

Abstract
Established strategies for discovering selective kinase inhibitors are target-centric as they often target certain structural or reactive features in the target kinase. In the absence of such prominent features, there is a lack of general methods for discovering selective inhibitors. Here we describe a new strategy that exploits conformational flexibility of kinases for achieving selective kinase inhibition. Through ring closure, we designed and synthesized a panel of isoquinoline-containing compounds as rigidified analogs of an amidophenyl-containing parent compound. These analogs potently inhibit kinases including Abl and BRAF but have diminished inhibition against some other kinases compared to the parent compound. Sequence analysis reveals that many of the kinases that are potently inhibited by the isoquonoline-containing compounds contain a long insertion within their catalytic domains. A crystal structure of one rigid compound bound to BRAF confirmed its binding mode. Our findings highlight the potential of a novel strategy of rigidification for improving the selectivity of kinase inhibitors.
Keywords: kinase inhibitors, selectivity, ring closure, rigidification, Abl, BRAF
Graphical abstract
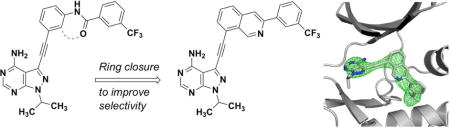
1. INTRODUCTION
Protein kinases play critical roles in numerous cellular processes including proliferation, differentiation, death, and migration [1]. Small-molecule inhibitors are highly attractive tools for dissecting the roles of protein kinases in these cellular processes because of their ability to acutely block the activity of protein kinases in cells [2]. In addition to serving as tool compounds for mapping kinase functions, small-molecule kinase inhibitors have been successfully developed as therapeutic agents for the treatment of human diseases, particularly the cancers whose proliferation is driven by the activity of oncogenic kinase [3]. With protein kinases established as a major class of drug targets, there are intensive efforts in discovering novel kinase inhibitors as therapeutic candidates.
The vast majority of kinase inhibitors have poor target selectivity because they bind to the active site that is highly conserved among the hundreds of human kinases [4, 5]. Structure-based design (SBD) has proven to be a powerful approach when a target kinase contains a rare structural feature (e.g. a special pocket) or a rare reactive feature (e.g. a cysteine residue) [6, 7]. In the absence of such distinct features, it is difficult to use SBD to design inhibitors that can distinguish between closely related kinases though.
Previous studies revealed protein conformational flexibility as one underexplored attribute of protein kinases [8]. It was demonstrated that highly homologous kinases could have distinct preferences for conformational states [9]. Small molecules could achieve selective inhibition by binding to conformational states that were only attainable by certain kinases [10].{Smith, 2009 #4} We thus reason that a more rigid inhibitor should be better accommodated by more flexible kinases giving rise to higher target selectivity. This notion is supported by a recently reported strategy that features the use of atropisomers, chiral molecules arising from hindered rotation around a single bond to increase the selectivity of kinase inhibitors. [11]. Gustafson and coworkers showed that rigidification of pyrrolopyrimidine-based inhibitors through an atropisomeric axis led to higher target selectivity in a panel of kinases [12, 13]. However, there have been no other investigations that focus on investigating the effects of rigidity on kinase inhibitors to the best of our knowledge. Our lab has previously developed novel pyrazolopyrimidine-based inhibitors of Bcr-Abl to overcome drug resistance caused by the T315I mutation [14]. While some of our alkyne-containing pyrazolopyrimidines exemplified by compound 1 potently inhibit Bcr-AblT315I, their selectivity for Abl kinase is poor, reflected by the fact that dozens of kinases were potently inhibited by compound 1. We hypothesize that rigidification of such promiscuous inhibitors through a ring-closure strategy would allow them to differentiate between kinases of different conformational flexibility and thus lead to improved selectivity. Here we report the design, synthesis and characterization of a series of alkyne-bridged isoquinolino-pyrazolo[3,4-d]pyrimidines as inhibitors of oncogenic kinases in vitro and in cells.
2. RESULTS AND DISCUSSION
2.1 Design
We have previously described 3-phenylacetylene substituted pyrazolo[3,4-d]pyrimidines as potent inhibitors of both the wild-type and a drug-resistant mutant of Bcr-Abl [14]. One of the most potent inhibitors in this series (1) contains a trifluoromethylphenyl group linked to a phenylacetylene moiety via an amide linker. Docking analysis predicts that the amide group is almost coplanar with the phenyl ring when 1 is bound to Abl [14]. We surmise that replacing the amide linker with a cyclized ring should maintain the active conformation of the inhibitor for binding to kinases like Abl while the increased rigidity may not be tolerated by other kinases thus leading to enhanced selectivity. The isoquinoline ring was chosen as the cyclized form of the amidophenyl group with the nitrogen atom mimicking the amide oxygen atom (Figure 1).
Figure 1.
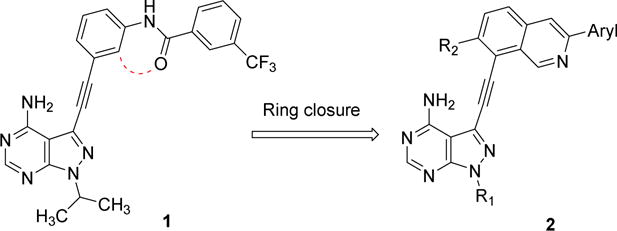
Cyclization of the amidobenzene group in a previously described Bcr-Abl inhibitor 1 yields the isoquinoline-containing compounds 2.
2.2. Chemistry
A panel of isoquinoline-containing candidate kinase inhibitors was designed based on the ring closure strategy. The synthesis of these compounds involved multiple cross-coupling reactions to connect three aryl rings together. Coupling of 3-iodopyrazolopyrimidines 3a or 3b with trimethylsilylacetylene followed by desilylation with K2CO3 produced 4a or 4b (Scheme 1) [15]. 8-Bromoisoquinolin-3-yl trifluoromethane sulfonate 5a was prepared under previously described conditions [16] and then reacted with different aryl boronic acids to generate 3-aryl-8-bromo-isoquinoline intermediates 6a–d. The Suzuki reactions proceeded with a good selectivity for the triflate over the bromine as the leaving group [17].
Scheme 1.
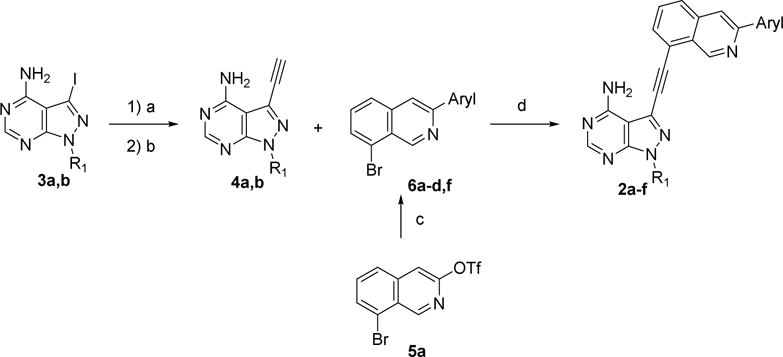
Reagents and conditions: (a) Trimethylsilylacetylene, (PPh3)4Pd, CuI, TEA, DMF, r.t., N2; (b) K2CO3, methanol; (c) arylboronic acid,(PPh3)4Pd, K2CO3, toluene/ethanol, 100 °C; (d) (PPh3)2PdCl2, CuI, DIPEA, DMF, 80 °C, N2.
Bromination of one resulting intermediate 6d and subsequent reaction with N-methylpiperazine yielded 6f (Scheme 2) [18].
Scheme 2.
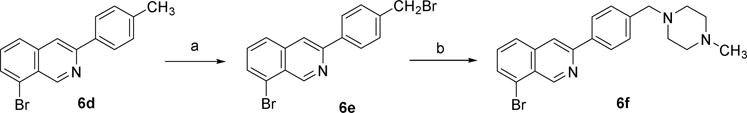
Reagents and conditions: (a) NBS, AIBN, CCl4, reflux; (b) N-methylpiperazine, Et3N, THF, 50 °C.
Finally, the intermediates 6 were reacted with pyrazolopyrimidines 4 to yield the target molecules 2a–f that were shown in Scheme 1. The synthesis of compound 2g required the development of a new route (Scheme 3). 2-Iodo-3-methylbenzonitrile was first reduced by AlH3 using a previously described method [19] to generate the benzyl amine intermediate 7, which was then converted into intermediate 5b using a route similar to that of 5a [16]. Finally, installation of a pyrazolopyrimidinoalkyne group and an aryl group on the isoquinoline ring with sequential cross-coupling reactions yielded 2g [15, 17].
Scheme 3.
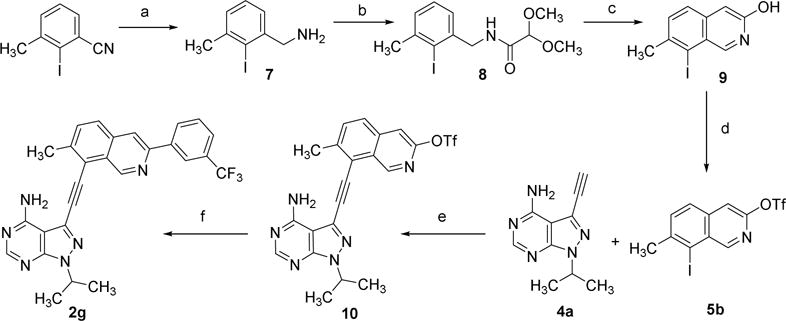
Reagents and conditions: (a) LiAlH4, H2SO4, THF; (b) methyl 2,2-dimethoxyacetate, 80 °C; (c) H2SO4, 40 °C; (d) (Tf)2O, pyridine; (e) (PPh3)4Pd, CuI, TEA, DMF, r.t., N2; (f) (3-(trifluoromethyl)phenyl)boronic acid, (PPh3)4Pd, K2CO3, toluene/ethanol, 100 °C.
2.3. Kinome profiling
We selected 2a out of the resulting panel of compounds and determined its kinome inhibition profile along with that of the parent compound 1 (Table S1). A panel of 244 kinases was selected for evaluation. Consistent with our previous profiling results against a smaller panel of kinases [14], 1 has poor selectivity reflected by the fact that at 1 μM it inhibited over 90% of the activity of 44 kinases in the panel. Compound 2a, on the other hand, inhibited 18 kinases by over 90% out of the same panel of kinases. Interestingly, the 18 primary targets of 2a are all shared with 1, suggesting that the cyclized isoquinoline ring in 2a is tolerated by a portion of the primary targets of 1. These dually sensitive targets include Abl, Arg, BRAF, FGFR, Kit, and RET, all of which are known to be highly sensitive to inhibition by type II kinase inhibitors [4, 20]. There are 26 kinases that were over 90% inhibited by 1 but not by 2a. The degrees of inhibition against these nonshared kinases vary dramatically between the two compounds as shown in the cases of Csk, Jak1, Ptk5, Slk, and Txk (Table S1), suggesting that these kinases may have difficulty accommodating the rigid ring structure introduced in 2a. We compared and analyzed the sequences of the dually sensitive kinases with those with the highest differential sensitivity to compounds 1 and 2a. While there is no obvious residue differences at positions predicted to line the inhibitor-binding site between the two groups, at least one third of the dually sensitive kinases contain long insertions within their kinase domains. For example, FGFR1, FGFR2, Klt4, Kdr, Kit, and Ret contain an insertion of over 14 amino acids between αD and αE helices (Figure 2). Such an insertion between the kinase N and C lobes may render these kinases conformationally more flexible and better at accommodating rigid inhibitors such as 2a. Additional factors such as differences in binding pockets and insertions elsewhere may contribute to the sensitivity to the rigid inhibitors reported here. Overall, the kinome profiling data suggest that the ring-closure strategy led to a moderate increase of kinase selectivity.
Figure 2.
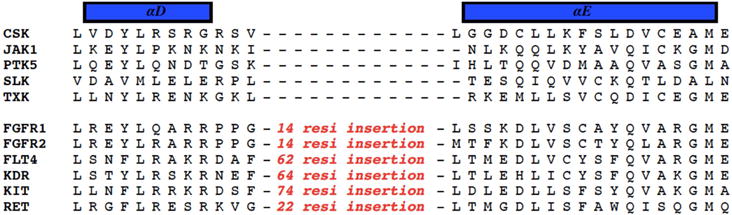
Sequence analysis reveals that most of the kinases that are sensitive to inhibition by both 1 and 2a (bottom group) contain a long insertion between their αD and αE helices compared to the kinases that are sensitive to 1 but not 2a (top group).
2.4. In vitro study
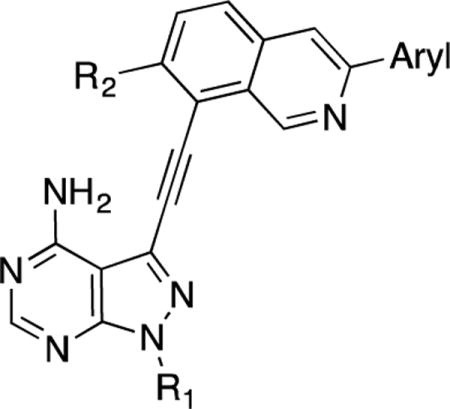
We then measured the potency of the panel of isoquinoline-containing compounds against a protein tyrosine kinase Abl in vitro using a phosphocellulose paper disk assay [21]. A screen of compounds 2a–g at a fixed concentration of 30 nM revealed their relative potency at inhibiting Abl kinase (Figure S1). We selected the more potent five compounds (2a, 2c, 2d, 2f and 2g) from the panel of isoquinoline-containing compounds and determined their IC50 values from dose-response studies. While these compounds inhibited Abl kinase quite potently, their potency is 5 to 36 fold lower than that of the parent compound 1 (Table 1). These results suggest that the introduction of a rigid isoquinoline ring is well tolerated by Abl kinase in vitro despite a small decrease of potency. We chose to also measure the inhibition of the isoquinoline-containing compounds against BRAF, a serine/threonine kinase potently inhibited by 2a based on the kinome profiling data. In particular, we tested the inhibitors against an oncogenic mutant (V600E) of BRAF that occurs in approximately half of advanced melanoma [22]. An in vitro assay involving phosphorylation of MEK by BRAF and immunoblot detection of phosphorylated MEK by a phospho-specific antibody was employed [23]. We determined the IC50 values of compounds 2a–g against BRAFV600E in dose-response studies (Table 1). The same assay revealed an IC50 value of 21 nM for vemurafenib, a FDA approved drug targeting BRAF enzyme, in agreement with previous reports [24]. Compounds 2a–g all have IC50 values less than 100 nM against BRAFV600E, exhibiting moderate decreases of potency in comparison with the parent compound 1 which displays an IC50 of 4 nM against BRAFV600E. Among them, 2d is the most potent inhibitor against BRAFV600E with an IC50 of 13 nM, indicating that an ethyl group is favored over an isopropyl group at the R1 position of the pyrazolopyrimidine for binding to BRAF kinase.
Table 1.
Inhibitory Activity as IC50 (nM) of Compounds 2a-f against BRAF-V600E and ABL in vitro and in cells.
| |||||||
|---|---|---|---|---|---|---|---|
| Compounds | Aryl | R1 | R2 |
in vitro
|
in cell
|
||
| ABLa | BRAF V600Ea | Ba/F3 BCR-ABLb | A375b | ||||
| 2a |
|
iPr | H | 3.0±0.3 | 65±15 | 942±119 | >10,000 |
| 2b |
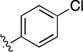
|
iPr | H | ndc | 46±11 | nd | nd |
| 2c |
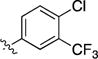
|
iPr | H | 22±6 | 87±12 | nd | nd |
| 2d |
|
Et | H | 3.5±0.4 | 13±1 | 1107±94 | >10,000 |
| 2e |
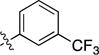
|
iPr | H | nd | 24±1 | nd | nd |
| 2f |
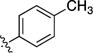
|
iPr | H | 7±1 | 51±3 | 354±84 | >10,000 |
| 2g |
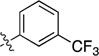
|
iPr | CH3 | 10±2 | 44±4 | 435±147 | 2300±23 |
| Vem | nd | 21±7 | nd | 127±3 | |||
| 1 | 0.6 | 4±2 | 7.5±0.5 | 97±2 | |||
IC50 values were determined by following the in vitro kinase assay protocols. The data represent the mean values of two independent experiments.
Growth inhibition was determined by MTT assay. Cells were plated in 96-well plates (1×103 cells/well for A375 and 2×104 cells/well for Ba/F3) and incubated with the inhibitors at different concentrations for 96 h and 48 h for A375 and Ba/F3 respectively. The data represent the mean values of two independent experiments, each carried out in triplicates.
not determined
2.5. Crystallography
Having confirmed the potent inhibition of two distinct oncogenic kinases by the isoquinoline-containing compounds 2a–g in vitro, we next set out to determine their binding mode to BRAF. To this end, we co-crystallized the representative inhibitor 2a bound to the BRAF kinase domain and solved its structure to 2.55Å resolution (Rfactor/Rfree=25.3/28.3) (see Table S2 for data collection and refinement statistics). Clear unbiased electron density was observed in the kinase ATP-pocket, allowing unambiguous modeling of 2a (Figure 3A). 2a engages the ATP-pocket of BRAF through key hydrogen bonds between the pyrazolo[3,4-d]pyrimidine of 2a with residues C532, Q530, and T529 of the hinge region and between the isoquinoline nitrogen of 2a with the backbone nitrogen of N594 (Figure 3B). Consistent with a type II inhibitor-binding mode, BRAF adopts a DFG-out conformation with the aryl group of 2a occupying the hydrophobic pocket formed by the displaced position of F595 (Figure 3C). Helix αC of the kinase N-lobe, resides in a productive-like inward position (Figure S2) that correlates with the kinase domain adopting a characteristic side-to-side dimer configuration reflective of the kinase-active conformation. Comparison of the binding mode of 2a with a non-cyclized inhibitor (72F, a close analogue of compound 1) bound to c-Src kinase domain shows that the position of the pyrazolo[3,4-d]pyrimidine of 2a resides in a near identical position of the pyrimidine rings of 72F inhibitor (Figure 3D). Also, the aryl group of both 2a and 72F reside in comparable positions in the hydrophobic pocket formed by the DFG-out conformation of the kinase domain (Figure 3D).
Figure 3.
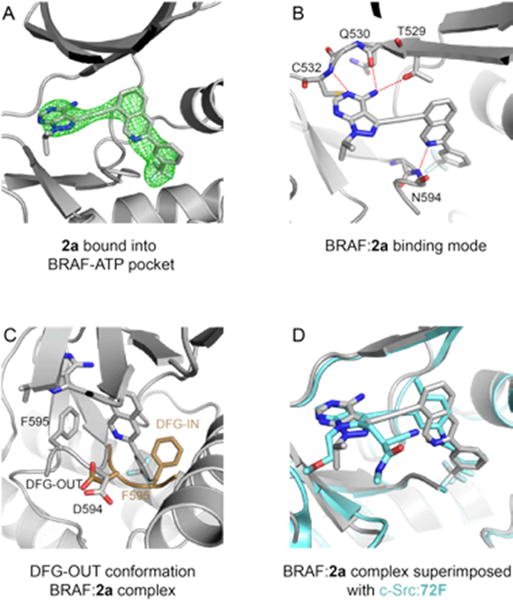
Crystal structure of 2a in complex with the BRAF kinase domain. (A). Unbiased Fo-Fc electron density maps contoured at 2.5σ in green showing 2a bound to the ATP binding pocket of BRAF kinase domain. (B). Binding mode of 2a with the BRAF ATP-binding pocket. Compound 2a and BRAF residues interacting with 2a are represented in stick. Red dash lines represent hydrogen bonds. (C). The BRAF protein adopts a DFG-out conformation when bound to 2a. The DFG loop of the BRAF:2a complex is colored in grey (adopting the DGF-out conformation). An example of a DFG loop of BRAF adopting the DFG-in conformation (PDB 4WO5) is superimposed in brown. (D). Superimposition of the BRAF:2a complex in grey with the c-SRC:72F complex in blue (PDB 5SYS).
Careful analysis of the structure did not show any evidence of distortion to the structure of BRAF to accommodate the rigidified compound 2a into the ATP binding pocket. These features confirm our design hypothesis that replacing the flexible amide linker in compound 1 with a cyclized ring in 2a–g will not perturb (but maintain) the overall binding mode to BRAF. These results also allow us to infer that any differences in sensitivity to inhibitors, likely reflects differences in the ability of the plastic kinase domain to accommodate a rigid inhibitor.
2.6. Antiproliferative effect
We next measured the inhibition of compounds 2 against Abl and BRAFV600E in cells taking advantage of the fact that Bcr-Abl and BRAFV600E are both established oncogenes that drive the proliferation of different cancer cells [25, 26]. Chemical inhibition of the oncogenic kinase should suppress the proliferation of such cancer cells. We first conducted MTT assays to evaluate the antiproliferative effects of the four most potent Abl inhibitors in vitro (2a, 2d, 2f and 2g) on Ba/F3 cells expressing Bcr-Abl. Surprisingly, all four inhibitors show relatively weak anti-proliferative potency against Bcr-Abl cell lines compared to what was observed with parent compound 1. The most potent compound in the series is 2f, displaying an IC50 value against Bcr-Abl Ba/F3 cells of 354 nM, is 44-fold less potent than that of the parent compound 1 (8 nM). The poor cellular efficacy of these compounds is rather unexpected in view of their potent inhibition against Abl kinase in vitro. We reason that the better potency of 2f compared to the other isoquinoline-containing compounds tested may reflect the increased bioavailability due to the incorporation of a piperazine group (Table 1).
We next measured the compounds’ IC50 values against A375 cells that harbor the oncogenic kinase BRAFV600E. Similar to what we observed with Bcr-Abl Ba/F3 cells, compounds 2a–g display rather poor efficacy at suppressing the proliferation of A375 cells (Table 1). The most effective compound of the series against BRAFV600E in vitro, namely 2d, has an IC50 value over 10 μM against A375 cells while the parent compound has an IC50 value of 97 nM. The low cellular efficacy of these compounds is in stark contrast with their potent inhibition of BRAFV600E in vitro.
In addition to the viability assays, we examined the effects of select compounds on the downstream signaling of the two oncogenic kinases, Bcr-Abl and BRAFV600E, in cells. Bcr-Abl activity leads to the autophosphorylation of Abl at Y412, thus phosphorylation level at this site is routinely employed as a marker of Bcr-Abl activity. Immunoblots revealed that 2f inhibited the phosphorylation of Abl at Y412 in BCR-ABL-expressing Ba/F3 cells with an IC50 value of 211 nM, in good agreement with that of the cell viability assay (Figure S3) [14]. In comparison, the potency of 2f is approximately 30-fold lower than that of compound 1 (IC50 = 7.5 nM) in the same assay. Phosphorylation levels of the downstream signaling components MEK and ERK are routinely measured to monitor the activity of oncogenic BRAF in cells. Vemurafenib abolished the phosphorylation of MEK and ERK at concentrations above 1 μM, confirming its potent inhibition of BRAFV600E in A375 cells (Figure 4). In contrast, the most potent in vitro inhibitor of BRAFV600E of the series, 2d, failed to suppress the phosphorylation of MEK at concentrations up to 100 μM.
Figure 4.
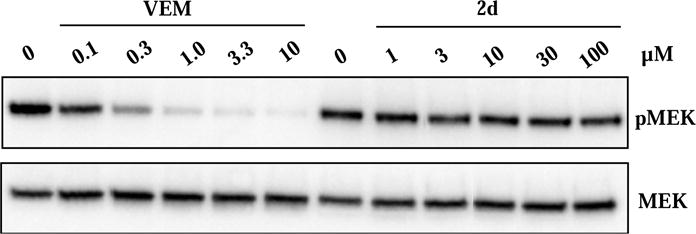
Effects of vemurafenib and compound 2d on downstream signaling of BRAFV600E in A375 cells. Vemurafenib (VEM) potently blocked phosphorylation of MEK while 2d had little effects at up to 100 μM. A375 cells were treated with vemurafenib or compound 2d at various concentrations before the cells were lysed, resolved by SDS-PAGE, transferred to PVDF membrane, and probed with antibodies for MEK and phospho-MEK (pMEK).
Taken together with the viability data shown above, these results suggest that compounds 2 have surprisingly low efficacy at engaging their targets in cells.
We reasoned that the lack of cell efficacy of the rigidified inhibitors might be due to diminished inhibition of full-length proteins, low solubility, poor membrane permeability, or a combination of the above factors. We first turned to immunoprecipitated kinase assays to determine whether the low cell efficacy is due to diminished binding of our rigid inhibitors to the full-length kinase. Full-length BRAFV600E was transfected into HEK293 cells, immunoprecipitated, and used to phosphorylate recombinant MEK in vitro. We found that 1 and 2a inhibited immunoprecipitated full-length BRAFV600E with IC50 values of 2 nM and 14 nM, respectively (Figure 5). These values are actually 2–4 folds better than their corresponding IC50 values against recombinant BRAFV600E kinase domain (Table 1), ruling out the possibility that lower cellular efficacy of 2a is due to a failure to inhibit full-length BRAFV600E.
Figure 5.
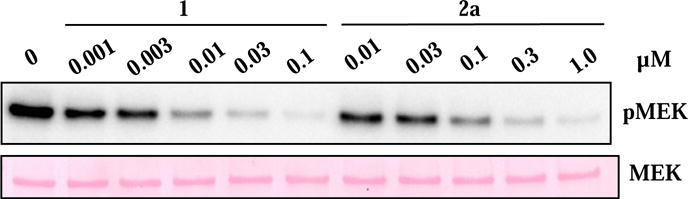
Compounds 1 and 2a caused potent, dose-dependent inhibition of full-length BRAFV600E in vitro. Full-length BRAFV600E was transfected into 293 cells, immunoprecipitated with a Flag antibody, and eluted with the Flag peptide before the activity was measured in the presence of various concentrations of either inhibitor.
We next tested whether the distinct efficacy of 1 and 2a is due to their solubility difference. The concentrations of saturated solutions of 1 and 2a in water at 37 °C were determined to be 161 and 60 μM, respectively (Tables S3 and S4). These data indicate that although less soluble than its parent compound, 2a should be sufficiently soluble at all the concentrations tested in the cell viability assays. Thus, the moderate solubility difference between 1 and 2a is unlikely to be responsible for the dramatically difference in their cell efficacy.
We then measured the permeability of 1 and 2a using the established Caco-2 assay [27]. The apparent permeability coefficient (Papp) and the efflux ratio defined as Papp(B−A)/Papp(A−B) were calculated following concentration measurements with LC-MS/MS (Table S5) [28]. Papp values for compounds 1 and 2a were determined to be 0.5 × 10−6 and 0.8 × 10−6 cm/s, respectively, indicating that the rigidified compound 2a has slightly higher apparent permeability than the parent compound presumably because of its greater hydrophobicity. The efflux ratios for 1 and 2a were determined to be 1.2 and 3.2, respectively, indicating that compound 2a is a better substrate of the well-established efflux pump P-glycoprotein (P-gp) than compound 1. These results suggest that compound 2a is actively pumped from inside to outside the cell membrane by efflux pumps like P-gp, which may account for the low cellular efficacy of the compound.
3. CONCLUSIONS
We have developed an innovative strategy for improving the selectivity of kinase inhibitors. The strategy of inhibitor rigidification that we report here is distinct from the common established approaches pursued to improve inhibitor selectivity in the field. Most of the previously reported strategies for inhibitor discovery are target-centric as they often target rare structural or reactive features in the target proteins. In contrast, our current strategy focuses on modifying rigidity, a physical property of the inhibitors. We demonstrate that rigidification through ring closure leads to a moderate but appreciable improvement in the selectivity of pyrazolopyrimidine-based kinase inhibitors. Depending on their sensitivity to the rigidified compounds, the kinase targets of the flexible parent compound can be classified into two groups: the tolerant group and the intolerant group. Sequence analysis reveals that many of the kinases that are potently inhibited by the isoquonoline-containing compounds contain a long insertion within their catalytic domains. This result is in agreement with our hypothesis that conformationally more flexible kinases are better at accommodating rigid small-molecule inhibitors.
The crystal structure of compound 2a in complex with BRAFV600E reveal that compound 2a is indeed a type II inhibitor targeting the DFG-out conformation of BRAF kinase. Interestingly, the nitrogen atom in the isoquinoline ring forms a hydrogen bond with the main-chain amide NH of G593. We believe that this feature of the binding mode of 2a is likely shared by most if not all the primary kinase targets of 2a because of the high structural conservation of the respective region among protein kinases.
We believe that the current strategy can be readily applied to type II kinase inhibitors, many of which contain rotatable amide or urea linkers between the adenine-pocket binding fragment and the allosteric pocket binding fragment. However, this strategy may be limited to improving selectivity for conformationally flexible kinases such as those containing a long insertion discovered here. Unfortunately, static crystal structures may not be the best tool to probe and validate this hypothesis in detail. To this end, the use of methods such as NMR and hydrogen–deuterium exchange, which provides direct measures of protein dynamics, may provide more insight. We note that many of the dually sensitive protein kinases that we found are implicated or proven to drive cancer growth, suggesting that there is great therapeutic utility for our strategy in the development of specific protein kinase inhibitors.
We found that rigidified compounds have surprisingly low cellular efficacy. Through a series of experiments, we demonstrate that this is due to the fact these compounds are good substrates for efflux pumps like P-gp. We suspect that the low permeability is peculiar for the current scaffold being used in this study. Indeed, an ongoing study in our lab shows that when applied to a different scaffold, the rigidification strategy yielded kinase inhibitors of high cellular efficacy (unpublished data). Taken together, these results suggest that inhibitor rigidification may be a useful strategy that could be used to improve selectivity for conformationally flexible targets.
4. EXPERIMENTAL SECTION
4.1 Chemistry
1H NMR and 13C NMR spectra were acquired on 400, 500, or 600 MHz Varian spectrometers. High-resolution mass was determined using a ThermoScientific™ Q Exactive™ hybrid quadrupole-Orbitrap mass spectrometer. Solubility and purities of final compounds were established by analytical HPLC by using Shimadzu HPLC system (model# DGU-20A5 CBM-20A LC-20AP SPD-M20A UV/vis). HPLC analysis conditions were Phenomenex C18(250 × 3.9 mm), flow rate of 1 mL/min, UV detection 254 nm, and 100% acetonitrile in isocratic mode (0.1% TFA was added into acetonitrile phase). All the target molecules (2a–f) were found to be >95% pure based on HPLC analysis. The Caco-2 permeability has been performed by SundiaMediTech Co., Ltd and based on their internal protocol.
4.1.1. 3-ethynyl-1-isopropyl-1H-pyrazolo[3,4-d] pyrimidin-4-amine (4a) and 1-ethyl-3-ethynyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (4b)
3-Iodo-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (3a) or 1-ethyl-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine (3b) was reacted with trimethylsilyl acetylene following a previously reported procedure before desilylation with potassium carbonate in methanol to yield the title compounds4a and 4b [15].
4.1.1.1 3-Ethynyl-1-isopropyl-1H-pyrazolo[3,4-d] pyrimidin-4-amine (4a)
Yield = 56%. 1H NMR (400 MHz, chloroform-d) δ 8.33 (s, 1H), 6.14 (s, 2H, NH2), 5.13 (hept, J = 6.7 Hz, 1H), 3.50 (s, 1H), 1.56 (d, J = 6.7 Hz, 6H).
4.1.1.2 1-Ethyl-3-ethynyl-1H-pyrazolo[3,4-d] pyrimidin-4-amine (4b)
Yield = 67%. 1H NMR (400 MHz, chloroform-d) δ 8.36 (s, 1H), 5.98 (s, 2H, NH2), 4.45 (q, J = 7.3 Hz, 2H), 1.51 (t, J = 7.3 Hz, 3H).
4.1.2. General procedure for synthesis of 8-Bromo-3-aryl-isoquinoline
A chemo selective Suzuki–Miyaura cross-coupling reaction was inspired by the previous reported method [17]. Briefly, different arylboronic acids (1 mmol) were added to a solution of compounds 5a that was prepared based on the previous literature [16] (1 mmol) in 10.0 mL of toluene/ethanol mixture (at a ratio of ¼, v/v,) and 1 mL of 2 M potassium carbonate aqueous solution. Palladium tetrakis (triphenyl phosphine) (0.05 mmol) was added to the above solution and was stirred for 2 h at 100 °C. After quenching the reaction with water, the resulting mixture was extracted with ethyl acetate and purified by silica column chromatography by using hexane and ethyl acetate as a mobile phase. Compounds 6a–e were synthesized with 43–58 % yield [17].
4.1.2.1. 8-Bromo-3-(3-(trifluoromethyl) phenyl) isoquinoline (6a)
1H NMR (400 MHz, chloroform-d) δ 9.64 (s, 1H), 8.36 (bs, 1H), 8.27 (d, J = 7.6 Hz, 1H), 8.00 (s, 1H), 7.84 – 7.75 (m, 2H), 7.66 – 7.53 (m, 2H), 7.49 (dd, J = 8.2, 7.5 Hz, 1H).
4.1.2.2. 8-Bromo-3-(4-chlorophenyl) isoquinoline (6b)
1H NMR (400 MHz, chloroform-d) δ 9.65 (s, 1H), 8.08 (d, J=8.5 Hz, 2H), 7.97 (s, 1H), 7.76-7.83 (m, 2H), 7.57 – 7.44 (m, 3H).
4.1.2.3. 8-Bromo-3-(4-chloro-3-(trifluoromethyl) phenyl) isoquinoline (6c)
1H NMR (400 MHz, chloroform-d) δ 9.69 (s, 1H), 8.51 (d, J = 2.3 Hz, 1H), 8.31 – 8.23 (m, 1H), 8.05 (s, 1H), 7.90 – 7.83 (m, 2H), 7.65 (d, J = 8.4 Hz, 1H), 7.57 (t, J = 7.9 Hz, 1H).
4.1.2.4. Bromo-3-(p-tolyl) isoquinoline (6d)
1H NMR (400 MHz, chloroform-d) δ 9.66 (s, 1H), 8.04 (d, 2H, J = 8 Hz), 7.97 (s,1H), 7.74-7.81 (m, 2H), 7.48 (dd, J = 8.2, 7.5 Hz, 1H), 7.33 (d, 2H, J = 8 Hz), 2.43 (s, 3H).
4.1.3. 8-Bromo-3-(4-(bromomethyl) phenyl) isoquinoline (6e)
To the solution of 1 mmol compound 6d in 10 ml CCl4, 1 mmol of NBS and 5 mg AIBN were added and the mixture was refluxed at 80 °C for about 24 h. The reaction mixture was diluted with water and extracted with ethyl acetate with the product purified by silica column chromatography using hexane and ethyl acetate as a mobile phase. [18]
Yield = 52%. 1H NMR (500 MHz, chloroform-d) δ 9.68 (s, 1H), 8.15 (d, 2H, J = 8 Hz), 8.01 (s, 1H), 7.86 – 7.79 (m, 2H), 7.58 – 7.49 (m, 3H), 4.59 (s, 2H).
4.1.4. 8-Bromo-3-(4-((4-methylpiperazin-1-yl) methyl) phenyl) isoquinoline (6f)
Compound 6e (1.0 mmol) was added in 2.0 mL anhydrous THF, and N-methyl piperazine (0.3 mL, 2.4 mmol) and triethylamine (0.5 mL, 3.0 mmol). The solution was stirred at 50 °C overnight. After evaporation of the solvent under the reduced pressure, the residue was diluted with water and extracted with ethyl acetate. The product was purified by silica column chromatography using DCM and CH3OH (at a ratio of 20:1) as the mobile phase.[18]
Yield = 73%. 1H NMR (400 MHz, DMSO-d6) δ 9.52 (s, 1H), 8.48 (s, 1H), 8.22 (d, 2H, J = 8 Hz), 8.06 (d, 1H, J = 8.2 Hz), 7.97 (d, J = 7.5 Hz, 1H), 7.69 (dd, J = 8.2, 7.5 Hz, 1H), 7.47 (d, J = 8.0 Hz, 2H), 3.60 (s, 2H), 3.14 – 2.77 (m, 8H), 2.56 (s, 3H).
4.1.5. General procedure for synthesis of compound 2a–f
A mixture of 4 (1mmol), 6 (1mmol), Cu(I) iodide (0.01mmol), and Pd(PPh3)2Cl2 (0.01mmol) were dissolved in 10 mL DMF in the sealed flask. The flask was evacuated and backfilled with dry nitrogen gas three times. DIPEA (4mmol) was then added before the reaction was stirred at 80°C overnight. The reaction mixture was diluted with 5% ammonium hydroxide solution and extracted with ethyl acetate. The residue was purified by flash column chromatography on silica gel using hexane and ethyl acetate as a mobile phase, with the exception of compound 6f which was purified using DCM and CH3OH (at a ratio of 20:1) as the mobile phase. The products were obtained with 30–45 % yield [29].
4.1.5.1. 1-Isopropyl-3-((3-(3-(trifluoromethyl)phenyl)isoquinolin-8-yl)ethynyl)-1H-pyrazolo[3,4 -d]pyrimidin-4-amine (2a)
1H NMR (600 MHz, DMSO-d6) δ 9.73 (s, 1H, H1-isoquinoline), 8.69 (s, 1H, H4-isoquinoline), 8.57 (bs, 1H, H2-phenyl), 8.54 (d, J= 8 Hz, 1H, H4-phenyl), 8.26 (s, 1H, H6-pyrazolopyrimidine), 8.14 (dd, J = 8.5, 1.0 Hz, 1H, isoquinoline), 8.12 (dd, J = 7.2, 1.1 Hz, 1H, isoquinoline), 7.87 (dd, J = 8.2, 7.2 Hz, 1H, H6-isoquinoline), 7.82 – 7.78 (m, 1H, phenyl), 7.77 (m, 1H, phenyl), 5.07 (hept, J = 6.6 Hz, 1H, CH), 1.49 (d, J = 6.7 Hz, 6H, CH3).13C NMR (151 MHz, DMSO-d6) δ 158.29, 156.72, 152.86, 150.81, 149.21, 139.82, 136.69, 133.31, 131.13, 130.84, 130.52, 130.22 (q, J = 31.40 Hz), 129.22, 127.07, 125.83, 125.12, 123.36, 120.03, 117.66, 101.36, 89.45, 87.63, 49.42, 22.19. HRMS(ESI) calculated for C26H19 F3N6 [M+H]+ is 473.1702, found 473.1708.
4.1.5.2. 3-((3-(4-chlorophenyl)isoquinolin-8-yl)ethynyl)-1-isopropyl-1H-pyrazolo[3,4-d]pyrimi din-4-amine (2b)
1H NMR (500 MHz, DMSO-d6) δ 9.73 (s, 1H, H1-isoquinoline), 8.58 (s, 1H, H4-isoquinoline), 8.29 (d, J = 8 Hz, 2H, phenyl), 8.28 (s, 1H, H6-pyrazolopyrimidine), 8.17 – 8.09 (m, 2H), 7.88 (dd, J = 8.0, 7.1, 1H, H6-isoquinoline), 7.61(d, J = 8 Hz, 2H, phenyl), 5.15 – 5.04 (hept, J = 6.7 Hz, 1H, CH), 1.52 (d, J = 6.7 Hz, 6H, CH3).13C NMR (126 MHz, DMSO-d6) δ 158.32, 156.75, 152.88, 150.71, 149.86, 137.68, 136.72, 134.22, 133.10, 131.05, 129.37, 129.16, 128.80, 126.86, 125.15, 120.01, 117.03, 109.99, 101.38, 89.54, 87.56, 49.44, 22.22. HRMS(ESI) calculated for C25H19 ClN6 [M+H]+ is 439.1438, found 439.1431.
4.1.5.3. 3-((3-(4-chloro-3-(trifluoromethyl)phenyl)isoquinolin-8-yl)ethynyl)-1-isopropyl-1H-pyr azolo[3,4-d]pyrimidin-4-amine (2c)
1H NMR (500 MHz, DMSO-d6) δ 9.73 (s, 1H, H1-isoquinoline),8.72 (d, 1H, H4-isoquinoline), 8.68 (d, J = 2.2 Hz, 1H, H2-phenyl),8.54 (dd, J = 8.5, 2.2 Hz, 1H, H4-phenyl), 8.28 (s, 1H, H6-pyrazolopyrimidine), 8.17 – 8.11 (m, 2H), 7.93 – 7.85 (m, 2H), 5.09 (hept, J = 6.7 Hz, 1H, CH), 1.51 (d, J = 6.7 Hz, 6H, CH3).13C NMR (126 MHz, DMSO-d6) δ 158.31, 156.74, 152.88, 150.90, 148.17, 138.33, 136.64, 133.50, 132.78, 132.19, 131.51, 131.27, 129.24, 127.63 (q, J = 30.60 Hz), 127.14, 126.64, 125.93, 125.11, 124.47, 122.30, 120.11, 117.88, 101.38, 89.40, 87.70, 49.44, 22.21, 14.54.HRMS(ESI) calculated for C26H18ClF3N6[M+H]+ is 507.1312, found 507.13202.
4.1.5.4. 1-Ethyl-3-((3-(3-(trifluoromethyl)phenyl)isoquinolin-8-yl)ethynyl)-1H-pyrazolo[3,4-d]p yrimidin-4-amine (2d)
1H NMR (500 MHz, DMSO-d6) δ 9.76 (s, 1H, H1-isoquinoline), 8.73 (s, 1H, H1-isoquinoline), 8.60 (bs, 1H, H2-phenyl), 8.58 (d, J = 8 Hz, 1H, H4-phenyl),8.29 (s, 1H, H6-pyrazolopyrimidine), 8.21 – 8.12 (m, 2H, isoquinoline), 7.90 (dd, J = 8.1, 7.2 Hz, 1H, H6-isoquinoline), 7.86 – 7.76 (m, 2H, phenyl), 4.41 (q, J = 7.3 Hz, 2H, CH2), 1.45 (t, J = 7.3 Hz, 3H, CH3).13C NMR (126 MHz, DMSO-d6) δ 158.32, 156.93, 153.28, 150.81, 149.24, 139.84, 136.72, 133.34, 131.17, 130.88, 130.56, 130.25 (q, J = 31.36 Hz), 129.27, 127.10, 125.89, 125.32, 123.67, 120.39 120.03, 117.71, 101.20, 89.54, 87.55, 42.50, 15.08.HRMS(ESI) calculated for C25H17F3N6 [M+H]+ is 459.1545, found 459.15527.
4.1.5.5. 1-Isopropyl-3-((3-(p-tolyl)isoquinolin-8-yl)ethynyl)-1H-pyrazolo[3,4-d]pyrimidin-4-am ine (2e)
1H NMR (600 MHz, DMSO-d6) δ 9.68 (s, 1H, H1-isoquinoline), 8.46 (s, 1H, H4-isoquinoline), 8.26 (s, 1H, H6-pyrazolopyrimidine), 8.13 (d, J = 8 Hz, 2H, phenyl), 8.09 (d, J = 8.4 Hz, 1H, isoquinoline), 8.06 (d, J = 7.2 Hz, 1H, isoquinoline), 7.82 (dd, J = 8.4, 7.2 Hz, 1H, H6-isoquinoline), 7.33 (d, 2H, phenyl), 5.06 (hept, J = 6.7 Hz, 1H, CH), 2.36 (s, 3H, p-methyl), 1.49 (d, J = 6.7 Hz, 6H, CH3).13C NMR (151 MHz, DMSO-d6) δ 158.30, 156.72, 152.86, 151.20, 150.47, 138.90, 136.78, 136.05, 132.66, 130.79, 129.91, 129.04, 126.95, 126.59, 125.17, 119.87, 116.16, 101.35, 89.64, 87.43, 49.41, 22.19, 21.28.HRMS(ESI) calculated for C26H22N6 [M+H]+ is 419.1984, found 419.19894.
4.1.5.6. 1-Isopropyl-3-((3-(4-((4-methylpiperazin-1-yl)methyl)phenyl)isoquinolin-8-yl)ethynyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (2f)
1H NMR (500 MHz, DMSO-d6) δ 9.72 (s, 1H, H1-isoquinoline), 8.51 (s, 1H, H4-isoquinoline), 8.28 (s, 1H, H6-pyrazolopyrimidine), 8.21 (d, J = 8.1 Hz, 2H, phenyl), 8.13 (d, J = 8.4 Hz, 1H, isoquinoline), 8.10 (d, J = 7.2 Hz, 1H, isoquinoline), 7.86 (dd, J = 8.4, 7.2 Hz, 1, H6-isoquinoline), 7.46 (d, J = 8.1 Hz, 2H, phenyl), 5.09 (hept, J = 6.8 Hz, 1H, CH), 3.54 (s, 2H, phenylCH2), 2.30-2.60 (bs, 8H, CH2), 2.22 (s, 3H, NCH3), 1.52 (d, J = 6.7 Hz, 6H, CH3).13C NMR (126 MHz, DMSO-d6) δ 158.33, 156.75, 152.88, 151.09, 150.56, 139.58, 137.60, 136.78, 132.81, 130.87, 129.78, 129.10, 126.94, 126.69, 125.19, 119.93, 116.53, 101.37, 89.63, 87.49, 62.06, 54.94, 52.65, 49.43, 45.77, 22.22. HRMS(ESI) calculated for C31H32N8 [M+H]+ is 517.2828, found 517.28473.
4.1.6. (2-iodo-3-methylphenyl) Methanamine (7)
1.05 g of concentrated sulfuric acid was added dropwise to 21 mL of 1M LiAlH4 suspension in THF and vigorously was stirred for 1 h at 0 °C. Then 2.43 g2-iodo-3-methylbenzonitrile (10 mmol) was dissolved in anhydrous THF and the mixture was stirred overnight. Finally, the reaction was quenched by addition of 15 mL of 2.4 M sodium hydroxide and then the organic phase was extracted with ethyl acetate. The final product was purified by flash chromatography using by silica gel flash column chromatography using DCM and CH3OH (at a ratio of 20:1) as the mobile phase.[19] The yield was 53%.1H NMR (400 MHz, chloroform-d) δ 7.24 – 7.18 (m, 1H), 7.18 – 7.11 (m, 2H), 3.91 (s, 2H, CH2), 2.48 (d, J = 0.6 Hz, 3H, CH3).
4.1.7. 8-Iodo-7-methylisoquinolin-3-ol (9)
Compound 7 (15 mmol) and methyl 2,2-dimethoxypropionate (30 mmol) were kept in a 10 mL screw-capped vial was stirred at 80 ° C overnight. The reaction mixture was loaded on silica and resulting dimethoxy acetamide intermediate 8 was purified by silica column chromatography using hexane and ethylacetate at a ratio of 2:1. The resulted compound without further purification was dissolved in 5 mL concentrated sulfuric acid and stirred at 40 °C overnight. Subsequently, the acidic solution poured into ice–water, and the precipitate was collected and washed with water and diluted sodium bicarbonate solution [16]. The yield was 42%.1H NMR (500 MHz, DMSO-d6) δ 8.91 (s, 1H, H1-isoquinoline), 7.64 (d, J = 8.4 Hz, 1H), 7.49 (d, J = 8.4 Hz, 1H), 6.87 (s, 1H, H4-isoquinoline), 5.74 (bs, 1H, OH), 2.54 (s, 3H, CH3).
4.1.8. 8-Iodo-7-methylisoquinolin-3-yl trifluoromethanesulfonate (5b)
The 1.8 mmol dried compound 10 was dissolved in 12.5 mL dry pyridine and stirred at 0 °C. Trifluoromethane sulfonic anhydride (0.5 mL) was added dropwise and the solution warmed to room temperature and stirred overnight. Subsequently, the solvent was reduced under reduced pressure and the product was purified by silica column chromatography using hexane and ethyl acetate at a ratio of 10:1.[16] The yield of this reaction is 72 %. 1H NMR (500 MHz, chloroform-d) δ 9.40 (s, 1H, H1-isoquinoline), 7.78 (d, J = 8.3 Hz, 1H), 7.64 (d, J = 8.3 Hz, 1H), 7.48 (s, 1H, H4-isoquinoline), 2.75 (s, 3H, CH3).
4.1.9. 8-((4-amino-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)ethynyl)-7-methylisoquinoli n-3-yl trifluoromethane sulfonate (10)
Compound 11 was prepared by a similar procedure that used to prepare compound 4a and 4b.[15] The reaction yield was 43%.1H NMR (500 MHz, chloroform-d) δ 9.64 (s, 1H, H1-isoquinoline), 8.44 (bs, 1H, H6-pyrazolopyrimidine), 7.87 (d, J = 8.6 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.59 (s, 1H), 5.84 (s, 1H, H4-isoquinoline), 5.22 (hept, J = 6.7 Hz, 1H, CH), 2.82 (s, 3H, CH3), 1.64 (d, J = 6.7 Hz, 6H, CH3). HRMS(ESI) calculated for C21H17F3N6O3S [M+H]+ is 491.11077, found 491.11072.
4.1.10. 1-Isopropyl-3-((7-methyl-3-(3-(trifluoromethyl)phenyl)isoquinolin-8-yl)ethynyl)-1H-pyr azolo[3,4-d]pyrimidin-4-amine (2g)
Compound 2g was prepared by a similar procedure that used to prepare compound 6 [17]. The reaction yield was 32%.1H NMR (500 MHz, DMSO-d6) δ 9.72 (s, 1H, H1-isoquinoline), 8.66 (s, 1H, H4-isoquinoline), 8.57 (bs, 1H, H2-phenyl), 8.55 (d, J = 8 Hz, 1H, H4-phenyl), 8.29 (s, 1H, H6-pyrazolopyrimidine), 8.07 (d, J = 8.4 Hz, 1H), 7.86 – 7.74 (m, 3H), 5.09 (hept, J = 6.9 Hz, 1H, CH), 2.74 (s, 3H, CH3), 1.52 (d, J = 6.7 Hz, 6H, CH3). 13C NMR (126 MHz, DMSO-d6) δ 158.38, 156.74, 152.88, 150.40, 148.48, 142.38, 139.91, 135.15, 133.66, 130.71, 130.51, 130.21 (q, J = 31.40 Hz), 128.67, 127.47, 125.70, 125.28, 123.25, 118.46, 117.51, 101.46, 91.66, 88.37, 49.50, 22.22, 21.46.HRMS(ESI) calculated for C27H21F3N6 [M+H]+ is 487.1858, found 487.18613.
4.2. Determination of compounds’ water solubility
Water solubility of compounds 1 and 2a were determined according to the previously literature [30]. Around 1 to 2 mg of each compound was added to 1 mL of deionized water. The suspensions were shaken at 37 °C for 24 hr. The mixture was centrifuged, and the supernatants were filtered through using Phenomenex PTFE membrane, 0.2 μL. The supernatant was diluted two folds with acetonitrile and injected into the HPLC. The solubility was calculated based on the calibration curve for five different concentrations. Each sample was injected at least three times.
4.3. Biology activity
4.3.1. In Vitro Kinase Assay
Abl kinase assays were conducted following a previously described procedure.[14] BRAF kinase assays were carried out by incubating 100 μM ATP, 0.3 nM of BRAFV600E, and 3 μM of MEKK97R with various concentrations of an inhibitor at 30°C for 20 min. The reaction mixture was then quenched with sample buffer, boiled, resolved by SDS-PAGE, and transferred to PVDF membrane. The membrane was finally probed with an antibody for phospho-MEK [23].
4.3.2. Cells and Reagents
A375 cells expressing BRAF V600E were purchased from ATCC. The cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM)(Corning) supplemented with 10% fetal bovine serum (v/v, HyClone, ThermoScientific) at 37 °C and 5.0% CO2. Vemurafinib was purchased from Selleckchem. The following antibodies were purchased from Cell Signaling (Boston, MA, USA): anti-pMEK, anti-pERK, anti-MEK, and anti-ERK. Anti-mouse secondary antibody was purchased from Jackson Immuno Research Laboratories (West Grove, PA, USA), whereas anti-rabbit secondary antibody was obtained from Abcam (Cambridge).
4.3.3. Western Blot Analysis
A375 cells expressing B-RAF V600E were treated with vehicle (DMSO), drug, or the inhibitors at various concentrations for 1 h. The cells were lysed, collected, and normalized using BCA protein assay kit (Bio-Rad). The samples were diluted in 4X SDS loading buffer (Bio-Rad), resolved by 4–20% SDS-PAGE (Bio-Rad) and then transferred onto PVDF membrane (Bio-Rad). Standard immunoblotting protocols were used. All Western blots were blocked with 5% nonfat milk in TBST (10 mMTris, 150 mMNaCl, 0.1% Tween-20, pH 8.0) for 1 h at room temperature. The primary antibodies were diluted to an appropriate concentration and incubated overnight at 4 °C. Subsequently, the blots were probed with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10 000) for 1 h at room temperature and then developed using enhanced chemiluminescence (ECL, Bio-Rad) and visualized by ChemiDoc XRS+ molecular imager (Bio-Rad).
4.3.4. MTT Assay
Cells were plated into 96-wells plates (1×103 cells/well for A375, and 2×104 cells/well for Ba/F3) and incubated with drugs or inhibitors at the indicated concentrations (96 h for A375 and 48 h for Ba/F3). Cell viability was then measured by adding MTT (Calbiochem) and incubating for 1 h. The reaction was quenched with formazan-solubilization reagent (50% DMF (v/v), 20% SDS w/v in H2O), and the absorbance was determined at 540 nm using SpectraFluoro Plus plate reader (Tecan). The reaction was carried out in triplicate, and the IC50 values were determined by using Graphpad Prism with a nonlinear regression method.
4.4. Protein expression, purification and crystallization
BRAF16mut was expressed with TEV protease cleavable His tags using a pProEx vector in BL21(DE3)-RIL bacterial expression cells and purified with nickel-affinity chromatography, TEV-cleavage overnight and purified through gel-filtration chromatography into a final buffer of 20 mM HEPES, pH 7.5, 200 mM NaCl, 1 mM TCEP, 5mM MgCl2 and 5% (v/v) glycerol. Following gel filtration, all protein fractions corresponding to greater than 95% purity were pooled and concentrated to 20 mg/mL, then flash frozen in liquid nitrogen. Protein concentration was estimated by UV-Vis absorption spectosctopy using a NanoDrop spectrophotometer (Thermo Scientific).
Braf16mut:2a complex was obtained by mixing Braf16mut at 155 μM (5g.L−1) with 2a at 300μM for 30 min at 4°C followed by centrifugation at 18000g for 5min at 4°C to remove precipitation. Braf16mut:2a complex was crystallized at 20˚C in hanging drop with 1:1μL mix in 30% PEG 3350, 0.1M Bis-Tris Propane pH 6.5, 0.2M Sodium Nitrate. X-ray diffraction data were collected on a flash-frozen crystal cryo-protected in mother liquor containing 25% ethylene glycol at 100K on station 24-ID-C, NE CAT beamline, Advanced Photon Source (APS). Data reduction was performed using XDS package [31]. The structure was solved by molecular replacement using PDB 4R5Y (REF) as a search model in Phaser [32]. Structure of 2a was build using elbow [33]. Model building and refinement was performed using COOT [34] and PHENIX [35]. The data statistics and refinement details are reported in Table S2 in Supporting Information.
Supplementary Material
Highlights.
Creation of a ring-closure strategy to leverage the different conformational flexibility of protein kinases for improving target selectivity.
Implementation of the strategy on an aminobenzene-containing inhibitor (1) afforded isoquinoline-based analogues (2) that exhibited a moderate increase of selectivity.
The crystal structure of compound 2a in complex with BRAFV600E was solved, which confirmed 2a as a type II inhibitor targeting the DFG-out conformation of BRAF kinase.
Acknowledgments
We thank Dr. N. A. Graham and Mr. A. Delfarah for their generous help with HR-MS data collection. This work was supported by the National Science Foundation (CHE-1455306 to C.Z.), the National Institute of Health (R01DE026003 to C.Z.), the Canadian Cancer Society Research Institute (704116 to F.S.), and by the Canadian Institutes for Health Research (FDN143277 to F.S.). P.M. was supported by a TD Bank postdoctoral fellowship. F.S. holds a Canada Research Chair (Tier 1) in Structural Biology of Cell Signaling. Diffraction work conducted at the Northeastern Collaborative Access Team beamlines was funded by the National Institute of Health (P41 GM103403 and S10 RR029205).
Appendix A. Supplementary data
1H-NMR and 13C-NMR spectra, and HPLC chromatograms of title compounds can be found in the Supporting Information.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession codes
The coordinates for the crystal structures of BRAF with compound 2a have been uploaded to Protein Data Bank with the accession code still pending.
Note
The authors declare no conflict of interest.
References
- 1.Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 2.Müller S, Chaikuad A, Gray NS, Knapp S. The ins and outs of selective kinase inhibitor development. Nature chemical biology. 2015;11:818–821. doi: 10.1038/nchembio.1938. [DOI] [PubMed] [Google Scholar]
- 3.Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discovery Today. 2016;21:5–10. doi: 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M. A small molecule–kinase interaction map for clinical kinase inhibitors. Nature biotechnology. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 5.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, Mclauchlan H, Klevernic I, Arthur JSC, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochemical Journal. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brear P, De Fusco C, Georgiou KH, Francis-Newton NJ, Stubbs CJ, Sore HF, Venkitaraman AR, Abell C, Spring DR, Hyvönen M. Specific inhibition of CK2a from an anchor outside the active site. 2016 doi: 10.1039/c6sc02335e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 9.Krishnamurty R, Brigham JL, Leonard SE, Ranjitkar P, Larson ET, Dale EJ, Merritt EA, Maly DJ. Active site profiling reveals coupling between domains in SRC-family kinases. Nature chemical biology. 2013;9:43–50. doi: 10.1038/nchembio.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong M, Seeliger MA. Targeting conformational plasticity of protein kinases. ACS chemical biology. 2014;10:190–200. doi: 10.1021/cb500870a. [DOI] [PubMed] [Google Scholar]
- 11.Clayden J, Moran WJ, Edwards PJ, LaPlante SR. The challenge of atropisomerism in drug discovery. Angewandte Chemie International Edition. 2009;48:6398–6401. doi: 10.1002/anie.200901719. [DOI] [PubMed] [Google Scholar]
- 12.Maddox S, Hecht D, Gustafson JL. Enhancing the selectivity of kinase inhibitors in oncology: a chemical biology perspective, in. Future Science. 2016 doi: 10.4155/fmc.15.193. [DOI] [PubMed] [Google Scholar]
- 13.Smith DE, Marquez I, Lokensgard ME, Rheingold AL, Hecht DA, Gustafson JL. Exploiting atropisomerism to increase the target selectivity of kinase inhibitors. Angewandte Chemie International Edition. 2015;54:11754–11759. doi: 10.1002/anie.201506085. [DOI] [PubMed] [Google Scholar]
- 14.Liu X, Kung A, Malinoski B, Prakash GS, Zhang C. Development of alkyne-containing pyrazolopyrimidines to overcome drug resistance of Bcr-Abl kinase. Journal of medicinal chemistry. 2015;58:9228–9237. doi: 10.1021/acs.jmedchem.5b01125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang CH, Zheng MW, Li YP, Lin XD, Huang M, Zhong L, Li GB, Zhang RJ, Lin WT, Jiao Y. Design, synthesis, and structure–activity relationship studies of 3-(Phenylethynyl)-1 H-pyrazolo [3, 4-d] pyrimidin-4-amine derivatives as a new class of Src inhibitors with potent activities in models of triple negative breast cancer. Journal of medicinal chemistry. 2015;58:3957–3974. doi: 10.1021/acs.jmedchem.5b00270. [DOI] [PubMed] [Google Scholar]
- 16.Durola F, Hanss D, Roesel P, Sauvage JP, Wenger OS. A new family of biisoquinoline chelates. European journal of organic chemistry. 2007;2007:125–135. [Google Scholar]
- 17.Hosoi S, Ozeki M, Nakano M, Arimitsu K, Kajimoto T, Kojima N, Iwasaki H, Miura T, Kimura H, Node M. Mechanistic aspects of asymmetric intramolecular Heck reaction involving dynamic kinetic resolution: flexible conformation of the cyclohexenylidene– benzene system. Tetrahedron. 2015;71:2317–2326. [Google Scholar]
- 18.Li Y, Shen M, Zhang Z, Luo J, Pan X, Lu X, Long H, Wen D, Zhang F, Leng F. Design, synthesis, and biological evaluation of 3-(1 H-1, 2, 3-triazol-1-yl) benzamide derivatives as potent pan Bcr-Abl inhibitors including the threonine315→ isoleucine315 mutant. Journal of medicinal chemistry. 2012;55:10033–10046. doi: 10.1021/jm301188x. [DOI] [PubMed] [Google Scholar]
- 19.Windhorst AD, Timmerman H, Worthington EA, Bijloo GJ, Nederkoorn PH, Menge WM, Leurs R, Herscheid JD. Characterization of the binding site of the histamine H3 Receptor. 2. Synthesis, in vitro pharmacology, and QSAR of a series of monosubstituted benzyl analogues of thioperamide. Journal of medicinal chemistry. 2000;43:1754–1761. doi: 10.1021/jm981106w. [DOI] [PubMed] [Google Scholar]
- 20.Vijayan R, He P, Modi V, Duong-Ly KC, Ma H, Peterson JR, Dunbrack RL, Jr, Levy RM. Conformational analysis of the DFG-out kinase motif and biochemical profiling of structurally validated type II inhibitors. Journal of medicinal chemistry. 2014;58:466–479. doi: 10.1021/jm501603h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Witt JJ, Roskoski R. Rapid protein kinase assay using phosphocellulose-paper absorption. Analytical biochemistry. 1975;66:253–258. doi: 10.1016/0003-2697(75)90743-5. [DOI] [PubMed] [Google Scholar]
- 22.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, Zhang J, Lin J, Ewing T, Matusow B. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015 doi: 10.1038/nature14982. [DOI] [PubMed] [Google Scholar]
- 25.Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proceedings of the National Academy of Sciences. 1988;85:9312–9316. doi: 10.1073/pnas.85.23.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Project CG, Jones CM, Marshall CJ, Springer CJ. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 27.Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology. 1989;96:736–749. [PubMed] [Google Scholar]
- 28.Yamashita S, Konishi K, Yamazaki Y, Taki Y, Sakane T, Sezaki H, Furuyama Y. New and better protocols for a short‐term Caco‐2 cell culture system. Journal of pharmaceutical sciences. 2002;91:669–679. doi: 10.1002/jps.10050. [DOI] [PubMed] [Google Scholar]
- 29.Ren X, Pan X, Zhang Z, Wang D, Lu X, Li Y, Wen D, Long H, Luo J, Feng Y. Identification of GZD824 as an Orally Bioavailable Inhibitor That Targets Phosphorylated and Nonphosphorylated Breakpoint Cluster Region–Abelson (Bcr-Abl) Kinase and Overcomes Clinically Acquired Mutation-Induced Resistance against Imatinib. Journal of medicinal chemistry. 2013;56:879–894. doi: 10.1021/jm301581y. [DOI] [PubMed] [Google Scholar]
- 30.Ye N, Zhu Y, Chen H, Liu Z, Mei FC, Wild C, Chen H, Cheng X, Zhou J. Structure– Activity Relationship Studies of Substituted 2-(Isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl Cyanide Analogues: Identification of Potent Exchange Proteins Directly Activated by cAMP (EPAC) Antagonists. Journal of medicinal chemistry. 2015;58:6033–6047. doi: 10.1021/acs.jmedchem.5b00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kabsch W. Xds. Acta Crystallographica Section D: Biological Crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of applied crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moriarty NW, Grosse-Kunstleve RW, Adams PD. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallographica Section D: Biological Crystallography. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D: Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D: Biological Crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.