml) for 15 and 30 minutes at 37°C. At end of treatment, cell lysates were prepared using RIPA buffer and immunoblotting was performed to detect the protein levels of (A) Iκβα and p-Iκβα (Ser 32) (B) p-P38 (Thr180/Tyr182), total p38, p-JNK (Thr183/Tyr185) and total JNK. β-actin was used as a control for equal loading. Immunoblot results are representatives of three blots performed on samples obtained from three individuals. (C) Fold change for the expression of p-ERK relative to total ERK was quantified by measuring specific signal intensities using ImageJ software. *p≤0.05, as compared to pcDNA.
Highlights.
Nrf2/ARE signaling is activated in OA cartilage compared with cartilage from healthy donors.
Nrf2 overexpression suppresses IL-1β-mediated ROS generation in OA chondrocytes.
Ectopic expression of Nrf2 inhibits apoptotic signaling in OA chondrocytes under pathological conditions.
Expression of Nrf2 suppresses mitochondrial dysfunction in IL-1β-stimulated OA chondrocytes.
Nrf2 exerts chondroprotection through activation of ERK1/2/ELK1-P70S6K-P90RSK signaling axis.
Acknowledgments
This work was supported by National Institute of Health grants (RO1-AT-005520; RO1-AT-007373; RO1-AR- 067056; R21-AR- 064890) and funds from the Northeast Ohio Medical University to TMH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goldring MB. The role of the chondrocyte in osteoarthritis. Arthritis Rheum. 2000;43(9):1916–26. doi: 10.1002/1529-0131(200009)43:9<1916::AID-ANR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 2.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697–707. doi: 10.1002/art.34453. Epub 2012 Mar 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haseeb A, Haqqi TM. Immunopathogenesis of osteoarthritis. Clin Immunol. 2013;146(3):185–96. doi: 10.1016/j.clim.2012.12.011. Epub 2013 Jan 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maneesh M, Jayalekshmi H, Suma T, Chatterjee S, Chakrabarti A, Singh TA. Evidence for oxidative stress in osteoarthritis. Indian J Clin Biochem. 2005;20(1):129–30. doi: 10.1007/BF02893057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ziskoven C, Jager M, Zilkens C, Bloch W, Brixius K, Krauspe R. Oxidative stress in secondary osteoarthritis: from cartilage destruction to clinical presentation? Orthop Rev (Pavia) 2010;2(2):e23. doi: 10.4081/or.2010.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lepetsos P, Papavassiliou AG. ROS/oxidative stress signaling in osteoarthritis. Biochim Biophys Acta. 2016;1862(4):576–91. doi: 10.1016/j.bbadis.2016.01.003. Epub 2016 Jan 6. [DOI] [PubMed] [Google Scholar]
- 7.Hwang HS, Kim HA. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int J Mol Sci. 2015;16(11):26035–54. doi: 10.3390/ijms161125943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuerwegh AJ, Dombrecht EJ, Stevens WJ, Van Offel JF, Bridts CH, De Clerck LS. Influence of pro-inflammatory (IL-1 alpha, IL-6, TNF-alpha, IFN-gamma) and anti-inflammatory (IL-4) cytokines on chondrocyte function. Osteoarthritis Cartilage. 2003;11(9):681–7. doi: 10.1016/s1063-4584(03)00156-0. [DOI] [PubMed] [Google Scholar]
- 9.Thomas CM, Fuller CJ, Whittles CE, Sharif M. Chondrocyte death by apoptosis is associated with cartilage matrix degradation. Osteoarthritis Cartilage. 2007;15(1):27–34. doi: 10.1016/j.joca.2006.06.012. Epub 2006 Jul 21. [DOI] [PubMed] [Google Scholar]
- 10.Chan K, Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci U S A. 1999;96(22):12731–6. doi: 10.1073/pnas.96.22.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26(2):175–82. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- 12.Lee JM, Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol. 2004;37(2):139–43. doi: 10.5483/bmbrep.2004.37.2.139. [DOI] [PubMed] [Google Scholar]
- 13.Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10(8):1343–74. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kotlo KU, Yehiely F, Efimova E, Harasty H, Hesabi B, Shchors K, Einat P, Rozen A, Berent E, Deiss LP. Nrf2 is an inhibitor of the Fas pathway as identified by Achilles’ Heel Method, a new function-based approach to gene identification in human cells. Oncogene. 2003;22(6):797–806. doi: 10.1038/sj.onc.1206077. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Hung AC, Ng PY, Nakayama K, Hu Y, Li B, Porter AG, Dhakshinamoorthy S. PKCdelta mediates Nrf2-dependent protection of neuronal cells from NO-induced apoptosis. Biochem Biophys Res Commun. 2009;386(4):750–6. doi: 10.1016/j.bbrc.2009.06.129. Epub 2009 Jun 27. [DOI] [PubMed] [Google Scholar]
- 16.Danilov CA, Chandrasekaran K, Racz J, Soane L, Zielke C, Fiskum G. Sulforaphane protects astrocytes against oxidative stress and delayed death caused by oxygen and glucose deprivation. Glia. 2009;57(6):645–56. doi: 10.1002/glia.20793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, Tornatore C, Sweetser MT, Yang M, Sheikh SI, Dawson KT. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 18.Abusarah J, Benabdoune H, Shi Q, Lussier B, Martel-Pelletier J, Malo M, Fernandes JC, de Souza FP, Fahmi H, Benderdour M. Elucidating the Role of Protandim and 6-Gingerol in Protection Against Osteoarthritis. J Cell Biochem. 2017;118(5):1003–1013. doi: 10.1002/jcb.25659. Epub 2017 Jan 5. [DOI] [PubMed] [Google Scholar]
- 19.Guo YX, Liu L, Yan DZ, Guo JP. Plumbagin prevents osteoarthritis in human chondrocytes through Nrf-2 activation. Mol Med Rep. 2017;15(4):2333–2338. doi: 10.3892/mmr.2017.6234. Epub 2017 Feb 22. [DOI] [PubMed] [Google Scholar]
- 20.Hosseinzadeh A, Jafari D, Kamarul T, Bagheri A, Sharifi AM. Evaluating the Protective Effects and Mechanisms of Diallyl Disulfide on Interlukin-1beta-Induced Oxidative Stress and Mitochondrial Apoptotic Signaling Pathways in Cultured Chondrocytes. J Cell Biochem. 2017;118(7):1879–1888. doi: 10.1002/jcb.25907. Epub 2017 Feb 15. [DOI] [PubMed] [Google Scholar]
- 21.Hsu DZ, Chu PY, Jou IM. Enteral sesame oil therapeutically relieves disease severity in rat experimental osteoarthritis. Food Nutr Res. 2016;60:29807. doi: 10.3402/fnr.v60.29807. eCollection 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan NM, Haseeb A, Ansari MY, Devarapalli P, Haynie S, Haqqi TM. Wogonin, a plant derived small molecule, exerts potent anti-inflammatory and chondroprotective effects through the activation of ROS/ERK/Nrf2 signaling pathways in human Osteoarthritis chondrocytes. Free Radic Biol Med. 2017;106:288–301. doi: 10.1016/j.freeradbiomed.2017.02.041. Epub 2017 Feb 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong P, Chen G, Jiang A, Wang Y, Song C, Zhuang J, Xi C, Wang G, Ji Y, Yan J. Sesamin inhibits IL-1beta-stimulated inflammatory response in human osteoarthritis chondrocytes by activating Nrf2 signaling pathway. Oncotarget. 2016;7(50):83720–83726. doi: 10.18632/oncotarget.13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xue EX, Lin JP, Zhang Y, Sheng SR, Liu HX, Zhou YL, Xu H. Pterostilbene inhibits inflammation and ROS production in chondrocytes by activating Nrf2 pathway. Oncotarget. 2017;30(10):16716. doi: 10.18632/oncotarget.16716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cai D, Yin S, Yang J, Jiang Q, Cao W. Histone deacetylase inhibition activates Nrf2 and protects against osteoarthritis. Arthritis Res Ther. 2015;17:269. doi: 10.1186/s13075-015-0774-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mankin HJ. Biochemical and metabolic abnormalities in osteoarthritic human cartilage. Fed Proc. 1973;32(4):1478–80. [PubMed] [Google Scholar]
- 27.Khan NM, Ansari MY, Haqqi TM. Sucrose, But Not Glucose, Blocks IL1-beta-Induced Inflammatory Response in Human Chondrocytes by Inducing Autophagy via AKT/mTOR Pathway. J Cell Biochem. 2017;118(3):629–639. doi: 10.1002/jcb.25750. Epub 2016 Oct 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan NM, Haseeb A, Ansari MY, Haqqi TM. A wogonin-rich-fraction of Scutellaria baicalensis root extract exerts chondroprotective effects by suppressing IL-1beta-induced activation of AP-1 in human OA chondrocytes. Sci Rep. 2017;7:43789. doi: 10.1038/srep43789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25(1):162–71. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ansari MY, Khan NM, Ahmad I, Haqqi TM. Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthritis Cartilage. 2017;8(17):31125–1. doi: 10.1016/j.joca.2017.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makki MS, Haseeb A, Haqqi TM. MicroRNA-9 promotion of interleukin-6 expression by inhibiting monocyte chemoattractant protein-induced protein 1 expression in interleukin-1beta-stimulated human chondrocytes. Arthritis Rheumatol. 2015;67(8):2117–28. doi: 10.1002/art.39173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan NM, Poduval TB. Bilirubin augments radiation injury and leads to increased infection and mortality in mice: molecular mechanisms. Free Radic Biol Med. 2012;53(5):1152–69. doi: 10.1016/j.freeradbiomed.2012.07.007. Epub 2012 Jul 20. [DOI] [PubMed] [Google Scholar]
- 33.Khan NM, Ahmad I, Ansari MY, Haqqi TM. Wogonin, a natural flavonoid, intercalates with genomic DNA and exhibits protective effects in IL-1beta stimulated osteoarthritis chondrocytes. Chem Biol Interact. 2017;274:13–23. doi: 10.1016/j.cbi.2017.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan NM, Sandur SK, Checker R, Sharma D, Poduval TB, Sainis KB. Pro-oxidants ameliorate radiation-induced apoptosis through activation of the calcium-ERK1/2-Nrf2 pathway. Free Radic Biol Med. 2011;51(1):115–28. doi: 10.1016/j.freeradbiomed.2011.03.037. Epub 2011 Apr 8. [DOI] [PubMed] [Google Scholar]
- 35.Rousset F, Hazane-Puch F, Pinosa C, Nguyen MV, Grange L, Soldini A, Rubens-Duval B, Dupuy C, Morel F, Lardy B. IL-1beta mediates MMP secretion and IL-1beta neosynthesis via upregulation of p22(phox) and NOX4 activity in human articular chondrocytes. Osteoarthritis Cartilage. 2015;23(11):1972–80. doi: 10.1016/j.joca.2015.02.167. [DOI] [PubMed] [Google Scholar]
- 36.Wei L, Sun XJ, Wang Z, Chen Q. CD95-induced osteoarthritic chondrocyte apoptosis and necrosis: dependency on p38 mitogen-activated protein kinase. Arthritis Res Ther. 2006;8(2):R37. doi: 10.1186/ar1891. Epub 2006 Jan 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buytaert E, Dewaele M, Agostinis P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim Biophys Acta. 2007;1776(1):86–107. doi: 10.1016/j.bbcan.2007.07.001. Epub 2007 Jul 6. [DOI] [PubMed] [Google Scholar]
- 38.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11(9):621–32. doi: 10.1038/nrm2952. Epub 2010 Aug 4. [DOI] [PubMed] [Google Scholar]
- 39.Zhou PH, Liu SQ, Peng H. The effect of hyaluronic acid on IL-1beta-induced chondrocyte apoptosis in a rat model of osteoarthritis. J Orthop Res. 2008;26(12):1643–8. doi: 10.1002/jor.20683. [DOI] [PubMed] [Google Scholar]
- 40.Checker R, Gambhir L, Sharma D, Kumar M, Sandur SK. Plumbagin induces apoptosis in lymphoma cells via oxidative stress mediated glutathionylation and inhibition of mitogen-activated protein kinase phosphatases (MKP1/2) Cancer Lett. 2015;357(1):265–78. doi: 10.1016/j.canlet.2014.11.031. Epub 2014 Nov 18. [DOI] [PubMed] [Google Scholar]
- 41.Cuadrado A, Martin-Moldes Z, Ye J, Lastres-Becker I. Transcription factors NRF2 and NF-kappaB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J Biol Chem. 2014;289(22):15244–58. doi: 10.1074/jbc.M113.540633. Epub 2014 Apr 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ganesh Yerra V, Negi G, Sharma SS, Kumar A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-kappaB pathways in diabetic neuropathy. Redox Biol. 2013;1:394–7. doi: 10.1016/j.redox.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soares MP, Seldon MP, Gregoire IP, Vassilevskaia T, Berberat PO, Yu J, Tsui TY, Bach FH. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol. 2004;172(6):3553–63. doi: 10.4049/jimmunol.172.6.3553. [DOI] [PubMed] [Google Scholar]
- 44.Haseeb A, Khan NM, Ashruf OS, Haqqi TM. A Polyphenol-rich Pomegranate Fruit Extract Suppresses NF-kappaB and IL-6 Expression by Blocking the Activation of IKKbeta and NIK in Primary Human Chondrocytes. Phytother Res. 2017;31(5):778–782. doi: 10.1002/ptr.5799. Epub 2017 Mar 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khan NM, Haseeb A, Ansari MY, Devarapalli P, Haynie S, Haqqi TM. Dataset of effect of Wogonin, a natural flavonoid, on the viability and activation of NF-kappaB and MAPKs in IL-1beta-stimulated human OA chondrocytes. Data Brief. 2017;12:150–155. doi: 10.1016/j.dib.2017.03.054. eCollection 2017 Jun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chowdhury TT, Salter DM, Bader DL, Lee DA. Signal transduction pathways involving p38 MAPK, JNK, NFkappaB and AP-1 influences the response of chondrocytes cultured in agarose constructs to IL-1beta and dynamic compression. Inflamm Res. 2008;57(7):306–13. doi: 10.1007/s00011-007-7126-y. [DOI] [PubMed] [Google Scholar]
- 47.Shi X, Ye H, Yao X, Gao Y. The involvement and possible mechanism of NR4A1 in chondrocyte apoptosis during osteoarthritis. Am J Transl Res. 2017;9(2):746–754. eCollection 2017. [PMC free article] [PubMed] [Google Scholar]
- 48.Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, Kunsch C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Physiol Heart Circ Physiol. 2006;290(5):H1862–70. doi: 10.1152/ajpheart.00651.2005. Epub 2005 Dec 9. [DOI] [PubMed] [Google Scholar]
- 49.Ghatan S, Larner S, Kinoshita Y, Hetman M, Patel L, Xia Z, Youle RJ, Morrison RS. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J Cell Biol. 2000;150(2):335–47. doi: 10.1083/jcb.150.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim SJ, Ju JW, Oh CD, Yoon YM, Song WK, Kim JH, Yoo YJ, Bang OS, Kang SS, Chun JS. ERK-1/2 and p38 kinase oppositely regulate nitric oxide-induced apoptosis of chondrocytes in association with p53, caspase-3, and differentiation status. J Biol Chem. 2002;277(2):1332–9. doi: 10.1074/jbc.M107231200. Epub 2001 Oct 31. [DOI] [PubMed] [Google Scholar]
- 51.Notoya K, Jovanovic DV, Reboul P, Martel-Pelletier J, Mineau F, Pelletier JP. The induction of cell death in human osteoarthritis chondrocytes by nitric oxide is related to the production of prostaglandin E2 via the induction of cyclooxygenase-2. J Immunol. 2000;165(6):3402–10. doi: 10.4049/jimmunol.165.6.3402. [DOI] [PubMed] [Google Scholar]
- 52.Li X, Feng K, Li J, Yu D, Fan Q, Tang T, Yao X, Wang X. Curcumin Inhibits Apoptosis of Chondrocytes through Activation ERK1/2 Signaling Pathways Induced Autophagy. Nutrients. 2017;9(4):E414. doi: 10.3390/nu9040414. (pii) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shakibaei M, Schulze-Tanzil G, de Souza P, John T, Rahmanzadeh M, Rahmanzadeh R, Merker HJ. Inhibition of mitogen-activated protein kinase kinase induces apoptosis of human chondrocytes. J Biol Chem. 2001;276(16):13289–94. doi: 10.1074/jbc.M010859200. Epub 2001 Jan 16. [DOI] [PubMed] [Google Scholar]
- 54.Lu Z, Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. 2006;58(11):621–31. doi: 10.1080/15216540600957438. [DOI] [PubMed] [Google Scholar]
- 55.Biswas SC, Greene LA. Nerve growth factor (NGF) down-regulates the Bcl-2 homology 3 (BH3) domain-only protein Bim and suppresses its proapoptotic activity by phosphorylation. J Biol Chem. 2002;277(51):49511–6. doi: 10.1074/jbc.M208086200. Epub 2002 Oct 17. [DOI] [PubMed] [Google Scholar]
- 56.Sturgill TW, Ray LB, Erikson E, Maller JL. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature. 1988;334(6184):715–8. doi: 10.1038/334715a0. [DOI] [PubMed] [Google Scholar]
- 57.Ho E, Karimi Galougahi K, Liu CC, Bhindi R, Figtree GA. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol. 2013;1:483–91. doi: 10.1016/j.redox.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mueller CF, Laude K, McNally JS, Harrison DG. ATVB in focus: redox mechanisms in blood vessels. Arterioscler Thromb Vasc Biol. 2005;25(2):274–8. doi: 10.1161/01.ATV.0000149143.04821.eb. Epub 2004 Oct 28. [DOI] [PubMed] [Google Scholar]
- 59.Yui Naoko KY, Fujiya Hiroto, Musha Haruki. Mechanical and oxidative stress in osteoarthritis. J Phys Fitness Sports Med. 2016;5(1):81–86. [Google Scholar]
- 60.Wruck CJ, Fragoulis A, Gurzynski A, Brandenburg LO, Kan YW, Chan K, Hassenpflug J, Freitag-Wolf S, Varoga D, Lippross S, Pufe T. Role of oxidative stress in rheumatoid arthritis: insights from the Nrf2-knockout mice. Ann Rheum Dis. 2011;70(5):844–50. doi: 10.1136/ard.2010.132720. Epub 2010 Dec 20. [DOI] [PubMed] [Google Scholar]
- 61.Takada T, Miyaki S, Ishitobi H, Hirai Y, Nakasa T, Igarashi K, Lotz MK, Ochi M. Bach1 deficiency reduces severity of osteoarthritis through upregulation of heme oxygenase-1. Arthritis Res Ther. 2015;17:285. doi: 10.1186/s13075-015-0792-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang L, Gai P, Xu R, Zheng Y, Lv S, Li Y, Liu S. Shikonin protects chondrocytes from interleukin-1beta-induced apoptosis by regulating PI3K/Akt signaling pathway. Int J Clin Exp Pathol. 2015;8(1):298–308. eCollection 2015. [PMC free article] [PubMed] [Google Scholar]
- 63.Xue H, Tu Y, Ma T, Liu X, Wen T, Cai M, Xia Z, Mei J. Lactoferrin Inhibits IL-1beta-Induced Chondrocyte Apoptosis Through AKT1-Induced CREB1 Activation. Cell Physiol Biochem. 2015;36(6):2456–65. doi: 10.1159/000430206. Epub 2015 Jul 27. [DOI] [PubMed] [Google Scholar]
- 64.Blanco FJ, Lopez-Armada MJ, Maneiro E. Mitochondrial dysfunction in osteoarthritis. Mitochondrion. 2004;4(5–6):715–28. doi: 10.1016/j.mito.2004.07.022. Epub 2004 Oct 1. [DOI] [PubMed] [Google Scholar]
- 65.Bonay M, Deramaudt TB. Nrf2: new insight in cell apoptosis. Cell Death Dis. 2015;6:e1897. doi: 10.1038/cddis.2015.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dong J, Yan D, Chen SY. Stabilization of Nrf2 protein by D3T provides protection against ethanol-induced apoptosis in PC12 cells. PLoS One. 2011;6(2):e16845. doi: 10.1371/journal.pone.0016845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu Y, Yang H, Wen Y, Li B, Zhao Y, Xing J, Zhang M, Chen Y. Nrf2 Inhibits Periodontal Ligament Stem Cell Apoptosis under Excessive Oxidative Stress. Int J Mol Sci. 2017;18(5):E1076. doi: 10.3390/ijms18051076. (pii) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi X, Li Y, Hu J, Yu B. Tert-butylhydroquinone attenuates the ethanol-induced apoptosis of and activates the Nrf2 antioxidant defense pathway in H9c2 cardiomyocytes. Int J Mol Med. 2016;38(1):123–30. doi: 10.3892/ijmm.2016.2605. Epub 2016 May 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Niture SK, Jaiswal AK. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J Biol Chem. 2012;287(13):9873–86. doi: 10.1074/jbc.M111.312694. Epub 2012 Jan 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88(Pt B):179–88. doi: 10.1016/j.freeradbiomed.2015.04.036. Epub 2015 May 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meng XB, Sun GB, Wang M, Sun J, Qin M, Sun XB. P90RSK and Nrf2 Activation via MEK1/2-ERK1/2 Pathways Mediated by Notoginsenoside R2 to Prevent 6-Hydroxydopamine-Induced Apoptotic Death in SH-SY5Y Cells. Evid Based Complement Alternat Med. 2013;2013:971712. doi: 10.1155/2013/971712. Epub 2013 Sep 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Chondrocytes phenotype was maintained in monolayer culture: Human OA chondrocytes were cultured in monolayer and total RNA was isolated and mRNA expression of COL2A1, ACAN, COL10A1 and COL1A1 were quantified using TaqMan assay. Gene expression was determined by the ΔCt method and results were normalized to β-actin expression.
Supplementary Figure 2. Treatment of OA chondrocytes with IL-1β activated Nrf2/ARE signaling: Human OA chondrocytes were stimulated with (A) IL-1β (1 ng/ml) for indicated time or (B–C) IL-1β (0–10 ng/ml) for 16 hours. At the end of treatment, chondrocytes were harvested and RNA was isolated for real time PCR analysis or cell lysates were prepared using RIPA buffer for immunoblot analysis. (A, B) Expression of Nrf2 and its downstream signaling targets HO-1, NQO1 and SOD2 was measured by quantitative PCR using the SYBR® green assay (Life Technologies). β-actin was used as endogenous expression control. Bar graph represents mean±SD from two subjects. *p≤0.05, as compared to control, # ≤0.05, as compared to IL-1β. (C) Protein expression of Nrf2, HO-1, NQO1 and SOD2 was investigated by immunoblotting using antibodies against indicated protein. β-actin was used as a control for equal loading. Immunoblot results are representatives of two blots performed on samples obtained from two individuals.
Supplementary Figure 3. Transfection efficacy of Nrf2 knockdown and over-expression in human OA chondrocytes: Human OA chondrocytes were transfected with siRNA specific for Nrf2 (siNrf2, 100 nM) or negative controls siRNA (siCNT, 100 nM) using (A, B) X-tremeGENE™ siRNA transfection reagent or (C, D) transfected with Nrf2 expression plasmid (pNrf2, 1μg) or pcDNA3.1 as control plasmid (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit on 4D on Amaxa Nucleofection System. Forty-eight hours after the transfection, RNA was isolated for real time PCR analysis or cell lysates were prepared using RIPA buffer for immunoblot analysis. Expression of Nrf2 and its downstream signaling targets HO-1, NQO1 and SOD2 was measured by (A, C) quantitative PCR using SYBR® green assay (Life Technologies) and (B, D) immunoblotting using antibodies against indicated protein. β-actin was used as endogenous expression control.
Supplementary Figure 4. Nrf2 over-expression inhibited apoptosis in OA chondrocytes under pathological conditions: Human OA chondrocytes were transfected with (A) siRNA specific for Nrf2 (siNrf2, 100 nM) or negative controls siRNA (siCNT, 100 nM) or (B) Nrf2 expression plasmid (pNrf2, 1μg) or pcDNA3.1 as control plasmid (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit on 4D on Amaxa Nucleofection System. Forty-eight hours after the transfection, OA chondrocytes were serum starved and then stimulated with or without IL-1β (10 ng/ml) for 48 h. (A) Cell viability was measured by MTT assays using method described in method sections. Unstimulated chondrocytes were served as control and viability was expressed relative to control cells. Data point was expressed as a mean ± SD of two replicates from a representative experiment and three such independent experiments were carried out. *p≤0.05, as compared to siCNT, # ≤0.05, as compared to siCNT+IL-1β. (B) Apoptosis was estimated by TUNEL assay as described in method section. Bar chart corresponding to Flow cytometric histograms showed percent TUNEL positive chondrocytes. Percent apoptosis was calculated using flow Jo software and expressed as a mean±SD of three replicates from a representative experiment and three such independent experiments were carried out. *p≤0.05, as compared to pcDNA, # ≤0.05, as compared to pcDNA+IL-1β.
Supplementary Figure 5. Nrf2 over-expression inhibited intrinsic apoptotic pathways in OA chondrocytes under pathological conditions: Human OA chondrocytes were transfected with Nrf2 expression plasmid (pNrf2, 1μg) or pcDNA3.1 as control plasmid (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit on 4D on Amaxa Nucleofection System. (A) Forty-eight hours after the transfection, OA chondrocytes were stained with MistoSOX red (5 μM) and then stimulated with IL-1β (1 ng/ml, 30 minutes) and the production of mitochondrial O2− was estimated by measuring the fluorescence emission at 580 nm following the excitation at 510 nm. Bar graph shows mean±SD of four replicates from a representative experiment and three such independent experiments were carried out. (B) Fold change relative to β-actin for the expression cytosolic cytochrome-c was quantified by measuring specific signal intensities using ImageJ software. *p≤0.05, as compared to pcDNA, # ≤0.05, as compared to pcDNA+IL-1β. (C) Forty-eight hours after the transfection, OA chondrocytes were serum starved and then stimulated with or without IL-1β (10 ng/ml) for 48 h and protein expression in the cell lysates prepared above was investigated by immunoblotting using antibodies against Bax, Bad, Bcl2, Bcl-xl and Mcl-1. β-actin was used as a control for equal loading. The β-actin in Figure 6A and supplementary Figure 5C are from the same experiment. Immunoblot results are representatives of three blots performed on samples obtained from three individuals with or without for 48 h.
Supplementary Figure 6. Nrf2 over-expression did not inhibit the activation of NF-κB, p38 MAPK and JNK in human OA chondrocytes under pathological conditions: Human OA chondrocytes were transfected with Nrf2 expression plasmid (pNrf2, 1μg) or pcDNA3.1 as control plasmid (1μg) using P3 Primary Cell 4D-Nucleofector™ X Kit on 4D on Amaxa Nucleofection System. Forty-eight hours after the transfection, OA chondrocytes were serum starved and then stimulated with or without IL-1β (10 ng/ml) for 15 and 30 minutes at 37°C. At end of treatment, cell lysates were prepared using RIPA buffer and immunoblotting was performed to detect the protein levels of (A) Iκβα and p-Iκβα (Ser 32) (B) p-P38 (Thr180/Tyr182), total p38, p-JNK (Thr183/Tyr185) and total JNK. β-actin was used as a control for equal loading. Immunoblot results are representatives of three blots performed on samples obtained from three individuals. (C) Fold change for the expression of p-ERK relative to total ERK was quantified by measuring specific signal intensities using ImageJ software. *p≤0.05, as compared to pcDNA.