

Abstract
The muscarinic acetylcholine receptors are a subfamily of G protein-coupled receptors that regulate numerous fundamental functions of the central and peripheral nervous system. The past few years have witnessed unprecedented new insights into muscarinic receptor physiology pharmacology and structure. These advances include the first structural views of muscarinic receptors in both inactive and active conformations, as well as a better understanding of the molecular underpinnings of muscarinic receptor regulation by allosteric modulators. These recent findings should facilitate the development of new muscarinic receptor subtype-selective ligands that could prove to be useful for the treatment of many severe pathophysiological conditions.
The muscarinic acetylcholine receptors (mAChRs) comprise a family of five related G protein-coupled receptors (GPCRs) belonging to the α-branch of class A GPCRs1. The mAChR family consists of five distinct subtypes, denoted M1 to M5 (and encoded by the genes CHRM1 to CHRM5). Three of these receptor subtypes (M1, M3 and M5) have been shown to couple to G proteins of the Gq/11 family, whereas the remaining two subtypes (M2 and M4) preferentially signal through the Gi/o family of G proteins2. The mAChRs have a central role in human physiology, regulating heart rate, smooth muscle contraction, glandular secretion and many fundamental functions of the central nervous system (CNS)3.
Currently, drugs targeting muscarinic receptors are used for the treatment of several pathophysiological conditions, including chronic obstructive pulmonary disease, overactive bladder and Sjögren's syndrome3. Despite the powerful and diverse pharmacological actions of mus-carinic receptor agonists and antagonists, the development of these drugs for other clinical applications has probably been held back, at least in part, by the lack of small-molecule ligands that can inhibit or activate specific mAChRs with high selectivity. As a result, the precise physiological and pathophysiological roles of the individual mAChR subtypes have, until recently, remained poorly defined. However, during the past 15 years the generation and phenotypic analysis of Chrm1- to Chrm5-knockout mice has been instrumental in improving our understanding of the biology of the individual mAChRs3.
Recent studies with novel mAChR mouse models have provided additional insights into the physiological roles of the different mAChR subtypes4. Moreover, during the past few years, several laboratories have succeeded in developing ligands that show high selectivity for specific mAChR subtypes5,6. In contrast to conventional muscarinic receptor ligands, which bind to the orthosteric receptor site, most of these new ligands bind to distinct allosteric sites. Such allosteric ligands can influence the potency and efficacy of orthosteric ligands, and they may possess agonistic or inverse agonist activity in their own right. Interestingly, bitopic muscarinic receptor ligands with preference for certain subtypes have also been developed recently7–9. Such agents, which can interact with both allosteric and orthosteric receptor sites simultaneously, offer new opportunities to target specific mAChR subtypes for therapeutic purposes.
The recent determination of the first mAChR structures (M2 and M3 subtypes) represents a milestone in the mAChR field10–12. These studies have provided the first molecular views of mAChRs in both their inactive10,11 and active conformations12, revealing the molecular nature of the binding sites for orthosteric muscarinic receptor ligands. Moreover, structural and computational studies have identified the mechanisms by which drug-like allosteric modulators bind to the M2 subtype12,13.
In this Review, we summarize and discuss a series of recent studies that have advanced our knowledge of mAChR biology, structure and pharmacology. In particular, we emphasize the potential therapeutic implications of these novel findings. As mAChRs are prototypic class A GPCRs, the topics covered herein have implications for other members of this receptor superfamily.
Novel mAChR mouse models
Since the late 1990s, studies with Chrm1- to Chrm5- knockout mice have elucidated many important physiological functions of the individual mAChR subtypes3. The outcome of this work suggested that modulating the activity of specific mAChR subtypes by selective ligands might prove to be beneficial for the treatment of many CNS disorders and other clinical conditions3. Until recently, essentially all phenotyping studies were carried out with conventional, constitutive mAChR-knockout mice, which lack one or more of the genes encoding mAChRs throughout development in all cells of the body. The proper interpretation of the phenotypes observed with conventional mAChR-knockout mice is often complicated because individual mAChR subtypes are expressed in many different tissues and cell types, and compensatory molecular or physiological changes can occur during development.
To circumvent these difficulties, novel conditional knockout mice have been created that lack specific mAChR subtypes only in certain tissues or cell types. For these studies, mice containing ‘floxed’ versions of specific genes encoding mAChRs were crossed with transgenic mice expressing Cre recombinase in a cell type- or tissue-selective fashion (TABLE 1). mAChR knock-in mice have also been reported, in which the native genes encoding M3 or M4 receptors — CHRM3 or CHRM4, respectively— were replaced with mutant versions (TABLE 1). Here, we summarize several recent studies that have used novel mAChR-mutant mice as well as conventional mAChR-knockout mice. These studies are discussed in the context of selected human diseases in which the development of novel classes of drugs targeting muscarinic receptors might be beneficial.
Table 1. Novel mAChR-mutant mouse models.
| Mouse strain | Major phenotypes | Refs |
|---|---|---|
| Chrm1-mutant mice selectively lacking M1 receptors in the forebrain or hippocampal CA3 pyramidal neurons (use of floxed M1 receptor mice) | Lack of mGluR-mediated LTD in the hippocampus, accompanied by changes in presynaptic neurotransmitter release | 116 |
| Mice carrying a floxed Chrm2 gene (floxed M2 receptor mice) | N/A | * |
| Chrm3-mutant mice selectively lacking M3 receptors in specific cell types (use of floxed M3 receptor mice) | Pancreatic β-cells: deficits in glucose tolerance, accompanied by impaired insulin release in vivo | 34 |
| Brain (neurons and glial cells): reduced body size; hypoplasia of the anterior pituitary gland; reduced levels of pituitary and serum growth hormone, as well as prolactin | 117 | |
| Brain (neurons and glial cells): low bone mass, probably owing to enhanced central sympathetic outflow | 118 | |
| Hepatocytes: none (no changes in glucose homeostasis and hepatic glucose fluxes) | 119 | |
| Osteoblasts: none (no changes in bone formation and bone mass) | 118 | |
| Smooth muscle cells (airways): restoration of normal lung function in obese mice | 120 | |
| Chrm3-knock-in mice containing 15 Ser→Ala point mutations within the i3 loop of the M3 receptor | Greatly reduced M3 receptor phosphorylation and impaired arrestin recruitment following M3 receptor activation; deficits in fear conditioning, learning and memory | 121 |
| Impaired glucose tolerance, accompanied by reduced M3 receptor-mediated insulin release from pancreatic islets | 40 | |
| Chrm4-mutant mice selectively lacking M4 receptors in D1 dopamine receptor-expressing neurons (use of floxed M4 receptor mice) | Increased dopamine efflux in the nucleus accumbens; modulation of dopamine-dependent behaviours | 24 |
| Strong reduction of the antipsychotic-like effects of xanomeline in two mouse models of psychosis | 25 | |
| Chrm5-knock-in mice containing an 18-amino-acid deletion within the M5 receptor-i3 loop (residues 369–386) | Reduction in evoked dopamine release from striatal slices | 122 |
LTD, long-term depression; mAChR, muscarinic acetylcholine receptor (five distinct subtypes, denoted M1 to M5, which are encoded by the genes Chrm1 to Chrm5 in mice); mGluR, metabotropic glutamate receptor.
J.W. & J. Jeon, unpublished observations.
Targeting mAChRs in disease
Alzheimer's disease
Alzheimer's disease, which is the most common form of dementia, is recognized as a major public health crisis14. At present, there are no drugs available that are highly effective in preventing Alzheimer's disease or slowing its progress. Based on behavioural, pharmacological, anatomical and neurochemical evidence, M1 receptor-selective agonists seem to have the potential to ameliorate the symptoms of Alzheimer's disease and related cognitive disorders15. In fact, several recent studies support the concept that M1 receptor-selective agonists or positive allosteric modulators (PAMs) may prove to be useful as cognition-enhancing drugs. Davis et al.16 demonstrated that M1 receptor deficiency increases amyloidogenic processing of amyloid precursor protein (APP) — a key feature of Alzheimer's disease — in a mouse model of Alzheimer's disease. In a related study, Medeiros et al.17 reported that the lack of M1 receptors further exacerbated cognitive processes and Alzheimer's disease-related pathological features in a mouse model of Alzheimer's disease. In addition, Chrm1-knockout mice exhibited an age-dependent cognitive decline in tasks that they performed normally at a younger age17. Most importantly, several recent studies have shown that compounds that act as PAMs at M1 receptors have cognition-enhancing activity in rodents and are able to improve impaired cognition in mouse models of Alzheimer's disease18,19. Taken together, these new findings provide a rational basis for the development of M1-selective drugs (PAMs or agents that can activate M1 receptors directly) for the treatment of Alzheimer's disease and related disorders.
Schizophrenia
Schizophrenia, which is a chronic disabling brain disorder that affects ∼1% of the general population20, is characterized by enhanced central dopa-minergic signalling. Studies with conventional mAChR-knockout mice have indicated that the lack of central M1 and/or M4 receptors leads to a ‘dopamine hypersen-sitivity phenotype’, which suggests that agents that can enhance signalling through these receptor subtypes may be endowed with antipsychotic activity3. In agreement with this concept, two clinical trials demonstrated that xanomeline, an M1- and M4-preferring muscarinic receptor agonist21, was effective in ameliorating psychosis-like symptoms22,23. A recent study with cell type-specific Chrm4-knockout mice strongly suggests that a distinct subpopulation of neuronal M4 receptors expressed by D1 dopamine receptor-expressing neurons may be of particular importance in mediating the antipsychotic actions triggered by M4 receptor activation24. In addition, Dencker et al.25 reported that the antipsychotic-like effects of xanomeline were almost completely abolished in two mouse models of psychosis in which mice lacked M4 receptors selectively in D1 receptor-expressing neurons. In agreement with these findings, several recent preclinical studies suggest that PAMs of M1 and/or M4 receptors may prove to be clinically useful for the treatment of schizophrenia (reviewed in REFS 18,26–28).
Drug addiction
Acetylcholine has a key role in modulating the neurochemical and behavioural CNS responses to drugs of abuse29. Phenotypic analysis of Chrm5-knockout mice suggested that centrally active M5 receptor blockers might prove to be useful for the treatment of drug abuse, including cocaine or opioid addiction3. More recent studies with other mAChR-knockout lines indicate that signalling through central M1 and M4 receptors may also modulate drug-seeking behaviour. For example, recent work demonstrated that allosteric M1 receptor agonists and xanomeline can attenuate the reinforcing and discriminative stimulus effects of cocaine30,31. These effects were greatly reduced or abolished in Chrm1-knockout mice or in mice that were deficient in both M1 and M4 receptors (Chrm1–/– Chrm4–/– mice). Moreover, Schmidt et al.32 reported that the reinforcing effects of chronic cocaine self-administration are significantly increased in Chrm4-knockout mice. These findings indicate that centrally active drugs that can selectively stimulate M1 and/or M4 receptors may prove to be useful in the treatment of drug addiction.
Type 2 diabetes
Type 2 diabetes has emerged as a major threat to public health worldwide33. One of the key patho-physiological features of type 2 diabetes is that pancreatic β-cells are unable to secrete sufficient insulin to overcome peripheral insulin resistance. Studies with β-cell-selective Chrm3-mutant mice34,35 showed that strategies aimed at enhancing the activity of β-cell M3 receptors may prove to be useful for the treatment of type 2 diabetes. To further test this hypothesis, two recent studies36,37 analysed transgenic mice that expressed an M3 receptor-based designer receptor (Gq DREADD; where DREADD stands for ‘designer receptor exclusively activated by a designer drug’) selectively in pancreatic β-cells. This modified M3 receptor is unable to bind acetylcholine, the endogenous ligand, but can be selectivity activated by clozapine N-oxide (CNO), a drug that is otherwise pharmacologically inert36,38. Systematic metabolic analysis of this mouse line confirmed that CNO-dependent activation of this designer M3 receptor has numerous beneficial effects on β-cell function and is able to prevent experimentally induced diabetes36,37. Although there is evidence that the ability of β-cell M3 receptors to promote insulin release depends on the presence of G proteins of the Gq family39, two recent studies suggest that M3 receptor-mediated activation of arrestin-dependent β-cell pathways may further enhance insulin secretion40,41. Currently, ligands that are able to selectively promote signalling through M3 receptors are not available, and the development of such agents would greatly facilitate studies aimed at testing the potential usefulness of targeting β-cell M3 receptors for the treatment of type 2 diabetes and related metabolic disorders.
Cancer
Accumulating evidence suggests that mAChR-dependent signalling pathways can promote cell proliferation and cancer progression42,43. Interestingly, Raufman et al.44 demonstrated that Chrm3-knockout mice displayed reduced epithelial cell proliferation, tumour number and size in a mouse model of colon neoplasia (azoxymethane-induced colon cancer). In a related study45, M3 receptor deficiency was associated with a pronounced reduction in tumour number and volume in a genetic model of intestinal neoplasia (ApcMin/+ mice). Magnon et al.46 reported that M1 receptor deficiency inhibited mAChR-mediated prostate cancer invasion and metastasis in two mouse models of prostate cancer. These findings support the concept that subtype-selective mAChR antagonists may prove to be clinically useful for the treatment of certain forms of cancer.
Novel mAChR pharmacology
A major shift in drug discovery has been the recognition that most — if not all — GPCRs possess spatially distinct allosteric sites that can be exploited by small molecules to modulate the activity of orthosteric ligands6,47,48. The mAChRs are arguably the pre-eminent models for contributing to our understanding of GPCR allostery5,49-51 (see BOX 1 for a representative list of muscarinic allosteric modulators).
Box 1. Selected allosteric modulators of mAChRs.
The allosteric modulators listed here have been used to provide important insights into muscarinic acetylcholine receptor (mAChR) allostery. Note that this list is by no means exhaustive. Some of the modulators bind to all five mAChR subtypes but are listed under the mAChR subtype for which they have been most extensively studied.
M1 receptor
Tacrine
MT7
Brucine
Staurosporine
BQCA
VU0119498
VU0029627
ML169
M2 receptor
Gallamine
Alcuronium
C7/3-phth
W84
DUO3
Tacrine
LY2033298
LY2119620
M3 receptor
N-chloromethyl brucine
VU0119498
M4 receptor
MT3
Thiochrome
LY2033298
LY2119620
VU0010010
VU0152100
M5 receptor
VU0119498
VU0238429
ML375
Pharmacological characteristics of allosteric modulators
Allosteric ligands promote conformational changes in the receptor that manifest as an alteration in the properties of a ligand bound to the classical, orthosteric site, and by the potential for direct activation of receptor signalling via the allosteric site52. PAMs enhance orthosteric activity, negative allosteric modulators (NAMs) inhibit it, and agents that occupy an allosteric site but do not change the activity of orthosteric ligands are referred to as neutral allosteric ligands (NALs). Allosteric compounds that directly activate GPCRs are called allosteric agonists. However, these designations are contextual, as they are dependent on the receptor, orthosteric ligand and the assay system used52–54. For example, the M1 receptor-selective modulator BQCA (REF. 55) acts solely as a PAM of acetylcholine activity when assayed in a cell line with low M1 receptor expression56 and/or at signalling pathways that are weakly coupled to the M1 receptor57, but it acts as both a full allosteric agonist and as a PAM in a system with a high M1 receptor reserve57.
Characteristically, the effects of allosteric modulators are saturable: that is, no further activity is observed above a certain limit, irrespective of modulator dose. This phenomenon is driven by the cooperativity between orthosteric and allosteric sites, and can range from subtle fine-tuning to very large modulation. For instance, N-chloromethyl brucine potentiates acetylcholine affinity by threefold at the M3 receptor58, whereas BQCA potentiates acetylcholine affinity at the M1 receptor by ∼100-fold57. A second property is probe dependence59, whereby the magnitude and direction of the allosteric effect mediated by the same modulator acting at the same receptor varies depending on the orthosteric ligand used to probe receptor function. A striking example is LY2033298, which is a PAM of oxotremorine-M signalling at the M2 receptor, but a NAM of xanomeline at the same receptor60.
The third characteristic of GPCR allostery is biased agonism, which refers to the ability of different ligands to stabilize a subset of functionally relevant GPCR conformations such that different signalling outputs are achieved at the exclusion of others54,61,62. Biased agonism can be imposed on the signalling of orthosteric agonists by co-bound modulators. One example is the compound VU0029767, which potentiates acetylcholine-mediated phospholipase C activity via the M1 receptor, but does not affect acetylcholine-induced phospholipase D activation63. Finally, a crucial property of allosteric modulators is potential mAChR-subtype selectivity. This can be attained either by targeting a less conserved site on the receptor or via selective cooperativity with the orthosteric ligand, even if the allosteric site is shared between subtypes64. Indeed, selective cooperativity is the major mechanism by which compounds such as thiochrome and LY2033298 show high selectivity for the M4 receptor64–66.
Bitopic muscarinic ligands
In recent years, research has turned to the rational design of hybrid molecules, which are designed to simultaneously bridge orthosteric and allosteric sites within a single receptor; such compounds have been termed ‘bitopic’, ‘dualsteric’ or ‘multivalent’7,67,68. As summarized in FIG. 1, each of the different modes of targeting mAChRs has its pros and cons. The orthosteric site can provide high affinity and is supported by decades of studies of structure–activity relationships (SARs), but mAChR-subtype selectivity remains largely unattainable. Allosteric sites provide the opportunity to achieve greater receptor selectivity and promote more physiological response patterns. However, high-affinity modulators have not been reported to date, and certain modulator scaffolds — like many orthosteric drugs — show minimal improvements in activity despite numerous modifications69. The bitopic ligand approach attempts to target the allosteric site to achieve selectivity and the orthosteric site to provide high affinity.
Figure 1. Modes of targeting mAChRs (GPCRs) by different classes of ligands.
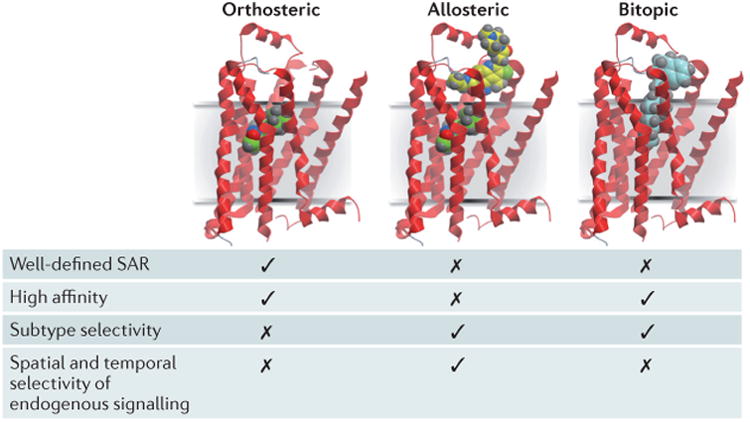
Orthosteric ligands (green) bind to the site recognized by the endogenous agonist (acetylcholine) for the receptor. Allosteric ligands (yellow) bind to a topographically distinct site. Bitopic ligands (blue) concomitantly interact with both orthosteric and allosteric sites. The key properties generally associated with each mode of receptor targeting are also indicated. GPCR, G protein-coupled receptor; mAChR, muscarinic acetylcholine receptor; SAR, structure–activity relationship.
Despite some challenges7,68,70, the pursuit of bitopic ligands has yielded interesting pharmacological findings. Disingrini et al.71 were the first to combine a nonselective, high-affinity orthosteric agonist (iperoxo) with an M2 receptor-selective allosteric modulator to generate an M2 receptor-selective agonist. This concept was extended by Mohr and colleagues72–74. Interestingly, although bitopic agonists with mAChR-subtype selectivity and signalling bias have been described, substantial gains in affinity were generally not observed7. By contrast, the bitopic muscarinic receptor antagonist THRX-160209 demonstrated remarkable gains in both affinity and selectivity for the M2 receptor75, which highlights that the appropriate combination of an orthosteric antagonist, NAM and linker can yield ligands with the desired pharmacological properties.
These findings raise the possibility that previously described selective and/or biased agonists may represent unappreciated bitopic ligands. Consistent with this concept, a study of the biased M2 receptor partial agonist McN-A-343 unmasked a pure orthosteric agonist and pure NAM of agonist efficacy following reverse engineering of McN-A-343 into fragments76. A more recent study of the M1 receptor-selective agonist TBPB also identified orthosteric and allosteric fragments of this molecule53. This latter study is of particular relevance because TBPB, together with other novel M1 receptor-selective agonists such as AC-42 and 77-LH-28-1, was originally classified as an allosteric agonist77–80. However, like McN-A-343, these agents display mixed modes of orthosteric or allosteric pharmacology depending on the experimental assay conditions48,53,81. It is thus likely that such compounds adopt novel poses within the mAChRs that bridge both orthosteric and allosteric sites, hence accounting for their unique pharmacology81.
Towards the molecular mechanisms of allostery
The simplest mechanism to account for allosteric behaviour is the Monod–Wyman–Changeux (MWC) model82,83, which postulates that receptors exist in an equilibrium between different states, and that orthosteric and allosteric ligands select one or more of these states over others84. Because this occurs via topographically distinct sites, the state that is stabilized by one class of ligand can present a modified binding surface for the other class, which is manifested as the allosteric interaction. If this mechanism were restricted to two states, it would be expected that any ligand favouring the active state would display some degree of agonism, whereas compounds stabilizing the inactive state would show some degree of inverse agonism. Moreover, allosteric ligands that favoured the active state would be PAMs for agonists but NAMs for inverse agonists; this is the simplest molecular explanation for probe dependence. Interestingly, these predictions were confirmed in a recent study of BQCA activity at the M1 receptor57, which indicates that some mAChR modulators can mediate their effects predominantly through a simple conformational selection mechanism.
However, this simple model does not explain the probe dependence observed with compounds such as LY2033298 at the M2 receptor, which can show PAM or NAM activity with different agonists60. It also does not explain biased allosteric modulation, or why compounds such as gallamine are NAMs for agonists and antagonists85. Thus, additional mechanisms seem to be involved, most obviously involving multiple active receptor states86. Prior studies have also shown that compounds such as staurosporine and WIN62577 interact with a second allosteric site on mAChRs that is distinct from the common site87,88. The location of this site remains unidentified, but it may be intracellular89. If so, one can envisage more complex scenarios that involve an allosteric mechanism with respect to the orthosteric site, but potentially a steric mechanism with respect to receptor–transducer interactions. Similarly, it has been postulated that gallamine may utilize a two-step binding process, involving a peripheral domain that is distinct from the common allosteric site, in its interaction with the receptor85. Finally, the simplest allosteric models do not explicitly incorporate receptor oligomers as the key functional units; a tetrameric arrangement has been proposed, at least for the M2 receptor, to account for the observed cooperative effects of both orthosteric and allosteric ligands90,91. Thus, despite the identification of a vast repertoire of behaviours for muscarinic allosteric ligands, the molecular mechanisms underlying these behaviours are only starting to be elucidated. It is particularly exciting, therefore, that recent structural and computational biology breakthroughs have shed new light on this matter.
Structural insights into ligand binding mode
Although mAChRs are among the most extensively studied GPCRs in terms of their pharmacology and biological function, the molecular structures of these receptors have, until recently, remained poorly understood. In 2012 the first mAChR structures were published10,11, revealing the molecular organization of the M2 and M3 receptor subtypes in inactive, antagonist (inverse agonist)-bound conformations. This work was made possible owing to advances in GPCR biochemistry and crystallography, principal among them the T4 lysozyme fusion method92, new detergents93, the lipidic cubic phase crystallization method94,95 and micro-focus X-ray diffraction data collection96.
The structures of the M2 and M3 receptors revealed that, like other biogenic amine receptors, members of the mAChR family share the seven-pass transmembrane topology and overall fold of other GPCRs, with a few features extending outside of the membrane plane (FIG. 2). The ligand-binding pocket is deeply buried within the membrane (FIGS 2, 3a), and is situated similarly to the orthosteric binding sites of the related histamine97, dopamine98, adrenaline99–101 and serotonin102,103 receptors. The ligands bound to the M2 and M3 receptors in the two crystal structures — quinuclidinyl benzilate (QNB) and tiotropium, respectively — are both antagonists (inverse agonists) with similar chemical structures. In each case, the ligand occupies a similar pose (FIG. 3b), engaging in extensive hydrophobic contact with the receptor. A smaller number of polar contacts are evident: a pair of hydrogen bonds between Asn6.52 and the ligand hydroxyl and ketone, and a charge–charge interaction between the cationic amine of the ligand and the conserved Asp3.32 (superscripts denote Ballesteros–Weinstein104 GPCR numbering) (FIG. 3b). The latter interaction is seen in all biogenic amine receptor structures solved to date and has been shown to make a major energetic contribution to binding105. The paired hydrogen bonding between the ligand and Asn6.52 seems to be a unique feature of the mAChR family and has been proposed to be an important factor in slow ligand dissociation from mAChRs106.
Figure 2. Overall structure of the M2 and M3 receptors.
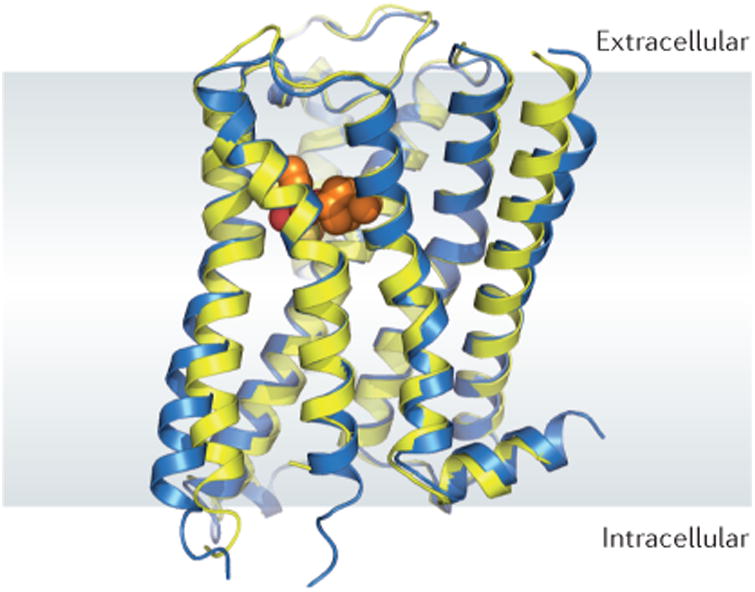
The structures of the inactive, antagonist-bound M2 (blue) and M3 (yellow) receptors are highly similar to each other in overall fold. For the sake of clarity, only the M3 receptor ligand (tiotropium) is shown. The overall architecture of the M2 and M3 muscarinic acetylcholine receptors (mAChRs) is very similar to that of other biogenic amine G protein-coupled receptors (GPCRs), with similar orthosteric ligand binding sites (orange spheres).
Figure 3. Structure of the orthosteric mAChR ligand binding site.
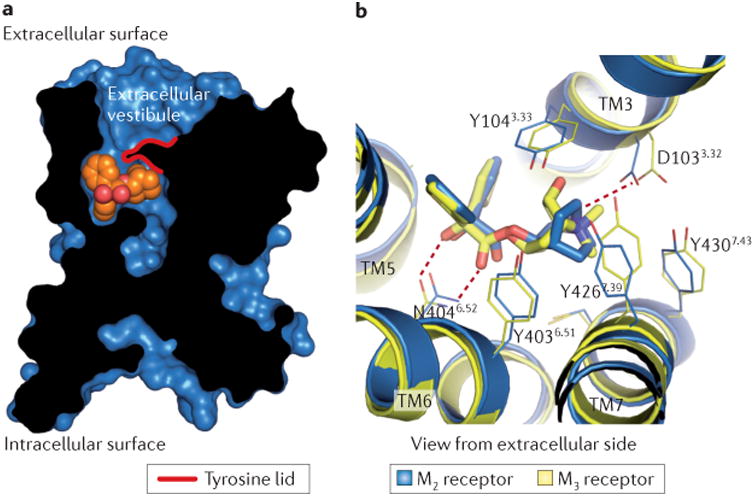
a | A cross-section through the M2 receptor structure reveals a large, solvent-accessible cavity extending through the receptor. The QNB ligand (orange spheres) is bound within the receptor transmembrane core (black, receptor protein; blue, receptor surface). b | The orthosteric ligand (antagonist)-binding sites for the M2 and M3 receptors are almost identical in both structure and sequence. Polar contacts (red dotted lines) between the receptor and bound antagonist (M2, QNB; M3, tiotropium) are identical for the two receptors. Residues are numbered according to the human M2 receptor sequence. mAChR, muscarinic acetylcholine receptor.
Comparison of the orthosteric ligand-binding pockets of the M2 and M3 receptors reveals a probable underlying factor for the limited success in the development of mAChR-subtype-selective ligands. The residues lining the orthosteric binding site are conserved absolutely in sequence and, moreover, are positioned almost identically in space in both receptors (FIG. 3b). Although there are a few subtle differences between the structures of the M2 and M3 receptors, attempts to exploit these minor structural variations to obtain ligands with increased mAChR-subtype selectivity have thus far been unsuccessful105.
Strikingly, in the structures of the M2 and M3 receptors, the orthosteric ligand is almost completely occluded from solvent, with a tyrosine ‘lid’ located directly above — that is, extracellular to — the ligand (FIG. 3). This lid divides a large, solvent-accessible cavity into two distinct regions, only one of which interacts with the bound antagonists (FIG. 3a). The upper portion of this cavity, termed the extracellular vestibule, is lined with residues that have been implicated in the binding of allosteric modulators107. As discussed below, LY2119620, which is a PAM of mAChRs, binds to this outer cavity12. Although the backbone fold of the structures surrounding this allosteric site is remarkably well conserved between the M2 and M3 receptors, the amino acid side chains lining it show more divergence10. Hence, the development of ligands that interact with non-conserved amino acids in the allosteric site provides a possible means by which subtype-selective therapeutics might be developed.
Molecular dynamics studies of the M3 receptor indicate that the allosteric site may have a role in facilitating the binding of orthosteric ligands. Simulations of tio-tropium binding to the M3 receptor suggested that this ligand may rapidly interact with the extracellular vestibule of the receptor, but in these simulations tiotropium failed to enter the orthosteric site10. This may be due to limitations in the timescale accessible to molecular dynamics simulations, as well as the slow binding kinetics of tiotropium10. Notably, it has been shown experimentally that orthosteric ligands can bind to an allosteric site on the M2 receptor, albeit with low affinity90. Interactions with the extracellular vestibule may initiate desolvation of the ligand, which is required for the occupation of the orthosteric pocket. If binding to the allosteric site is indeed a metastable state for orthosteric muscarinic ligands, deliberate exploitation of this feature could potentially facilitate the development of muscarinic ligands with increased mAChR-subtype selectivity and/or improved kinetic properties.
Molecular basis for NAM function at the M2 receptor
Recently, the molecular mechanism of allosteric modulator binding to the inactive state of the M2 receptor was investigated by computational molecular dynamics and site-directed mutagenesis studies13. Using the inactive crystal structure of the M2 receptor as a starting model, the binding of a series of structurally diverse cationic small-molecule modulators was simulated using long-timescale molecular dynamics. These simulations suggested that modulator binding to the extracellular vestibule may be driven largely by interactions between cationic amines on the modulators and aromatic residues on the receptor. The allosteric pocket appears to contain two cation-π centres: one including Trp4227.35 and Tyr177ECL2 (extracellular loop 2), and the other bounded by Tyr802.61 and Tyr832.64. The importance of these regions in modulator binding was confirmed by site-directed mutagenesis experiments, which showed that affinity for the NAM heptane-1,7-bis(dimethyl-3′-phthalimido-propyl)ammonium bromide (C7/3-phth) could be enhanced by the addition of more aromatic residues or acidic residues to the extracellular vestibule13.
Dror et al.13 also examined the interaction between the modulators and orthosteric antagonists to obtain insights into cooperativity. Two key mechanisms were proposed. The first was electrostatic repulsion between the modulator and the antagonist. The second was conformational coupling between orthosteric and allosteric sites. Specifically, molecular dynamics simulations indicated that bulky modulators (for example, alcuronium) are likely to stabilize an open extracellular vestibule conformation reminiscent of that seen in the crystal structure of the M2 receptor bound to QNB11. By contrast, smaller modulators (for example, C7/3-phth) may preferentially stabilize a closed conformation of the extracellular vestibule13, similar to that seen in molecular dynamics simulations of the unliganded M3 receptor10. Thus, in the inactive state the smaller modulators generally exhibit negative cooperativity with antagonists (that is, they destabilize antagonist binding), whereas the bulkier modulators typically enhance antagonist binding. Consistent with this model, the addition of bulky substituents to C7/3-phth improved the binding of this NAM to the M2 receptors occupied by radiolabelled 3H-N-methylscopolamine13. Taken together, these data support the concept that orthosteric antagonist binding can be enhanced or diminished by a modulator that preferentially stabilizes a particular conformation of the extracellular vestibule.
Activation and positive allosteric modulation of the M2 receptor
The initial structures of the M2 and M3 receptors were obtained in complex with high-affinity antagonists (inverse agonists) and consequently represent inactive receptor conformations. Obtaining crystals of active GPCRs has thus far proved to be extremely challenging; this is probably due in large part to the conformational heterogeneity induced by agonist binding108. To date, these problems have been surmounted in the case of rhodopsin109–111, the β2-adrenergic receptor (β2-AR)112,113 and, more recently, the M2 receptor12. In the last two cases, the first active-state structures have been obtained with the aid of conformationally selective antibody fragments, which mimic G proteins and stabilize the active conformation of the receptors to which they bind. This approach should prove to be useful for obtaining active-state structures of other GPCRs in the future.
The most striking feature of the active-state M2 receptor structure, like that of active β2-AR and rhodopsin, is the outward rotation of transmembrane domain 6 to create a G protein-binding cavity on the intracellular surface of the receptor (FIG. 4a,b). The active-state structure of the M2 receptor shows substantial similarity to the active states of both β2-AR and rhodopsin, with a slightly closer resemblance to the latter structure.
Figure 4. Activation and allosteric modulation of the M2 receptor.
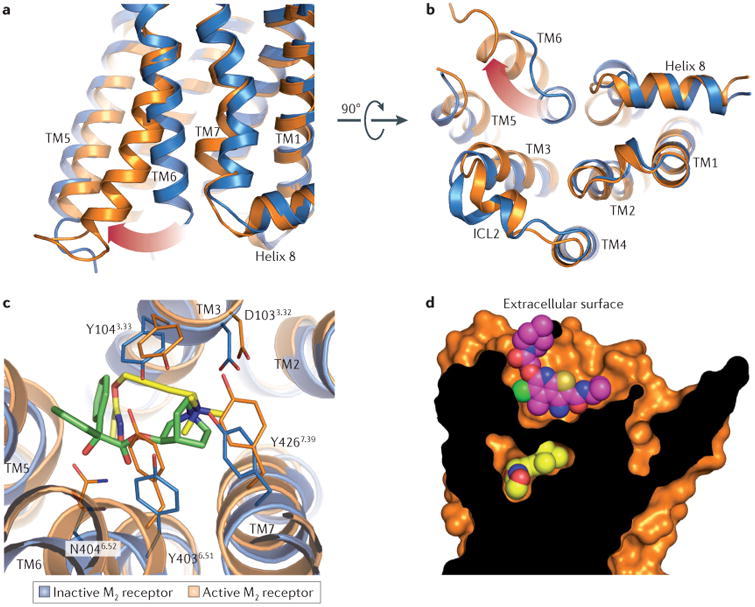
As shown in part a and part b, the intracellular tip of transmembrane domain 6 (TM6) rotates outwards in the active M2 receptor structure (orange) relative to the inactive state (blue). As shown in part c, the orthosteric binding site contracts upon M2 receptor activation, enclosing the agonist iperoxo (yellow) in a smaller binding site, as compared to the antagonist (QNB; green) binding cavity. Residues are numbered according to the human M2 receptor sequence. As shown in part d, LY2119620 (magenta), a muscarinic positive allosteric modulator, binds to the extracellular vestibule of the M2 receptor directly above the orthosteric agonist iperoxo (yellow). ICL2, intracellular loop 2.
One of the most intriguing structural features of active β2-AR and rhodopsin is that both receptors show far more subtle rearrangements in the ligand-binding pocket and extracellular surface than on the intracellular side. This observation suggests that these regions are relatively fixed conformationally, with few changes associated with agonist binding or activation. By contrast, the active structure of the M2 receptor revealed large changes throughout the receptor, including an unexpected contraction of the extracellular vestibule, in addition to changes on the surface of the intracellular receptor that closely parallel those seen in β2-AR and rhodopsin (FIG. 4c,d). This extracellular rearrangement results in a narrower allosteric site and a smaller orthosteric site that is occluded entirely from solvent (FIG. 4d). The much smaller size of the orthosteric site is reflected by the lower molecular weight of muscarinic agonists compared to antagonists and inverse agonists.
In the active state of the M2 receptor, the small orthosteric agonist iperoxo engages Asp1033.32 with its cationic head group, whereas the polar isoxazoline tail contacts Asn4046.52 with a single hydrogen bond, paralleling the interactions of the inactive receptor with the antagonist QNB (FIG. 4c). These two interactions are bridged by an acetylene moiety in iperoxo, which runs perpendicular to transmembrane domain 3. Within the binding site, iperoxo exhibits a high degree of shape complementarity to the active conformation, filling the pocket almost entirely (FIG. 4d). By contrast, in the inactive conformation the M2 receptor presents a large orthosteric site that is better suited to the binding of larger antagonists and inverse agonists. The exceptional complementarity of iperoxo with the active — but not inactive — orthosteric site of the receptor may account for its agonistic activity.
In addition to the substantial contraction of the orthosteric site upon activation, the extracellular vestibule is similarly much smaller in the active conformation of the M2 receptor, even in the absence of a bound allosteric modulator. Isomorphous crystals were grown in the presence of LY2119620, a PAM of mAChRs12, and inspection of electron density maps revealed binding of this ligand to the extracellular vestibule (FIG. 4d). Strikingly, structures of the active M2 receptor with and without LY2119620 were highly similar, even in the extracellular vestibule12. Indeed, the only substantial change in this pocket was a reorientation of Trp4227.35, which directly interacts with the bound LY2119620. In addition, Phe181ECL2 was poorly resolved in the allosteric complex, which suggests that this residue may become more mobile upon binding of the receptor to this modulator. As Phe181ECL2 is a leucine residue in all other mAChR subtypes, interaction with this residue may allow modulators to have different effects on M2 receptors compared to other mAChR subtypes.
The remarkable similarity of the extracellular vestibule conformation in the active M2 receptor with and without bound LY2119620 suggests a mechanism for allosteric modulation. PAMs may achieve their effects by preferentially binding to and stabilizing the closed (that is, active) conformation of the extracellular vestibule. In doing so, PAMs may shift the receptor conformational equilibrium towards active conformations not only of the allosteric site but also of the receptor as a whole (FIG. 5). Analogously, NAMs may preferentially bind the open conformation of the extracellular vestibule seen in the inactive structures, thereby skewing the conformational ensemble of the receptor in favour of inactive states.
Figure 5. Hypothetical mechanism for allosteric modulation of the M2 receptor by a PAM.
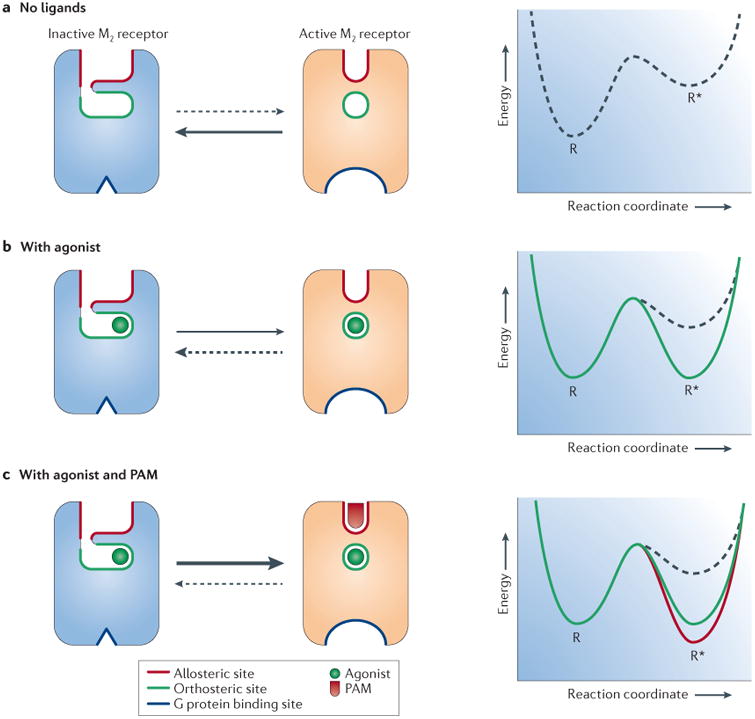
a | Scheme of the M2 receptor with the orthosteric and allosteric binding sites highlighted in green and red, respectively. In the absence of a ligand, the receptor adopts inactive conformations, which are relatively more stable. b | Agonist binding shifts the equilibrium in favour of active receptor conformations. c | Binding of a positive allosteric modulator (PAM) such as LY2119620 to the active -state receptor enhances the affinity of the receptor for the agonist and shifts the equilibrium to further favour active receptor conformations.
In total, the mAChRs contain at least three binding sites that have been defined pharmacologically and structurally: the extracellular vestibule, the orthosteric binding pocket and the G protein-binding site (FIG. 5). Comparison of the active- and inactive-state structures of the M2 receptor shows that all three sites can undergo substantial conformational rearrangements upon receptor activation. However, it is important to recognize that the crystal structures present only static views of a single receptor conformation and thus provide, at best, an incomplete picture.
The structures solved to date are suggestive of a concerted conformational change throughout all three sites, but it remains to be seen whether changes in each site can occur independently of one another. Such a possibility is suggested by structures of the A2A adenosine receptor bound to agonists114,115. Conformational changes in the orthosteric site are indicative of receptor activation, but the intracellular surface remains in a closed, inactive state. Activation of the mAChRs might similarly involve separable conformational changes at each of the three sites rather than a single concerted activation throughout the entire receptor. Spectroscopic and other biophysical assays will probably be required to definitively establish the mechanism of receptor activation and the means through which ligands stabilize active receptor conformations.
Conclusions and future challenges
The past few years have witnessed unprecedented progress in our understanding of the biology, pharmacology and structure of mAChRs. The use of novel mutant mouse models, including mAChR knock-in mice with altered signalling properties and mutant mice in which distinct mAChR subtypes can be deleted in a temporally and spatially controlled fashion, should provide more detailed insights into mAChR biology.
From a structural point of view, the most obvious future goal is to obtain high-resolution structures for all five mAChR subtypes, both in their inactive and active conformations, as well as in complex with different classes of ligands (orthosteric, allosteric or bitopic ligands). Such studies may reveal a broad spectrum of mAChR conformations depending on the receptor subtype and activation state, as well as receptor-bound ligands. Identifying the structural basis that underlies the ability of mAChRs, as well as other GPCRs, to interact with G proteins, arrestins, GPCR kinases (GRKs) and other GPCR-associated signalling molecules should also be of great general interest. Taken together, the outcome of these studies should considerably facilitate the development of novel muscarinic receptor agents and tools that show a high degree of mAChR subtype selectivity and hopefully cause fewer side effects. Finally, lessons learned from work in the mAChR field may be applicable to GPCRs in general, offering new insights into how to design better therapeutic agents targeting this important class of cell surface receptors.
Acknowledgments
We apologize to all investigators whose important contribu-tions could not be acknowledged owing to space limitations. The work of A.C.K. and B.K.K. was supported by a US National Science Foundation Graduate Research Fellowship (A.C.K.) and by the National Science Foundation grant CHE-1223785 and US National Institutes of Health (NIH) grant U19GM106990 (B.K.K.). A.C. and P.M.S. received funds from Program Grant No. APP1055134 of the National Health and Medical Research Council (NHMRC) of Australia. A.C. and P.M.S. are NHMRC Principal Research Fellows. The research of D.G. and J.W. was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) at the NIH. We thank all our co-workers and collaborators for their invaluable contributions to the work summarized in this Review.
Footnotes
Competing interests statement: The authors declare no competing interests.
References
- 1.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 2.Hulme EC, Birdsall NJ, Buckley NJ. Muscarinic receptor subtypes. Annu Rev Pharmacol Toxicol. 1990;30:633–673. doi: 10.1146/annurev.pa.30.040190.003221. [DOI] [PubMed] [Google Scholar]
- 3.Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nature Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 4.Wess J. Novel muscarinic receptor mutant mouse models. Handb Exp Pharmacol. 2012;208:95–117. doi: 10.1007/978-3-642-23274-9_6. [DOI] [PubMed] [Google Scholar]
- 5.Conn PJ, Jones CK, Lindsley CW. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol Sci. 2009;30:148–155. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nature Rev Drug Discov. 2013;12:630–644. doi: 10.1038/nrd4052. [DOI] [PubMed] [Google Scholar]
- 7.Lane JR, Sexton PM, Christopoulos A. Bridging the gap: bitopic ligands of G-protein-coupled receptors. Trends Pharmacol Sci. 2013;34:59–66. doi: 10.1016/j.tips.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Bock A, Mohr K. Dualsteric GPCR targeting and functional selectivity: the paradigmatic M2 muscarinic acetylcholine receptor. Drug Discov Today Technol. 2013;10:e245–e252. doi: 10.1016/j.ddtec.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 9.De Amici M, Dallanoce C, Holzgrabe U, Tränkle C, Mohr K. Allosteric ligands for G protein-coupled receptors: a novel strategy with attractive therapeutic opportunities. Med Res Rev. 2010;30:463–549. doi: 10.1002/med.20166. [DOI] [PubMed] [Google Scholar]
- 10.Kruse AC, et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482:552–556. doi: 10.1038/nature10867. This study reports the first high-resolution structure of the M3 receptor in complex with tiotropium, a clinically used muscarinic antagonist and inverse agonist. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haga K, et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482:547–551. doi: 10.1038/nature10753. In this study, the authors present the first high-resolution structure of the M2 receptor in complex with an orthosteric muscarinic antagonist and inverse agonist — QNB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kruse AC, et al. Activation and allosteric modulationof a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. This study provides the first high-resolution structural information of an agonist-activated mAChR (the M2 subtype) and reveals how a PAM interacts with the M2 receptor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dror RO, et al. Structural basis for modulation of aG-protein-coupled receptor by allosteric drugs. Nature. 2013;503:295–299. doi: 10.1038/nature12595. This study represents a computational biology breakthrough in delineating the molecular mechanisms governing the allosteric modulation of the M2 receptor. [DOI] [PubMed] [Google Scholar]
- 14.Ballard C, et al. Alzheimer's disease. Lancet. 2011;377:1019–1031. doi: 10.1016/S0140-6736(10)61349-9. [DOI] [PubMed] [Google Scholar]
- 15.Langmead CJ, Watson J, Reavill C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther. 2008;117:232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 16.Davis AA, Fritz JJ, Wess J, Lah JJ, Levey AI. Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. J Neurosci. 2010;30:4190–4196. doi: 10.1523/JNEUROSCI.6393-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medeiros R, et al. Loss of muscarinic M1 receptor exacerbates Alzheimer's disease-like pathology and cognitive decline. Am J Pathol. 2011;179:980–991. doi: 10.1016/j.ajpath.2011.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melancon BJ, Tarr JC, Panarese JD, Wood MR, Lindsley CW. Allosteric modulation of the M1 muscarinic acetylcholine receptor: improving cognition and a potential treatment for schizophrenia and Alzheimer's disease. Drug Discov Today. 2013;18:1185–1199. doi: 10.1016/j.drudis.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davie BJ, Christopoulos A, Scammells PJ. Development of M1 mAChR allosteric and bitopic ligands: prospective therapeutics for the treatment of cognitive deficits. ACS Chem Neurosci. 2013;4:1026–1048. doi: 10.1021/cn400086m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Os J, Kapur S. Schizophrenia. Lancet. 2009;374:635–645. doi: 10.1016/S0140-6736(09)60995-8. [DOI] [PubMed] [Google Scholar]
- 21.McKinzie DL, Bymaster FP. Muscarinic mechanisms in psychotic disorders. Handb Exp Pharmacol. 2012;213:233–265. doi: 10.1007/978-3-642-25758-2_9. [DOI] [PubMed] [Google Scholar]
- 22.Bodick NC, et al. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol. 1997;54:465–473. doi: 10.1001/archneur.1997.00550160091022. [DOI] [PubMed] [Google Scholar]
- 23.Shekhar A, et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- 24.Jeon J, et al. A subpopulation of neuronal M4 muscarinic acetylcholine receptors plays a critical role in modulating dopamine-dependent behaviors. J Neurosci. 2010;30:2396–2405. doi: 10.1523/JNEUROSCI.3843-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dencker D, et al. Involvement of a subpopulation of neuronal M4 muscarinic acetylcholine receptors in the antipsychotic-like effects of the M1/M4 preferring muscarinic receptor agonist xanomeline. J Neurosci. 2011;31:5905–5908. doi: 10.1523/JNEUROSCI.0370-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foster DJ, Jones CK, Conn PJ. Emerging approaches for treatment of schizophrenia: modulation of cholinergic signaling. Discov Med. 2012;14:413–420. [PMC free article] [PubMed] [Google Scholar]
- 27.Jones CK, Byun N, Bubser M. Muscarinic and nicotinic acetylcholine receptor agonists and allosteric modulators for the treatment of schizophrenia. Neuropsychopharmacology. 2012;37:16–42. doi: 10.1038/npp.2011.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dencker D, et al. Muscarinic acetylcholine receptor subtypes as potential drug targets for the treatment of schizophrenia, drug abuse and Parkinson's disease. ACS Chem Neurosci. 2012;3:80–89. doi: 10.1021/cn200110q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sofuoglu M, Mooney M. Cholinergic functioning in stimulant addiction: implications for medications development. CNS Drugs. 2009;23:939–952. doi: 10.2165/11310920-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomsen M, et al. Attenuation of cocaine's reinforcing and discriminative stimulus effects via muscarinic M1 acetylcholine receptor stimulation. J Pharmacol Exp Ther. 2010;332:959–969. doi: 10.1124/jpet.109.162057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomsen M, et al. Contribution of both M1 and M4 receptors to muscarinic agonist-mediated attenuation of the cocaine discriminative stimulus in mice. Psychopharmacol. 2012;220:673–685. doi: 10.1007/s00213-011-2516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt LS, et al. Increased cocaine self-administration in M4 muscarinic acetylcholine receptor knockout mice. Psychopharmacol. 2011;216:367–378. doi: 10.1007/s00213-011-2225-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lam DW, LeRoith D. The worldwide diabetes epidemic. Curr Opin Endocrinol Diabetes Obes. 2012;19:93–96. doi: 10.1097/MED.0b013e328350583a. [DOI] [PubMed] [Google Scholar]
- 34.Gautam D, et al. A critical role for β cell M3 muscarinic acetylcholine receptors in regulating insulin release and blood glucose homeostasis in vivo. Cell Metab. 2006;3:449–461. doi: 10.1016/j.cmet.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 35.Gautam D, et al. Beneficial metabolic effects caused by persistent activation of β-cell M3 muscarinic acetylcholine receptors in transgenic mice. Endocrinology. 2010;151:5185–5194. doi: 10.1210/en.2010-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guettier JM, et al. A chemical-genetic approach to study G protein regulation of β cell function in vivo. Proc Natl Acad Sci USA. 2009;106:19197–19202. doi: 10.1073/pnas.0906593106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jain S, et al. Chronic activation of a designer Gq-coupled receptor improves β cell function. J Clin Invest. 2013;123:1750–1762. doi: 10.1172/JCI66432. This study shows that chronic, exogenous ligand-induced activation of an M3 receptor-derived designer receptor expressed by pancreatic β-cells prevents diabetes in different mouse models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci USA. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sassmann A, et al. The Gq/G11 -mediated signaling pathway is critical for autocrine potentiation of insulin secretion in mice. J Clin Invest. 2010;120:2184–2193. doi: 10.1172/JCI41541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong KC, et al. M3-muscarinic receptor promotes insulin release via receptor phosphorylation/arrestindependent activation of protein kinase D1. Proc Natl Acad Sci USA. 2010;107:21181–21186. doi: 10.1073/pnas.1011651107. This analysis of phosphorylation-deficient M3 receptor knock-in mice strongly suggests that arrestin-dependent signalling pathways contribute to M3 receptor-stimulated insulin release. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakajima K, Wess J. Design and functional characterization of a novel, arrestin-biased designer G protein-coupled receptor. Mol Pharmacol. 2012;82:575–582. doi: 10.1124/mol.112.080358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shah N, Khurana S, Cheng K, Raufman JP. Muscarinic receptors and ligands in cancer. Am J Physiol Cell Physiol. 2009;296:C221–C232. doi: 10.1152/ajpcell.00514.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spindel ER. Muscarinic receptor agonists and antagonists: effects on cancer. Handb Exp Pharmacol. 2012;208:451–468. doi: 10.1007/978-3-642-23274-9_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raufman JP, et al. Genetic ablation of M3 muscarinic receptors attenuates murine colon epithelial cell proliferation and neoplasia. Cancer Res. 2008;68:3573–3578. doi: 10.1158/0008-5472.CAN-07-6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raufman JP, et al. Muscarinic receptor subtype-3 gene ablation and scopolamine butylbromide treatment attenuate small intestinal neoplasia in Apcmin/+ mice. Carcinogenesis. 2011;32:1396–1402. doi: 10.1093/carcin/bgr118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magnon C, et al. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361. doi: 10.1126/science.1236361. This study reports that M1 receptor deficiency inhibits mAChR-mediated prostate cancer invasion and metastasis in two mouse models of prostate cancer. [DOI] [PubMed] [Google Scholar]
- 47.Christopoulos A. Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nature Rev Drug Discov. 2002;1:198–210. doi: 10.1038/nrd746. [DOI] [PubMed] [Google Scholar]
- 48.May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 49.Christopoulos A, Lanzafame A, Mitchelson F. Allosteric interactions at muscarinic cholinoceptors. Clin Exp Pharmacol Physiol. 1998;25:185–194. doi: 10.1111/j.1440-1681.1998.t01-4-.x. [DOI] [PubMed] [Google Scholar]
- 50.Birdsall NJ, Lazareno S. Allosterism at muscarinic receptors: ligands and mechanisms. Mini Rev Med Chem. 2005;5:523–543. doi: 10.2174/1389557054023251. [DOI] [PubMed] [Google Scholar]
- 51.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keov P, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology. 2011;60:24–35. doi: 10.1016/j.neuropharm.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 53.Keov P, et al. Reverse engineering of the selective agonist TBPB unveils both orthosteric and allosteric modes of action at the M1 muscarinic acetylcholine receptor. Mol Pharmacol. 2013;84:425–437. doi: 10.1124/mol.113.087320. [DOI] [PubMed] [Google Scholar]
- 54.Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nature Rev Drug Discov. 2013;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 55.Ma L, et al. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci USA. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shirey JK, et al. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J Neurosci. 2009;29:14271–14286. doi: 10.1523/JNEUROSCI.3930-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Canals M, et al. A Monod–Wyman–Changeux mechanism can explain G protein-coupled receptor (GPCR) allosteric modulation. J Biol Chem. 2012;287:650–659. doi: 10.1074/jbc.M111.314278. This study presents a chemical biology framework with which to study and classify the simplest allosteric ligand behaviours. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lazareno S, Birdsall NJ. Detection, quantitation,and verification of allosteric interactions of agentswith labeled and unlabeled ligands at G protein-coupled receptors: interactions of strychnine and acetylcholine at muscarinic receptors. Mol Pharmacol. 1995;48:362–378. [PubMed] [Google Scholar]
- 59.Kenakin T. New concepts in drug discovery: collateral efficacy and permissive antagonism. Nature Rev Drug Discov. 2005;4:919–927. doi: 10.1038/nrd1875. [DOI] [PubMed] [Google Scholar]
- 60.Valant C, Felder CC, Sexton PM, Christopoulos A. Probe dependence in the allosteric modulation of a G protein-coupled receptor: implications for detection and validation of allosteric ligand effects. Mol Pharmacol. 2012;81:41–52. doi: 10.1124/mol.111.074872. This study highlights the importance of probe dependence in the study of the effects of allosteric modulators. [DOI] [PubMed] [Google Scholar]
- 61.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nature Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stallaert W, Christopoulos A, Bouvier M. Ligand functional selectivity and quantitative pharmacology at G protein-coupled receptors. Expert Opin Drug Discov. 2011;6:811–825. doi: 10.1517/17460441.2011.586691. [DOI] [PubMed] [Google Scholar]
- 63.Marlo JE, et al. Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol Pharmacol. 2009;75:577–588. doi: 10.1124/mol.108.052886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lazareno S, Dolezal V, Popham A, Birdsall NJ. Thiochrome enhances acetylcholine affinity at muscarinic M4 receptors: receptor subtype selectivity via cooperativity rather than affinity. Mol Pharmacol. 2004;65:257–266. doi: 10.1124/mol.65.1.257. [DOI] [PubMed] [Google Scholar]
- 65.Chan WY, et al. Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc Natl Acad Sci USA. 2008;105:10978–10983. doi: 10.1073/pnas.0800567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suratman S, et al. Impact of species variability and ‘probe-dependence’ on the detection and in vivo validation of allosteric modulation at the M4 muscarinic acetylcholine receptor. Br J Pharmacol. 2011;162:1659–1670. doi: 10.1111/j.1476-5381.2010.01184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Valant C, Sexton PM, Christopoulos A. Orthosteric/allosteric bitopic ligands: going hybrid at GPCRs. Mol Interv. 2009;9:125–135. doi: 10.1124/mi.9.3.6. [DOI] [PubMed] [Google Scholar]
- 68.Mohr K, et al. Rational design of dualsteric GPCR ligands: quests and promise. Br J Pharmacol. 2010;159:997–1008. doi: 10.1111/j.1476-5381.2009.00601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Melancon BJ, et al. Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. J Med Chem. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Valant C, Robert Lane J, Sexton PM, Christopoulos A. The best of both worlds? Bitopic orthosteric/allosteric ligands of G protein-coupled receptors Annu Rev Pharmacol Toxicol. 2012;52:153–178. doi: 10.1146/annurev-pharmtox-010611-134514. [DOI] [PubMed] [Google Scholar]
- 71.Disingrini T, et al. Design, synthesis, and action of oxotremorine-related hybrid-type allosteric modulators of muscarinic acetylcholine receptors. J Med Chem. 2006;49:366–372. doi: 10.1021/jm050769s. [DOI] [PubMed] [Google Scholar]
- 72.Antony J, et al. Dualsteric GPCR targeting: a novel route to binding and signaling pathway selectivity. FASEB J. 2009;23:442–450. doi: 10.1096/fj.08-114751. [DOI] [PubMed] [Google Scholar]
- 73.Kebig A, Kostenis E, Mohr K, Mohr-Andra M. An optical dynamic mass redistribution assay reveals biased signaling of dualsteric GPCR activators. J Recept Signal Transduct Res. 2009;29:140–145. doi: 10.1080/10799890903047437. [DOI] [PubMed] [Google Scholar]
- 74.Bock A, et al. The allosteric vestibule of a seven transmembrane helical receptor controls G-protein coupling. Nature Commun. 2012;3:1044. doi: 10.1038/ncomms2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Steinfeld T, Mammen M, Smith JA, Wilson RD, Jasper JR. A novel multivalent ligand that bridges the allosteric and orthosteric binding sites of the M2 muscarinic receptor. Mol Pharmacol. 2007;72:291–302. doi: 10.1124/mol.106.033746. [DOI] [PubMed] [Google Scholar]
- 76.Valant C, et al. A novel mechanism of G protein-coupled receptor functional selectivity. Muscarinic partial agonist McN-A-343 as a bitopic orthosteric/ allosteric ligand. J Biol Chem. 2008;283:29312–29321. doi: 10.1074/jbc.M803801200. This is the first study to show that functionally selective ligands may mediate their behaviour via a bitopic mechanism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spalding TA, et al. Discovery of an ectopic activation site on the M1 muscarinic receptor. Mol Pharmacol. 2002;61:1297–1302. doi: 10.1124/mol.61.6.1297. [DOI] [PubMed] [Google Scholar]
- 78.Langmead CJ, et al. Probing the molecular mechanism of interaction between 4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl]-piperidine (AC-42) and the muscarinic M1 receptor: direct pharmacological evidence that AC-42 is an allosteric agonist. Mol Pharmacol. 2006;69:236–246. doi: 10.1124/mol.105.017814. [DOI] [PubMed] [Google Scholar]
- 79.Jones CK, et al. Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J Neurosci. 2008;28:10422–10433. doi: 10.1523/JNEUROSCI.1850-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gregory KJ, Hall NE, Tobin AB, Sexton PM, Christopoulos A. Identification of orthosteric and allosteric site mutations in M2 muscarinic acetylcholine receptors that contribute to ligand-selective signaling bias. J Biol Chem. 2010;285:7459–7474. doi: 10.1074/jbc.M109.094011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Avlani VA, et al. Orthosteric and allosteric modes of interaction of novel selective agonists of the M1 muscarinic acetylcholine receptor. Mol Pharmacol. 2010;78:94–104. doi: 10.1124/mol.110.064345. [DOI] [PubMed] [Google Scholar]
- 82.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 83.Changeux JP. Allosteric receptors: from electric organ to cognition. Annu Rev Pharmacol Toxicol. 2010;50:1–38. doi: 10.1146/annurev.pharmtox.010909.105741. [DOI] [PubMed] [Google Scholar]
- 84.Canals M, Sexton PM, Christopoulos A. Allostery in GPCRs: ‘MWC’ revisited. Trends Biochem Sci. 2011;36:663–672. doi: 10.1016/j.tibs.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 85.Ehlert FJ, Griffin MT. Two-state models and the analysis of the allosteric effect of gallamine at the M2 muscarinic receptor. J Pharmacol Exp Ther. 2008;325:1039–1060. doi: 10.1124/jpet.108.136960. [DOI] [PubMed] [Google Scholar]
- 86.Abdul-Ridha A, Lane JR, Sexton PM, Canals M, Christopoulos A. Allosteric modulation of a chemogenetically modified G protein-coupled receptor. Mol Pharmacol. 2013;83:521–530. doi: 10.1124/mol.112.083006. [DOI] [PubMed] [Google Scholar]
- 87.Lazareno S, Popham A, Birdsall NJ. Allosteric interactions of staurosporine and other indolocarbazoles with N-[methyl-3H]scopolamine and acetylcholine at muscarinic receptor subtypes: identification of a second allosteric site. Mol Pharmacol. 2000;58:194–207. doi: 10.1124/mol.58.1.194. [DOI] [PubMed] [Google Scholar]
- 88.Lazareno S, Popham A, Birdsall NJ. Analogs of WIN 62,577 define a second allosteric site on muscarinic receptors. Mol Pharmacol. 2002;62:1492–1505. doi: 10.1124/mol.62.6.1492. [DOI] [PubMed] [Google Scholar]
- 89.Espinoza-Fonseca LM, Trujillo-Ferrara JG. The existence of a second allosteric site on the M1 muscarinic acetylcholine receptor and its implications for drug design. Bioorg Med Chem Lett. 2006;16:1217–1220. doi: 10.1016/j.bmcl.2005.11.097. [DOI] [PubMed] [Google Scholar]
- 90.Redka DS, Pisterzi LF, Wells JW. Binding of orthosteric ligands to the allosteric site of the M2 muscarinic cholinergic receptor. Mol Pharmacol. 2008;74:834–843. doi: 10.1124/mol.108.048074. [DOI] [PubMed] [Google Scholar]
- 91.Shivnaraine RV, Huang XP, Seidenberg M, Ellis J, Wells JW. Heterotropic cooperativity within and between protomers of an oligomeric M2 muscarinic receptor. Biochemistry. 2012;51:4518–4540. doi: 10.1021/bi3000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rosenbaum DM, et al. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 93.Chae PS, et al. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nature Methods. 2010;7:1003–1008. doi: 10.1038/nmeth.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Landau EM, Rosenbusch JP. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci USA. 1996;93:14532–14535. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nature Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Smith JL, Fischetti RF, Yamamoto M. Micro-crystallography comes of age. Curr Opin Struct Biol. 2012;22:602–612. doi: 10.1016/j.sbi.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shimamura T, et al. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chien EY, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Warne T, et al. Structure of a β1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rasmussen SG, et al. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 101.Cherezov V, et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wacker D, et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang C, et al. Structural basis for molecular recognition at serotonin receptors. Science. 2013;340:610–614. doi: 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ballesteros J, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 105.Kruse AC, et al. Muscarinic receptors as model targets and antitargets for structure-based ligand discovery. Mol Pharmacol. 2013;84:528–540. doi: 10.1124/mol.113.087551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tautermann CS, et al. Molecular basis for the long duration of action and kinetic selectivity of tiotropium for the muscarinic M3 receptor. J Med Chem. 2013;56:8746–8756. doi: 10.1021/jm401219y. [DOI] [PubMed] [Google Scholar]
- 107.Gregory KJ, Sexton PM, Christopoulos A. Allosteric modulation of muscarinic acetylcholine receptors. Curr Neuropharmacol. 2007;5:157–167. doi: 10.2174/157015907781695946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nygaard R, et al. The dynamic process of β2-adrenergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Scheerer P, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 110.Choe HW, et al. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 111.Standfuss J, et al. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature. 2011;471:656–660. doi: 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rasmussen SG, et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rasmussen SG, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. This crystal structure represents the first high-resolution view of the active-state ternary complex composed of an agonist-occupied GPCR (β2-AR) and a G protein (nucleotide-free Gs heterotrimer) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lebon G, et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xu F, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kamsler A, McHugh J, Gerber D, Huang SY, Tonegawa S. Presynaptic M1 muscarinic receptors are necessary for mGluR long-term depression in the hippocampus. Proc Natl Acad Sci USA. 2010;107:1618–1623. doi: 10.1073/pnas.0912540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gautam D, et al. Neuronal M3 muscarinic acetylcholine receptors are essential for somatotroph proliferation and normal somatic growth. Proc Natl Acad Sci USA. 2009;106:6398–6403. doi: 10.1073/pnas.0900977106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shi Y, et al. Signaling through the M3 muscarinic receptor favors bone mass accrual by decreasing sympathetic activity. Cell Metab. 2010;11:231–238. doi: 10.1016/j.cmet.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li JH, et al. Hepatic muscarinic acetylcholine receptors are not critically involved in maintaining glucose homeo stasis in mice. Diabetes. 2009;58:2776–2787. doi: 10.2337/db09-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Arteaga-Solis E, et al. Inhibition of leptin regulation of parasympathetic signaling as a cause of extreme body weight-associated asthma. Cell Metab. 2013;17:35–48. doi: 10.1016/j.cmet.2012.12.004. This study reports that leptin signalling in the brain promotes bronchodilation by inhibiting parasympathetic signalling through airway smooth muscle M3 receptors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Poulin B, et al. The M3-muscarinic receptor regulates learning and memory in a receptor phosphorylation/ arrestin-dependent manner. Proc Natl Acad Sci USA. 2010;107:9440–9445. doi: 10.1073/pnas.0914801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bendor J, et al. AGAP1/AP-3-dependent endocytic recycling of M5 muscarinic receptors promotes dopamine release. EMBO J. 2010;29:2813–2826. doi: 10.1038/emboj.2010.154. [DOI] [PMC free article] [PubMed] [Google Scholar]