


Abstract
Homochirality is a general feature of biological macromolecules, and Nature includes few examples of heterochiral proteins. Herein, we report on the design, chemical synthesis, and structural characterization of heterochiral proteins possessing loops of amino acids of chirality opposite to that of the rest of a protein scaffold. Using the protein Ecballium elaterium trypsin inhibitor II, we discover that selective β-alanine substitution favors the efficient folding of our heterochiral constructs. Solution nuclear magnetic resonance spectroscopy of one such heterochiral protein reveals a homogeneous global fold. Additionally, steered molecular dynamics simulation indicate β-alanine reduces the free energy required to fold the protein. We also find these heterochiral proteins to be more resistant to proteolysis than homochiral l-proteins. This work informs the design of heterochiral protein architectures containing stretches of both d- and l-amino acids.
Graphical abstract
Heterochirality is rare among biological molecules such as proteins; in Nature, proteins are folded from polypeptides comprised primarily of l-amino acids and achiral glycine.1 Despite this bias, their enantiomers, d-amino acids, are occasionally post-translationally incorporated into biomolecules such as peptidoglycan,2 nonribosomal peptides,3 and even some animal proteins.4 Nature’s utility for such heterochiral polypeptides and proteins is not fully understood. However, it has been suggested that the selective incorporation of d-amino acids may confer proteolytic stability and enhanced protein function.5 Still, there are few heterochiral proteins possessing more than a single d-amino acid.
The best characterized heterochiral proteins have been synthetic α-helical bundles;6,7 Baker, Gellman, and co-workers have designed helical assemblies that exhibit tertiary and quaternary structure, respectively. Before these, heterochiral protein syntheses largely involved targeted d-amino acid substitutions in structure–function studies of existing l-proteins.8–10 Recently, d-amino acid scans have revealed that they may be more tolerated in natural proteins.11 Horne and co-workers have also demonstrated that in addition to d-amino acids, other types of unnatural amino acids, including β-amino acids, can be incorporated into proteins while preserving tertiary folding.12
We were motivated by this body of work and set out to design a heterochiral protein possessing a loop of amino acids with inverted chirality [i.e., l-protein with a d-amino acid loop (Figure 1)]. Loops are important structural features that often facilitate natural and engineered protein–protein interactions.13,14 Understanding how a stretch of d-amino acids may be engineered into a loop of a l-protein, or vice versa, could expand upon the conformational space that a protein scaffold might access.
Figure 1.

Ecballium elaterium trypsin inhibitor II (EETI-II) variants explored in the design and folding of heterochiral proteins. Ramachandran plots, solution nuclear magnetic resonance structure ensembles, and primary sequences with disulfide connectivity (yellow) for (A) wild-type EETI-II (Protein Data Bank entry 2IT7), (B) integrin binding EETI-II 2.5F, and (C) a heterochiral EETI-II 2.5F analogue in which the chirality of the amino acids in the protein loop is inverted relative to the chirality of the rest of the scaffold. Uppercase letters denote l-amino acids or achiral amino acids, and lowercase and underlined letters denote d-amino acids; β denotes β-alanine.
We began our work with a model protein, Ecballium elaterium trypsin inhibitor II [EETI-II (Figure 1A)].15 Wild-type EETI-II (WT EETI-II) is a monomeric protein 28 amino acids in length, with three disulfide bonds arranged in a defined topology.16–18 Disulfide rich miniproteins, such as EETI-II, have been developed to interact with an array of different protein targets.19 EETI-II was chosen because of its synthetic accessibility and its well-characterized folding pathway. Folding of WT EETI-II initially involves formation of two disulfide bonds and the protein core (Cys9–Cys21 and Cys15–Cys27). This is followed by formation of the final disulfide bond between the binding loop and the core (Cys2–Cys19). EETI-II is also a clinically relevant protein scaffold; its trypsin binding loop (Pro3–Arg8) has been expanded and retargeted toward cell-surface integrin receptors for brain tumor imaging [EETI-II 2.5F (Figure 1B)].20,21
Using sequences of both trypsin and integrin binding EETI-II variants, we investigated whether it was possible to invert the chirality of their binding loops while maintaining their global protein fold. Based on previous folding investigations of WT EETI-II,22–24 we hypothesized that a protein core with two disulfide bonds would form regardless of loop chirality. If formation of the final disulfide bond and tertiary folding were to be adversely affected by the chiral inversion, it could be evaluated, in part, by using liquid chromatography and mass spectrometry (LC–MS) to monitor disulfide bond formation over time.
RESULTS AND DISCUSSION
Homo- and heterochiral EETI-II constructs were chemically synthesized and purified on multimilligram scales (Table S8).25,26 These included several analogues with amino acid substitutions, such as β-alanine (β-Ala), that would change the conformational properties of the loop. The invariant EETI-II protein core (through the second cysteine) and then the variable loop regions were synthesized on approximately 0.025 mmol scales. This afforded several dozen milligrams of crude, reduced polypeptide. These peptides were purified and then lyophilized with 20–40% isolated yields.
We used a thiol-containing redox buffer to facilitate disulfide bond formation and protein folding of our constructs. This entailed a two-step procedure at room temperature. First, purified peptides were dissolved in buffer containing 50 mM TRIS (pH 7.8), 5.7 M Gu·HCl as a chaotropic agent, and 5.0 mM TCEP as a reducing agent; this ensured that different polypeptides were brought to identical denatured and reduced states prior to folding. Second, denatured peptides were diluted into an oxidative folding buffer containing 50 mM TRIS (pH 7.8), 0.47 mM cystine, and 1 mM cysteine; this solution enabled protein disulfide bond formation via thiol–disulfide exchange. As oxidative folding progressed, reactions were quenched with acid at various time points and analyzed by reverse-phase LC–MS.
Surprisingly, we discovered that β-Ala facilitated efficient folding of a heterochiral EETI-II 2.5F analogue (Figure 2). Homochiral EETI-II 2.5F rapidly formed a two-disulfide bond intermediate that eventually yielded a predominant three-disulfide product (Figure 2A). This was inferred by an observed 6 Da loss of mass. Leftward retention time shifts of the intermediate and final products also indicated burial of protein hydrophobic patches and weaker affinity for the reverse-phase column.11,27 In contrast, a heterochiral EETI-II 2.5F analogue with part of its binding loop inverted to d-amino acids (Pro3–Pro10) yielded a three-disulfide product with a broader peak width and a smaller retention shift (Figure 2B). This indicated possible product heterogeneity, improper folding, or distortion of the global fold relative to folded homochiral EETI-II 2.5F. The heterochiral analogue was able to recapture the peak profile and retention shift of homochiral EETI-II 2.5F only when β-Ala was substituted at positions where the amino acids underwent the transition from l to d (Figure 2C).
Figure 2.

β-Alanine enables efficient folding of a heterochiral EETI-II 2.5F analogue. LC–MS (total ion current vs time) monitoring for the oxidative folding of (A) homochiral EETI-II 2.5F, (B) a heterochiral EETI-II 2.5F analogue with a stretch containing seven d-amino acids and one Gly in the binding loop, and (C) a heterochiral EETI-II 2.5F analogue containing five d-amino acids, one Gly, and two β-Ala substitutions. Protein folding and disulfide bond formation were mediated using a buffer containing a thiol redox pair (pH 7.8, 0.47 mM cystine, 1 mM cysteine). Charge-state series represent the major product (asterisks) at time zero, 2 min, and 16 h; observed masses represent monoisotopic masses.
Additional folding experiments indicated that the position of β-Ala as well as the overall loop length affected the folding of heterochiral EETI-II proteins. With another set of constructs based on the integrin binder EETI-II 2.5D, β-Ala again rescued the folding of a heterochiral EETI-II 2.5D analogue (Figures S5–S8). However, this was not the case for heterochiral constructs based on WT EETI-II (Figures S9–S12). Additionally, for heterochiral EETI-II 2.5F, changing the position of one β-Ala substitution (i.e., Thr13 vs Pro10) resulted in a well-defined peak but did not shift the retention time as much as homochiral EETI-II 2.5F did (Figures S13–S15). We were also able to fold variants with d-amino acid cores, l-amino acid loops, and β-Ala substitutions (Figures S16–S18).
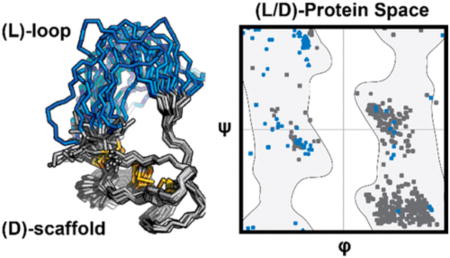
The solution structure of one such EETI-II 2.5F analogue revealed a global fold similar to that of WT EETI-II (Figure 3). This analogue possessed a d-amino acid core, a loop of five l-amino acids, one Gly, and two β-Ala substitutions. Structural calculations and explicit solvent refinement were performed using CYANA and YASARA, respectively.28–30 Homonuclear distance restraints were derived from nuclear Overhauser effect (NOE) cross-peaks and mass spectrometry data indicating disulfide bond formation. The 20 lowest-CYANA target function conformers were all in agreement with respect to the global fold of the heterochiral protein [average backbone root-mean-square deviation of 1.82 ± 0.26 Å (Table S11)]. This suggested that the folding product observed by LC–MS was indeed homogeneous. Calculations were also performed for all possible three-disulfide bond permutations. The d-Cys2–d-Cys24, d-Cys14–d-Cys26, d-Cys20–d-Cys32 arrangement was in strongest agreement with experimentally observed NOE cross-peaks. This indicated that the disulfide bonds formed in the isolated heterochiral EETI-II 2.5F folding product were the same as those in WT EETI-II.16
Figure 3.

Solution nuclear magnetic resonance (NMR) reveals a homogeneous fold for a heterochiral EETI-II 2.5F analogue. Solution NMR structural ensemble of a heterochiral EETI-II 2.5F analogue possessing a scaffold core (gray) comprised of d-amino acids and a loop (blue) comprised of five l-amino acids, one Gly, and two β-Ala substitutions. This ensemble represents the 20 lowest-CYANA target function conformers after explicit solvent refinement using YASARA. Distance restraints used in calculations were derived from NOE measurements and the detection of disulfide bond formation through high-resolution mass spectrometry.
Although the loop of this heterochiral protein exhibited conformational flexibility, the core was well-defined and similar to that of WT EETI-II (Figure 4). Relatively few NOE cross-peaks were assigned to the loop. The core possessed both identical topology and structure as would be expected for WT EETI-II, albeit a mirror image. In our structure, the presence of β-sheet and distorted helix secondary structure (panels A and B of Figure 4, respectively) was further corroborated by the proximity and potential for hydrogen bonding within these features (Gly33 N–H⋯O═C d-Val25, Gly27 N–H⋯O═C d-Phe31, and d-Asp19 N–H⋯O═C d-Gln16); these hydrogen bonds correspond with those that have been previously measured and reported in solution structures of EETI-II.16,31 Other long-range hydrogen bonds could also be inferred on the basis of proximity (d-Cys14 N–H⋯O═C Gly30 and d-Cys32 N–H⋯O═C d-Leu12).
Figure 4.

Heterochiral EETI-II 2.5F retains a secondary structure similar to that of WT EETI-II. (A) Backbone alignment and residual mean square of inverted WT EETI-II and heterochiral 2.5F. Backbone trace of a heterochiral EETI-II 2.5F analogue emphasizing (B) the β-sheet and turn region (d-Val25–Gly33) and (C) the 310-helix region (d-Asp17–d-Cys20). White, blue, gray, and red atoms represent hydrogen, nitrogen, carbon, and oxygen, respectively. Green dashes represent hydrogen bonds that were identified on the basis of proximity and were reported previously in solution structures of EETI-II variants.
In addition to β-Ala, we discovered that other substitutions, including Gly (Figure 5), facilitated folding of heterochiral EETI-II proteins. We synthesized and attempted to fold several EETI-II 2.5F analogues with alternative substitutions (Figures S19–S28). These included dual Gly, single β-Ala, and β-Ala paired with l- or d-β-amino acids. With the exception of single-β-alanine substitutions, all of these analogues folded into a predominant three-disulfide product with a retention time shift matching that of their homochiral counterparts.
Figure 5.

Glycine also enables efficient folding of a heterochiral EETI-II 2.5F analogue. LC–MS (total ion current vs time) monitoring for the oxidative protein folding of a heterochiral EETI-II 2.5F analogue possessing a scaffold core comprised of l-amino acids and a loop containing a stretch of five d-amino acids, one Gly, and two additional Gly substitutions. Charge-state series represent the major product (asterisks) at time zero, 2 min, and 16 h; observed masses represent monoisotopic masses.
Steered molecular dynamics simulation indicated that EETI-II 2.5F analogues incorporating β-Ala or Gly substitutions required less free energy to form their final disulfide bond compare to analogues without substitutions (Figure 6). On the basis of LC–MS and nuclear magnetic resonance (NMR) data, we modeled a rigid two-disulfide EETI-II folding intermediate (Cys14–Cys26 and Cys20–Cys32) with a flexible loop; using Jarzynski’s equality,32 we estimated the work required to bring Cys2 of the loop into the proximity of Cys24 of the core for formation of the third disulfide bond. Analogues without β-Ala or Gly substitutions required substantially more free energy (~8 kcal/mol) to form the Cys2–Cys24 disulfide bond. This energetic cost paired with the observation of shorter LC–MS retention time shifts (i.e., Figure 2B) suggests that the final disulfide bonds of these analogues can more easily be achieved by decreasing the rigidity of the core and the compactness of the global fold.
Figure 6.

Heterochiral EETI-II 2.5F proteins containing β-Ala and Gly substitutions require less free energy to fold. Steered molecular dynamics (SMD) simulations of homochiral EETI-II 2.5F (1) and several heterochiral EETI-II 2.5F analogues without (2 and 3) and with (4–6) β-Ala or Gly substitutions. Potentials of mean force (PMF) as a function of the distance between the two Sγ atoms of residues Cys2 and Cys24. For each system, the 100 SMD trajectories were separated into 20 groups with five trajectories in each group. The PMF for each group was estimated using Jarzynski’s equality. The results from the 20 groups were then averaged and error bars (standard errors of mean) calculated.
d-amino acids selectively shielded a region of a heterochiral protein from proteolytic degradation. Using stability assays, we studied several homo- and heterochiral EETI-II 2.5F analogues that differed only in the chirality of their loop or core (Figure 7). These proteins were incubated at 37 °C with the nonspecific protease, proteinase K. We observed a trend of an increasing rate of degradation that correlated to the amount of l-amino acids in the protein. We also studied proteolytic stability in the presence of trypsin. Trypsin hydrolyzed the Arg6–Gly7 amide bond in the loops of proteins consisting of l-amino acids but not of d-amino acids (Figures S30–S33).
Figure 7.

Heterochiral EETI-II 2.5F exhibits unique proteolysis resistance compared to that of homochiral EETI-II 2.5F. Proteinase K induced degradation of several homochiral (1 and 4) and heterochiral (2 and 3) EETI-II 2.5F analogues as measured by LC–MS extracted ion current for the remaining intact peptide. The stretches of d-amino acids of each protein are underlined. All measurements were performed under identical conditions and in triplicate; error bars are too small to be discerned.
The use of β-amino acids throughout this work demonstrates another means by which they can be leveraged in protein engineering. Gellman,33 Schepartz,34,35 and Horne36 demonstrated β-amino acids may be utilized alone, or in conjunction with α-amino acids, to create novel protein architectures with medicinal and functional importance. We demonstrate an additional scenario in which β-amino acids may be used to facilitate folding of heterochiral polypeptides. We hope that this work will continue to inspire the design and synthesis of novel proteins that make use of noncanonical amino acids that otherwise cannot be accessed by Nature’s translational machinery.
In summary, we have successfully designed, synthesized, and demonstrated tertiary folding of a heterochiral protein based on the EETI-II protein scaffold. When Pauling and Corey first laid the groundwork for thinking about protein structure at a molecular level, they were principally concerned with l-amino acids.37 Ramachandran built upon their work and extended their analysis to define favorable and unfavorable peptide dihedral angles for natural proteins.38 Since their time, synthetic peptide chemistry has come a long way to challenge the classical ways of imagining protein structure. Continued efforts in the area of unnatural protein design may lead to discovery of proteins with novel folds and function for practical applications.
Supplementary Material
Acknowledgments
Funding
We graciously acknowledge our external funding sources: National Institutes of Health (NIH)/National Institute of General Medical Sciences (NIGMS) Biotechnology Training Program (5T32GM008334-25, S.K.M.) and Defense Advanced Research Projects Agency (Grant 023504-001, B.L.P.). We also are thankful for the support of a Tufts start-up fund and the Knez Family Faculty Investment Fund (Y.-S.L.) This study made use of the National Magnetic Resonance Facility at Madison, which is supported by NIH Grant P41GM103399 (old number, P41RR002301). Equipment was purchased with funds from the University of Wisconsin—Madison, NIH Grants P41GM103399, S10RR02781, S10RR08438, S10RR023438, S10RR025062, and S10RR029220, the National Science Foundation (DMB-8415048, OIA-9977486, and BIR-9214394), and the U.S. Department of Agriculture.
Footnotes
ASSOCIATED CONTENT
- All materials and experimental procedures are detailed at length, including information pertaining to the reagents used; chemical synthesis, purification, oxidative folding, and LC–MS analysis of EETI-II analogues; NMR sample preparation, data acquisition, and analysis; molecular dynamics simulations; and proteolysis stability assays (PDF)
The authors declare no competing financial interest.
References
- 1.Mitchell JBO, Smith J. D-amino acid residues in peptides and proteins. Proteins: Struct., Funct., Genet. 2003;50:563–571. doi: 10.1002/prot.10320. [DOI] [PubMed] [Google Scholar]
- 2.Holtje J-V. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 1998;62:181–203. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finking R, Marahiel MA. Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 2004;58:453–488. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]
- 4.Kreil G. D-amino acids in animal peptides. Annu. Rev. Biochem. 1997;66:337–345. doi: 10.1146/annurev.biochem.66.1.337. [DOI] [PubMed] [Google Scholar]
- 5.Heck SD, Siok CJ, Krapcho KJ, Kelbaugh PR, Thadeio PF, Welch MJ, Williams RD, Ganong AH, Kelly ME, Lanzetti AJ, Gray WR, Phillips D, Parks TN, Jackson H, Ahlijanian MK, Saccomano NA, Volkmann RA. Functional consequences of posttranslational isomerization of Ser46 in a calcium hannel toxin. Science. 1994;266:1065–1068. doi: 10.1126/science.7973665. [DOI] [PubMed] [Google Scholar]
- 6.Bhardwaj G, Mulligan VK, Bahl CD, Gilmore JM, Harvey PJ, Cheneval O, Buchko GW, Pulavarti SVSRK, Kaas Q, Eletsky A, Huang P-S, Johnsen WA, Greisen PJ, Rocklin GJ, Song Y, Linsky TW, Watkins A, Rettie SA, Xu X, Carter LP, Bonneau R, Olson JM, Coutsias E, Correnti CE, Szyperski T, Craik DJ, Baker D. Accurate de novo design of hyperstable constrained peptides. Nature. 2016;538:329–335. doi: 10.1038/nature19791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mortenson DE, Steinkruger JD, Kreitler DF, Perroni DV, Sorenson GP, Huang L, Mittal R, Yun HG, Travis BR, Mahanthappa MK, Forest KT, Gellman SH. High-resolution structures of a heterochiral coiled coil. Proc. Natl. Acad. Sci. U. S. A. 2015;112:13144–13149. doi: 10.1073/pnas.1507918112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobayashi M, Ohgaku S, Iwasaki M, Maegawa H, Shigeta Y, Inouye K. Supernormal Insulin: [D-PheB24]-insulin with increased affinity for insulin receptors. Biochem. Biophys. Res. Commun. 1982;107:329–336. doi: 10.1016/0006-291x(82)91708-9. [DOI] [PubMed] [Google Scholar]
- 9.Anil B, Song B, Tang Y, Raleigh DP. Exploiting the right side of the ramachandran plot: substitution of glycines by D-alanine can significantly increase protein stability. J. Am. Chem. Soc. 2004;126:13194–13195. doi: 10.1021/ja047119i. [DOI] [PubMed] [Google Scholar]
- 10.Valiyaveetil FI, Sekedat M, MacKinnon R, Muir TW. Glycine as a D-amino acid surrogate in the K+ -selectivity filter. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17045–17049. doi: 10.1073/pnas.0407820101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon MD, Maki Y, Vinogradov AA, Zhang C, Yu H, Lin Y-S, Kajihara Y, Pentelute BL. D-amino acid scan of two small proteins. J. Am. Chem. Soc. 2016;138:12099–12111. doi: 10.1021/jacs.6b03765. [DOI] [PubMed] [Google Scholar]
- 12.Reinert ZE, Lengyel GA, Horne WS. Protein-like tertiary folding behavior from heterogeneous backbones. J. Am. Chem. Soc. 2013;135:12528–12531. doi: 10.1021/ja405422v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papaleo E, Saladino G, Lambrughi M, Lindorff-Larsen K, Gervasio FL, Nussinov R. The role of protein loops and linkers in conformational dynamics and allostery. Chem. Rev. 2016;116:6391–6423. doi: 10.1021/acs.chemrev.5b00623. [DOI] [PubMed] [Google Scholar]
- 14.Gavenonis J, Sheneman BA, Siegert TR, Eshelman MR, Kritzer JA. Comprehensive analysis of loops at protein-protein interfaces for macrocycle design. Nat. Chem. Biol. 2014;10:716–723. doi: 10.1038/nchembio.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Favel A, Mattras H, Coletti-Previero MA, Zwilling R, Robinson EA, Castro B. Protease inhibitors from Ecballium elaterium seeds. Int. J. Pept. Protein Res. 1989;33:202–208. doi: 10.1111/j.1399-3011.1989.tb00210.x. [DOI] [PubMed] [Google Scholar]
- 16.Heitz A, Chiche L, Le-Nguyen D, Castro B. 1H 2D NMR and distance geometry study of the folding of Ecballium elaterium trypsin inhibitor, a member of the squash inhibitors family. Biochemistry. 1989;28:2392–2398. doi: 10.1021/bi00432a009. [DOI] [PubMed] [Google Scholar]
- 17.Chiche L, Gaboriaud C, Heitz A, Mornon J-P, Castro B, Kollman PA. Use of restrained molecular dynamics in water to determine three-dimensional protein structure: prediction of the three-dimensional structure of Ecballium elaterium trypsin inhibitor II. Proteins: Struct., Funct., Genet. 1989;6:405–417. doi: 10.1002/prot.340060407. [DOI] [PubMed] [Google Scholar]
- 18.Kräzner R, Debreczeni JÉ, Pape T, Schneider TR, Wentzel A, Kolmar H, Sheldrick GM, Uson I. Structure of Ecballium elaterium trypsin inhibitor II (EETI-II): A rigid molecular scaffold. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2005;61:1255–1262. doi: 10.1107/S0907444905021207. [DOI] [PubMed] [Google Scholar]
- 19.Craik DJ, Du J. Cyclotides as drug design scaffolds. Curr. Opin. Chem. Biol. 2017;38:8–16. doi: 10.1016/j.cbpa.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 20.Kimura RH, Levin AM, Cochran FV, Cochran JR. Engineered cystine knot peptides that bind αvβ3, αvβ5, and α5β1 integrins with low-nanomolar affinity. Proteins: Struct., Funct., Genet. 2009;77:359–369. doi: 10.1002/prot.22441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore SJ, Hayden Gephart MG, Bergen JM, Su YS, Rayburn H, Scott MP, Cochran JR. Engineered knottin peptide enables noninvasive optical imaging of intracranial medulloblastoma. Proc. Natl. Acad. Sci. U. S. A. 2013;110:14598–14603. doi: 10.1073/pnas.1311333110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le-Nguyen D, Heitz A, Chiche L, Hajji MEl, Castro B. Characterization and 2D NMR study of the stable [9–21, 15–27] 2 disulfide intermediate in the folding of the 3 disulfide trypsin inhibitor EETI II. Protein Sci. 1993;2:165–174. doi: 10.1002/pro.5560020205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heitz A, Chiche L, Le-Nguyen D, Castro B. Folding of the squash trypsin inhibitor EETI II: evidence of native and non-native local structural preferences in a linear analogue. Eur. J. Biochem. 1995;233:837–846. doi: 10.1111/j.1432-1033.1995.837_3.x. [DOI] [PubMed] [Google Scholar]
- 24.Heitz A, Le-Nguyen D, Castro B, Chiche L. Conformational study of a native monodisulfide bridge analogue of EETI II. Lett. Pept. Sci. 1997;4:245–249. [Google Scholar]
- 25.Simon MD, Heider PL, Adamo A, Vinogradov AA, Mong SK, Li X, Berger T, Policarpo RL, Zhang C, Zou Y, Liao X, Spokoyny AM, Jensen KF, Pentelute BL. Rapid flow-based peptide synthesis. ChemBioChem. 2014;15:713–720. doi: 10.1002/cbic.201300796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mong SK, Vinogradov AA, Simon MD, Pentelute BL. Rapid total synthesis of DARPin pE59 and barnase. ChemBioChem. 2014;15:721–733. doi: 10.1002/cbic.201300797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mant CT, Zhou NE, Hodges RS. Correlation of protein retention times in reversed-phase chromatography with polypeptide chain length and hydrophobicity. J. Chromatogr. 1989;476:363–375. doi: 10.1016/s0021-9673(01)93882-8. [DOI] [PubMed] [Google Scholar]
- 28.Gütert P, Buchner L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR. 2015;62:453–471. doi: 10.1007/s10858-015-9924-9. [DOI] [PubMed] [Google Scholar]
- 29.Lee W, Tonelli M, Markley JL. NMRFAMSPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2015;31:1325–1327. doi: 10.1093/bioinformatics/btu830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krieger E, Vriend G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015;36:996–1007. doi: 10.1002/jcc.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nielsen KJ, Alewood D, Andrews J, Kent SBH, Craik DJ. An 1H NMR determination of the three-dimensional structures of mirror-image forms of a Leu-5 variant of the trypsin inhibitor form Ecballium elaterium (EETI-II) Protein Sci. 1994;3:291–302. doi: 10.1002/pro.5560030213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park S, Khalili-Araghi F, Tajkhorshid E, Schulten K. Free energy calculation from steered molecular dynamics simulations using Jarzynski’s equality. J. Chem. Phys. 2003;119:3559–3566. [Google Scholar]
- 33.Cheloha RW, Maeda A, Dean T, Gardella TJ, Gellman SH. Backbone modification of a polypeptide drug alters duration of action in vivo. Nat. Biotechnol. 2014;32:653–655. doi: 10.1038/nbt.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang PSP, Schepartz A. β-Peptide bundles: Design. Build. Analyze. Biosynthesize. Chem. Commun. 2016;52:7420–7432. doi: 10.1039/c6cc01546h. [DOI] [PubMed] [Google Scholar]
- 35.Wang PSP, Nguyen JB, Schepartz A. Design and high-resolution structure of a β3-peptide bundle catalyst. J. Am. Chem. Soc. 2014;136:6810–6813. doi: 10.1021/ja5013849. [DOI] [PubMed] [Google Scholar]
- 36.Reinert ZE, Horne WS. Protein backbone engineering as a strategy to advance foldamers toward the frontier of protein-like tertiary structure. Org. Biomol. Chem. 2014;12:8796–8802. doi: 10.1039/c4ob01769b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pauling L, Corey RB. Configurations of polypeptide chains with favored orientations around single bonds: two new pleated sheets. Proc. Natl. Acad. Sci. U. S. A. 1951;37:729–40. doi: 10.1073/pnas.37.11.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.