

ABSTRACT
Membrane protein turnover and degradation are required for the function and health of all cells. Neurons may live for the entire lifetime of an organism and are highly polarized cells with spatially segregated axonal and dendritic compartments. Both longevity and morphological complexity represent challenges for regulated membrane protein degradation. To investigate how neurons cope with these challenges, an increasing number of recent studies investigated local, cargo‐specific protein sorting, and degradation at axon terminals and in dendritic processes. In this review, we explore the current answers to the ensuing questions of where, what, and when membrane proteins are degraded in neurons. © 2017 The Authors Developmental Neurobiology Published by Wiley Periodicals, Inc. Develop Neurobiol 78: 283–297, 2018
Keywords: neuronal maintenance, membrane degradation, endosome, lysosome, autophagy
INTRODUCTION
Continuous synthesis and degradation through homeostatic regulation of protein turnover ensure a functional pool of proteins. Neuronal longevity and morphological complexity represent challenges for both cytosolic and membrane protein turnover (Steward and Schuman, 2003; Wang and Hiesinger, 2012; Bezprozvanny and Hiesinger, 2013; Alvarez‐Castelao and Schuman, 2015). Cytosolic proteins are predominantly subject to proteasomal degradation (Ciechanover, 2005; Yi and Ehlers, 2007; Tai and Schuman, 2008; Bhattacharyya et al., 2014; Cohen‐Kaplan et al., 2016). In contrast, membrane proteins are either degraded through an endo‐lysosomal mechanism or autophagy (Klionsky and Emr, 2000; Luzio et al., 2007; Eskelinen and Saftig, 2009; Schulze et al., 2009; Tooze et al., 2014; Huber and Teis, 2016; Galluzzi et al., 2017). Defects in cytosolic and membrane protein degradation typically result in protein accumulation and neuronal dysfunction. Such defects can occur at synapses prior to defects in the cell body and are hallmarks of many neurodegenerative diseases (Arendt, 2009; Shankar and Walsh, 2009; Wong and Cuervo, 2010; Morales et al., 2015; Soto and Kerschensteiner, 2015).
Local protein synthesis and degradation via the proteasome have long been described in neurons (Steward and Schuman, 2003). Recent whole‐proteome analyses in yeast suggest distinct subcellular localization of protein synthesis and degradation pathways, which might indicate an evolutionary base for the compartmentalized regulation of these events in morphologically more complex cells, including neurons (Shao and Hegde, 2014). Interestingly, a recent proteomics study in neurons based on inhibition of the ubiquitin‐proteasome system indicated that only a minority of synaptic proteins depend on proteasomal degradation under basal conditions (Hakim et al., 2016). However, many of the proteins for which local mRNA deposits have been found in dendrites are membrane proteins, including the NMDA and inositol 1,4,5‐triphosphate (InsP3) receptors (Steward and Schuman, 2003). Here, we focus on membrane degradation and refer readers to excellent reviews on the degradation of cytoplasmic proteins by the ubiquitin‐proteasome system (Bingol and Schuman, 2005; Yi and Ehlers, 2007; Tai and Schuman, 2008; Alvarez‐Castelao and Schuman, 2015; Kaushik and Cuervo, 2015; Labbadia and Morimoto, 2015; Cohen and Ziv, 2017).
Membrane protein turnover is of particular importance to the maintenance of neuronal function. At the presynaptic terminal the synaptic vesicle cycle poses a formidable challenge to membrane protein turnover. Recent work has provided evidence for how dysfunctional or aging synaptic proteins are sorted for degradation (Uytterhoeven et al., 2011; Fernandes et al., 2014; Sheehan et al., 2016). Similarly, the postsynaptic compartment requires continuous cycles of endocytosis/exocytosis of membrane proteins, such as neurotransmitter receptors (Coussen, 2009; Santos et al., 2009). However, where the actual degradation occurs, with what cargo‐specificity, and when during the development, function, and aging of neurons remain challenging questions for all known mechanisms.
Canonical endolysosomal degradation and autophagy are responsible for degradation of (but not limited to) membrane proteins. In both pathways, proteins are delivered to highly acidic, degradative organelles where degradation is initiated by acidification‐activated proteases (Kaur and Debnath, 2015; Xu and Ren, 2015; Luzio et al., 2007; Schink et al., 2016; Takats et al., 2016; Lorincz et al., 2017). Canonical endolysosomal degradation requires maturation of early endosome to late endosome or multivesicular body (MVB), followed by fusion with lysosomes for degradation. Ubiquitin attachment to membrane proteins serves as a signal for the endocytic internalization from the plasma membrane and is the signal for trafficking of protein from early endocytic vesicles to MVBs. Endosomal sorting complexes required for transport (ESCRT) is the core protein machinery to recognize ubiquitinated proteins in endosomes and sort them to MVBs (Katzmann et al., 2001; Henne et al., 2011).
In (macro‐) autophagy, a double membrane called phagophore forms around “to‐be‐degraded” cargo, such as vesicles containing membrane proteins, cytosolic proteins, protein aggregates, and organelles. After cargo engulfment, autophagosomes fuse with lysosomal vesicles to form degradative autolysosomes (Xie and Klionsky, 2007; Kraft and Martens, 2012; Coutts and La Thangue, 2016; Nishimura et al., 2017; Wang et al., 2017). Despite the abundance of transmembrane proteins in both presynaptic and postsynaptic compartments, progress has only recently been made to address what membrane proteins are degraded by either mechanism in neurons (Ashrafi and Schwarz, 2013; Huber and Teis, 2016; Mancias and Kimmelman, 2016; Zaffagnini and Martens, 2016; Vijayan and Verstreken, 2017). More studies have focused on the dysregulation of these degradative pathways in relation to neurodegenerative diseases than their wildtype maintenance function (Hara et al., 2006; Komatsu et al., 2006; Menzies et al., 2017; Moors et al., 2017). In the following sections, we will focus on membrane degradation via the autophagosomal and endolysosomal system, and our current understanding of where, what, and when these mechanisms degrade membrane proteins in neurons independent of disease‐specific neurotoxic insults.
WHERE ARE MEMBRANE PROTEINS DEGRADED IN NEURONS?
The distances between dendrites, the cell body, and the axon tip raise questions about the spatial regulation of membrane protein sorting and degradation. Three ways to remove synaptic membrane proteins from distal axons and dendrites have been proposed (Fig. 1): (1) synaptic membrane proteins may be sorted into endosomes and autophagosomes for retrograde trafficking back to the cell body, (2) membrane proteins may be sorted and degraded locally, and (3) membrane proteins may be secreted and taken up by neighboring cells for degradation. In this section, we will review current evidence for these three possibilities. How the implicated endolysosomal or autophagosomal mechanisms may operate in the different neuronal compartments will be discussed in the subsequent section.
Figure 1.
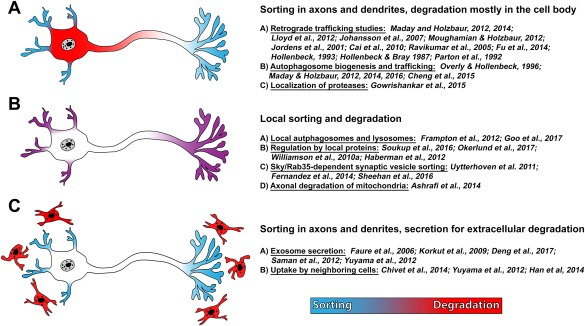
Three models for where neuronal membrane proteins sort and degrade. (A) Synaptic membrane proteins are sorted for degradation locally, and then undergo retrograde axonal trafficking to the cell body for degradation. (B) Sorting and degradation of synaptic membrane proteins occur locally in axon terminals and dendrites. (C) Neurons release synaptic membrane proteins outside via extracellular vesicles, which are taken by neighboring cells for degradation.
Retrograde Transport Back to Cell Body
Neurons employ robust microtubule‐dependent transport machinery to traffic proteins and organelles between the cell body, dendrites and distal axons (Maday et al., 2014). Microtubule‐based axonal transport utilizes two main types of molecular motors: kinesin for plus‐end directed anterograde transport (from cell body to axon terminals) and dynein‐dynactin complex for minus‐end directed retrograde transport (from axon terminals to cell body) (Schnapp and Reese, 1989; Pilling et al., 2006; Encalada et al., 2011). Live imaging of late endosomes, lysosomes, and autophagosomes in axons revealed net retrograde trafficking, suggesting that degradation may happen in the cell body [Fig. 1(A)] (Hollenbeck and Bray, 1987; Parton et al., 1992; Hollenbeck, 1993; Maday et al., 2014; Cheng et al., 2015). Defective retrograde transport causes dramatic accumulations of late endosomes and autophagosomes in axons as well as impaired autophagic and lysosomal degradation (Ravikumar et al., 2005; Cai et al., 2010; Lloyd et al., 2012). Such defects have been linked to late‐onset, progressive motor neuron degeneration such as amyotrophic lateral sclerosis (ALS) or spinal muscular atrophy (SMA) (Hafezparast et al., 2003; Puls et al., 2003; Munch et al., 2004; Levy et al., 2006; Lai et al., 2007; Chevalier‐Larsen et al., 2008; Laird et al., 2008; Strom et al., 2008; Hirokawa et al., 2010).
Autophagosomes form in the distal axons, initially exhibit bidirectional movements, but eventually switch to robust retrograde transport for degradation in the cell body (Maday and Holzbaur, 2012, 2014; Maday et al., 2014). This initial bidirectional transport is carried out when both kinesin‐1 and dynein motors tightly bind to autophagosomes, but on interaction between LC3 on autophagosome and JNK‐interacting protein 1 (JIP1), kinesin‐1 activation is blocked, resulting in the robust retrograde transport back to cell body (Fu et al., 2014; Maday et al., 2014). Dynein motors are recruited to autophagosomes after fusion with late endosomes (Cheng et al., 2015). In some studies, autophagosome maturation and degradation were observed only during or after retrograde transport following autophagosome biogenesis at the axon terminal (Maday and Holzbaur, 2012), consistent with the earlier report of the progressive increase in the proportion of acidic endocytic organelles along axons closer to the soma (Overly and Hollenbeck, 1996). These findings support the prevalent idea that degradation may occur preferentially in the cell body or en route to the cell body [Fig. 1(A)] (Maday and Holzbaur, 2016).
Late endosomes and lysosomes exhibit retrograde transport behavior distinct from autophagosomes, most likely resulting from different molecular interactions with adaptors and motor proteins. Kinesin‐2 is the primary motor protein to regulate anterograde movement of late endosomes and lysosomes (Brown et al., 2005). Retrograde transport of late endosomes and lysosomes requires direct interaction between Rab7 effector protein Rab7 Interacting Lysosomal Protein (RILP) and the C‐terminal region of a dynactin subunit p150 (Glued) (Jordens et al., 2001; Johansson et al., 2007; Maday et al., 2014). The highly conserved CAP‐Gly domain of p150 initiates the retrograde transport from distal axons (Lloyd et al., 2012; Moughamian and Holzbaur, 2012). Snapin, a neuronal SNARE‐binding protein, also regulates retrograde transport of late endosomes by tethering them to dynein (Cai et al., 2010). Unlike autophagosomes that exhibit robust retrograde transport, late endosomes, and lysosomes have been reported to traffic bidirectionally with frequent pauses and directional changes in a constant tug‐of‐war between the opposing motors that are simultaneously present on the organelles (Deacon et al., 2003; Bananis et al., 2004; Muller et al., 2008; Hendricks et al., 2010; Lloyd et al., 2012; Maday et al., 2014).
Lysosomes containing proteases are preferentially enriched in the cell body, whereas lysosomes at distal axons have been reported to lack luminal proteases and, therefore, lack degradative capacity (Gowrishankar et al., 2015). In this case, late endosomes, lysosomes, and autophagosomes would require to retrogradely traffic back to cell body for degradation [Fig. 1(A)]. Highly acidic pH is required for lysosomal function, and less acidic pH can, therefore, reduce the degradative capacity of lysosomes. A recent study reported that lysosomal pH depends on intracellular positioning, with lysosomes closer to the cell periphery being less acidic (Johnson et al., 2016), although it is not known if this is also the case for lysosomes at synapses. These studies support the need for retrograde transport of degradative organelles and degradation in the cell body.
In some vertebrates, synapses can be separated from the cell body through axons over a meter in length. Considering these distances, even if the cargos are transported at the fastest reported axonal speed (∼10 cm/day), it would take days for synaptic proteins to reach the cell body for degradation (Grafstein and Forman, 1980; Miller and Heidemann, 2008). Such long distances may not be an obstacle for retrograde trafficking‐dependent degradation if dysfunctional cargoes do not affect normal function and health of a neuron. Alternatively, degradation and protein synthesis are often closely linked through feedback mechanisms that ensure protein homeostasis (Alvarez‐Castelao and Schuman, 2015; Cajigas et al., 2010). In this case, long trafficking distances would represent a challenge for mechanisms of local protein homeostasis in neurons.
Local Membrane Protein Degradation at Synapses
Neurons have to rapidly and homeostatically respond to varying conditions both during developmental axonal and dendritic growth as well as neuronal activity. Ample evidence for local protein synthesis and proteasomal degradation in dendrites strongly suggest the autonomous regulation of protein turnover at synapses (Pierce et al., 2001; Wang et al., 2010; Ramirez and Couve, 2011; Holt and Schuman, 2013). Most studies on protein degradation in neurons have focused on proteasomal degradation, and several studies have demonstrated that proteins are degraded locally through proteasomal degradation at postsynaptic terminals (Speese et al., 2003; Yi and Ehlers, 2007; Hamilton and Zito, 2013). However, proteasomal degradation may not be responsible for the degradation of the majority of synaptic proteins in axon terminals (Hakim et al., 2016).
Similar to the proteasomal degradation machinery, membrane degradation machinery has been observed both at axon terminals and near or in dendritic spines (Frampton et al., 2012; Goo et al., 2017). Frampton et al. (2012) showed the abundance of proteins lysosomal‐associated membrane protein 2 (LAMP2) and microtubule‐associated protein 1 light chain 3 (LC3) in axons and axon terminals, suggesting that lysosomes and autolysosomes are located at axon terminals. Using compartmentalized rat superior cervical ganglion (SCG) neuronal cultures, they observed a significant increase of LAMP2 protein exclusively in the distal axons on nerve growth factor (NGF) stimulation (Frampton et al., 2012). Recruitment of lysosomes to dendritic spines in an activity‐dependent manner was also recently reported (Goo et al., 2017). These observations suggest that the abundance of lysosomes, and perhaps lysosomal degradation, is regulated locally at distal axon terminals, and possibly independently from lysosomes at the cell body [Fig. 1(B)].
Several recent studies characterized mechanisms for the local regulation of autophagosome formation by synaptic proteins at axon terminals. Soukup et al. (2016) demonstrated an unexpected role of the synaptically enriched protein Endophilin A (EndoA), which was previously characterized during synaptic vesicle endocytosis (Song and Zinsmaier, 2003). On phosphorylation by the Parkinson's disease‐associated kinase LRRK2, EndoA recruits Atg3 to the growing membrane of autophagosomes and causes it to colocalize with Atg8 (LC3) (Soukup et al., 2016). In addition, Okerlund et al. (2017) showed that the presynaptic active zone protein Bassoon selectively inhibits autophagy by interacting with Atg5 [Fig. 2(B)]. The CC2 domain of Bassoon interacts with Atg5, possibly regulating the formation of the Atg5‐Atg12‐Atg16 complex. Loss of Bassoon function triggers synaptic autophagy (Okerlund et al., 2017). The idea of autophagosome formation at the axon terminals is consistent with findings by Maday and Holzbaur (2012, 2014, 2016). Although local degradation of membrane proteins via autophagy has not been directly demonstrated, local degradation of mitochondria through PINK/PARKIN‐mediated mitophagy at axon terminals has been reported (Ashrafi et al., 2014). As shown in this study, selective induction of mitochondrial damage by mitochondrial KillerRed (mt‐Kr), a genetically encoded photosensitizer targeted to mitochondria, leads to the recruitment of autophagosomes and lysosomes to damaged mitochondria for degradation locally in the axons.
In addition to autophagy, two neuron‐specific proteins that predominantly function at synaptic terminals have been reported to constitute a local, neuron‐specific branch of the endolysosomal degradation system: the vesicular ATPase component V100 and the vesicle SNARE neuronal Synaptobrevin (n‐Syb) (Williamson et al., 2010a; Haberman et al., 2012; Wang and Hiesinger, 2012; Bezprozvanny and Hiesinger, 2013). Both proteins were initially characterized as synaptic vesicle proteins in synaptic vesicle exocytosis (Perin et al., 1991; DiAntonio et al., 1993; Deitcher et al., 1998; Schoch et al., 2001; Hiesinger et al., 2005). Loss of function of either V100 or n‐Syb causes local sorting and degradation defects at axon terminals, which eventually lead to adult‐onset neurodegeneration in Drosophila photoreceptors (Williamson et al., 2010a; Haberman et al., 2012). After the initial cargo overload in endosomes in both mutants, autophagy is activated as an apparent secondary, compensatory effect. It is interesting to note that both proteins are almost exclusively located at axon terminals, indicating that neurons implement an additional, neuron‐specific endolysosomal sorting, and degradation mechanism locally at axon terminals to meet a high or specialized demand for membrane protein turnover.
Presence of membrane protein degradation machinery and identification and characterization of synaptic autophagy and endolysosomal degradation are strong indicators that membrane proteins are sorted for degradation locally at synapses [Fig. 1(B)]. However, direct demonstration of where membrane proteins are degraded in neurons remains unclear because protein degradation is mostly assayed in the entire neuron without separation of distal axons and dendrites from cell bodies (Cohen et al., 2013; Price et al., 2010; Sheehan et al., 2016; Cohen and Ziv, 2017). Protein half‐lives in neurons are studied using isotopic labeling with amino acids, such as stable isotope labelling with amino acids in cell culture (SILAC), followed by mass spectrometry (MS) in mouse or rat cortical neurons (Price et al., 2010; Cohen et al., 2013). Activity‐dependent protein degradation in neurons has been demonstrated biochemically with western blot analyses before and after neural activation in cultured rat hippocampal or cortical neuron (Shehata et al., 2012; Widagdo et al., 2015; Sheehan et al., 2016). Separation of neuronal compartments can be achieved using microfluidic culture devices (Taylor et al., 2005; Park et al., 2006; Taylor et al., 2006; Taylor and Jeon, 2010; Taylor et al., 2010). Such methods have been used to demonstrate local proteasomal degradation in growth cones (Deglincerti et al., 2015), and local degradation of damaged mitochondria by autophagy in axons (Ashrafi et al., 2014).
Release into Neighboring Cells
Neurons can rid themselves of membrane proteins and other cargo through the release of extracellular vesicles (EVs) followed by uptake in a neighboring cell [Fig. 1(C)] (Simons and Raposo, 2009; Raposo and Stoorvogel, 2013; Budnik et al., 2016). Two major types of EVs are exosomes and microvesicles. Exosomes are intraluminal vesicles (ILVs) inside multivesicular bodies (MVBs) that are released into the extracellular space on direct fusion of MVBs with the plasma membrane. Microvesicles are vesicles that form by an outward budding of the plasma membrane. As a result, microvesicles are typically larger (100 nm to 1 um in diameter) than exosomes (30–100 nm in diameter) (Colombo et al., 2014). The content that can be released as EVs is not limited to membrane proteins and also includes mRNA, cytosolic proteins, and lipids. Two general functions of exosome secretion have been proposed: degradation and intercellular communication. Here, we focus on what is known about degradation of membrane proteins by neighboring cells, while other functions of exosomes are presented and comprehensively reviewed elsewhere (Lotvall and Valadi, 2007; Simons and Raposo, 2009; Fruhbeis et al., 2012; Raposo and Stoorvogel, 2013; Budnik et al., 2016).
Exosome secretion from neurons was first reported in mature cortical neurons as well as in vivo at the Drosophila neuromuscular junction (NMJ) (Faure et al., 2006; Korkut et al., 2009). Since then, many studies have focused on the release of disease‐associated proteins and aggregates, such as tau, amyloid‐beta peptides, huntingtin, and superoxide dismutase‐1 (SOD‐1), from neurons via exosomes (Saman et al., 2012; Yuyama et al., 2012; Deng et al., 2017). It was recently demonstrated that the synaptic vesicle protein cysteine string protein (CSPα) mediates the release of exosomes containing polyglutamine expanded protein 72Q huntingtin and superoxide dismutase‐1 (SOD‐1) (Deng et al., 2017). Consequently, loss‐of‐function of CSPα in Drosophila and C. elegans mutants demonstrate uncoordinated movements, neurodegeneration, and early lethality possibly due to defective release of 72Q huntingtin and SOD‐1 (Zinsmaier et al., 1994; Kashyap et al., 2014).
EVs released from a neuron can be taken up by another neuron, glia or any other neighboring cells. Synaptic activity was shown to bias the binding of exosomes to neighboring neurons rather than glial cells (Chivet et al., 2014). The EVs can then be degraded in any of these recipient cells by fusing directly with the plasma membrane followed by endocytosis and endolysosomal degradation. Amyloid‐beta peptides released via neuronal exosomes were reported to be cleared via microglia (Yuyama et al., 2012). Exosomes may also be released into other neighboring cells, including muscles at the neuromuscular junction (NMJ) (Budnik et al., 2016) and epithelial cells in the case of Drosophila sensory class IV dendritic arborization (da) neurons (Han et al., 2014). Furthermore, clearance of degenerating dendrite fragments of Drosophila class IV da neuron was reported for the neighboring epidermal cells (Han et al., 2014). In contrast, it is less clear whether synaptic membrane proteins are degraded in neighboring cells.
WHAT MEMBRANE PROTEINS AND MECHANISMS ARE IMPLICATED IN NEURONAL DEGRADATION
Autophagosomes and endolysosomal compartments are available in distal axons and dendrites. Recent studies have started to identify what cargos may be degraded by these mechanisms, but the list of membrane proteins tested so far is relatively short (Fig. 2). In this section, we will discuss what is currently known about what cargoes are degraded by what membrane protein degradation mechanism in neurons.
Figure 2.
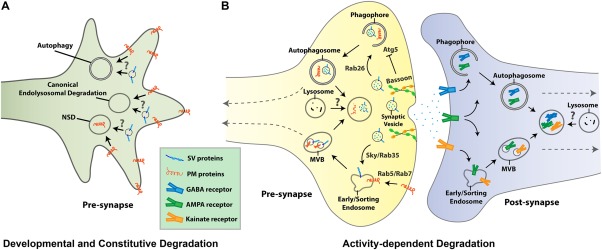
Schematic overviews of cargo‐sorting and degradation mechanisms operating in developing and mature neurons. (A) Developmental and constitutive degradation by autophagy, canonical endolysosomal degradation, and neuron‐specific endolysosomal degradation (Neuronal Sort and Degrade; NSD). Plasma membrane (PM) proteins such as guidance receptors are degraded through NSD (Williamson et al., 2010b). It has not been shown what membrane proteins are degraded through autophagy and canonical endolysosomal degradation in a developing neuron. (B) Mechanisms of activity‐dependent cargo‐sorting and degradation in presynaptic and postsynaptic terminals through endolysosomal degradation or autophagy.
Synaptic Autophagy
Autophagy is highly conserved from yeast to mammals, and most of our knowledge is derived from studies in non‐neuronal cells or neuronal cell bodies (Ohsumi, 2014). Unlike non‐neuronal cells, primary cultured neurons may not upregulate autophagy in response to starvation, but instead employ constitutive autophagy to maintain protein homeostasis (Maday and Holzbaur, 2016). Several recent studies focused on the molecular characterization of neuronal autophagy, particularly at presynaptic terminals (Soukup et al., 2016; Okerlund et al., 2017; Vanhauwaert et al., 2017; Vijayan and Verstreken, 2017).
Macroautophagy is well characterized for its ability to bulk degrade membrane proteins, organelles, cytosolic proteins, and protein aggregates. The recent observation of synaptic vesicles in pre‐autophagosomal structures in Drosophila NMJ and cultured rat hippocampal neuron cell bodies by Binotti et al. (2015) suggests that synaptic vesicle proteins may be degraded as bulk cargo, although degradation was not directly shown [Fig. 2(B)]. They demonstrated that a small Rab GTPase Rab26, which is predominantly localized to synaptic regions in Drosophila (Chan et al., 2011), interacts with Atg16L and directs synaptic vesicles to pre‐autophagosomal structures (Binotti et al., 2015). Rab26 overexpression resulted in accumulation of synaptic vesicles in preautophagosomes in Drosophila NMJ. This suggests a mechanism that directly engulfs the entire synaptic vesicles for degradation without more specific protein sorting.
Postsynaptic membrane proteins such as GABA and AMPA receptor subunits are reported to be degraded through autophagy, although where these receptors are degraded still remains unknown [Fig. 2(B)] (Rowland et al., 2006; Shehata et al., 2012; Widagdo et al., 2015). Degradation of AMPA receptor subunits GluA1 and GluA2, assayed by western blot analyses of cultured rat hippocampal or cortical neurons, was correlated with the autophagosomal or lysosomal markers LC3‐II and LAMP1, respectively (Shehata et al., 2012; Widagdo et al., 2015). Moreover, inhibition of autophagy with Wortmannin prevented AMPA receptor degradation (Shehata et al., 2012). In C. elegans neuromuscular system, internalized GABA receptors were reported to colocalize with autophagosomes (Rowland et al., 2006). Loss of both GABA and acetylcholine motor neuron innervations in the postsynapse, the dorsal muscle, caused internalization and sorting of only GABA, but not acetylcholine, receptors to autophagosomes in the dorsal muscle. This suggests that autophagy may employ cargo‐specific sorting for degradation postsynaptically.
Canonical Endolysosomal Degradation
In addition to autophagy, neurons share canonical endolysosomal machinery with non‐neuronal cells. In endolysosomal degradation, membrane proteins are internalized through endocytosis and subsequently progress through late endosomal and lysosomal stages. Genetic studies in yeast, worms, flies, and mammals have identified a set of conserved and essential proteins that function in the endolysosomal progressions and lysosome biogenesis. The two ubiquitously expressed small Rab GTPases, Rab5 and Rab7, are key regulators of endocytosis and endosomal maturation, respectively [Fig. 2(B)] (Bucci et al., 1992; Vitelli et al., 1997). The ESCRT pathway is the core machinery that sort and direct ubiquitinated proteins for degradation (Katzmann et al., 2001; Henne et al., 2011; Schuh and Audhya, 2014). Membrane proteins can be ubiquitinated by the sequential action of E1 (ubiquitin‐activating enzymes), E2 (ubiquitin‐carrier/conjugating enzymes), and E3 (ubiquitin ligases). Ubiquitinated membrane proteins are then recognized by the ESCRT‐0 complex, which results in subsequent recruitment of ESCRT‐I, ‐II, and –III for delivery of the ubiquitinated protein into MVB by intraluminal vesicle (ILV) formation (Henne et al., 2011; Schuh and Audhya, 2014). Numerous regulatory and tethering complexes are required for endolysosomal progression, including the CORVET and HOPS, which are reviewed in detail elsewhere (Balderhaar and Ungermann, 2013; Solinger and Spang, 2013; Luzio et al., 2014).
Rab7 is expressed ubiquitously in all cell types of an organism, but its early expression is strongly enriched in the nervous system in Drosophila (Chan et al., 2011). Missense mutations in rab7 cause the neuropathy Charcot–Marie–Tooth type 2B (CMT2B) in patients (Verhoeven et al., 2003). Studies of the disease‐associated mutations suggested toxic gain‐of‐function effects in cell culture (Spinosa et al., 2008; Cogli et al., 2010; McCray et al., 2010; Cogli et al., 2013; Zhang et al., 2013). In contrast, a study in Drosophila revealed no toxic effects of the mutant proteins, but indicated partial loss of function as the underlying mechanism (Cherry et al., 2013). It is intriguing that mutations in rab7, which is ubiquitously expressed, cause the first observable defect in some of the longest neurons in the human body. As discussed above, neurons may be more sensitive to defects in membrane protein degradation because of their longevity as well as morphological complexity. Hence, neurons, and particularly synapses, may have particularly high or specialized demands on endolysosomal degradation. However, it remains to be investigated what membrane proteins are degraded by canonical endolysosomal degradation as opposed to autophagy and neuron‐specific mechanisms in neurons (Fig. 2).
Skywalker/Rab35‐Dependent Sorting and Degradation Is Cargo‐Specific
At axon terminals, synaptic vesicles undergo continuous exo‐/endocytic recycling (Heuser and Reese, 1973; Sudhof, 2004; Rizzoli, 2014; Kononenko and Haucke, 2015). Endocytosed membrane and membrane proteins are either sorted for synaptic vesicle recycling or degradation. Sorting stations have been proposed as “sorting endosome” or “early vacuoles.” Key regulators of this sorting mechanism are Rab35 and its GTPase activating protein (GAP), Skywalker [Fig. 2(B)] (Uytterhoeven et al., 2011; Sheehan et al., 2016). In sky mutants, Uytterhoeven et al. (2011) observed that synaptic vesicle recycling through the sorting endosome was increased. Correspondingly, degradation of dysfunctional (ubiquitinated) n‐Syb was increased, and this resulted in an increased readily releasable pool (RRP) and synaptic neurotransmission at the Drosophila NMJ. Mutations in skywalker were also reported to cause epilepsy and DOORS syndrome in human (Fischer et al., 2016). Using a fluorescence timer tagged n‐Syb, Fernandes et al. (2014) reported an increased protein turnover through lysosomal degradation in the sky mutant, which resulted in a younger pool of proteins compared to wild‐type (Fernandes et al., 2014). These seminal studies show how local synaptic vesicle protein turnover affects the pools of functional synaptic vesicles. Sorting and degradation may require recognition of dysfunctional proteins, as indicated by ubiquitination. Alternatively, the sky‐dependent mechanism may function to continuously turn over synaptic vesicle proteins independent of their functional status. It remains to be investigated whether and how functional and dysfunctional proteins residing on the same vesicle could be sorted out and whether their degradation ultimately occurs locally.
Sheehan et al. (2016) report the first evidence of differential specificities for different synaptic vesicle proteins through the Rab35‐dependent mechanism in vertebrate neurons. On neuronal activation, the Skywalker/Rab35 and ESCRT pathway selectively sorts and degrades neuronal Synaptobrevin (n‐Syb)/VAMP2 and SV2, but not Synaptotagmin 1 (Syt1) and Syntaxin (Syx). This suggests that individual SV proteins are recognized and sorted for degradation. How Sky/Rab35 recognizes these specific proteins for degradation remains an open question. It also remains unknown whether Sky/Rab35 specifically sorts SV proteins or also other non‐SV proteins in the presynaptic terminal, whether this sorting mechanism also exists in the postsynaptic terminals, and whether retrograde trafficking of degradative vesicles plays a role.
Neuron‐Specific Endolysosomal Degradation
Autophagy and the Sky/Rab35‐dependent endolysosomal mechanism exist in all cells, but are likely to function in a specialized manner at synaptic terminals (Fernandes et al., 2014; Vijayan and Verstreken, 2017). In addition, at least two neuron‐specific integral membrane proteins function in endolysosomal sorting and degradation based on findings in Drosophila: the vesicle SNARE neuronal Synaptobrevin (n‐Syb) and the vesicle ATPase component V100 (Williamson et al., 2010a; Haberman et al., 2012). Both proteins have previously been characterized as synaptic vesicle proteins (Perin et al., 1991; Schoch et al., 2001). Loss of either protein in Drosophila photoreceptor neurons leads to endolysosomal membrane accumulations at axon terminals, indicating a link between the synaptic vesicle cycle and synaptic membrane turnover (Williamson et al., 2010a; Haberman et al., 2012; Wang and Hiesinger, 2012; Bezprozvanny and Hiesinger, 2013). However, it is currently unclear whether this neuron‐specific branch of the endolysosmal system has a cargo‐specificity that differs from the canonical endolysosomal degradation. Several plasma membrane receptors have been shown to accumulate in the v100 mutant brains (Williamson et al., 2010b). In the v100 mutant photoreceptor neurons, different membrane receptors accumulated in cell bodies versus axon terminals, suggesting that the neuron‐specific branch of the endolysosomal system may sort and degrade different membrane proteins in axon terminals versus in the cell body.
WHEN ARE MEMBRANE PROTEINS DEGRADED IN NEURONS?
Membrane protein turnover plays roles during neuronal development, function, and maintenance. Constitutive membrane protein degradation may occur during all stages of a neuron's lifetime and is not regulated by neuronal activity [Fig. 2(A)]. In contrast, activity‐dependent turnover and degradation is a direct function of neuronal activity levels and is relevant to function and maintenance [Fig. 2(B)]. In this last section, we highlight the differences between these mechanisms during the life of a neuron.
Constitutive and Developmental Degradation
Activity‐independent, constitutive protein degradation has been highlighted recently (Maday and Holzbaur, 2016; Cohen and Ziv, 2017). Synaptic vesicle proteins may undergo constitutive turnover and degradation already during development [Fig. 2(A)]. Synaptotagmin 1 (Syt1), a calcium sensor necessary for synaptic vesicle release, and n‐Syb are already present at axon terminals prior to synaptogenesis (Hiesinger et al., 1999; Williamson et al., 2010b). While synaptic autophagy has been reported as a constitutive degradation mechanism (Maday and Holzbaur, 2016), it remains unclear whether this mechanism degrades synaptic vesicle proteins during development.
Membrane protein degradation has been implicated in neural development, including axonal growth, synapse elimination, and pruning (Yogev and Shen, 2014; Wojnacki and Galli, 2016). Spatiotemporal control of developmentally required membrane receptor availability on the surface of axon terminals is regulated through protein turnover. Defects in the endolysosomal system lead to accumulations of undegraded membrane proteins before and after synaptogenesis (Wang et al., 2013). Mutations in the ubiquitous endosomal maturation factor rab7 in Drosophila surprisingly do not affect embryo and larval development (Cherry et al., 2013). However, membrane proteins slowly accumulate during development and autophagy is upregulated as a consequence. During later adult stages, the impaired clearance of membrane proteins in the developing organism causes neurodegeneration. Drosophila photoreceptor neurons deficient for rab7 complete development and function normally as long as they are not stimulated (Cherry et al., 2013). These findings indicate that constitutive rab7‐dependent endolysosomal turnover is not required for development of these cells. Similarly, mutations in the neuron‐specific genes n‐Syb and V100 lead to accumulation of early endosomes and autophagosomes during development, but do not affect photoreceptor neuron development (Williamson et al., 2010a, 2010b; Haberman et al., 2012; Cherry et al., 2013). Timing may be critical: since Drosophila photoreceptor neurons develop over a timespan of only a few days, “debris” accumulation may not affect these fast developing neurons as profoundly as neurons with longer development.
Developmental elimination of excess synapses through pruning is reported to occur via autophagy. Components of autophagic machinery begin to express and localize to axons in early development (Song et al., 2008). Blocking autophagosome formation by Atg7 knockdown causes overextension of axons, whereas activation of autophagy by rapamycin suppresses axonal extension (Ban et al., 2013). In developing motor neuron axon terminals, the NMJ, excessive synapses are eliminated through engulfment of retracting axon tips by the surrounding glial cells, and the degrading axonal membranes were associated with LC3‐positive autolysosomes (Song et al., 2008). On the postsynaptic site, hyperactivated mTOR, hence, impaired autophagy, has been linked to reduced developmental dendritic pruning causing autism‐like neurodevelopmental disorders. mTOR inhibition by rapamycin corrects developmental spine pruning defects in mice mutant for autism‐causing Tsc2 (Tang et al., 2014).
Activity‐Dependent Degradation
Activity levels of neurons have a substantial impact on the turnover rate of SV proteins and transmembrane receptors on both the presynaptic and postsynaptic side. The Sky/Rab35 mechanism is activity‐dependent, while a possible constitutive role has not yet been shown. As described above, sky was originally discovered in Drosophila (Uytterhoeven et al., 2011) and the Rab35 mechanism was recently shown to function activity‐dependently in rat hippocampal neurons as well (Sheehan et al., 2016; Sheehan and Waites, 2017). Sheehan et al. compared protein levels before and after treatment of cultured neurons with either activity enhancer or blocker. Neuronal activity induced Rab35 activation and binding to the ESCRT‐0 protein Hrs, which they identified as a novel Rab35 effector. Their findings demonstrate that the Rab35/ESCRT pathway facilitates the activity‐dependent removal of SV proteins, to maintain presynaptic protein homeostasis (Sheehan et al., 2016; Sheehan and Waites, 2017). It remains to be shown to what extent individual proteins or organelles are recognized as dysfunctional, or, alternatively, whether continuous turnover of proteins or organelles irrespective of their functional state may suffice to ensure neuronal health.
Prolonged neuronal activity also induces autophagy at both presynaptic and postsynaptic sites. Soukup et al. (2016) induced action potentials in motor neurons by the overexpression of transient receptor potential cation channel A1 (TrpA1) and observed increased formation of Atg8‐positive autophagosomes and LAMP2‐positive lysosomes at presynaptic terminals (Soukup et al., 2016). Conversely, neuronal stimulation in rat hippocampal neurons induces autophagosome formation both presynaptically and postsynaptically and possibly regulates the degradation of GABA and AMPA receptors (Shehata et al., 2012; Widagdo et al., 2015).
Activity‐dependent regulation of the density and number of AMPA and kainate receptors on the postsynaptic membrane is a key feature of long‐term changes on synaptic strength. Also, long‐term memory formation from unstable short‐term memory traces depends on rapid spatiotemporal changes of synaptic protein composition (Fioravante and Byrne, 2011; Jarome and Helmstetter, 2014). Activity‐dependent sorting of AMPAR to lysosomes has been reported (Ehlers, 2000; Schwarz et al., 2010; Widagdo and Anggono, 2015; Widagdo et al., 2015). More specifically, activity‐dependent ubiquitination of AMPAR results in sorting into LAMP1‐positive lysosomes for degradation (Ehlers, 2000; Schwarz et al., 2010; Widagdo et al., 2015). In a similar manner, intense activation of kainate receptors causes a PKC‐dependent, but Ca2+‐independent, internalization into lysosomes for degradation (Martin and Henley, 2004). Intriguingly, recruitment of lysosomes to dendritic spines was reported to be activity‐dependent (Goo et al., 2017). In sum, neuronal activity regulates the intracellular trafficking and degradation of postsynaptic membrane proteins to allow rapid spatiotemporal changes of synaptic protein composition, which has been implicated in long term memory formation (Fioravante and Byrne, 2011; Jarome and Helmstetter, 2014). However, it remains unclear in most cases, whether degradation occurs locally at the synapse or during and after retrograde trafficking back to the cell body.
CONCLUSION
In this review, we highlighted recent advances and open questions on neuronal membrane protein degradation. Recent studies have beautifully shown that specific membrane proteins are sorted for degradation by different mechanisms, at different places in neurons and during different times. However, our survey of the where, what, and when of neuronal membrane protein degradation highlights key open questions:
Where? While local sorting has been demonstrated for all neuronal compartments, the question of local degradation versus retrograde trafficking remains largely unanswered.
What? The cargo‐specificity of both autophagy and endolysosomal degradation mechanism is largely unknown and may vary in the different neuronal compartments.
When? Both endolysosomal and autophagic degradation have been proposed as constitutive and activity‐dependent mechanisms in various contexts. When these mechanisms are activated in neurons will require experimental evidence from both in vitro and in vivo studies.
Answers to these questions are complicated by interdependencies: axonal and dendritic terminals are likely to employ different membrane sorting and degradation mechanisms as a function of developmental and functional stages. Conversely, the same mechanism, for example, autophagy, has been proposed to function differently, and possibly with altered cargo‐specificity, in different cellular compartments. Biochemical analyses of such spatiotemporally segregated and interdependent process are difficult and are dependent on technological advances such as microfluidic chambers and isolation of the different neuronal compartments. Primary neuronal culture is an excellent choice for cell biological studies using both biochemistry and imaging, but may not always reflect normal developmental and functional context. Finally, in vivo systems are typically less accessible. As always, a combination of these techniques is most likely to yield meaningful solutions, as long as each system is interpreted relative to other approaches and its own limitations.
We would like to thank all members of the Hiesinger lab for discussions.
LITERATURE CITED
- Alvarez‐Castelao B, Schuman EM. 2015. The regulation of synaptic protein turnover. J Biol Chem 290:28623–28630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T. 2009. Synaptic degeneration in Alzheimer's disease. Acta Neuropathol 118:167–179. [DOI] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. 2014. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 206:655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schwarz TL. 2013. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balderhaar HJ, Ungermann C. 2013. CORVET and HOPS tethering complexes—Coordinators of endosome and lysosome fusion. J Cell Sci 126:1307–1316. [DOI] [PubMed] [Google Scholar]
- Ban BK, Jun MH, Ryu HH, Jang DJ, Ahmad ST, Lee JA. 2013. Autophagy negatively regulates early axon growth in cortical neurons. Mol Cell Biol 33:3907–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bananis E, Nath S, Gordon K, Satir P, Stockert RJ, Murray JW, Wolkoff AW. 2004. Microtubule‐dependent movement of late endocytic vesicles in vitro: Requirements for Dynein and Kinesin. Mol Biol Cell 15:3688–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Hiesinger PR. 2013. The synaptic maintenance problem: Membrane recycling, Ca2+ homeostasis and late onset degeneration. Mol Neurodegener 8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Yu H, Mim C, Matouschek A. 2014. Regulated protein turnover: Snapshots of the proteasome in action. Nat Rev Mol Cell Biol 15:122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. 2005. Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol 15:536–541. [DOI] [PubMed] [Google Scholar]
- Binotti B, Pavlos NJ, Riedel D, Wenzel D, Vorbruggen G, Schalk AM, Kuhnel K, et al. 2015. The GTPase Rab26 links synaptic vesicles to the autophagy pathway. Elife 4:e05597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CL, Maier KC, Stauber T, Ginkel LM, Wordeman L, Vernos I, Schroer TA. 2005. Kinesin‐2 is a motor for late endosomes and lysosomes. Traffic 6:1114–1124. [DOI] [PubMed] [Google Scholar]
- Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, Zerial M. 1992. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 70:715–728. [DOI] [PubMed] [Google Scholar]
- Budnik V, Ruiz‐Canada C, Wendler F. 2016. Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci 17:160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Lu L, Tian JH, Zhu YB, Qiao H, Sheng ZH. 2010. Snapin‐regulated late endosomal transport is critical for efficient autophagy‐lysosomal function in neurons. Neuron 68:73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajigas IJ, Will T, Schuman EM. 2010. Protein homeostasis and synaptic plasticity. EMBO J 29:2746–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CC, Scoggin S, Wang D, Cherry S, Dembo T, Greenberg B, Jin EJ, et al. 2011. Systematic discovery of Rab GTPases with synaptic functions in Drosophila. Curr Biol 21:1704–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng XT, Zhou B, Lin MY, Cai Q, Sheng ZH. 2015. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol 209:377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry S, Jin EJ, Ozel MN, Lu Z, Agi E, Wang D, Jung WH, et al. 2013. Charcot‐Marie‐Tooth 2B mutations in rab7 cause dosage‐dependent neurodegeneration due to partial loss of function. Elife 2:e01064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier‐Larsen ES, Wallace KE, Pennise CR, Holzbaur EL. 2008. Lysosomal proliferation and distal degeneration in motor neurons expressing the G59S mutation in the p150Glued subunit of dynactin. Hum Mol Genet 17:1946–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivet M, Javalet C, Laulagnier K, Blot B, Hemming FJ, Sadoul R. 2014. Exosomes secreted by cortical neurons upon glutamatergic synapse activation specifically interact with neurons. J Extracell Vesicles 3:24722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. 2005. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin‐proteasome system and onto human diseases and drug targeting. Cell Death Differ 12:1178–1190. [DOI] [PubMed] [Google Scholar]
- Cogli L, Progida C, Lecci R, Bramato R, Kruttgen A, Bucci C. 2010. CMT2B‐associated Rab7 mutants inhibit neurite outgrowth. Acta Neuropathol 120:491–501. [DOI] [PubMed] [Google Scholar]
- Cogli L, Progida C, Thomas CL, Spencer‐Dene B, Donno C, Schiavo G, Bucci C. 2013. Charcot‐Marie‐Tooth type 2B disease‐causing RAB7A mutant proteins show altered interaction with the neuronal intermediate filament peripherin. Acta Neuropathol 125:257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LD, Ziv NE. 2017. Recent insights on principles of synaptic protein degradation. F1000Res 6:675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LD, Zuchman R, Sorokina O, Muller A, Dieterich DC, Armstrong JD, Ziv T, et al. 2013. Metabolic turnover of synaptic proteins: Kinetics, interdependencies and implications for synaptic maintenance. PLoS One 8:e63191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen‐Kaplan V, Livneh I, Avni N, Cohen‐Rosenzweig C, Ciechanover A. 2016. The ubiquitin‐proteasome system and autophagy: Coordinated and independent activities. Int J Biochem Cell Biol 79:403–418. [DOI] [PubMed] [Google Scholar]
- Colombo M, Raposo G, Thery C. 2014. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30:255–289. [DOI] [PubMed] [Google Scholar]
- Coussen F. 2009. Molecular determinants of kainate receptor trafficking. Neuroscience 158:25–35. [DOI] [PubMed] [Google Scholar]
- Coutts AS, La Thangue NB. 2016. Regulation of actin nucleation and autophagosome formation. Cell Mol Life Sci 73:3249–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon SW, Serpinskaya AS, Vaughan PS, Lopez Fanarraga M, Vernos I, Vaughan KT, Gelfand VI. 2003. Dynactin is required for bidirectional organelle transport. J Cell Biol 160:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deglincerti A, Liu Y, Colak D, Hengst U, Xu G, Jaffrey SR. 2015. Coupled local translation and degradation regulate growth cone collapse. Nat Commun 6:6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL. 1998. Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene neuronal‐synaptobrevin. J Neurosci 18:2028–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Koutras C, Donnelier J, Alshehri M, Fotouhi M, Girard M, Casha S, et al. 2017. Neurons export extracellular vesicles enriched in cysteine string protein and misfolded protein cargo. Sci Rep 7:956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAntonio A, Burgess RW, Chin AC, Deitcher DL, Scheller RH, Schwarz TL. 1993. Identification and characterization of Drosophila genes for synaptic vesicle proteins. J Neurosci 13:4924–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. 2000. Reinsertion or degradation of AMPA receptors determined by activity‐dependent endocytic sorting. Neuron 28:511–525. [DOI] [PubMed] [Google Scholar]
- Encalada SE, Szpankowski L, Xia CH, Goldstein LS. 2011. Stable kinesin and dynein assemblies drive the axonal transport of mammalian prion protein vesicles. Cell 144:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen EL, Saftig P. 2009. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta 1793:664–673. [DOI] [PubMed] [Google Scholar]
- Faure J, Lachenal G, Court M, Hirrlinger J, Chatellard‐Causse C, Blot B, Grange J, et al. 2006. Exosomes are released by cultured cortical neurones. Mol Cell Neurosci 31:642–648. [DOI] [PubMed] [Google Scholar]
- Fernandes AC, Uytterhoeven V, Kuenen S, Wang YC, Slabbaert JR, Swerts J, Kasprowicz J, et al. 2014. Reduced synaptic vesicle protein degradation at lysosomes curbs TBC1D24/sky‐induced neurodegeneration. J Cell Biol 207:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, Byrne JH. 2011. Protein degradation and memory formation. Brain Res Bull 85:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B, Luthy K, Paesmans J, De Koninck C, Maes I, Swerts J, Kuenen S, et al. 2016. Skywalker‐TBC1D24 has a lipid‐binding pocket mutated in epilepsy and required for synaptic function. Nat Struct Mol Biol 23:965–973. [DOI] [PubMed] [Google Scholar]
- Frampton JP, Guo C, Pierchala BA. 2012. Expression of axonal protein degradation machinery in sympathetic neurons is regulated by nerve growth factor. J Neurosci Res 90:1533–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruhbeis C, Frohlich D, Kramer‐Albers EM. 2012. Emerging roles of exosomes in neuron‐glia communication. Front Physiol 3:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu MM, Nirschl JJ, Holzbaur EL. 2014. LC3 binding to the scaffolding protein JIP1 regulates processive dynein‐driven transport of autophagosomes. Dev Cell 29:577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo‐San Pedro JM, Cecconi F, Choi AM, et al. 2017. Molecular definitions of autophagy and related processes. EMBO J 36:1811–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goo MS, Sancho L, Slepak N, Boassa D, Deerinck TJ, Ellisman MH, Bloodgood BL, et al. 2017. Activity‐dependent trafficking of lysosomes in dendrites and dendritic spines. J Cell Biol 216:2499–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar, S. , Yuan, P. , Wu, Y. , Schrag, M. , Paradise, S. , Grutzendler, J. , De Camilli, P. , et al. 2015. Massive accumulation of luminal protease‐deficient axonal lysosomes at Alzheimer's disease amyloid plaques. Proc Natl Acad SciUSA 112:E3699–E3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafstein B, Forman DS. 1980. Intracellular transport in neurons. Physiol Rev 60:1167–1283. [DOI] [PubMed] [Google Scholar]
- Haberman A, Williamson WR, Epstein D, Wang D, Rina S, Meinertzhagen IA, Hiesinger PR. 2012. The synaptic vesicle SNARE neuronal Synaptobrevin promotes endolysosomal degradation and prevents neurodegeneration. J Cell Biol 196:261–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad‐Annuar A, Bowen S, Lalli G, et al. 2003. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 300:808–812. [DOI] [PubMed] [Google Scholar]
- Hakim V, Cohen LD, Zuchman R, Ziv T, Ziv NE. 2016. The effects of proteasomal inhibition on synaptic proteostasis. EMBO J 35:2238–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton AM, Zito K. 2013. Breaking it down: The ubiquitin proteasome system in neuronal morphogenesis. Neural Plast 2013:196848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Song Y, Xiao H, Wang D, Franc NC, Jan LY, Jan YN. 2014. Epidermal cells are the primary phagocytes in the fragmentation and clearance of degenerating dendrites in Drosophila. Neuron 81:544–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki‐Migishima R, Yokoyama M, et al. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889. [DOI] [PubMed] [Google Scholar]
- Hendricks AG, Perlson E, Ross JL, Schroeder HW III, Tokito M, Holzbaur EL. 2010. Motor coordination via a tug‐of‐war mechanism drives bidirectional vesicle transport. Curr Biol 20:697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henne WM, Buchkovich NJ, Emr SD. 2011. The ESCRT pathway. Dev Cell 21:77–91. [DOI] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. 1973. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol 57:315–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiesinger PR, Fayyazuddin A, Mehta SQ, Rosenmund T, Schulze KL, Zhai RG, Verstreken P, et al. 2005. The v‐ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell 121:607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiesinger PR, Reiter C, Schau H, Fischbach KF. 1999. Neuropil pattern formation and regulation of cell adhesion molecules in Drosophila optic lobe development depend on synaptobrevin. J Neurosci 19:7548–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y. 2010. Molecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron 68:610–638. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ. 1993. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol 121:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ, Bray D. 1987. Rapidly transported organelles containing membrane and cytoskeletal components: Their relation to axonal growth. J Cell Biol 105:2827–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt CE, Schuman EM. 2013. The central dogma decentralized: New perspectives on RNA function and local translation in neurons. Neuron 80:648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber LA, Teis D. 2016. Lysosomal signaling in control of degradation pathways. Curr Opin Cell Biol 39:8–14. [DOI] [PubMed] [Google Scholar]
- Jarome TJ, Helmstetter FJ. 2014. Protein degradation and protein synthesis in long‐term memory formation. Front Mol Neurosci 7:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Rocha N, Zwart W, Jordens I, Janssen L, Kuijl C, Olkkonen VM, et al. 2007. Activation of endosomal dynein motors by stepwise assembly of Rab7‐RILP‐p150Glued, ORP1L, and the receptor betalll spectrin. J Cell Biol 176:459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DE, Ostrowski P, Jaumouille V, Grinstein S. 2016. The position of lysosomes within the cell determines their luminal pH. J Cell Biol 212:677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordens I, Fernandez‐Borja M, Marsman M, Dusseljee S, Janssen L, Calafat J, Janssen H, et al. 2001. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein‐dynactin motors. Curr Biol 11:1680–1685. [DOI] [PubMed] [Google Scholar]
- Kashyap SS, Johnson JR, McCue HV, Chen X, Edmonds MJ, Ayala M, Graham ME, et al. 2014. Caenorhabditis elegans dnj‐14, the orthologue of the DNAJC5 gene mutated in adult onset neuronal ceroid lipofuscinosis, provides a new platform for neuroprotective drug screening and identifies a SIR‐2.1‐independent action of resveratrol. Hum Mol Genet 23:5916–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann DJ, Babst M, Emr SD. 2001. Ubiquitin‐dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT‐I. Cell 106:145–155. [DOI] [PubMed] [Google Scholar]
- Kaur J, Debnath J. 2015. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 16:461–472. [DOI] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM. 2015. Proteostasis and aging. Nat Med 21:1406–1415. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD. 2000. Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kominami E, Tanaka K. 2006. Autophagy and neurodegeneration. Autophagy 2:315–317. [DOI] [PubMed] [Google Scholar]
- Kononenko NL, Haucke V. 2015. Molecular mechanisms of presynaptic membrane retrieval and synaptic vesicle reformation. Neuron 85:484–496. [DOI] [PubMed] [Google Scholar]
- Korkut C, Ataman B, Ramachandran P, Ashley J, Barria R, Gherbesi N, Budnik V. 2009. Trans‐synaptic transmission of vesicular Wnt signals through Evi/Wntless. Cell 139:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C, Martens S. 2012. Mechanisms and regulation of autophagosome formation. Curr Opin Cell Biol 24:496–501. [DOI] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI. 2015. The biology of proteostasis in aging and disease. Annu Rev Biochem 84:435–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C, Lin X, Chandran J, Shim H, Yang WJ, Cai H. 2007. The G59S mutation in p150(glued) causes dysfunction of dynactin in mice. J Neurosci 27:13982–13990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, Farah MH, Ackerley S, Hoke A, Maragakis N, Rothstein JD, Griffin J, et al. 2008. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci 28:1997–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JR, Sumner CJ, Caviston JP, Tokito MK, Ranganathan S, Ligon LA, Wallace KE, et al. 2006. A motor neuron disease‐associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J Cell Biol 172:733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd TE, Machamer J, O'Hara K, Kim JH, Collins SE, Wong MY, Sahin B, et al. 2012. The p150(Glued) CAP‐Gly domain regulates initiation of retrograde transport at synaptic termini. Neuron 74:344–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz P, Mauvezin C, Juhasz G. 2017. Exploring autophagy in Drosophila. Cells 6:pii E22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotvall J, Valadi H. 2007. Cell to cell signalling via exosomes through esRNA. Cell Adh Migr 1:156–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Hackmann Y, Dieckmann NM, Griffiths GM. 2014. The biogenesis of lysosomes and lysosome‐related organelles. Cold Spring Harb Perspect Biol 6:a016840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Pryor PR, Bright NA. 2007. Lysosomes: Fusion and function. Nat Rev Mol Cell Biol 8:622–632. [DOI] [PubMed] [Google Scholar]
- Maday S, Holzbaur EL. 2012. Autophagosome assembly and cargo capture in the distal axon. Autophagy 8:858–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Holzbaur EL. 2014. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell 30:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Holzbaur EL. 2016. Compartment‐specific regulation of autophagy in primary neurons. J Neurosci 36:5933–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur EL. 2014. Axonal transport: Cargo‐specific mechanisms of motility and regulation. Neuron 84:292–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Kimmelman AC. 2016. Mechanisms of selective autophagy in normal physiology and cancer. J Mol Biol 428:1659–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Henley JM. 2004. Activity‐dependent endocytic sorting of kainate receptors to recycling or degradation pathways. EMBO J 23:4749–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCray BA, Skordalakes E, Taylor JP. 2010. Disease mutations in Rab7 result in unregulated nucleotide exchange and inappropriate activation. Hum Mol Genet 19:1033–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, et al. 2017. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 93:1015–1034. [DOI] [PubMed] [Google Scholar]
- Miller KE, Heidemann SR. 2008. What is slow axonal transport? Exp Cell Res 314:1981–1990. [DOI] [PubMed] [Google Scholar]
- Moors TE, Hoozemans JJ, Ingrassia A, Beccari T, Parnetti L, Chartier‐Harlin MC, van de Berg WD. 2017. Therapeutic potential of autophagy‐enhancing agents in Parkinson's disease. Mol Neurodegener 12:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales I, Sanchez A, Rodriguez‐Sabate C, Rodriguez M. 2015. The degeneration of dopaminergic synapses in Parkinson's disease: A selective animal model. Behav Brain Res 289:19–28. [DOI] [PubMed] [Google Scholar]
- Moughamian AJ, Holzbaur EL. 2012. Dynactin is required for transport initiation from the distal axon. Neuron 74:331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller MJ, Klumpp S, Lipowsky R. 2008. Tug‐of‐war as a cooperative mechanism for bidirectional cargo transport by molecular motors. Proc Natl Acad Sci USA 105:4609–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, et al. 2004. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63:724–726. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Tamura N, Kono N, Shimanaka Y, Arai H, Yamamoto H, Mizushima N. 2017. Autophagosome formation is initiated at phosphatidylinositol synthase‐enriched ER subdomains. EMBO J 36:1719–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsumi Y. 2014. Historical landmarks of autophagy research. Cell Res 24:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okerlund ND, Schneider K, Leal‐Ortiz S, Montenegro‐Venegas C, Kim SA, Garner LC, Gundelfinger ED, et al. 2017. Bassoon controls presynaptic autophagy through Atg5. Neuron 93:897–913.e7. [DOI] [PubMed] [Google Scholar]
- Overly CC, Hollenbeck PJ. 1996. Dynamic organization of endocytic pathways in axons of cultured sympathetic neurons. J Neurosci 16:6056–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Vahidi B, Taylor AM, Rhee SW, Jeon NL. 2006. Microfluidic culture platform for neuroscience research. Nat Protoc 1:2128–2136. [DOI] [PubMed] [Google Scholar]
- Parton RG, Simons K, Dotti CG. 1992. Axonal and dendritic endocytic pathways in cultured neurons. J Cell Biol 119:123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perin MS, Fried VA, Stone DK, Xie XS, Sudhof TC. 1991. Structure of the 116‐kDa polypeptide of the clathrin‐coated vesicle/synaptic vesicle proton pump. J Biol Chem 266:3877–3881. [PubMed] [Google Scholar]
- Pierce JP, Mayer T, McCarthy JB. 2001. Evidence for a satellite secretory pathway in neuronal dendritic spines. Curr Biology 11:351–355. [DOI] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM. 2006. Kinesin‐1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell 17:2057–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JC, Guan S, Burlingame A, Prusiner SB, Ghaemmaghami S. 2010. Analysis of proteome dynamics in the mouse brain. Proc Natl Acad Sci USA 107:14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, et al. 2003. Mutant dynactin in motor neuron disease. Nat Genet 33:455–456. [DOI] [PubMed] [Google Scholar]
- Ramirez OA, Couve A. 2011. The endoplasmic reticulum and protein trafficking in dendrites and axons. Trends Cell Biol 21:219–227. [DOI] [PubMed] [Google Scholar]
- Raposo G, Stoorvogel W. 2013. Extracellular vesicles: Exosomes, microvesicles, and friends. J Cell Biol 200:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Acevedo‐Arozena A, Imarisio S, Berger Z, Vacher C, O'Kane CJ, Brown SD, et al. 2005. Dynein mutations impair autophagic clearance of aggregate‐prone proteins. Nat Genet 37:771–776. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO. 2014. Synaptic vesicle recycling: Steps and principles. EMBO J 33:788–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland AM, Richmond JE, Olsen JG, Hall DH, Bamber BA. 2006. Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J Neurosci 26:1711–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, et al. 2012. Exosome‐associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem 287:3842–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos SD, Carvalho AL, Caldeira MV, Duarte CB. 2009. Regulation of AMPA receptors and synaptic plasticity. Neuroscience 158:105–125. [DOI] [PubMed] [Google Scholar]
- Schink KO, Tan KW, Stenmark H. 2016. Phosphoinositides in control of membrane dynamics. Annu Rev Cell Dev Biol 32:143–171. [DOI] [PubMed] [Google Scholar]
- Schnapp BJ, Reese TS. 1989. Dynein is the motor for retrograde axonal transport of organelles. Proc Natl Acad Sci USA 86:1548–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch S, Deak F, Konigstorfer A, Mozhayeva M, Sara Y, Sudhof TC, Kavalali ET. 2001. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294:1117–1122. [DOI] [PubMed] [Google Scholar]
- Schuh AL, Audhya A. 2014. The ESCRT machinery: From the plasma membrane to endosomes and back again. Crit Rev Biochem Mol Biol 49:242–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze H, Kolter T, Sandhoff K. 2009. Principles of lysosomal membrane degradation: Cellular topology and biochemistry of lysosomal lipid degradation. Biochim Biophys Acta 1793:674–683. [DOI] [PubMed] [Google Scholar]
- Schwarz LA, Hall BJ, Patrick GN. 2010. Activity‐dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J Neurosci 30:16718–16729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Walsh DM. 2009. Alzheimer's disease: Synaptic dysfunction and Abeta. Mol Neurodegener 4:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Hegde RS. 2014. Cell Biology. Local synthesis and disposal. Science 346:701–702. [DOI] [PubMed] [Google Scholar]
- Sheehan P, Waites CL. 2017. Coordination of synaptic vesicle trafficking and turnover by the Rab35 signaling network. Small GTPases 27:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan P, Zhu M, Beskow A, Vollmer C, Waites CL. 2016. Activity‐dependent degradation of synaptic vesicle proteins requires Rab35 and the ESCRT pathway. J Neurosci 36:8668–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehata M, Matsumura H, Okubo‐Suzuki R, Ohkawa N, Inokuchi K. 2012. Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long‐term depression. J Neurosci 32:10413–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Raposo G. 2009. Exosomes–vesicular carriers for intercellular communication. Curr Opin Cell Biol 21:575–581. [DOI] [PubMed] [Google Scholar]
- Solinger JA, Spang A. 2013. Tethering complexes in the endocytic pathway: CORVET and HOPS. FEBS J 280:2743–2757. [DOI] [PubMed] [Google Scholar]
- Song JW, Misgeld T, Kang H, Knecht S, Lu J, Cao Y, Cotman SL, et al. 2008. Lysosomal activity associated with developmental axon pruning. J Neurosci 28:8993–9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Zinsmaier KE. 2003. Endophilin and synaptojanin hook up to promote synaptic vesicle endocytosis. Neuron 40:665–667. [DOI] [PubMed] [Google Scholar]
- Soto F, Kerschensteiner D. 2015. Synaptic remodeling of neuronal circuits in early retinal degeneration. Front Cell Neurosci 9:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukup SF, Kuenen S, Vanhauwaert R, Manetsberger J, Hernandez‐Diaz S, Swerts J, Schoovaerts N, et al. 2016. A LRRK2‐dependent EndophilinA phosphoswitch is critical for macroautophagy at presynaptic terminals. Neuron 92:829–844. [DOI] [PubMed] [Google Scholar]
- Speese SD, Trotta N, Rodesch CK, Aravamudan B, Broadie K. 2003. The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr Biol 13:899–910. [DOI] [PubMed] [Google Scholar]
- Spinosa MR, Progida C, De Luca A, Colucci AM, Alifano P, Bucci C. 2008. Functional characterization of Rab7 mutant proteins associated with Charcot‐Marie‐Tooth type 2B disease. J Neurosci 28:1640–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Schuman EM. 2003. Compartmentalized synthesis and degradation of proteins in neurons. Neuron 40:347–359. [DOI] [PubMed] [Google Scholar]
- Strom AL, Gal J, Shi P, Kasarskis EJ, Hayward LJ, Zhu H. 2008. Retrograde axonal transport and motor neuron disease. J Neurochem 106:495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. 2004. The synaptic vesicle cycle. Annu Rev Neurosci 27:509–547. [DOI] [PubMed] [Google Scholar]
- Tai HC, Schuman EM. 2008. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci 9:826–838. [DOI] [PubMed] [Google Scholar]
- Takats S, Boda A, Csizmadia T, Juhasz G. 2016. Small GTPases controlling autophagy‐related membrane traffic in yeast and metazoans. Small GTPases 22:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, et al. 2014. Loss of mTOR‐dependent macroautophagy causes autistic‐like synaptic pruning deficits. Neuron 83:1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Blurton‐Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. 2005. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods 2:599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Dieterich DC, Ito HT, Kim SA, Schuman EM. 2010. Microfluidic local perfusion chambers for the visualization and manipulation of synapses. Neuron 66:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Jeon NL. 2010. Micro‐scale and microfluidic devices for neurobiology. Curr Opin Neurobiol 20:640–647. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Rhee SW, Jeon NL. 2006. Microfluidic chambers for cell migration and neuroscience research. Methods Mol Biol 321:167–177. [DOI] [PubMed] [Google Scholar]
- Tooze SA, Abada A, Elazar Z. 2014. Endocytosis and autophagy: Exploitation or cooperation?. Cold Spring Harb Perspect Biol 6:a018358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uytterhoeven V, Kuenen S, Kasprowicz J, Miskiewicz K, Verstreken P. 2011. Loss of skywalker reveals synaptic endosomes as sorting stations for synaptic vesicle proteins. Cell 145:117–132. [DOI] [PubMed] [Google Scholar]
- Vanhauwaert R, Kuenen S, Masius R, Bademosi A, Manetsberger J, Schoovaerts N, Bounti L, et al. 2017. The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J 36:1392–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer‐Grumbach M, Kwon JM, FitzPatrick D, et al. 2003. Mutations in the small GTP‐ase late endosomal protein RAB7 cause Charcot‐Marie‐Tooth type 2B neuropathy. Am J Hum Genet 72:722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayan V, Verstreken P. 2017. Autophagy in the presynaptic compartment in health and disease. J Cell Biol 216:1895–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitelli R, Santillo M, Lattero D, Chiariello M, Bifulco M, Bruni CB, Bucci C. 1997. Role of the small GTPase Rab7 in the late endocytic pathway. J Biol Chem 272:4391–4397. [DOI] [PubMed] [Google Scholar]
- Wang D, Chan CC, Cherry S, Hiesinger PR. 2013. Membrane trafficking in neuronal maintenance and degeneration. Cell Mol Life Sci 70:2919–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Hiesinger PR. 2012. Autophagy, neuron‐specific degradation and neurodegeneration. Autophagy 8:711–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DO, Martin KC, Zukin RS. 2010. Spatially restricting gene expression by local translation at synapses. Trends Neurosci 33:173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Davis S, Zhu M, Miller EA, Ferro‐Novick S. 2017. Autophagosome formation: Where the secretory and autophagy pathways meet. Autophagy 13:973–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widagdo J, Anggono V. 2015. Ubiquitin signals the demise of AMPA receptors. Oncotarget 6:15718–15719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widagdo J, Chai YJ, Ridder MC, Chau YQ, Johnson RC, Sah P, Huganir RL, et al. 2015. Activity‐dependent ubiquitination of GluA1 and GluA2 regulates AMPA receptor intracellular sorting and degradation. Cell Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson WR, Wang D, Haberman AS, Hiesinger PR. 2010a. A dual function of V0‐ATPase a1 provides an endolysosomal degradation mechanism in Drosophila melanogaster photoreceptors. J Cell Biol 189:885–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson WR, Yang T, Terman JR, Hiesinger PR. 2010b. Guidance receptor degradation is required for neuronal connectivity in the Drosophila nervous system. PLoS Biol 8:e1000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojnacki J, Galli T. 2016. Membrane traffic during axon development. Dev Neurobiol 76:1185–1200. [DOI] [PubMed] [Google Scholar]
- Wong E, Cuervo AM. 2010. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 13:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Klionsky DJ. 2007. Autophagosome formation: Core machinery and adaptations. Nat Cell Biol 9:1102–1109. [DOI] [PubMed] [Google Scholar]
- Xu H, Ren D. 2015. Lysosomal physiology. Annu Rev Physiol 77:57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. 2007. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev 59:14–39. [DOI] [PubMed] [Google Scholar]
- Yogev S, Shen K. 2014. Cellular and molecular mechanisms of synaptic specificity. Annu Rev Cell Dev Biol 30:417–437. [DOI] [PubMed] [Google Scholar]
- Yuyama K, Sun H, Mitsutake S, Igarashi Y. 2012. Sphingolipid‐modulated exosome secretion promotes clearance of amyloid‐beta by microglia. J Biol Chem 287:10977–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaffagnini G, Martens S. 2016. Mechanisms of selective autophagy. J Mol Biol 428:1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Fishel Ben Kenan R, Osakada Y, Xu W, Sinit RS, Chen L, Zhao X, et al. 2013. Defective axonal transport of Rab7 GTPase results in dysregulated trophic signaling. J Neurosci 33:7451–7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinsmaier KE, Eberle KK, Buchner E, Walter N, Benzer S. 1994. Paralysis and early death in cysteine string protein mutants of Drosophila. Science 263:977–980. [DOI] [PubMed] [Google Scholar]