

Abstract
Sickle cell anemia (SCA) is an autosomal recessive disorder caused by mutation in the β-globin gene. Pulmonary hypertension (PH), a complication of SCA, results in severe morbidity and mortality. PH is a multifactorial disease: systemic vasculopathy, pulmonary vasoconstriction, and endothelial dysfunction and remodeling. Placenta growth factor (PlGF), an angiogenic growth factor, elaborated from erythroid cells, has been shown to contribute to inflammation, pulmonary vasoconstriction and airway hyper-responsiveness (AH) in mouse models of sickle cell disease. In this review, we summarize the cell-signaling mechanism(s) by which PlGF regulates the expression of genes involved in inflammation, PH and AH in cell culture and corroborate these findings in mouse models of SCA and in individuals with SCA. The role of microRNAs (miRNAs) in the post-transcriptional regulation of these genes is presented and how these miRNAs located in their host genes are transcriptionally regulated. An understanding of the transcriptional regulation of these miRNAs provides a new therapeutic approach to ameliorate the clinical manifestations of SCA.
Keywords: Sickle cell anemia, Pulmonary hypertension, Placenta growth factor, Hypoxia-inducible factor, microRNAs, transcriptional and post-transcriptional regulation
1. Sickle Cell Anemia
Sickle cell anemia (SCA) and its variants are genetic disorders of the hemoglobin molecule. These disorders include sickle cell disease (HbSS), sickle β-thalassemia syndromes (HbS-thal) and hemoglobin S associated with other hemoglobinopathies. The hemoglobin S (HbS) molecule contains a mutation in the β-globin chain, wherein the glutamic acid residue at the 6th position is replaced by a non-polar valine residue. At low oxygen tensions polymerization of HbS occurs forming long fibers, resulting in the sickled morphology of RBCs and contributing to reduced deformability of sickle RBC (SS RBC). These features of SS RBCs result in shortened red cell survival, culminating in chronic hemolytic anemia and vaso-occlusion (1–3). Vaso-occlusion of the microvasculature occurs by trapping of the less deformable SS RBCs, adhesion of SS RBCs to the vascular endothelium, activation and adhesion of neutrophils to endothelium, leukocytosis, inflammation and thrombosis (4–9). The clinical manifestations of SCA include chronic anemia, episodic vaso-occlusive painful events, acute chest syndrome, splenic sequestration, chronic leg ulcers, stroke, pulmonary hypertension and asthma/reactive airway disease (3, 6, 7, 10). Recurrent vaso-occlusions cause ischemia/reperfusion injury that ultimately results in tissue/organ damage and the chronic morbidity and foreshortened life-span associated with SCA (3). In fact, cardiopulmonary complications are the most common cause of adult mortality in SCA.
1.1 Pulmonary hypertension in SCA
Pulmonary hypertension (PH), a complication of SCA, results in severe morbidity and mortality (11). The pathogenesis of PH is likely multi-factorial: including hemolysis-mediated impaired nitric oxide bioavailability, chronic thromboembolic disease from a pro-coagulant state, systemic vasculopathy from hypoxia and inflammation, pulmonary vasoconstriction, and endothelial dysfunction and remodeling (8, 10, 12, 13).
Nitric oxide (NO) and endothelin-1 are opposing vasoactive factors that regulate the pulmonary vascular tone (14, 15). During the last decade a number of studies, both experimental and clinical, have focused on the depletion of nitric oxide by elevated levels of hemoglobin in plasma in the clinical manifestations of PH in SCASCA (10, 12, 16–19). However, the role of NO in the pathophysiology of PH in SCA has been challenged (13). Studies show plasma levels of endothelin-1 are significantly higher in SCA patients(20, 21), and elevated levels of ET-1 are associated with endothelial dysfunction(22). Our studies show high levels of placenta growth factor (PlGF) in plasma of SCA patients is associated with elevated plasma ET-1 levels, and elevated pulmonary artery pressure, reflective of pulmonary hypertension (23). In this article, we will address how placenta growth factor (PlGF) regulates transcription of ET-1 and the role of microRNAs (miRNAs) in regulating expression of ET-1. Recent comprehensive reviews summarize the biosynthesis of endothelin, the structure and function of endothelin receptors, genetic endothelin knockout mouse models, and the role of endothelin in human pharmacology (24).
1.2 Asthma and Reactive Airway disease in SCA
Asthma is a disease of the airways characterized by airway obstruction, airway hyper-responsiveness/reactivity, and inflammation leading to intermittent respiratory symptoms (25). Leukotrienes (LT) are lipid mediators that play an important role in the pathophysiology of asthma, allergic inflammation and innate immunity (26). Since inflammation has an important role in the pathogenesis of vaso-occlusion and asthma augments the pro-inflammatory state, a vicious cycle of inflammation, asthma and vaso-occlusion develops in SCA (27). Asthma among children and adults with SCA is associated with increased incidence of SCA-related morbidity and mortality (27, 28). Studies show elevated levels of leukotrienes in plasma in SCA individuals, which correlates with increased vaso-occlusion and acute chest syndrome (ACS) (29, 30).
The biosynthesis of leukotrienes predominantly occurs in leukocytes, and the first step of the process is activation of membrane phospholipids by phospholipase A2 to yield arachidonic acid (AA; C20-fatty acid). Next, AA is converted to leukotriene A4 (LTA4) by 5-lipoxygenase (5-LO) in concert with 5-lipoxygenase activating protein (FLAP) (31, 32). LTA4 is converted either to leukotriene B4 (LTB4) by LTA4 hydrolase or conjugated with reduced glutathione by leukotriene C4 (LTC4) synthase to yield LTC4 (33). LTC4 is subsequently converted to LTD4 and then to LTE4; these three LTs are collectively referred to as cysteinyl leukotrienes (cyst LTs). Leukotriene B4 is amongst the most potent inflammatory and chemotactic molecule for neutrophils, while Cyst LTs promote airway hyper-responsiveness (AHR). We show placenta growth factor induces the synthesis of leukotrienes in vitro and in mouse models of SCA, and correlates with airway hyper-responsiveness in SCA (34, 35). We will address how enzymes involved in LT synthesis are transcriptionally and post-transcriptionally regulated in this article. The biochemical and clinical aspects of leukotrienes are reviewed elsewhere (36–38).
1.3 Placenta Growth Factor in the pathophysiology of SCA
Placenta growth factor (PlGF), an angiogenic growth factor, belonging to the vascular endothelial growth factor (VEGF) family, was originally discovered in 1991 (39). The human PlGF gene (PGF) encodes four isoforms PlGF 1–4, which are generated by alternative mRNA splicing (40). PlGF was originally identified in the placenta, where it regulates growth and differentiation of trophoblasts. It is also expressed in umbilical vein endothelial cells and other non-placental tissues, like the thyroid gland and developing lung tissue (41, 42). Tordjman et al. discovered erythroid cells produce angiogenic growth factors, including PlGF (43). Our studies show increased plasma levels of PlGF in SCA individuals is a consequence of increased erythropoiesis, resulting from hemolytic anemia seen in these patients (44). The expression of PlGF is enhanced by erythropoietin (Epo) in CD34+ progenitor cells of bone marrow (Figure 1) (44). PlGF binds exclusively to VEGFR-1 receptor, while VEGF binds to both VEGFR-1 and VEGFR-2 receptors. PlGF has pleiotropic effects on different cell types and regulates various biological activities, e.g. cell proliferation and migration (41, 45, 46). Studies utilizing PlGF gain of function and loss of function approaches in mouse models, revealed the roles of PlGF in specific tissues and cells. These effects contribute to disease pathophysiology as described in the review by Dewerchin and Carmeliet (41). For example, PlGF-deficient mice show reduced angiogenesis and inflammation of the infarcted myocardium (47) and delivery of either PlGF gene or protein in infarcted mice led to cardiac improvement (48). Our studies show overexpression of PlGF in erythroid cells via lentiviral vector delivery in C57 mice (normal mice) to the PlGF levels seen in Berkeley Sickle mouse (BK-SS) resulted in augmented ET-1 plasma levels. This was manifested by increased right ventricular hypertrophy and right ventricular pressures, both features of pulmonary hypertension (23). Furthermore, overexpression of PlGF in normal mice induces PAI-1 levels and promoted a prothrombic state (49). Thus unregulated PlGF expression is conducive to several pathophysiologic changes associated with SCA. A brief review summarizes the role of PlGF in the pathobiology of blood diseases, including hemoglobinopathies and hematologic malignancies (50).
Figure 1. PlGF-mediated activation of HiF-1α and downstream activation of target genes in monocytes and endothelial cells.
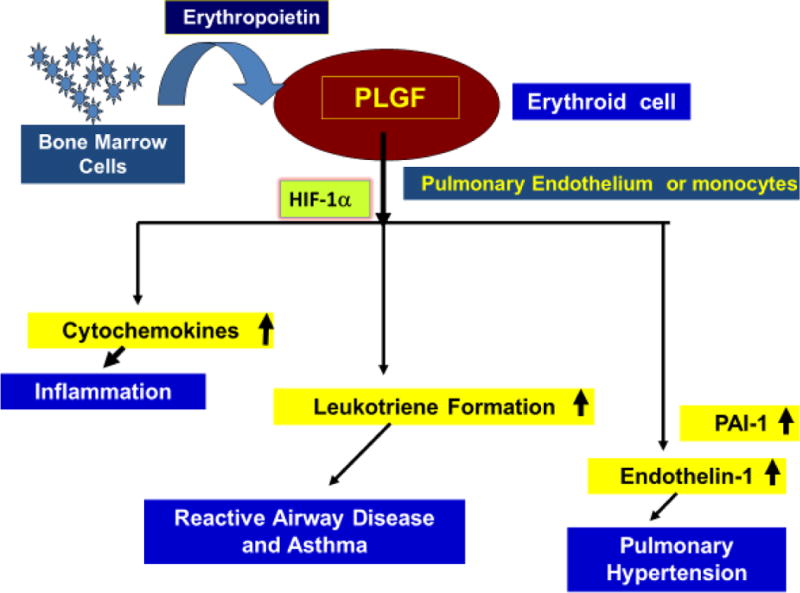
Due to increased erythropoiesis in SCD as a compensatory response to severe hemolytic anemia, Erythropoietin (Epo) is released at high levels which targets erythroid cells to secrete PlGF. Released PlGF in circulation can target blood cells, e.g. MNCs and vascular endothelium (endothelial cells). PlGF causes activation of HIF-1α, independent of hypoxia and stimulates expression of cytochemokines, leukotriene formation via activation of lipoxygenase enzymes, and expression of ET-1 and PAI-1, the latter is involved in pulmonary hypertension in SCD.
2. PlGF-mediated transcription of cytochemokines and inflammation in sickle cell disease
Vascular occlusion leads to recurrent episodes of painful crises and damage to the various end organs and is the major cause of morbidity and mortality in SCA (4, 51, 52). The classical studies of Hebbel and his coworkers (53) show that the extent of endothelial cell adherence by SS RBCs to cultured endothelial cells appears to be comparable to the clinical severity of vaso-occlusive events in SCA. However, SCA individuals with an identical alteration in the β-globin genes show distinct variability in the severity and frequency of vaso-occlusive episodes, suggesting that factors other than sickle RBCs may be playing a role in the pathophysiology of vaso-occlusion in SCA.
A salient feature of SCA is leukocytosis, which occurs even in the absence of infection or inflammation. The activation of monocytes, neutrophils, endothelial cells and coagulation cascade observed in SCA at steady state is presumed to be secondary to the sickling phenomena (4, 54–56). Although activated leukocytes can promote vaso-occlusion (57), the mechanistic activation of monocytes and neutrophils, and leukocytosis remained enigmatic. Studies by Kalra and Malik, and their coworkers (44, 58) show mononuclear cells (MNCs) from SCA patients at steady state are highly activated as these MNCs exhibit higher mRNA levels of cytochemokines (IL-1β, IL-8, MIP-1β, MCP-1 and VEGF) compared to MNCs isolated from healthy normal individuals. The MNCs of normal individuals show an absence or trace mRNA levels of these same cytochemokines (44).
Since SCA patients have higher circulating levels of PlGF, it has been hypothesized that PlGF may be responsible for activation of MNCs (44). Studies of MNCs isolated from normal healthy individuals (AA) upon treatment with PlGF show significantly increased mRNA levels of pro-inflammatory cytochemokines, as observed in MNCs of SCA individuals (Figure 1)(44). Peripheral blood monocytes isolated from SCA patients are in an activated state for inflammation as exemplified by increased mRNA levels of cytochemokines (TNF-α, IL-1β, MIP-1b, MCP-1, IL-8), compared to monocytes isolated from healthy individuals (58). The activation of monocytes by PlGF is mediated by VEGFR-1 and occurs via activation of PI-3 kinase/AKT and ERK-1/2 pathways (58). These studies show mononuclear cells/monocytes from SCA patients are activated for expression of cytochemokines as these cells are in the milieu of circulating PlGF, thus contributing to the inflammatory state observed in SCA patients. These cytochemokines released by monocytes have a potential to augment expression of adhesion molecules (VCAM-1, ICAM-1, P-selectin and E-selectin) in the vascular endothelium. These adhesion molecules are involved in adhesion of sickle RBCs and monocytes via corresponding surface ligands which lead to vaso-occlusion in small capillaries and ischemia in SCA.
2.2 PlGF-mediated transcription of ET-1, and post-transcriptional regulation of ET-1 by miRNAs
Since erythropoiesis is expanded in SCA individuals, resulting in elevated plasma PlGF (44), it has been hypothesized that PlGF may be a key mediator in activation of endothelial cells to augment expression of ET-1, a potent vasoconstrictor. Studies show PlGF increases mRNA and protein expression of ET-1 in cultured human pulmonary microvascular endothelial cells (HPMVEC) via activation of PI-3 Kinase and NADPH-oxidase, the latter generates reactive oxygen species (ROS) (59). Other studies show ET-1 expression is induced by hypoxia via activation of hypoxia inducible factor-1α (HIF-1α) (60, 61). By contrast the PlGF-mediated expression of ET-1 in HPMVEC involves activation of HIF-1α, independent of hypoxia (Figure 1) (59). Similarly in monocytes, PlGF increases mRNA and protein expression of endothelin-B receptor (ET-BR) via activation of HIF-1α, independent of hypoxia (59).
PlGF-mediated signaling was observed to increase expression of HIF-1α mRNA and protein. This hypoxia-independent induction of HIF-1α by PlGF is a distinguishing feature of SCA. ET-1 gene transcription results from HIF-1α binding to hypoxia response elements (HRE) in the promoter of ET-1 (Figure 1). This was demonstrated experimentally in promoter reporter assays by mutation of the HREs which abrogated PlGF induction of the reporter gene and was further validated by chromatin immunoprecipitation (ChIP) analysis (59).
When PlGF-treated monocytic cells are treated with ET-1, these cells show an augmentation of MCP-1 and IL-8 mRNA expression, indicating that PlGF-primed cells are more responsive to ET-1 with respect to cytochemokines expression (58). This leads to further inflammation and likely exacerbates vaso-occlusion. Thus, intrinsically raised PlGF levels due to increased turnover of red cells in SCA and in other chronic hemolytic anemias (e.g., thalassemia) may contribute to pulmonary hypertension (PH) and inflammation seen in these diseases.
To determine whether elevated PlGF in SCA contributes to PH, mouse models of SCA, viz. Berkeley Sickle mice (BKSS) and the knock in sickle mice (SS mice) were utilized. BKSS mice carry two copies of a transgene carrying the human α –and sickle β-globin genes (SS) and are knockouts for the endogenous α and β-globin genes. In the second model, knock-in sickle (SS) and normal (AA) mice were created where the human α and wt or sickle β-globin loci replace the corresponding murine loci by homologous recombination. These mice were developed by Dr. Timothy Townes (University of Alabama, Birmingham, AL) (62). The BKSS mice have significantly elevated levels of PlGF and endothelin-1 and develop right ventricular hypertrophy and dilation (23), as previously observed for right ventricular hypertrophy and increased right ventricular pressures associated with high pulmonary artery pressure in BKSS mice (14). Since anemia, hypoxia and hemolysis are confounding factors in development of PH in SCA, we used a novel strategy to show the role of PlGF in PH in these genetic mouse models (23). We overexpressed PlGF in normal mice to levels seen in BKSS mice with a lentiviral vector expressing PlGF in erythroid cells; this led to abnormally elevated ET-1 levels. These mice exhibited signs of PH, as seen in BKSS mice (23). These results were corroborated in human studies, wherein 123 patients with SCA were examined for plasma levels of PlGF and ET-1. Patients were stratified into three groups based on peak pulmonary artery pressure, defined as tricuspid regurgitant jet velocity (TRV). TRV < 2.5 m/sec was defined as normal, TRV of 2.5–2.9 m/s as mildly elevated (mild PH) and TRV> 3 m/s as severe PH. SCA patients with the highest TRV have the highest PlGF and ET-1 levels, and correlate with severity of PH (23). PlGF levels also correlated with markers of hemolysis, bilirubin, RBC count, hemoglobin and LDH (63). Furthermore, other work shows hemin upregulates the expression of PlGF in erythroid cells (64). These studies show markers of hemolysis, erythropoiesis and PlGF release are intrinsically linked via common pathways, which contribute to PH and inflammation in SCA. Thus, both in vitro and in vivo studies show PlGF induces the expression of ET-1, which may play a significant role in development of PH in SCA.
3. miRNAs in post-transcriptional regulation of ET-1
miRNAs are endogenous small (20–25 nt) noncoding RNAs that bind to the 3′-UTR (untranslated region) of target mRNA sequences and inhibit the expression of target genes by repressing translation and/or stability of target mRNA (65). The role of miRNAs as biomarkers and regulators of gene expression and their role in physiological and pathological processes have been addressed in several reviews (66–70). In the context of SCA, studies show PlGF attenuates levels of miR-648, with seed sequence complementary to the 3′-UTR of ET-1, in cultured endothelial cells (71). To examine the transcription of miR-648, a bioinformatics analysis of the gene encompassing miR-648 was examined in UCSC browser. miR-648 is located in a 5′-proximal intron of MICAL3 gene (a member of the MICAL family of flavoprotein monoxygenases) (72). Studies show both MICAL3 and pre-miR648 are co-transcribed, and PAX5 transcription factor is involved in the transcription of MICAL3 (71). Upon synthesis of MICAL3 pre-mRNA, excision of the intron containing pre-miR648 occurs and concomitantly leads to maturation of miRNA as miR-648. It is pertinent to note human miR-648 does not have a corresponding ortholog in the mouse. Thus, this has precluded study in animal models of SCA, e.g. BKSS mice. Since miR-648 post-transcriptionally modulated ET-1 expression, the plasma levels of miR-648 were examined in SCA patients and healthy matched controls. These data show miR-648 levels are ~4-times lower in SCA patients compared to unaffected controls (71). These studies in vivo support the inverse relationship between miR-648 and ET-1 observed in cultured endothelial cells in response to PlGF (71).
3.1 miRNAs in PlGF-mediated expression of HIF-1α
Since PlGF upregulates hypoxia independent, mRNA and protein expression of HIF-1α, the stability of HIF-1α was examined. PlGF treatment of cultured endothelial cells shows significantly reduced levels of miR-199a-5-p, and the predicted miRNA recognition element (MRE) for miR-199-5p is located at +16 to +38 nts of the 3′-UTR of HIF-1α mRNA as depicted in the schematic (73) (Figure. 2). Studies utilizing luciferase reporter for HIF-1α-3′UTR show exogenous miR-199a-5p mimic reduces luciferase activity, while anti-miR-199a-5p antagonized endogenous miR-199a-5p and concomitantly resulted in an increase in luciferase activity, in the absence or presence of PlGF. Mutation of the MRE site in the 3′UTR-HIF-1α reporter rendered the reporter unresponsive to change in luciferase activity upon co-transfection with either miR-199a-5p or anti-miR-199a-5p mimic. This demonstrated that miR-199a-5p binds to the predicted MRE site in the 3′UTR of HIF-1α mRNA leading to a reduction of HIF-1α expression (73). Moreover, transfection with miR-199a-5p mimic reduced basal HIF-1α mRNA and conversely anti-miR-199a-5p antagonized endogenous miR-199a, as reflected by increased HIF-1α mRNA levels. Similar responses were seen for PlGF induced HIF-1α mRNA expression in response to miR-199a-5p mimic and anti-miR-199a-5p (73). These results convincingly showed miR-199a-5p was involved in modulating HIF-1α biosynthesis.
Figure 2. Schematic representation of microRNA (miR) targets in the 3′untranslated (UTR) region of HIF-1α, ET-1, PAI-1 and PlGF mRNAs.
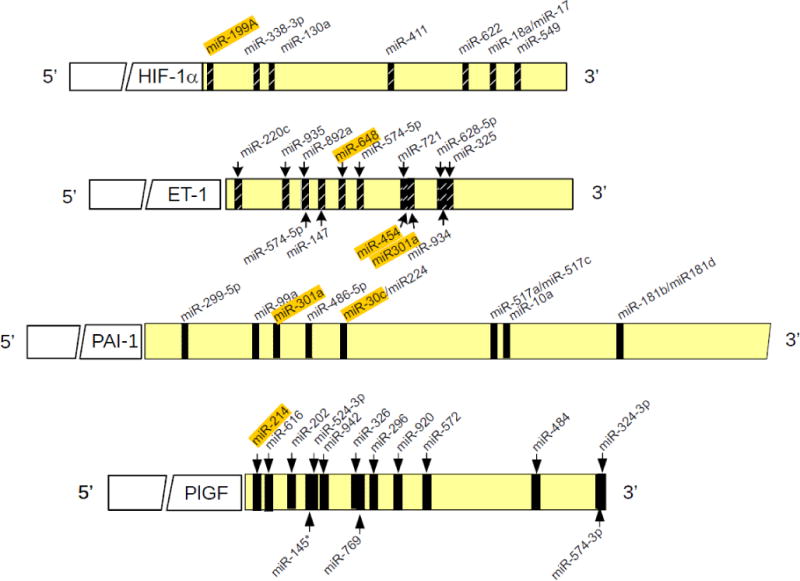
miRs highlighted in color have been characterized in response to PlGF and Epo treatment of cells.
3.2 Origin of miR-199a-5p and miR-199a2/miR-214 cluster in the DNM3 opposite strand (DNM3os) transcription unit
Genome annotation established that the murine miR-199a2/miR-214 cluster is expressed from the reverse strand of Dynamin 3 (DNM3); thus this locus is referred to as Dynamin 3 opposite strand (DNM3os) (74). The mouse DNM3os gene produces a ~6kb primary transcript that includes pre-miR-199a2 and pre-miR-214 (74). The orthologous human DNM3os gene also produces a transcript that includes pre-miR-199a2 and pre-miR-214, as depicted in the schematic of Figure 3 (73). The PlGF regulation of miR-199a-5p raised the question as to the origin of this miRNA since an identical miR-199a is expressed from the miR-199a1 gene locus in DNM2os (Figure 3) (73). It should be noted that the DNM family is comprised of three genes: DNM3, DNM2 and DNM1. Each has an intronic opposite strand locus that encodes a miR-199 homolog. DNM1 encodes miR-199b which is similar but no identical in nucleotide sequence to miR-199a1 and miR-199a2; and has a different seed sequence (73). For these reasons the expression of pre-miR-199b, pre-miR-199a1 and pre-miR-199a2 was analyzed in cultured endothelial cells, in response to PlGF (Figure 3). The data show PlGF reduced pre-miR-199a2 mRNA expression with no change in pre-miR-199a1 mRNA expression, while expression of pre-miR-199b expression increased (73). These results show PlGF selectively attenuates pre-miR-199a2 expression, which accounts for the observed changes in miR-199a-5p(73).
Figure 3. Origin and processing of miR-199-5-p (miR-199a2)/miR-214 located in the host gene DNM3os.
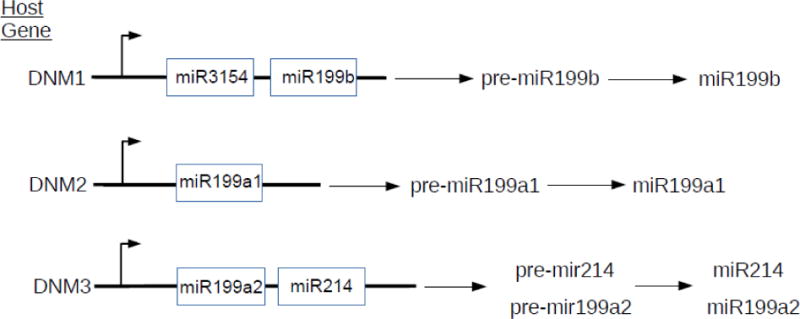
miR-199b, miR-199a1 and miR-199a2 are located in the introns of host genes DNM1, DNM2 and DNM3, respectively. These miRNAs are transcribed from the opposite strand of their respective host gene loci. Both miR-199a1 and miR-199a2 are identical in sequence and recognize the same mRNA target sites. The miRNA encoded by miR-199b is similar in nucleotide sequence to those encoded by miR-199a1 and miR-199a2, but has a different seed sequence. Upon PlGF treatment only expression of pre-miR-199a2 was reduced without affecting pre-miR-199a1 expression, the former major precursor of miR-199a2 is in the host gene DNM3os. Adapted and modified(73).
Next, studies were conducted to determine the co-regulation of pre-miR-199a2-5-p (referred to as miR-199a2) and pre-miR-214 located in DNM3os, and the DNM3os gene by PlGF (73). PlGF treatment of HMEC cells showed significant reduction in DNM3os RNA with a concomitant decrease in pre-miR-199a2 and pre-miR-214 expression, indicating those pre-miR-199a2/pre-miR214 clusters are in the same transcription unit of DNM3os. Furthermore, hypoxia-mediated signaling affected DNM3os RNA expression in a manner similar to PlGF-mediated signaling, namely hypoxia attenuated transcription of DNM3os, pre-miR-199a2 and pre-miR-214 in a coordinated manner(73). These studies show pre-miRs or mature miRs located in the host gene transcription unit are co-regulated.
3.3. DNM3os transcription is regulated via peroxisome proliferator-activated receptor-α (PPAR-a)
Bioinformatics analysis of the 2 kb −5′-flanking region of human DNM3os showed the presence of three PPARα binding sites proximal to the transcription start site. The role of PPARα in DNM3os transcription was established by use of PPARα shRNA and pharmacological agonists of PPARα, such as fenofibrate. Studies showed PPARα shRNA attenuated DNM3os transcription while fenofibrate augmented basal transcription of DNM3os and mature miR-199a2 and miR-214 in HMEC in endothelial cells (Figure 4) (73). This effect was specific, as a PPARγ agonist was ineffective in increasing DNM3os transcription. Thus miR-199a2 and miR-214 co-located in DNM3os are indeed co-transcriptionally regulated by PPARα.
Figure 4. Transcriptional regulation of miR-199a2 located in the intron of DNM3os gene by PPARα transcription factor and its pharmacological agonist Fenofibrate.
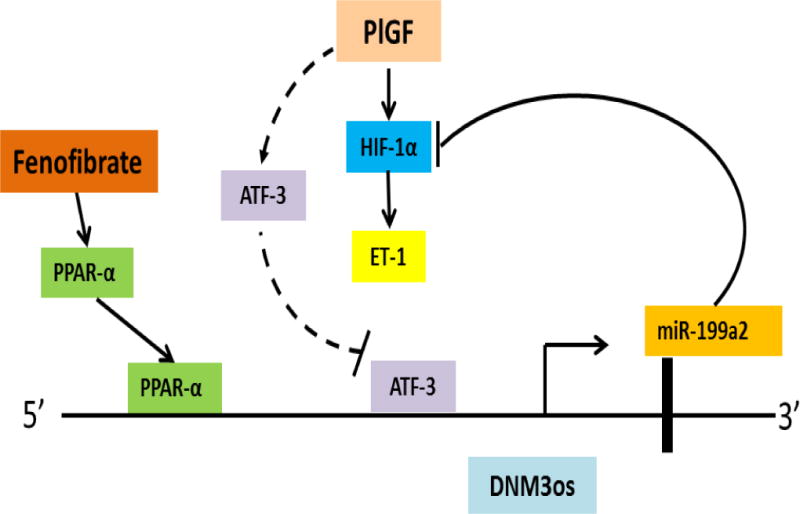
miR-199a2, which targets HIF-1α mRNA, is localized in an intron of DNM3os. Transcription factor, PPARα, which regulates DNM3os transcription concordantly regulates miR-199a2 expression as miR-199a2 is located in the DNM3os transcription unit. In the schematic, Fenofibrate, a PPARα agonist, augments the transcription of DNM3os and miR-199a2. As a result of increased expression of miR-199a2, there is a decrease in HIF-1α levels and concomitantly of ET-1, indicating therapeutic potential of Fenofibrate in PH. Adapted and modified (73).
To determine which of three PPARα binding sites in the DNM3os promoter were conducive to PlGF mediated transcription of DNM3os, two different approaches were utilized for readout of DNM3os transcription using a luciferase reporter and chromatin immunoprecipitation (ChIP) analysis. Studies show PPARα sites 1 and 2, among the three PPARa binding sites, in the DNM3os promoter were regulated by PlGF (Figure 4). These studies establish PPARα transcription factor binds to proximal PPARα cis-binding elements of the DNM3os promoter to coregulate transcription of DNM3os and the pre-miR-199a2/pre-mir-miR-214 cluster (73).
4. PPARα agonists augment miR-199a2, and reduce HIF-1a and ET-1 in vitro and in vivo
Fenofibrate, an FDA approved drug for cardiovascular disease, was next examined for its effect on HIF-1α expression as miR-199a2 targets the 3′UTR of HIF-1α mRNA. Fenofibrate reduces HIF-1α activity and concomitantly down regulates a target gene endothelin 1 (SERPINE1) relevant to pulmonary hypertension in SCA (73). The effect of Fenofibrate primarily occurred by upregulation of mature miR-199a2 culminating in inhibition of translation or turnover of HIF-1α mRNA (Figure 4). By contrast, studies by Staels and coworkers (75) have shown Fenofibrate inhibits endothelin-1 expression by a PPARα dependent induction of Krüppel-like factor 11 (KLF-11) which in turn represses ET-1 transcription by inhibiting AP-1 and SMAD co-activation of ET-1 transcription in endothelial cells. Previous studies show fibrates reduce HIF-1α activation in cancer cells in response to hypoxia (76). From work reported by the Kalra laboratory, both hypoxia and PlGF mediated expression of HIF-1α mRNA and protein are attenuated by Fenofibrate, likely by upregulation of miR-199a2, which targets the 3′UTR of HIF-1α mRNA (Figure 4)(73). Thus these reports clearly demonstrate that both transcriptional and post-transcriptional regulatory mechanisms regulate ET-1 expression.
To corroborate the in vitro results, the levels of mature miR-199a2 were examined in the plasma of SCA patients and unaffected siblings as controls. Plasma levels of miR-199a2 (n=11) was 5-fold lower in SCA compared to controls. Since there is an inverse relationship between miIR-199a2 levels to ET-1 levels, the data also show an increase in ET-1 plasma levels in SCA individuals vs. controls (73).
The direct association of plasma levels of miR-199a2 and ET-1 in patients with SCA were also confirmed in sickle mice. BkSS mice given oral Fenofibrate for 10 weeks showed increased pre-miR199a2 and concomitant reduction in ET-1 mRNA levels in lung tissues and plasma(73).
Thus miR-199a2 has a significant role in modulation of HIF-1α activity, and PPARα transcription factor is capable of regulating the expression of miR-199a2 via transcription of its host gene DNM3os (Figure 4). Fibrates are approved by the Food and Drug Administration for the treatment of dyslipidemia and flow mediated dilation of bronchial arteries in type 2 diabetic patients (77), and diabetic retinopathy (78). Thus fibrates have a therapeutic potential in alleviating ET-1 levels observed during pulmonary hypertension in SCA individuals.
4.1 Mechanism of PlGF-mediated repression of miR-199a2/miR-214 cluster in DNM3os gene
Since PlGF reduces the expression of miR-199a2 located in the DNM3os transcriptional unit, studies are warranted to delineate the molecular mechanism of repression of miR-199a2. PlGF treatment of cultured endothelial cells increased expression of activating transcription factor-3 (ATF3), a stress inducible gene (79). ATF3 has been shown to play an important role in the pathophysiology of diseases, including cancer, immunity and sepsis (80–82). Studies showed ATF3 binds to DNM3os promoter proximal cis-binding elements, located at −42/−35 nt and −816/−809 nt), as demonstrated by gain and loss of function of ATF3, DNM3os promoter-luciferase reporter assay and ChiP analysis (Figure 5)(79). Other studies have shown ATF3 can act either as a repressor or activator of transcription by recruitment of accessory proteins to the promoter element of genes (83).
Figure 5. Schematic of PlGF-induced ATF3 expression and its role in DNM3os/miR-199a2 repression.
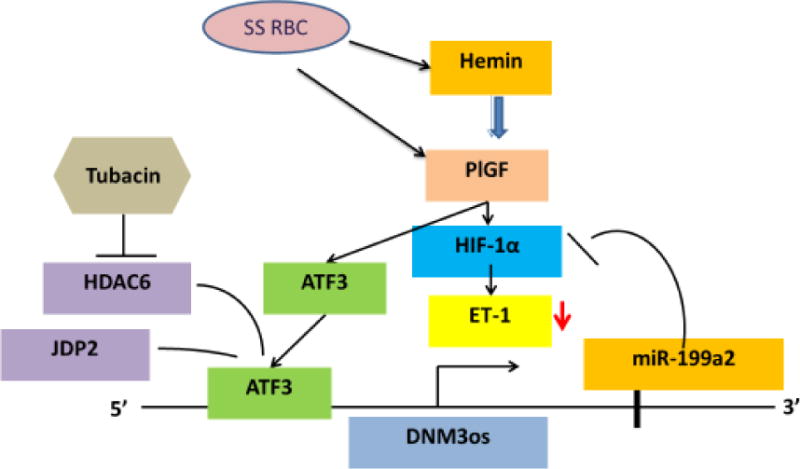
Due to increased erythropoiesis in SCD, hemin is released which induces the expression of PlGF in erythroid cells. Epo can also induce expression of PlGF from erythroid cells. PlGF increases expression of ATF3, which acts as repressor in association with JDP2 and HDAC6 on the promoter of the DNM3os/miR-199a2 transcription unit. Tubacin, a selective inhibitor of HDAC6, is permissive for expression of DNM3os/miR-199a2 both in vitro and in vivo. Increased levels of miR-199a2 attenuate HIF-1α with concomitant attenuation of downstream expression of target gene ET-1. Adapted and modified (79).
Analysis of ATF3 function on the DNM3os promoter was analyzed for association with other proteins. Following PlGF induction, proteomic analysis of endothelial cell lysates immunoprecipitated with antibody to ATF3 identified ATF3-interacting proteins (JDP2, ATF2 and HDACs). Since histone deacetylases (HDACs) are known to participate in gene repression by chromatin condensation following histone deacetylation (84), studies examined possible roles of HDACs 1,3,4,6,7,8,9 and 11 in PlGF-mediated repression of DNM3os utilizing commercially available HDAC shRNAs (79). Both HDAC6 and HDAC7 bound to the promoter of DNM3os along with JDP2 to cause repression of DNM3os transcription including pre-miR-199a2 (Figure 5).
The role of HDAC6 in DNM3os repression was validated by utilization of pharmacological inhibitor selective for HDAC6. Tubacin a selective inhibitor of HDAC6, Mocetonostat a potent inhibitor of HDAC1 but not HDAC6, and Trichostatin A (TSA) a broad spectrum class 1 and 2 deacetylase inhibitor were examined. Both Tubacin and TSA reversed PlGF-mediated repression of DNM3os transcription. The effect of Tubacin on derepression of DNM3os was locus specific as no effect was observed in transcription of DNM3 (sense strand) (79). Since histone marks, H3K27Ac and H3K9Ac are associated with chromatin remodeling at promoter and enhancer sites (85), studies examined the presence of such marks in the promoter of DNM3os proximal to the identified ATF3 sites. Indeed, PlGF treatment reduced H3K27Ac and H3K9Ac histone marks at the DNM3os locus, consistent with chromatin condensation. Conversely, Tubacin antagonizes this change to maintain an open chromatin state at the promoter region of DNM3os. Moreover, PlGF induced secretion of ET-1 is antagonized by Tubacin treatment and HDAC6 shRNA, indicating that chromatin changes are associated with transcription of DNM3os/miR-199a2(Figure 5)(79).
Tubacin has been shown to be effective in reducing ET-1 and PlGF plasma levels in BKSS mice (79). Specifically, subcutaneous pump delivery of Tubacin compared to Niltubacin (an inactive Tubacin congener)(86) to BKSS mice for 30 days significantly reduced plasma levels of ET-1 and PlGF. ET-1 mRNA levels in lung tissues of BKSS mice were also significantly reduced. These studies show Tubacin, a selective inhibitor of HDAC6, reduces ET-1 and PlGF levels in BKSS mice by increasing DNM3os/miR-199a2/miR-214 transcription(Figure 5)(79). Tubacin is currently in clinical trials for cancer patients, therefore its use in SCA patients is conceivable since safety issues have been addressed (87).
4.2 miR-214 targets 3′UTR of PlGF mRNA and its transcriptional regulation
PlGF is produced by erythroid cells and its plasma levels are high in mouse models of SCA and in SCA individuals. PlGF activates monocytes and endothelial cells leading to increased expression of cytochemokines and ET-1, respectively. Studies in BKSS mice and SCA patients support the role of PlGF in increasing plasma ET-1 levels and pulmonary artery pressure and hypertrophy, indicative of pulmonary hypertension(23). Thus, therapeutic modalities that block PlGF-mediated signaling can be beneficial to SCA patients.
Erythropoietin (Epo) is required for stimulating erythropoiesis in anemia, including SCA. Studies show Epo increases expression of PlGF in erythroid cells and K562 cells via activation of HIF-1α (88). Epo stabilizes the expression of HIF-1α protein, independent of hypoxia (88). There are two hypoxia response element (HRE) sites in the ~2.6kb PlGF-promoter region, located at −854 to −850 nt and −904 to −901 nt. PlGF-reporter luciferase assays and ChIP analysis validated the role of HIF-1α in Epo mediated induction of PlGF expression in erythroid cells(88).
In silico analysis of the 3′UTR of human PlGF mRNA show the presence of two complementary binding sites for miR-214 at +55/+78 nt and +99/+118 nt, and these target sites are well conserved among several mammalian species (Figure 2). Transfection with miR-214 mimic reduced Epo-mediated PlGF mRNA expression, indicating miR-214 attenuates PlGF expression. Luciferase translation reporter assays utilizing the 3′-UTR of PlGF also show reduction in Epo-mediated luciferase activity. Upon mutation of microRNA recognition elements (MRE) sites located at +55/+78 nt and +99/+118 nt in the PlGF 3′-UTR, this rendered the reporter refractory to the effect of miR-214 mimic in the reporter assay, thus showing a role for miR-214 in post-transcriptional regulation of PlGF (88).
The miR-199a2/miR-214 cluster located within an intron of the DNM3os locus. This locus encodes a long non-coding transcript that is also induced by Epo to transcribe the DNM3os gene with and concurrent synthesis of miR-199a2 and miR-214. As previously shown (73), PPARα binds to the promoter of DNM3os to mediate transcription of DNM3os. Upon mutation of these cis-binding recognition elements for PPARα Epo-mediated DNM3os reporter luciferase activity is abrogated. These studies show PPARα regulates transcription of DNM3os in erythroid cells as observed for PlGF-induced transcription of DNM3os. Furthermore, Fenofibrate, a PPARα agonist, induced DNM3os and pre-miR-214/pre-miR-199a2 expression in erythroid cells (K562). Fenofibrate treatment of erythroid cells shows a significant increase in expression of mature miR-214, as determined by Northern Blot analysis; this expression was reduced by PPARα antagonist GW6417 (88). These studies show Fenofibrate mediated upregulation of miR-214 reduces PlGF expression (Figure 6).
Figure 6. Transcriptional regulation of miRNAs targeting HIF-1α, ET-1, PAI-1 and PlGF mRNAs.
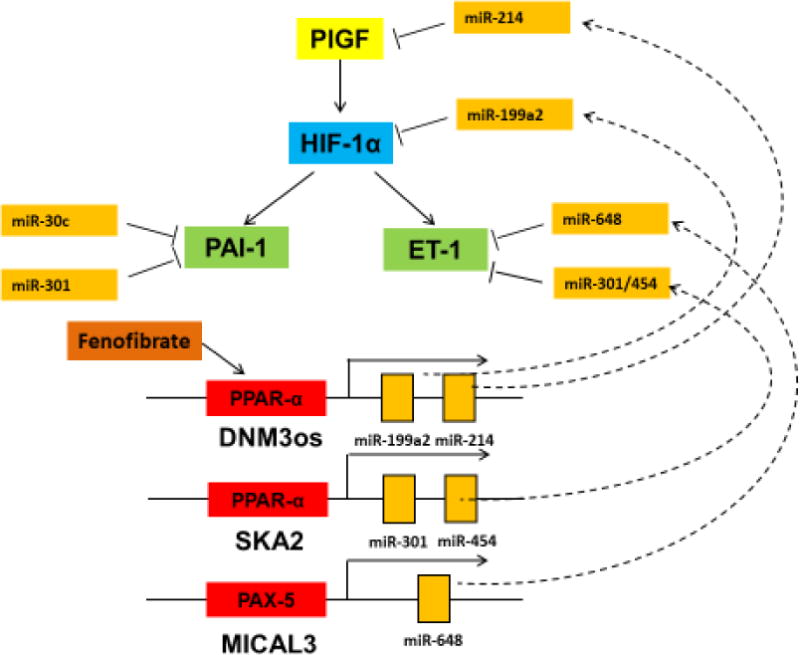
PlGF activates HIF-1α and upregulates expression of ET-1 and PAI-1. Fenofibrate activates PPARα sites located in host genes DNM3os and SKA2. As a result, there is increased expression of miRs, which target PlGF, HIF-1α and ET-1 mRNAs, thus attenuating their expression in cells and tissues. miR-30c and miR-301a target PAI-1 mRNA expression; the latter is located within and intron of its SKA2 host gene. PAX5 transcription factor regulates transcription of MICAL3 host gene wherein miR-648 is located. miR-648 targets directly the 3′UTR of ET-1 mRNA.
Next, we examined the plasma levels of erythropoietin (Epo) in BKSS mice and show Epo levels are ~64-fold higher in BKSS mice compared to control mice. This indicated RBC sickling correlates with an increase in the expression of Epo. The lungs of BKSS mice exhibit 4-fold higher PlGF mRNA and significantly low levels of pre-miR-214 compared to control mice. Thus both in vitro and in vivo studies show miR-214 is inversely correlated to post-transcriptional expression of PlGF (88). Fenofibrate, a PPARα agonist, has been shown to upregulate the expression of miR-214 and concomitantly reduce levels of PlGF. These studies thus provide a novel therapeutic approach for the use of fibrates, an FDA approved drug, to ameliorate the pathological consequences of dysfunctional PlGF mediated inflammation and pulmonary hypertension in SCA.
4.3 The roles of miR-30c /miR-301a in PAI-1 induction in endothelial cells
SCA patients have increased levels of activated coagulation factors, including elevated plasma levels of tissue factor and plasminogen activator inhibitor-1 (PAI-1); this leads to a prothrombotic state predisposing such individuals to pulmonary hypertension and stroke (89). Plasma PAI-1 levels are high in SCA patients and mouse models (BKSS) of SCA. Immunolabeling of lung tissues of BKSS mice shows strong signals in alveolar macrophages and endothelial cells for PAI-1 (49). Treatment of human pulmonary microvascular endothelial cells (HPMVEC) with PlGF results in significant induction of PAI-1 mRNA and protein. PlGF mediated upregulation of PAI-1 involves activation of HIF-1α, as observed for ET-1 expression (49). This role of PlGF in PAI-1 induction was validated in mouse models of SCA (BKSS) and PlGF−/− mice, which showed PAI-1 induction and null expression, respectively. Administration of an adenoviral vector encoding PlGF into control C57BL/6 mice resulted in increased plasma levels of both PlGF and PAI-1, thus showing PlGF is responsible for increased expression of PAI-1(49).
To examine the post-transcriptional mechanism of PlGF-induced PAI-1 mRNA stability, a bioinformatics approach showed potential miRNA target sites in the 3′UTR of PAI-1 mRNA (90). Among several miRNAs, miR-30c and miR-301a targeted the 3′UTR of PAI-1 mRNA as demonstrated by the effect of respective miRNAs mimics on PlGF-mediated PAI-1 mRNA expression and luciferase translation reporter containing the 3′UTR of PAI-1(90). These studies show PlGF-mediated induction of PAI-1 is post-transcriptionally regulated by both miR-30c and miR-301a, which have complementary target sequences in the 3′UTR of PAI-1 mRNA (Figure 2). Finally, other studies show reduced levels of miR-30c and miR-301a in plasma of SCA patients compared to normal controls, thus providing correlation of a post-transcriptional regulatory mechanism explaining the increased levels of PAI-1 seen in SCA patients and BKSS mouse model(90).
4.4 miR-301 located in the intron of host gene SKA2 is transcriptionally regulated by PPARα
Genome sequence information shows miR-301a is located in the first intron of the SKA2 (Spindle and kinetochore associated protein 2) gene, and is co-localized with miR-454 (Figure 6) (91). PlGF treatments of HPMEC show a time dependent increase in SKA2 RNA expression with a maximum increase at 4 hr post addition, while pre-miR-301a and pre-miR-454 maximal expression occurs at 2 hr post-addition, followed by a decline, indicating pre-miR transcription precedes SKA2 synthesis. PlGF-mediated transcription of SKA2 involves PPARα as demonstrated by shRNA knockdown of this transcription factor, by PPARα agonist effect, and by PPARαChIP analysis(91). Both miR-301 and miR-454 target the 3′UTR of ET-1 and PAI-1 (91). As previously shown, miR-301 levels are significantly reduced with corresponding higher levels of PAI-1 in SCA patients. We also observed reduced expression of miR-454 in plasma from these SCA patients. These studies indicate PlGF-mediated transcription of PAI-1 is post-transcriptionally regulated by miR-301a/miR-454, and the transcription of these miRNAs located in the host gene SKA2 is regulated by PPARα (Figure 6)(91).
4.5 PlGF-mediated activation of 5-lipoxygenase activating protein involved in leukotriene formation in SCA
Patients with SCA exhibit increased inflammation, a high incidence of airway hyper-responsiveness (AHR) and increased circulating leukotrienes (29, 35, 92, 93). In SCA, the prevalence of AHR is higher than that seen in clinically symptomatic asthma. Biomarkers of hemolysis show a strong correlation with AHR in SCA subjects (94). Thus, the etiology of the high incidence of AHR in SCA may be related to erythroid cells and hemin released during hemolysis. Erythroid cells generate PlGF which are high in SCA subjects. Studies show mononuclear cells (MNCs) from SCA individuals are highly activated in steady state illness compared to MNCs obtained from healthy controls. MNCs from SCA show significant chronic increases in the expression of 5-lipoxygenase (5-LO) and 5-lipoxygenase activating protein (FLAP), which are key enzymes involved in leukotriene synthesis (34). Similarly MNCs from control subjects upon treatment with PlGF show an increase in 5-LO and FLAP as seen in SCA. As discussed above, the prothrombotic state and vascular complications associated with PlGF occur in addition to activation of FLAP via activation of HIF-1α. PlGF increased binding of HIF-1α to the FLAP promoter as demonstrated by FLAP promoter reporter luciferase assay and ChIP(34). We show PlGF, that is elevated in SCA, mediates AHR. In allergen-exposed mice, loss of PlGF reduces inflammation and eisonophilia, and attenuates AHR, and also reduces Th2 cytokine IL-13 and enzymes i.e. 5-LO and LTC4 synthase involved in leukotriene synthesis. Conversely, PlGF−/− mice treated with leukotrienes exhibited similar response as wild type response to allergen exposure. Thus, the intrinsically elevated levels of PlGF from erythroid cells in SCA contribute to increased circulating levels of leukotrienes (LTs) which mediate inflammation and AHR(35). Additionally, Th2 IL-13 activates STAT6, which in turn increases PlGF production from alveolar macrophages, eosinophils and pulmonary airway epithelial cells. Thus, intrinsically generated PlGF from erythroid cells in SCA and allergen induced PlGF via activation of IL-13 pathway are the key effectors of AHR in both SCA mouse models and in patients with asthma. These studies show PlGF exacerbates AHR and links Th2 pathways and leukotrienes in asthma. In the SCA mouse model where higher leukotriene levels and increased AHR are observed, these symptoms can be dampened by 5-LO inhibitor, Zileuton or by an antibody to PlGF(35).
Hypoxia mediates upregulation of FLAP via activation of HIF-1α and NF-κB(95). miR-135a and miR-199a-5p, which target the 3′UTR of FLAP mRNA show attenuated levels in response to hypoxia (95). miR-199a2 has been shown to regulate 5-LO in macrophages(96), while miR-19a-3p and miR-125b-5p target 5-LO in a cell specific manner (97). Overall, these studies show that miRNAs play significant roles in the post-transcriptional regulation of 5-LO and FLAP, the key enzymes involved in LT synthesis.
5. Conclusions and future directions
The pathogenesis of PH is multi-factorial and exhibits: impaired nitric oxide bioavailability, pulmonary vasoconstriction, and endothelial dysfunction and remodeling. Here, we focus our studies on the role of placenta growth factor (PlGF), elaborated from erythroblast cells, in the pathogenesis of inflammation, pulmonary hypertension and airway-hyper responsive (AH) disease in SCA. Due to increased compensatory erythropoiesis in response to increased destruction (hemolysis) of sickle RBCs, there are increased levels of circulating PlGF in SCA individuals compared to healthy controls. This PlGF elaboration by erythroid precursors causes activation of monocytes to upregulate the expression of inflammatory cytochemokines in SCA. Moreover, PlGF causes increased expression of endothelin-1, a potent vasoconstrictor, in cultured endothelial cells via activation of hypoxia inducible factor-1α, independent of hypoxia. Mouse models of SCA, such as Berkeley sickle mice, genetic models of PlGF null mice, and mice expressing ectopically high levels of PlGF, show a correlation between high plasma levels of PlGF to increased levels of ET-1 and associated clinical features of PH. Results from in vitro and mouse model studies have been further validated in SCA patients with PH. Moreover, PlGF mediated activation of enzymes (5-LO and FLAP) in monocytes leading to high levels of leukotrienes, has been validated in the SCA mouse model culminating in AH. These studies show PlGF plays an important role in activation of monocytes and endothelial cells in upregulating the expression of genes involved in PH and leukotriene synthesis.
Non-coding miRNAs have been identified that post-transcriptionally regulate the PlGF-mediated expression of ET-1, HIF-1α, PAI-1 and FLAP (Figure 6). PlGF mediated signaling involves activation of HIF-1α, independent of hypoxia, with concomitant upregulation of target genes, e.g. ET-1, PAI-1 and FLAP. PlGF mediated stability of HIF-1α mRNA is regulated by repressing levels of miR-199a-5-p (miR-199a2), which targets the 3′UTR of HIF-1α mRNA. The miR-199a2/miR-214 cluster is located in an intron of host gene DNM3os. miR-214 has been identified to target the 3′UTR of PlGF mRNA, thus miR-214 post-transcriptionally regulates the expression of PlGF. Since these miRNAs reside in the introns of host genes, we identified the mode of transcriptional regulation of the host gene transcription unit and concomitant synthesis of pre-miRNAs. We identified transcription factors, which regulate the expression of miRNAs and their effect on target genes. In particular we discovered PPARα binds to the promoters of DNM3os and SKA2 genes, and regulates the transcription of miR-199a2/miR-214 cluster and miR-301/miR-454, respectively (Figure 6). Pharmacological agonists of PPARα such as Fenofibrate were found to augment the expression of these indicated miRs, which target key genes associated with clinical manifestations of SCA (i.e., HIF-1α, PlGF, ET-1 and PAI-1). Consequently, agonist dependent synthesis of these miRNAs reduces their respective mRNA targets as illustrated in Figure 6. The identification of transcription factors which regulate the expression of host genes for miRNAs presents a novel therapeutic approach to modulate the expression of miRNA-target genes involved in disease pathophysiology. However, there remain concerns in the safety issues associated with miRNA based therapies as miRNAs can have off-target effects, unanticipated consequences of viral vectors employed for delivery, and RNA-mediated immunostimulation. The role of non-coding RNAs (lncRNAs) in regulation of gene expression is a novel area of investigation, which needs to be addressed in future studies.
Practice points.
Angiogenic placenta growth factor (PlGF), released from erythroid cells, has an important role in inflammation, pulmonary hypertension and airway hyper-responsiveness in sickle cell disease (SCD).
miRNAs bind cognate sequences in the 3′ untranslated regions (3′UTR) of mRNA and post-transcriptionally repress gene expression.
Specific miRNAs regulate expression of PlGF and effector target genes, i.e., endothelin-1 (ET-1), hypoxia inducible factor–1α (HIF-1α), plasminogen activator inhibitor-1 (PAI-1) and 5-lipoxygenase activating protein (FLAP). These genes promote pulmonary hypertension (PH), hyper coagulation, and airway hyper-responsiveness in SCD.
The expression of miRNAs is transcriptionally regulated. Insight into miRNA regulation provides novel ways to modulate miRNA expression, thereby altering expression of miRNA target genes: e.g., attenuation of ET-1 and PAI-1 expression to ameliorate PH in SCD.
PPAR-α agonist, Fenofibrate, an FDA approved drug, upregulates expression of miR-199a2 and miR-214, which target the 3′UTRs of HIF-1α and PlGF mRNAs, and decreases ET-1 (a HIF1α regulated gene) and PlGF, attenuating PH in mice with SCD. These agents have therapeutic potential in SCD patients with PH.
Research Agenda.
Since miRNAs target mRNAs of several genes, they can have off-target effects. However, long non coding RNAs (lncRNAs), > 200 nts in length, with no capacity to be translated to proteins, have recently emerged as important regulators of gene expression.
Endogenous anti-sense transcripts (AST), transcribed from the opposite DNA strand, have also emerged as modulators of sense RNA. For example, BC036851 (ET-1AS) within and adjacent to the ET-1 locus has the potential to regulate ET-1 expression and thus PH in SCD.
An understanding of transcriptional regulation of endogenous AST has potential for developing lncRNA-based treatments.
The identification of SNPs in AST alleles of patients with SCA and correlation with susceptibility to or severity of PH will pave a pathway for personalized medicine.
Acknowledgments
Due to paucity of space, the authors apologize to investigators whose work was not included in this review. This work was supported in part by Heart Lung and Blood Institute of National Institute of Health HLBI-RO1-111372 and RO1 HL079916 (VKK and PM). The authors thank all the postdoctoral fellows and graduate students in our laboratories who contributed extensively to the work quoted in this review.
Role of the Funding Source
The content is solely the responsibility of authors and does not necessarily represent the official views of the NHLBI or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts
The authors declare no conflicts of interest
References
- 1.Hebbel R. Beyond hemoglobin polymerization: the red blood cell membrane and sickle disease pathophysiology. Blood. 1991;77(2):214–37. [PubMed] [Google Scholar]
- 2.Francis RB, Jr, Johnson CS. Vascular occlusion in sickle cell disease: current concepts and unanswered questions. Blood. 1991;77(7):1405–14. [PubMed] [Google Scholar]
- 3.Kassim AA, DeBaun MR. Sickle Cell Disease, Vasculopathy, and Therapeutics. Annu Rev Med. 2013;64(1):451–66. doi: 10.1146/annurev-med-120611-143127. [DOI] [PubMed] [Google Scholar]
- 4.Bunn HF. Pathogenesis and treatment of sickle cell disease. New Engl J Med. 1997;337(11):762–9. doi: 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- 5.Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Op Hematol. 2002;9(2):101–6. doi: 10.1097/00062752-200203000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest. 2007;117(4):850–8. doi: 10.1172/JCI30920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang D, Xu C, Manwani D, Frenette PS. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. 2016;127(7):801–9. doi: 10.1182/blood-2015-09-618538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ataga KI, Moore CG, Hillery CA, Jones S, Whinna HC, Strayhorn D, et al. Coagulation activation and inflammation in sickle cell disease-associated pulmonary hypertension. Haematologica. 2008;93:20–6. doi: 10.3324/haematol.11763. [DOI] [PubMed] [Google Scholar]
- 9.Kato GJ, Hebbel RP, Steinberg MH, Gladwin MT. Vasculopathy in sickle cell disease: Biology, pathophysiology, genetics, translational medicine, and new research directions. Am J Hematol. 2009;84(9):618–25. doi: 10.1002/ajh.21475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127(3):750–60. doi: 10.1172/JCI89741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, et al. Pulmonary Hypertension as a Risk Factor for Death in Patients with Sickle Cell Disease. New Engl J Med. 2004;350(9):886–95. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 12.Potoka KP, Gladwin MT. Vasculopathy and pulmonary hypertension in sickle cell disease. Am J Physiol-Lun Cell Mol Physiol. 2015;308:L314–L324. doi: 10.1152/ajplung.00252.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bunn HF, Nathan DG, Dover GJ, Hebbel RP, Platt OS, Rosse WF, et al. Pulmonary hypertension and nitric oxide depletion in sickle cell disease. Blood. 2010;116(5):687–92. doi: 10.1182/blood-2010-02-268193. [DOI] [PubMed] [Google Scholar]
- 14.Hsu LL, Champion HC, Campbell-Lee SA, Bivalacqua TJ, Manci EA, Diwan BA, et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood. 2007;109(7):3088–98. doi: 10.1182/blood-2006-08-039438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ergul S, Brunson CY, Hutchinson J, Tawfik A, Kutlar A, Webb RC, et al. Vasoactive factors in sickle cell disease: In vitro evidence for endothelin-1-mediated vasoconstriction. Am J Hematol. 2004;76(3):245–51. doi: 10.1002/ajh.20107. [DOI] [PubMed] [Google Scholar]
- 16.Machado RF, Gladwin MT. Chronic sickle cell lung disease: new insights into the diagnosis, pathogenesis and treatment of pulmonary hypertension. Br J Haematol. 2005;129(4):449–64. doi: 10.1111/j.1365-2141.2005.05432.x. [DOI] [PubMed] [Google Scholar]
- 17.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: Reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21(1):37–47. doi: 10.1016/j.blre.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris CR, Suh JH, Hagar W, Larkin S, Bland DA, Steinberg MH, et al. Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease. Blood. 2008;111(1):402–10. doi: 10.1182/blood-2007-04-081703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gladwin MT, Vichinsky E. Pulmonary Complications of Sickle Cell Disease. New Engl J Med. 2008;359(21):2254–65. doi: 10.1056/NEJMra0804411. [DOI] [PubMed] [Google Scholar]
- 20.Graido-Gonzalez E, Doherty JC, Bergreen EW, Organ G, Telfer M, McMillen MA. Plasma Endothelin-1, Cytokine, and Prostaglandin E2Levels in Sickle Cell Disease and Acute Vaso-Occlusive Sickle Crisis. Blood. 1998;92(7):2551–5. [PubMed] [Google Scholar]
- 21.Rybicki AC, Benjamin LJ. Increased levels of endothelin-1 in plasma of sickle cell anemia patients. Blood. 1998;92(7):2594–6. [PubMed] [Google Scholar]
- 22.Ataga KI, Derebail VK, Caughey M, Elsherif L, Shen JH, Jones SK, et al. Albuminuria Is Associated with Endothelial Dysfunction and Elevated Plasma Endothelin-1 in Sickle Cell Anemia. PLoS One. 2016;11(9):e0162652. doi: 10.1371/journal.pone.0162652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sundaram N, Tailor A, Mendelsohn L, Wansapura J, Wang X, Higashimoto T, et al. High levels of placenta growth factor in sickle cell disease promote pulmonary hypertension. Blood. 2010;116(1):109–12. doi: 10.1182/blood-2009-09-244830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, et al. Endothelin. Pharmacol Rev. 2016;68(2):357–418. doi: 10.1124/pr.115.011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okunishi K, Peters-Golden M. Leukotrienes and airway inflammation. Biochim Biophys Acta. 2011;1810(11):1096–102. doi: 10.1016/j.bbagen.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hallstrand TS, Henderson WR. An update on the role of leukotrienes in asthma. Curr Opin Allergy Clin Immunol. 2010;10(1):60–6. doi: 10.1097/ACI.0b013e32833489c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knight-Perry J, DeBaun MR, Strunk RC, Field JJ. Leukotriene pathway in sickle cell disease: a potential target for directed therapy. Expert Rev Hematol. 2009;2(1):57–68. doi: 10.1586/17474086.2.1.57. [DOI] [PubMed] [Google Scholar]
- 28.DeBaun MR, Strunk RC. The intersection between asthma and acute chest syndrome in children with sickle-cell anaemia. Lancet. 2016;387(10037):2545–53. doi: 10.1016/S0140-6736(16)00145-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Setty BN, Stuart MJ. Eicosanoids in sickle cell disease: potential relevance of neutrophil leukotriene B4 to disease pathophysiology. J Lab Clin Med. 2002;139(2):80–9. doi: 10.1067/mlc.2002.121200. [DOI] [PubMed] [Google Scholar]
- 30.Jennings JE, Ramkumar T, Mao J, Boyd J, Castro M, Field JJ, et al. Elevated urinary leukotriene E(4) levels are associated with hospitalization for pain in children with sickle cell disease. Am J Hematol. 2008;83:640–3. doi: 10.1002/ajh.21199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peters-Golden M, Brock TG. 5-Lipoxygenase and FLAP. Prostaglandins, Leukot Essent Fatty Acids. 2003;69(2–3):99–109. doi: 10.1016/s0952-3278(03)00070-x. [DOI] [PubMed] [Google Scholar]
- 32.Bigby TD. The Yin and the Yang of 5-Lipoxygenase Pathway Activation. Mol Pharmacol. 2002;62(2):200–2. doi: 10.1124/mol.62.2.200. [DOI] [PubMed] [Google Scholar]
- 33.Zhao J-l, Austen KF, Lam BK. Cell-specific Transcription of Leukotriene C4Synthase Involves a Kruppel-like Transcription Factor and Sp1. J Biol Chem. 2000;275(12):8903–10. doi: 10.1074/jbc.275.12.8903. [DOI] [PubMed] [Google Scholar]
- 34.Patel N, Gonsalves CS, Yang M, Malik P, Kalra VK. Placenta growth factor induces 5-lipoxygenase–activating protein to increase leukotriene formation in sickle cell disease. Blood. 2009;113(5):1129–38. doi: 10.1182/blood-2008-07-169821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eiymo Mwa Mpollo M-S, Brandt EB, Shanmukhappa SK, Arumugam PI, Tiwari S, Loberg A, et al. Placenta growth factor augments airway hyperresponsiveness via leukotrienes and IL-13. J Clin Invest. 2016;126(2):571–84. doi: 10.1172/JCI77250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peters-Golden M, Henderson WRJ. Leukotrienes. New Engl J Med. 2007;357(18):1841–54. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 37.Lewis RA, Austen KF, Soberman RJ. Leukotrienes and other products of the 5-Lipoxygenase pathway;biochemistry and relation to pathobiology of human diseases. New Engl J Med. 1990;323:645–55. doi: 10.1056/NEJM199009063231006. [DOI] [PubMed] [Google Scholar]
- 38.Funk CD. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science. 2001;294(5548):1871–5. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 39.Maglione D, Guerriero V, Viglietto G, Delli-Bovi P, Persico MG. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci USA. 1991;88(20):9267–71. doi: 10.1073/pnas.88.20.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao Y, Ji W-R, Qi P, Rosin Å, Cao Y. Placenta Growth Factor: Identification and Characterization of a Novel Isoform Generated by RNA Alternative Splicing. Biochem Biophys Res Commun. 1997;235(3):493–8. doi: 10.1006/bbrc.1997.6813. [DOI] [PubMed] [Google Scholar]
- 41.Dewerchin M, Carmeliet P. PlGF: A Multitasking Cytokine with Disease-Restricted Activity. Cold Spring Harb Perspect Med. 2012;2(8):a011056. doi: 10.1101/cshperspect.a011056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oura H, Bertoncini J, Velasco P, Brown LF, Carmeliet P, Detmar M. A critical role of placental growth factor in the induction of inflammation and edema formation. Blood. 2003;101(2):560–7. doi: 10.1182/blood-2002-05-1516. [DOI] [PubMed] [Google Scholar]
- 43.Tordjman R, Delaire S, Plouët J, Ting S, Gaulard P, Fichelson S, et al. Erythroblasts are a source of angiogenic factors. Blood. 2001;97(7):1968–74. doi: 10.1182/blood.v97.7.1968. [DOI] [PubMed] [Google Scholar]
- 44.Perelman N, Selvaraj SK, Batra S, Luck LR, Erdreich-Epstein A, Coates TD, et al. Placenta growth factor activates monocytes and correlates with sickle cell disease severity. Blood. 2003;102(4):1506–14. doi: 10.1182/blood-2002-11-3422. [DOI] [PubMed] [Google Scholar]
- 45.De Falco S. The discovery of placenta growth factor and its biological activity. Exp Mol Med. 2012;44:1–9. doi: 10.3858/emm.2012.44.1.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim K-J, Cho C-S, Kim W-U. Role of placenta growth factor in cancer and inflammation. Exp Mol Med. 2012;44(1):10–19. doi: 10.3858/emm.2012.44.1.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7(5):575–83. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- 48.Luttun AERN, Tjwa MARC, Carmeliet PETE. Placental Growth Factor (PlGF) and Its Receptor Flt-1 (VEGFR-1): Novel Therapeutic Targets for Angiogenic Disorders. Ann N Y Acad Sci. 2002;979(1):80–93. doi: 10.1111/j.1749-6632.2002.tb04870.x. [DOI] [PubMed] [Google Scholar]
- 49.Patel N, Sundaram N, Yang M, Madigan C, Kalra VK, Malik P. Placenta growth factor (PlGF), a novel inducer of plasminogen activator inhibitor-1 (PAI-1) in sickle cell disease (SCD) J Biol Chem. 2010;285(22):16713–22. doi: 10.1074/jbc.M110.101691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newell LF, Holtan SG. Placental growth factor: What hematologists need to know. Blood Rev. 2017;31(1):57–62. doi: 10.1016/j.blre.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hebbel RP, Mohandas N. Sickle Cell Adherence. In: Embury SH, Hebbel RP, Mohandas N, Steinberg MH, editors. Sickle Cell Disease- Basic principles and clinical practice. New York: Raven Press; 1994. pp. 217–30. [Google Scholar]
- 52.Buchanan GR. In: Sickle Cell Disease: basic principles and clinical practice. Embury SH, Hebbel RP, Mohandas N, Steinberg MH, editors. NY: Raven Press Ltd; 1994. pp. 576–87. [Google Scholar]
- 53.Hebbel RP, Boogaerts MAB, Eaton JW, Steinberg MH. Erythrocyte Adherence to Endothelium in Sickle-Cell Anemia. New Engl J Med. 1980;302(18):992–5. doi: 10.1056/NEJM198005013021803. [DOI] [PubMed] [Google Scholar]
- 54.Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96(7):2451–9. [PubMed] [Google Scholar]
- 55.Sultana C, Shen Y, Rattan V, Johnson C, Kalra VK. Interaction of sickle erythrocytes with endothelial cells in the presence of endothelial cell conditioned medium induces oxidant stress leading to transendothelial migration of monocytes. Blood. 1998;92(10):3924–35. [PubMed] [Google Scholar]
- 56.Lard LR, Mul FP, de Haas M, Roos D, Duits AJ. Neutrophil activation in sickle cell disease. J Leukocyte Biol. 1999;66(3):411–15. doi: 10.1002/jlb.66.3.411. [DOI] [PubMed] [Google Scholar]
- 57.Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: A new paradigm. Proc Natl Acad Sci USA. 2002;99(5):3047–51. doi: 10.1073/pnas.052522799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Selvaraj SK, Giri RK, Perelman N, Johnson C, Malik P, Kalra VK. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood. 2003;102(4):1515–24. doi: 10.1182/blood-2002-11-3423. [DOI] [PubMed] [Google Scholar]
- 59.Patel N, Gonsalves CS, Malik P, Kalra VK. Placenta growth factor augments endothelin-1 and endothelin-B receptor expression via hypoxia-inducible factor-1 alpha. Blood. 2008;112(3):856–65. doi: 10.1182/blood-2007-12-130567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu J, Discher DJ, Bishopric NH, Webster KA. Hypoxia Regulates Expression of the Endothelin-1 Gene through a Proximal Hypoxia-Inducible Factor-1 Binding Site on the Antisense Strand. Biochem Biophys Res Commun. 1998;245(3):894–9. doi: 10.1006/bbrc.1998.8543. [DOI] [PubMed] [Google Scholar]
- 61.Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular Regulation of the Endothelin-1 Gene by Hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, and p300/CBP. J Biol Chem. 2001;276(16):12645–53. doi: 10.1074/jbc.M011344200. [DOI] [PubMed] [Google Scholar]
- 62.Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J, Townes TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood. 2006;108(4):1183–8. doi: 10.1182/blood-2006-02-004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brittain JE, Hulkower B, Jones SK, Strayhorn D, De Castro L, Telen MJ, et al. Placenta growth factor in sickle cell disease: association with hemolysis and inflammation. Blood. 2010;115:2014–20. doi: 10.1182/blood-2009-04-217950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang X, Mendelsohn L, Rogers H, Leitman S, Raghavachari N, Yang Y, et al. Heme-bound iron activates placenta growth factor in erythroid cells via erythroid Kruppel-like factor. Blood. 2014;124(6):946–54. doi: 10.1182/blood-2013-11-539718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell. 2004;116(2):281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 66.Gurtan AM, Sharp PA. The role of miRNAs in regulating gene expression networks. J Mol Biol. 2013;425(19):3582–600. doi: 10.1016/j.jmb.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou G, Chen T, Raj JU. MicroRNAs in Pulmonary Arterial Hypertension. Am J Resp Cell Mol Biol. 2015;52(2):139–51. doi: 10.1165/rcmb.2014-0166TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rothman AMK, Chico TJA, Lawrie A. MicroRNA in Pulmonary Vascular Disease. In: Timothy JAC, editor. Progress in Molecular Biology and Translational Science. Vol. 124. Academic Press; 2014. pp. 43–63. [DOI] [PubMed] [Google Scholar]
- 69.Lawrie CH. MicroRNAs in hematological malignancies. Blood Rev. 2013;27(3):143–54. doi: 10.1016/j.blre.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 70.Gosline SJC, Gurtan AM, JnBaptiste CK, Bosson A, Milani P, Dalin S, et al. Elucidating microRNA regulatory networks using transcriptional, post-transcriptional and histone modification measurements. Cell Rep. 2016;14(2):310–19. doi: 10.1016/j.celrep.2015.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li C, Gonsalves CS, Eiymo Mwa Mpollo MS, Malik P, Tahara SM, Kalra VK. MicroRNA 648 targets ET-1 mRNA and is cotranscriptionally regulated with MICAL3 by PAX5. Mol Cell Biol. 2015;35(3):514–28. doi: 10.1128/MCB.01199-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grigoriev I, Yu KL, Martinez-Sanchez E, Serra-Marques A, Smal I, Meijering E, et al. Rab6, Rab8, and MICAL3 Cooperate in Controlling Docking and Fusion of Exocytotic Carriers. Curr Biol. 2011;21(11):967–74. doi: 10.1016/j.cub.2011.04.030. [DOI] [PubMed] [Google Scholar]
- 73.Li C, Mpollo M-SEM, Gonsalves CS, Tahara SM, Malik P, Kalra VK. Peroxisome Proliferator-activated Receptor-α-mediated transcription of miR-199a2 attenuates endothelin-1 expression via hypoxia-inducible Factor-1α. J Biol Chem. 2014;289(52):36031–47. doi: 10.1074/jbc.M114.600775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee Y-B, Bantounas I, Lee D-Y, Phylactou L, Caldwell MA, Uney JB. Twist-1 regulates the miR-199a/214 cluster during development. Nucleic Acids Res. 2009;37(1):123–8. doi: 10.1093/nar/gkn920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Glineur C, Gross B, Neve B, Rommens C, Chew GT, Martin-Nizard F, et al. Fenofibrate Inhibits endothelin-1 expression by Peroxisome Proliferator–Activated Receptor α–dependent and independent mechanisms in human endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33(3):621–8. doi: 10.1161/ATVBAHA.112.300665. [DOI] [PubMed] [Google Scholar]
- 76.Zhou J, Zhang S, Xue J, Avery J, Wu J, Lind SE, et al. Activation of Peroxisome Proliferator-activated Receptor alpha (PPARalpha) suppresses hypoxia-inducible factor-1alpha (HIF-1alpha) signaling in cancer cells. J Biol Chem. 2012;287(42):35161–9. doi: 10.1074/jbc.M112.367367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lalloyer F, Staels B. Fibrates, Glitazones, and Peroxisome Proliferator-Activated Receptors. Arterioscler Thromb Vasc Biol. 2010;30(5):894–9. doi: 10.1161/ATVBAHA.108.179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Knickelbein JE, Abbott AB, Chew EY. Fenofibrate and Diabetic Retinopathy. Curr Diabetes Rep. 2016;16(10):90. doi: 10.1007/s11892-016-0786-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li C, Zhou Y, Loberg A, Tahara SM, Malik P, Kalra VK. Activated Transcription Factor 3 in association with histone deacetylase 6 negatively regulates microRNA 199a2 transcription by chromatin remodeling and reduces endothelin-1 expression. Mol Cell Biol. 2016;36(22):2838–54. doi: 10.1128/MCB.00345-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hai T, Wolford CC, Chang Y-S. ATF3, a Hub of the Cellular Adaptive-Response Network, in the Pathogenesis of Diseases: Is Modulation of Inflammation a Unifying Component? Gene Expr. 2010;15(1):1–11. doi: 10.3727/105221610x12819686555015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wolford CC, McConoughey SJ, Jalgaonkar SP, Leon M, Merchant AS, Dominick JL, et al. Transcription factor ATF3 links host adaptive response to breast cancer metastasis. J Clin Invest. 2013;123(7):2893–906. doi: 10.1172/JCI64410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hoetzenecker W, Echtenacher B, Guenova E, Hoetzenecker K, Woelbing F, Bruck J, et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat Med. 2012;18(1):128–34. doi: 10.1038/nm.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Darlyuk-Saadon I, Weidenfeld-Baranboim K, Yokoyama KK, Hai T, Aronheim A. The bZIP repressor proteins, c-Jun dimerization protein 2 and activating transcription factor 3, recruit multiple HDAC members to the ATF3 promoter. Biochim Biophys Acta. 2012;1819(11–12):1142–53. doi: 10.1016/j.bbagrm.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Narlikar GJ, Fan H-Y, Kingston RE. Cooperation between Complexes that Regulate Chromatin Structure and Transcription. Cell. 2002;108(4):475–87. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 85.Jenuwein T, Allis CD. Translating the Histone Code. Science. 2001;293(5532):1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 86.de Zoeten EF, Wang L, Butler K, Beier UH, Akimova T, Sai H, et al. Histone Deacetylase 6 and Heat Shock Protein 90 Control the Functions of Foxp3+ T-Regulatory Cells. Mol Cell Biol. 2011;31(10):2066–78. doi: 10.1128/MCB.05155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124(1):30–9. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gonsalves Caryn S, Li C, Mpollo M-Sandrine Eiymo M, Pullarkat V, Malik P, Tahara Stanley M, et al. Erythropoietin-mediated expression of placenta growth factor is regulated via activation of hypoxia-inducible factor-1α and post-transcriptionally by miR-214 in sickle cell disease. Biochem J. 2015;468(3):409–23. doi: 10.1042/BJ20141138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hillery CA, Panepinto JA. Pathophysiology of Stroke in Sickle Cell Disease. Microcirculation. 2004;11(2):195–208. doi: 10.1080/10739680490278600. [DOI] [PubMed] [Google Scholar]
- 90.Patel N, Tahara SM, Malik P, Kalra VK. Involvement of miR-30c and miR-301a in immediate induction of plasminogen activator inhibitor-1 by placenta growth factor in human pulmopnary endothelial cells. Biochem J. 2011;434:473–82. doi: 10.1042/BJ20101585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gonsalves Caryn S, Li C, Malik P, Tahara Stanley M, Kalra Vijay K. Peroxisome proliferator-activated receptor-α-mediated transcription of miR-301a and miR-454 and their host gene SKA2 regulates endothelin-1 and PAI-1 expression in sickle cell disease. Biosci Rep. 2015;35(6):e00275. doi: 10.1042/BSR20150190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boyd JH, Macklin EA, Strunk RC, DeBaun MR. Asthma is associated with Increased mortality in individuals with sickle cell anemia. Haematologica. 2007;92(8):1115–8. doi: 10.3324/haematol.11213. [DOI] [PubMed] [Google Scholar]
- 93.Ozbek OY, Malbora B, Sen N, Yazici AC, Ozyurek E, Ozbek N. Airway hyperreactivity detected by methacholine challenge in children with sickle cell disease. Pediatr Pulmonol. 2007;42(12):1187–92. doi: 10.1002/ppul.20716. [DOI] [PubMed] [Google Scholar]
- 94.Field JJ, Stocks J, Kirkham FJ, Rosen CL, Dietzen DJ, Semon T, et al. Airway Hyperresponsiveness in Children With Sickle Cell Anemia. Chest. 2011;139(3):563–8. doi: 10.1378/chest.10-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gonsalves CS, Kalra VK. Hypoxia-Mediated expression of 5-Lipoxygenase-Activating Protein involves HIF-1{alpha} and NF-{kappa}B and MicroRNAs 135a and 199a-5p. J Immunol. 2010;184(7):3878–88. doi: 10.4049/jimmunol.0902594. [DOI] [PubMed] [Google Scholar]
- 96.Fredman G, Li Y, Dalli J, Chiang N, Serhan CN. Self-Limited versus Delayed Resolution of Acute Inflammation: Temporal Regulation of Pro-Resolving Mediators and MicroRNA. Sci Rep. 2012;2:639. doi: 10.1038/srep00639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Busch S, Auth E, Scholl F, Huenecke S, Koehl U, Suess B, et al. 5-Lipoxygenase Is a Direct Target of miR-19a-3p and miR-125b-5p. J Immunol. 2015;194(4):1646–53. doi: 10.4049/jimmunol.1402163. [DOI] [PubMed] [Google Scholar]