

Abstract
In this work, we report the evaluation of the residue-residue contacts predicted by our three different methods in the CASP12 experiment, focusing on studying the impact of multiple sequence alignment, residue coevolution and machine learning on contact prediction. The first method (MULTICOM-NOVEL) uses only traditional features (sequence profile, secondary structure and solvent accessibility) with deep learning to predict contacts and serves as a baseline. The second method (MULTICOM-CONSTRUCT) uses our new alignment algorithm to generate deep multiple sequence alignment to derive coevolution-based features, which are integrated by a neural network method to predict contacts. The third method (MULTICOM-CLUSTER) is a consensus combination of the predictions of the first two methods. We evaluated our methods on 94 CASP12 domains. On a subset of 38 free-modeling domains, our methods achieved an average precision of up to 41.7% for top L/5 long-range contact predictions. The comparison of the three methods shows that the quality and effective depth of multiple sequence alignments, coevolution-based features, and machine learning integration of coevolution-based features and traditional features drive the quality of predicted protein contacts. On the full CASP12 dataset, the coevolution-based features alone can improve the average precision from 28.4% to 41.6%, and the machine learning integration of all the features further raises the precision to 56.3%, when top L/5 predicted long-range contacts are evaluated. And the correlation between the precision of contact prediction and the logarithm of the number of effective sequences in alignments is 0.66.
Keywords: Protein contact prediction, CASP, multiple sequence alignment, coevolution, deep learning, machine learning
1. INTRODUCTION
In the absence of homologous structural templates, a key input for successful ab initio protein structure prediction is residue-residue contacts [1, 2]. If a sufficient number of contacts can be predicted accurately, they alone can be used to reconstruct near native models for most proteins with accuracy of 2 Å RMSD [3, 4]. Among all the contacts, long-range contacts, which are generally harder to predict [5–8], but much more useful for structure reconstruction [2]. Hence, recent contact prediction methods focus on the prediction and evaluation of long-range contacts, and so do the CASP experiments. When the contact prediction category was introduced in the CASP experiments, in the initial rounds, methods like SVMcon [6] and DNCON [5] that use support vector machines and deep learning networks with traditional features such as sequence profile, secondary structure and solvent accessibility, were often the top performers demonstrating that machine learning techniques were useful for contact prediction. Recent methods like PconsC2 [9], MetaPSICOV [8] and RaptorX method [10] show that including contact predictions from coevolution-based methods like CCMpred [11], PSICOV [12], and FreeContact [13] as additional features can significantly improve the performance. Often, when sufficient homologous sequences can be found, these methods can predict top L/5 or L/10 long-range contacts with pretty high precision [8, 10, 11], where L is the length of the protein sequence. All these recently successful methods highlight that, besides machine learning techniques, coevolution-based features are important for accurate contact prediction.
Realizing the importance of coevolution-based features, which are entirely dependent upon the availability of homologous sequences, we developed a method for reliably generating deep multiple sequence alignments and coevolution-based features for accurate contact prediction, and participated in the recent CASP 12 experiment with three automated contact prediction methods - MULTICOM-NOVEL, MULTICOM-CONSTRUCT, and MULTICOM-CLUSTER. Our first method, MULTICOM-NOVEL, predicts contacts based on a deep learning contact prediction method - DNCON [5] that uses only traditional features such as sequence profile, secondary structure, and solvent accessibility. Our second method, MULTICOM-CONSTRUCT, relies on our deep multiple sequence alignment generation algorithm to predict coevolution-based features, which are used by a consensus method MetaPSICOV [8] as input to make contact prediction. Our third method, MULTICOM-CLUSTER, combines the predictions from the first two methods by choosing their common highly ranked contacts. Our second and third predictors mainly rely on our deep alignment generation algorithm to make predictions. In this paper, we discuss the performance of our methods in the CASP12 experiment, primarily focusing on identifying the major factors influencing contact prediction accuracy. Since predicted contacts are most useful for protein sequences for which homologous structural templates cannot be found, we emphasize our analysis on free modeling (template-free) targets, although we also include our analysis for all CASP12 targets to assess the benefits of combining traditional features and coevolution-based features with machine learning.
Overall, our contact prediction methods were successful mainly because of our deep alignment generation algorithm, which generates high-quality alignments when sufficient homologous alignments can be found, and at least some alignments (if possible) when homologous sequences are hard to find. We find that multiple sequence alignments, coevolution-based features, and machine learning integration are the key factors for successful protein contact prediction. In addition to the analysis on predicted contacts, we also discuss some findings of building 3D structural models using the CONFOLD method [14] with our predicted contacts as input.
2. MATERIALS AND METHODS
2.1 Generating deep multiple sequence alignments to derive coevolution-based features
Multiple sequence alignments (MSAs) play a central role for the success of a protein contact prediction method because the quality of multiple sequence alignment (MSA) entirely decides the accuracy of the coevolution-based contact prediction features, which largely determines the accuracy of overall contact prediction. Hence, it is crucial to have a reliable algorithm for producing high quality multiple sequence alignments. For reliability, it is important that the algorithm generates at least some sequences when homologous sequences are hard to find in sequence databases, and generates smaller but more useful alignments when an excessively large number of homologous sequences is available. On one hand, in the absence of any homologous sequences in the multiple sequence alignments or when there are just a few sequences, coevolution-based methods fail to make any predictions. On the other hand, when the size of alignment is too large (e.g. more than fifty thousand) and the input protein sequence is long, some methods like PSICOV [12] may take too long to converge and sometimes do not produce any results even in a few days. Based on this understanding, we designed an alignment generation algorithm that attempts to generate high coverage alignments at first, and when sufficient homologous sequences are not found, relies on various sequence similarity cut-off thresholds to increase the depth of search to generate at least some sequences whenever possible.
For generating MSAs, we start by assuming sufficient homologous sequences covering most of our input sequence are available. Then we gradually switch towards choosing the settings that allow us to search deeply to generate at least some sequences. Using HHblits [15], we first generate alignments that cover 75% of a target sequence and check if the alignment has at least 2.5L sequences, where L is the length of the query sequence. If at least 2.5L sequences are not obtained, the coverage threshold is lowered, at first to 68% and then to 60% if needed. If none of these coverage thresholds deliver at least 2.5L sequences, we switch to using JackHMMER [16] to find remotely homologous sequences. Once again, we assume that sufficient significant hits can be found and start alignment search with a very stringent e-value cut-off threshold of 1E−40 to find homologous sequences. If this threshold fails to generate at least 2.5L sequences, we increase the e-value threshold to 1E−30, 1E−20, 1E−10, 1E−4, and 1, step by step, and conclude when more than 2.5L sequences are generated. If none of the thresholds leads to an alignment with more than 2.5L sequences, the alignments generated with high e-value threshold of 1 are used as the final alignment. A range of e-value thresholds is required because, for some input protein sequences, a stringent e-value criterion (like 1E−40) produces too few sequences (just a hundred or so) whereas a looser criterion (like 1E−4) generates many sequences. We used the ‘UNIPROT20-2016’ and ‘UNIREF90’ sequence databases for HHblits and JackHMMER search respectively.
2.2 MULTICOM contact prediction methods
Our first method, MULTICOM-NOVEL, is based on our method DNCON, an ab initio contact prediction method trained using deep belief networks and boosting [5, 17]. Unlike recent contact prediction methods that use co-evolutionary features as key features, it does not use any co-evolutionary information. To make contact predictions, DNCON uses an ensemble of deep belief networks, each consisting of three layers of Restricted Boltzman Machines (RBM), which were trained and tested on a large dataset consisting of 1426 proteins. For each input pair of residues, DNCON predicts medium- and long-range contact probabilities using seven sets of deep belief networks trained using sequence window sizes of 7, 9, 11, 13, 15, 17, and 19 residues. This ‘ensembling’ of predictions from networks trained using different window sizes is one of the key technique that contributes to DNCON’s performance. The second key contributor of DNCON’s performance is the boosting technique that gradually increases the weight of misclassified examples during training. For each window size, instead of using a single deep belief network, DNCON uses ensembles of classifiers trained using the boosting technology [18]. For each classification (i.e. predicting whether a residue pair is in contact), 35 different networks were trained serially using 35 rounds of boosting, and the networks were assigned individual weights. For each input residue pair, the final prediction probability is the weighted average of the probabilities predicted by individual networks (For more details of DNCON, see [5] and [17]).
DNCON’s results from the training and testing experiments show that an ensemble of models trained at 7 window sizes (window sizes of 7, 9, 11, 13, 15, 17, and 19) delivers an accuracy of 34%, compared to 24% to 28% of individual models, on a test dataset of 196 proteins, when top L/5 long-range contacts are evaluated. DNCON was the top performer in the CASP10 contact prediction category [17] and therefore serves as a good benchmark (baseline) to study where the improvement of contact prediction comes from in CASP12.
Our second method, MULTICOM-CONSTRUCT, primarily relies on our deep alignment generation algorithm to generate multiple sequence alignments, which are supplied as input to the three standard coevolution-based methods, PSICOV [12], CCMpred [11], and FreeContact [13] to generate two-dimensional co-evolution features to be combined by MetaPSICOV [8] with traditional features to make contact prediction. During the development of the method, we found that some coevolution-based methods like PSICOV sometime could not converge within a reasonable time limit when there were too many or too few sequences in alignments. To guarantee to generate predictions from such methods within a certain time limit, tweaking their convergence parameters is needed. Specifically, to get around the convergence issue of PSICOV, we run it with three convergence parameters (‘d = 0.03’, ‘r = 0.001’, and ‘r = 0.01’) in parallel and wait for a maximum of five hours. The ‘d’ parameter selects the glasso exact algorithm and is expected to produce more accurate results but is slow. The ‘rho’ parameter (r) controls how quickly the programs converges and higher values tend to speed up the convergence but at the loss of prediction accuracy. We pick the job that finishes within the five-hour time limit according to the order (‘d = 0.03’, ‘r = 0.001’, and ‘r = 0.01’). In this way we are always able to have some prediction produced within the limited time. Such a shorter time limit was used during the CASP 12 experiment because our ab initio structure prediction methods used these predicted contacts as input to build 3D models, which themselves needed up to two days to build models.
Our third method, MULTICOM-CLUSTER, is a meta-predictor that combines contacts predicted by the first two methods. When at least 50 homologous sequences are found, this method uses the predictions made by MULTICOM-CONSTRUCT, otherwise combines the predictions of MULITICOM-CONSTRUCT and MULTICOM-NOVEL. As the first step for contact combination, we select two sets of up to 5L long-range contacts - one set from MULTICOM-NOVEL and another set from MULTICOM-CONSTRUCT. For each target, we first select top 5L contacts predicted by MULTICOM-NOVEL filtering out all the contacts not predicted by MULTICOM-CONSTRUCT, and then select up to top 5L contacts predicted by MULTICOM-CONSTRUCT which are present in the top 5L contacts from MULTICOM-NOVEL. This new set of contacts by MULTICOM-CONSTRUCT (having at most 5L contacts), and the set of contacts by MULTICOM-NOVEL are then updated by replacing their confidence values with the ranks, i.e. integer numbers starting from ‘5L’ for the most confident contact prediction and ending at ‘1’ for the least confident one. At this point, both sets have same contacts but different rankings. Then, the ranks for the MULTICOM-CONSTRUCT’s set are updated as the sum of the ranks in the two sets and are normalized by 10L. This new rank scores are then used to sort the contacts, as confidence scores, and used as input for MULTICOM-CLUSTER predictions. The final step is to scale the confidence values into a meaningful range between 0 and 1. Ideally, if we knew the number of long-range contacts in the target structure (Nc), we would normalize the confidence values such that the top Nc predictions have confidence more than 0.5. In the absence of such knowledge in reality, we normalize the confidence scores such that top L predicted contacts have confidence values more than 0.5, and submitted these contacts as MULTICOM-CLUSTER predictions.
2.3 Datasets and evaluation metrics
Out of the 90 targets released during the CASP 12 season, CASP12’s official contact evaluations released at CASP’s website were carried out on 70 targets (i.e. corresponding to 94 domains), excluding domains ‘T0865-D1’ and ‘T0880-D1’ because they do not have any long-range contacts. In this work, we consider all these 94 structural domains and its subset of 38 free-modeling domains for evaluation and comparison of our three methods, MULTICOM-NOVEL, MULTICOM-CONSTRUCT, and MULTICOM-CLUSTER. In this set of 94 domains, the native structures of 84 of them were available for our assessments. Hence, for some of our own evaluations, like evaluating the precision of co-evolutionary features, we use these 84 domains only. And to maintain consistency with the CASP released evaluations, we focus our analysis and evaluation at the domain level, although all predictions were made for the whole targets during the CASP12 experiment. Finally, before the CASP12 experiment, we used the dataset of CASP11 free-modeling domains to benchmark our methods. The results on CASP11 are also reported as a comparison with those on CASP12.
In addition to using our ConEVA contact evaluation toolkit [19] to do evaluation, we also referred to the evaluations published by CASP (released at http://predictioncenter.org/). We focus our evaluations on top L/5 and L/2 predicted long-range contacts and use precision as the primary evaluation metric, which is the fraction (ratio) of correct predictions in top predicted contacts. One important factor influencing the precision of contact prediction is the number of effective sequences in multiple sequence alignment, Meff [20], which is calculated at the domain or target level using the following equation:
where N is the number of sequences in the multiple sequence alignment and ni is the number of sequences which have at least 62% sequence identity with the ith sequence [8]. If all sequences in the alignment are very different, ni is 1 for each sequence and hence Meff sums to N, and on the contrary, if all sequences are very similar, ni is equal to N for all sequences and the sum of 1/N for N sequences gives 1, i.e. the Meff is just 1. For calculating Meff at the domain level, we trim the multiple sequence alignment column-wise, removing all the columns for which the reference native structure of a domain does not have any residues defined, so that the width of the alignment (number of columns) is same as the number of residues in the native structure of the domain.
3. Results and Discussion
3.1 Initial benchmark on CASP11 free-modeling dataset before CASP12 experiment
Prior to the CASP 12 experiment, we evaluated MULTICOM-CONSTRUCT that uses our new deep alignment generation algorithm to generate co-evolution features for contact prediction, on the dataset of 30 free-modeling structural domains of the CASP 11 experiment [21]. Following MULTICOM-CONSTRUCT’s pipeline, we generated alignments and coevolution-based features for the 24 protein targets (with full targets as input) containing the 30 free-modeling domains, predicted contacts for the targets, and evaluated the predictions at the domain level. For comparison, on the same dataset, we also predicted contacts using the publicly available MetaPSICOV method with default options, where alignments were generated using HHblits [15] with the coverage threshold parameter set to 60%. Moreover, we compared our results with the best performing group in the CASP11 contact prediction category, CONSIP2 [22], on the same dataset. The mean precisions of top L/5 long-range contacts predicted by MetaPSICOV, CONSIP2, and MULTICOM-CONSTRUCT are 29%, 29%, and 34.4% respectively (see Table 1). The improvement of our method is significant according to paired t-test of the difference in precision (p-value = 0.03). It is important to note that the same protein sequence database was used with MetaPSICOV and our method for a fair comparison. On average, our method can increase the number of sequences (N) in the alignment to 1546 (from 152), and the number of effective sequences (Meff) to 222 (from 69), which is probably the primary contributor for the improvement (Table1). For these free-modeling domains, the Pearson’s correlation coefficient between the precision of top L/5 long-range contacts predicted by MULTICOM-CONSTRUCT and the logarithm of the number of effective sequences (log(Meff)) in alignments is 0.60, which highlights the importance of the depth of multiple sequence alignments for contact quality. It is also important to note that the number of effective sequences was calculated at the domain level. Pearson’s correlation, when calculated using the number of effective sequences for the whole target alignment, gives much lower coefficients. This is because a high effective sequence number at whole target level does not guarantee a high number of effective sequences for each domain of a multi-domain target, as a sequence in an alignment may only cover a portion of the target.
Table 1.
Top L/5 long-range contacts predicted by MULTICOM-CONSTRUCT method compared with the top L/5 contacts predicted using the default MetaPSICOV method and the CONSIP2 method, on the 30 CASP11 free-modeling domains. Ntarget is the number of sequence in the alignment which is generated with the target sequence as input. Meffdomain is number of effective sequences in the alignment when alignments are trimmed to match the residues of the native structural domain. PL/5 refers to the precision of top L/5 long-range contacts.
| Domain | MetaPSICOV
|
CONSTRUCT
|
CONSIP2
|
||||
|---|---|---|---|---|---|---|---|
| Ntarget | Meffdomain | PL/5 | Ntarget | Meffdomain | PL/5 | PL/5 | |
| T0761-D1 | 1 | 1 | 0.0 | 4 | 2 | 0.0 | 5.6 |
| T0761-D2 | 1 | 1 | 13.0 | 4 | 2 | 13.0 | 8.7 |
| T0763-D1 | 3 | 2 | 30.8 | 7 | 3 | 15.4 | 46.2 |
| T0767-D2 | 109 | 58 | 66.7 | 774 | 88 | 66.7 | 58.3 |
| T0771-D1 | 9 | 4 | 26.7 | 32 | 11 | 16.7 | 10.0 |
| T0777-D1 | 55 | 25 | 15.9 | 747 | 41 | 18.8 | 23.2 |
| T0781-D1 | 2 | 2 | 10.0 | 40 | 15 | 2.5 | 5.0 |
| T0785-D1 | 1 | 1 | 4.6 | 6 | 2 | 4.6 | 18.2 |
| T0789-D1 | 274 | 133 | 44.8 | 2465 | 484 | 62.1 | 51.7 |
| T0789-D2 | 274 | 139 | 44.0 | 2465 | 522 | 60.0 | 28.0 |
| T0790-D1 | 276 | 140 | 44.4 | 1829 | 440 | 59.3 | 44.4 |
| T0790-D2 | 276 | 136 | 26.9 | 1829 | 455 | 69.2 | 26.9 |
| T0791-D1 | 265 | 109 | 63.3 | 2488 | 401 | 66.7 | 53.3 |
| T0791-D2 | 265 | 118 | 35.7 | 2488 | 481 | 75.0 | 42.9 |
| T0794-D2 | 258 | 121 | 52.9 | 1653 | 176 | 38.2 | 26.5 |
| T0806-D1 | 766 | 369 | 62.8 | 1130 | 306 | 70.6 | 84.3 |
| T0808-D2 | 121 | 29 | 27.8 | 1257 | 92 | 27.8 | 35.2 |
| T0810-D1 | 49 | 24 | 21.7 | 8669 | 1147 | 21.7 | 17.4 |
| T0814-D1 | 118 | 106 | 25.9 | 1404 | 145 | 48.2 | 37.0 |
| T0814-D2 | 118 | 107 | 69.6 | 1404 | 174 | 73.9 | 82.6 |
| T0820-D1 | 1 | 1 | 5.6 | 1 | 1 | 5.6 | 5.6 |
| T0824-D1 | 79 | 32 | 36.4 | 1257 | 254 | 72.7 | 45.5 |
| T0827-D2 | 680 | 229 | 26.7 | 3164 | 558 | 20.0 | 10.0 |
| T0831-D2 | 189 | 100 | 10.3 | 4659 | 242 | 7.7 | 7.7 |
| T0832-D1 | 5 | 2 | 2.4 | 83 | 26 | 4.8 | 2.4 |
| T0834-D1 | 42 | 21 | 0.0 | 269 | 48 | 0.0 | 5.0 |
| T0834-D2 | 42 | 16 | 5.9 | 269 | 45 | 5.9 | 17.7 |
| T0836-D1 | 223 | 26 | 36.6 | 2627 | 167 | 68.3 | 43.9 |
| T0837-D1 | 32 | 8 | 37.5 | 132 | 10 | 37.5 | 29.2 |
| T0855-D1 | 11 | 7 | 21.7 | 3234 | 322 | 0.0 | 17.4 |
|
| |||||||
| Average | 152 | 69 | 29.0 | 1546 | 222 | 34.4 | 29.7 |
3.2 Performance on CASP12 dataset
Table 2 summarizes the performance of our three methods on the subset of 38 CASP12 domains classified as free modeling. The mean precision of top L/5 long-range contacts predicted by our three methods MULTICOM-NOVEL, MULTICOM-CONSTRUCT, and MULTICOM-CLUSTER are 25.4%, 41.6% and 41.7% respectively. MULTICOM-CONSTRUCT and MULTICOM-CLUSTER, which rely on our deep multiple sequence alignment generation algorithm and coevolution-based features, have much higher mean precision compared to the baseline sequence-based machine learning method MULTICOM-NOVEL without using coevolution features, suggesting the enhanced co-evolution features is a major contributor to the improved precision. On this free-modeling dataset, our contact combination method, MULTICOM-CLUSTER, has improved performance on two domains T0869-D1 and T0923-D1, although, on average, its performance is similar to the MULTICOM-CONSTRUCT method. For 23 out of these 38 domains, our deep alignment generation algorithm concluded with alignments generated by JackHMMER at high e-value threshold of 1, suggesting that most of the domains in the free-modeling dataset did not have sufficient significantly homologous sequences with high coverage. The low-quality alignments, generated by JackHMMER at e-value threshold of 1, have the number of effective sequences ranging from 1 to 1331 (with mean as 107 and median as 31), and the precision of MULTICOM-CONSTRUCT’s contact predictions for these domains ranges from 3% to 95%. This suggests that high e-value thresholds do not always necessarily generate poor alignments, but rather lead to alignments of variable quality, some of which are useful for contact prediction.
Table 2.
Comparison of top L/5 long-range contacts predicted by our three methods, MULTICOM-NOVEL, MULTICOM-CONSTRUCT, and MULTICOM-CLUSTER for the 38 free-modeling structural domains using precision measure. L, Ntarget, and Meffdomain stand for the length of the target sequence, number of sequence in the alignment for the whole target sequence, and the number of effective sequences in the alignment when alignments are trimmed to match the residues of the native structural domain, respectively. The last three columns show the precision of top L/5 long-range contacts for the three methods. The ‘Alignment’ column shows the method and parameter used to generate the alignment, where ‘jhm’ stands for JackHMMER and ‘hhb’ stands for HHblits.
| Target | FM Domain | L | Alignment | Ntarget | Meffdomain | CONSTRUCT | CLUSTER | NOVEL |
|---|---|---|---|---|---|---|---|---|
| T0859 | T0859-D1 | 133 | jhm-e-0 | 2 | 1 | 4.4 | 4.4 | 0.0 |
| T0862 | T0862-D1 | 239 | jhm-e-0 | 163 | 31 | 26.3 | 26.3 | 21.1 |
| T0863 | T0863-D1 | 670 | jhm-e-0 | 453 | 73 | 2.6 | 2.6 | 5.1 |
| T0863 | T0863-D2 | 670 | jhm-e-0 | 453 | 54 | 4.2 | 4.2 | 4.2 |
| T0864 | T0864-D1 | 246 | jhm-e-0 | 526 | 134 | 64.0 | 64.0 | 32.0 |
| T0866 | T0866-D1 | 183 | hhb-cov75 | 1388 | 560 | 100.0 | 100.0 | 14.3 |
| T0869 | T0869-D1 | 120 | jhm-e-0 | 17 | 12 | 42.9 | 52.4 | 47.6 |
| T0870 | T0870-D1 | 138 | jhm-e-0 | 137 | 81 | 16.0 | 16.0 | 40.0 |
| T0878 | T0878-D1 | 358 | jhm-e-0 | 856 | 250 | 42.0 | 42.0 | 26.1 |
| T0880 | T0880-D2 | 193 | jhm-e-0 | 2 | 1 | 25.0 | 21.9 | 18.8 |
| T0886 | T0886-D1 | 346 | jhm-1e-40 | 3013 | 1182 | 78.6 | 78.6 | 7.1 |
| T0886 | T0886-D2 | 346 | jhm-1e-40 | 3013 | 1837 | 88.5 | 88.5 | 23.1 |
| T0888 | T0888-D1 | 121 | jhm-e-0 | 2 | 1 | 8.0 | 0.0 | 0.0 |
| T0890 | T0890-D2 | 191 | jhm-e-0 | 70 | 17 | 13.6 | 13.6 | 9.1 |
| T0892 | T0892-D2 | 193 | jhm-e-0 | 579 | 202 | 54.6 | 54.6 | 63.6 |
| T0894 | T0894-D1 | 324 | jhm-e-0 | 438 | 61 | 11.1 | 11.1 | 55.6 |
| T0896 | T0896-D3 | 486 | jhm-e-0 | 2295 | 7 | 12.1 | 12.1 | 9.1 |
| T0897 | T0897-D1 | 285 | jhm-e-0 | 130 | 10 | 7.1 | 7.1 | 17.9 |
| T0897 | T0897-D2 | 285 | jhm-e-0 | 130 | 57 | 52.0 | 52.0 | 20.0 |
| T0898 | T0898-D1 | 169 | jhm-1e-4 | 50000 | 389 | 4.6 | 4.6 | 13.6 |
| T0899 | T0899-D1 | 423 | jhm-1e-10 | 6580 | 125 | 71.2 | 71.2 | 40.4 |
| T0899 | T0899-D2 | 423 | jhm-1e-10 | 6580 | 31 | 44.4 | 44.4 | 33.3 |
| T0900 | T0900-D1 | 106 | jhm-e-0 | 16243 | 1331 | 95.2 | 95.2 | 71.4 |
| T0901 | T0901-D2 | 328 | hhb-cov50 | 5167 | 127 | 64.3 | 64.3 | 42.9 |
| T0904 | T0904-D1 | 341 | jhm-1e-10 | 23741 | 609 | 72.6 | 72.6 | 29.4 |
| T0905 | T0905-D1 | 353 | jhm-1e-10 | 8623 | 346 | 79.6 | 79.6 | 63.3 |
| T0905 | T0905-D2 | 353 | jhm-1e-10 | 8623 | 88 | 42.9 | 42.9 | 42.9 |
| T0907 | T0907-D3 | 315 | jhm-e-0 | 219 | 1 | 79.2 | 79.2 | 41.7 |
| T0912 | T0912-D3 | 624 | jhm-1e-20 | 7240 | 426 | 42.9 | 42.9 | 4.8 |
| T0914 | T0914-D1 | 337 | jhm-e-0 | 325 | 70 | 6.3 | 6.3 | 31.3 |
| T0914 | T0914-D2 | 337 | jhm-e-0 | 325 | 33 | 6.1 | 6.1 | 15.2 |
| T0915 | T0915-D1 | 161 | jhm-e-0 | 34 | 21 | 48.4 | 45.2 | 29.0 |
| T0918 | T0918-D1 | 546 | jhm-1e-20 | 3517 | 356 | 77.3 | 77.3 | 40.9 |
| T0918 | T0918-D2 | 546 | jhm-1e-20 | 3517 | 487 | 88.0 | 88.0 | 20.0 |
| T0918 | T0918-D3 | 546 | jhm-1e-20 | 3517 | 513 | 66.7 | 66.7 | 0.0 |
| T0923 | T0923-D1 | 409 | jhm-e-0 | 10 | 7 | 12.1 | 19.0 | 22.4 |
| T0941 | T0941-D1 | 470 | jhm-e-0 | 3 | 1 | 2.9 | 2.9 | 1.5 |
| T0946 | T0946-D1 | 292 | hhb-cov50 | 3170 | 80 | 25.0 | 25.0 | 6.3 |
|
| ||||||||
| Average | 253 | 41.6 | 41.7 | 25.4 | ||||
On the full dataset consisting of all 94 CASP12 domains, the mean precision of top L/5 long-range contacts for MULTICOM-NOVEL, MULTICOM-CONSTRUCT, and MULTICOM-CLUSTER are 25.8%, 50.3% and 50.1% respectively (see Table S1 for detailed results). Higher precisions on the complete dataset is due to the fact that the mean Meff for all the targets is 1619, greater than 253 for the free-modeling targets. Finally, the same as on CASP11 free-modeling dataset, we observed a Pearson’s correlation coefficient of 0.66 between the precision of top L/5 long-range contacts predicted by MULTICOM-CONSTRUCT and the logarithm of the number of effective sequences (Meff) on the CASP12 full dataset. Since it is relevant to compare the performance of all three methods on the target domains for which no sufficient number of sequences in alignments were found, we selected six free-modeling domains for which our method generated less than 20 sequences in the alignments (see Table S2). For these targets, while MULTICOM-NOVEL and MULTICOM-CONSTRUCT have average precision of 15% and 15.9% respectively, the contact combination made by MULTICOM-CLUSTER has average precision of 16.7%, showing a slight improvement.
3.3 Significance of coevolution-based features and machine learning integration
If reliable and deep multiple sequence alignments are available, two-dimensional pairwise features (contact probabilities or scores) predicted by coevolution-based methods are a key factor for high accuracy in final contact prediction. To study the significance of these features, we evaluated the precision of the coevolution-based contacts predicted by PSICOV, CCMpred and FreeContact separately, and compared them with the final prediction made by MULTICOM-CONSTRUCT. Excluding some targets for which PSICOV failed to converge within the five-hour time limit and some additional targets for which no more than 5 homologous sequences could be found, on the remaining 70 structural domains, CCMpred, FreeContact, and PSICOV have mean precision of 41.6%, 36.3%, and 34.1% respectively, for top L/5 long-range contacts. When top L/2 contacts are evaluated, the similar trend is observed for the three methods with the mean precision of 32.6% for CCMpred, 28.3% for FreeContact, and 25.4% for PSICOV, suggesting that the most accurate single coevolution-based predictor is CCMpred followed by FreeContact and PSICOV (see Table 3). These precisions are much higher than the average precision (25.4%) of our baseline MULTICOM-NOVEL method that does not use any coevolution-based features as input. These results indicate that the coevolution-based features are crucial for accurate contact prediction.
Table 3.
Precision of top L/5 and L/2 contacts predicted for CASP12 structural domains using PSICOV, FreeContact, and CCMpred, the maximum precision of the three methods, and the MULTICOM-CONSTRUCT method of using machine learning to integrate multiple co-evolution features. This dataset excludes the cases in where PSICOV failed to generate any results within the time limit.
| Domain | PSICOV | FreeContact | CCMpred | Maximum | MULTICOM-CONSTRUCT | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| L/5 | L/2 | L/5 | L/2 | L/5 | L/2 | L/5 | L/2 | L/5 | L/2 | |
| T0861-D1 | 79.0 | 54.5 | 79.0 | 66.0 | 83.9 | 77.6 | 83.9 | 77.6 | 85.5 | 81.4 |
| T0862-D1 | 0.0 | 0.0 | 0.0 | 0.0 | 15.8 | 6.4 | 15.8 | 6.4 | 26.3 | 12.8 |
| T0863-D1 | 2.6 | 3.1 | 0.0 | 0.0 | 2.6 | 2.1 | 2.6 | 3.1 | 2.6 | 7.2 |
| T0863-D2 | 1.4 | 1.7 | 0.0 | 0.0 | 0.0 | 0.6 | 1.4 | 1.7 | 4.2 | 3.4 |
| T0864-D1 | 20.4 | 14.6 | 42.9 | 25.2 | 55.1 | 26.8 | 55.1 | 26.8 | 65.3 | 45.5 |
| T0866-D1 | 95.2 | 63.5 | 81.0 | 63.5 | 95.2 | 71.2 | 95.2 | 71.2 | 100.0 | 78.9 |
| T0868-D1 | 13.0 | 10.3 | 4.4 | 5.2 | 17.4 | 13.8 | 17.4 | 13.8 | 82.6 | 60.3 |
| T0869-D1 | 14.3 | 12.5 | 0.0 | 3.9 | 19.1 | 13.5 | 19.1 | 13.5 | 42.9 | 36.5 |
| T0870-D1 | 16.0 | 9.7 | 12.0 | 6.5 | 16.0 | 9.7 | 16.0 | 9.7 | 16.0 | 8.1 |
| T0871-D1 | 73.4 | 50.0 | 57.8 | 38.8 | 81.3 | 61.3 | 81.3 | 61.3 | 93.8 | 79.4 |
| T0872-D1 | 27.8 | 18.2 | 33.3 | 18.2 | 33.3 | 22.7 | 33.3 | 22.7 | 66.7 | 31.8 |
| T0873-D1 | 43.5 | 26.8 | 66.3 | 55.4 | 66.3 | 58.9 | 66.3 | 58.9 | 82.6 | 70.6 |
| T0877-D1 | 10.7 | 7.0 | 7.1 | 5.6 | 10.7 | 8.5 | 10.7 | 8.5 | 17.9 | 21.1 |
| T0878-D1 | 27.5 | 18.6 | 39.1 | 21.5 | 36.2 | 20.4 | 39.1 | 21.5 | 42.0 | 29.7 |
| T0879-D1 | 81.8 | 70.0 | 75.0 | 70.9 | 77.3 | 73.6 | 81.8 | 73.6 | 97.7 | 85.5 |
| T0881-D1 | 5.0 | 4.0 | 2.5 | 5.0 | 5.0 | 5.0 | 5.0 | 5.0 | 0.0 | 3.0 |
| T0882-D1 | 6.3 | 5.0 | 12.5 | 7.5 | 18.8 | 10.0 | 18.8 | 10.0 | 6.3 | 10.0 |
| T0884-D1 | 14.3 | 13.9 | 7.1 | 5.6 | 7.1 | 8.3 | 14.3 | 13.9 | 7.1 | 13.9 |
| T0885-D1 | 56.5 | 35.1 | 39.1 | 33.3 | 47.8 | 33.3 | 56.5 | 35.1 | 95.7 | 61.4 |
| T0886-D1 | 71.4 | 57.1 | 78.6 | 68.6 | 78.6 | 77.1 | 78.6 | 77.1 | 78.6 | 77.1 |
| T0886-D2 | 80.0 | 50.0 | 88.0 | 60.9 | 92.0 | 60.9 | 92.0 | 60.9 | 88.0 | 82.8 |
| T0889-D1 | 89.6 | 78.3 | 87.5 | 80.8 | 87.5 | 80.8 | 89.6 | 80.8 | 95.8 | 90.8 |
| T0890-D1 | 25.0 | 24.4 | 12.5 | 9.8 | 12.5 | 7.3 | 25.0 | 24.4 | 43.8 | 22.0 |
| T0890-D2 | 19.1 | 11.3 | 0.0 | 0.0 | 0.0 | 5.7 | 19.1 | 11.3 | 14.3 | 11.3 |
| T0891-D1 | 63.6 | 41.1 | 59.1 | 42.9 | 68.2 | 46.4 | 68.2 | 46.4 | 90.9 | 87.5 |
| T0892-D1 | 21.4 | 14.3 | 42.9 | 22.9 | 50.0 | 25.7 | 50.0 | 25.7 | 35.7 | 28.6 |
| T0892-D2 | 22.7 | 16.4 | 31.8 | 18.2 | 18.2 | 14.6 | 31.8 | 18.2 | 54.6 | 49.1 |
| T0893-D1 | 0.0 | 2.7 | 0.0 | 5.4 | 6.7 | 8.1 | 6.7 | 8.1 | 6.7 | 8.1 |
| T0893-D2 | 91.2 | 80.0 | 91.2 | 80.0 | 94.1 | 83.5 | 94.1 | 83.5 | 97.1 | 89.4 |
| T0894-D1 | 11.1 | 11.1 | 27.8 | 15.6 | 22.2 | 13.3 | 27.8 | 15.6 | 11.1 | 13.3 |
| T0894-D2 | 18.2 | 18.5 | 27.3 | 14.8 | 36.4 | 18.5 | 36.4 | 18.5 | 54.6 | 33.3 |
| T0895-D1 | 4.2 | 5.0 | 4.2 | 5.0 | 12.5 | 5.0 | 12.5 | 5.0 | 33.3 | 30.0 |
| T0897-D1 | 0.0 | 0.0 | 0.0 | 1.5 | 0.0 | 0.0 | 0.0 | 1.5 | 7.1 | 7.3 |
| T0897-D2 | 24.0 | 22.6 | 20.0 | 14.5 | 32.0 | 24.2 | 32.0 | 24.2 | 52.0 | 25.8 |
| T0898-D1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 4.8 | 7.6 |
| T0898-D2 | 0.0 | 0.0 | 9.1 | 14.3 | 9.1 | 3.6 | 9.1 | 14.3 | 9.1 | 10.7 |
| T0899-D1 | 26.9 | 20.8 | 26.9 | 19.2 | 28.9 | 15.4 | 28.9 | 20.8 | 71.2 | 49.2 |
| T0899-D2 | 16.7 | 13.6 | 5.6 | 6.8 | 11.1 | 11.4 | 16.7 | 13.6 | 44.4 | 36.4 |
| T0900-D1 | 50.0 | 43.1 | 50.0 | 45.1 | 65.0 | 56.9 | 65.0 | 56.9 | 95.0 | 80.4 |
| T0901-D1 | 62.2 | 46.4 | 62.2 | 47.3 | 60.0 | 48.2 | 62.2 | 48.2 | 84.4 | 67.0 |
| T0901-D2 | 14.3 | 11.4 | 7.1 | 5.7 | 0.0 | 2.9 | 14.3 | 11.4 | 64.3 | 40.0 |
| T0902-D1 | 67.4 | 56.0 | 73.9 | 60.3 | 67.4 | 65.5 | 73.9 | 65.5 | 93.5 | 88.8 |
| T0903-D1 | 27.7 | 22.8 | 1.5 | 4.3 | 27.7 | 18.5 | 27.7 | 22.8 | 98.5 | 80.9 |
| T0904-D1 | 50.0 | 31.8 | 24.0 | 16.7 | 70.0 | 44.4 | 70.0 | 44.4 | 74.0 | 48.4 |
| T0905-D1 | 33.3 | 23.1 | 41.7 | 26.5 | 41.7 | 26.5 | 41.7 | 26.5 | 79.2 | 54.6 |
| T0905-D2 | 30.8 | 24.2 | 0.0 | 6.1 | 7.7 | 12.1 | 30.8 | 24.2 | 46.2 | 48.5 |
| T0909-D1 | 27.0 | 15.1 | 20.3 | 19.7 | 44.3 | 28.4 | 44.3 | 28.4 | 43.1 | 32.7 |
| T0911-D1 | 65.9 | 50.5 | 78.1 | 64.7 | 72.0 | 69.1 | 78.1 | 69.1 | 86.6 | 76.0 |
| T0912-D1 | 20.5 | 20.2 | 84.3 | 69.1 | 78.3 | 63.8 | 84.3 | 69.1 | 91.6 | 82.1 |
| T0912-D2 | 29.4 | 18.5 | 58.8 | 42.9 | 58.8 | 47.6 | 58.8 | 47.6 | 41.2 | 33.3 |
| T0912-D3 | 0.0 | 0.0 | 14.3 | 15.4 | 23.8 | 17.3 | 23.8 | 17.3 | 42.9 | 28.9 |
| T0913-D1 | 48.5 | 34.3 | 64.7 | 48.5 | 79.4 | 57.4 | 79.4 | 57.4 | 69.1 | 62.1 |
| T0914-D1 | 6.3 | 5.1 | 3.1 | 2.5 | 6.3 | 2.5 | 6.3 | 5.1 | 6.3 | 8.9 |
| T0914-D2 | 9.4 | 7.4 | 3.1 | 2.5 | 6.3 | 2.5 | 9.4 | 7.4 | 6.3 | 4.9 |
| T0915-D1 | 6.5 | 6.5 | 6.5 | 2.6 | 0.0 | 2.6 | 6.5 | 6.5 | 48.4 | 27.3 |
| T0917-D1 | 82.1 | 70.4 | 76.9 | 66.3 | 89.7 | 79.6 | 89.7 | 79.6 | 97.4 | 84.7 |
| T0918-D1 | 40.9 | 27.8 | 50.0 | 40.7 | 59.1 | 50.0 | 59.1 | 50.0 | 77.3 | 59.3 |
| T0918-D2 | 48.0 | 29.0 | 60.0 | 48.4 | 68.0 | 58.1 | 68.0 | 58.1 | 88.0 | 71.0 |
| T0918-D3 | 25.0 | 15.3 | 75.0 | 47.5 | 75.0 | 52.5 | 75.0 | 52.5 | 66.7 | 45.8 |
| T0920-D1 | 85.9 | 65.8 | 87.5 | 75.8 | 89.1 | 78.3 | 89.1 | 78.3 | 93.8 | 88.8 |
| T0920-D2 | 0.0 | 0.0 | 2.3 | 1.8 | 4.6 | 2.7 | 4.6 | 2.7 | 22.7 | 18.2 |
| T0921-D1 | 64.3 | 34.8 | 60.7 | 46.4 | 57.1 | 39.1 | 64.3 | 46.4 | 96.4 | 76.8 |
| T0922-D1 | 26.7 | 27.0 | 33.3 | 29.7 | 33.3 | 32.4 | 33.3 | 32.4 | 53.3 | 46.0 |
| T0928-D1 | 52.9 | 36.3 | 72.1 | 43.3 | 66.2 | 51.5 | 72.1 | 51.5 | 79.4 | 63.2 |
| T0944-D1 | 70.6 | 44.1 | 62.8 | 49.6 | 68.6 | 58.3 | 70.6 | 58.3 | 88.2 | 66.9 |
| T0945-D1 | 29.3 | 25.0 | 46.7 | 28.2 | 73.3 | 53.2 | 73.3 | 53.2 | 86.7 | 64.9 |
| T0946-D1 | 12.5 | 10.0 | 0.0 | 2.5 | 25.0 | 17.5 | 25.0 | 17.5 | 25.0 | 30.0 |
| T0946-D2 | 66.7 | 52.8 | 66.7 | 50.9 | 61.9 | 57.6 | 66.7 | 57.6 | 81.0 | 73.6 |
| T0947-D1 | 57.1 | 36.4 | 65.7 | 46.6 | 77.1 | 50.0 | 77.1 | 50.0 | 80.0 | 67.1 |
| T0948-D1 | 3.3 | 4.0 | 16.7 | 9.3 | 6.7 | 6.7 | 16.7 | 9.3 | 6.7 | 10.7 |
|
| ||||||||||
| Mean | 34.1 | 25.4 | 36.3 | 28.3 | 41.6 | 32.6 | 44.2 | 34.1 | 56.3 | 46.2 |
In Table 3 MULTICOM-CONSTRUCT has much higher mean precision of 56.3% for top L/5 long-range predictions (46.2% for top L/2), compared to each of the three individual coevolution-based features above. If we selected the best contact predictions made by the three coevolution-based predictions to evaluate for each domain, the mean precision (called maximum in Table 3) is 44.2% for top L/5 contacts, which is only slightly (2.6%) better than the performance of the best individual coevolution-based feature predictor CCMpred, but is still much lower than the mean precision 56.3% of MULTICOM-CONSTRUCT. These results indicate that, in addition to coevolution-based features being important, the machine learning approaches of integrating these coevolution-based features with the traditional sequence-based features are also very important. Analyzing the predictions made by MULTICOM-CONSTRUCT, we only found 2 out of 70 domains (T0918-D3 and T0912-D2) for which the machine learning integration had failed to perform better than an individual coevolution-based feature. Upon inspecting the three-dimensional structures of these two domains, however, we find both of them have the middle region of the structure missing, which might cause the failure of the machine learning integration. Generally speaking, in MULTICOM-CONSTRUCT, the neural network-based combination of the multiple co-evolution features and traditional features almost always performs better than individual coevolution-based features. Taking domain T0868-D1 as an example, when top L/5 long-range contacts are evaluated, the predictions by PSICOV, CCMpred, and FreeContact have precision of 13%, 4.4%, and 17.4% respectively, the final prediction made by MULTICOM-CONSTRUCT, however, boosts the precision to 82.6%. As shown Figure 1, the contacts predicted by MULTICOM-CONSTRUCT (Figure 1(A)) are much more near-native compared to the individual coevolution-based predictions.
Figure 1.
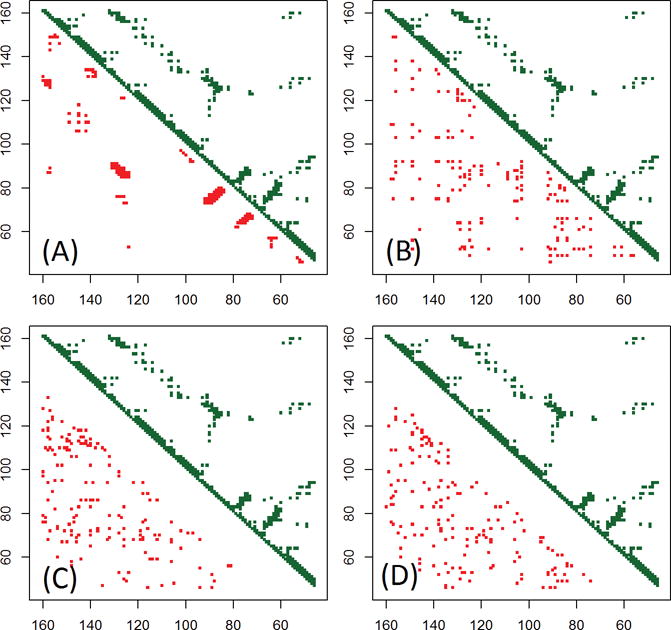
Contact map visualization of top L contacts predicted by MULTICOM-CONSTRUCT (A), PSICOV (B), FreeContact (C), and CCMpred (D) for the target domain T0868-D1. Green dots in upper triangles represent contacts in the native structure and red dots in lower triangles denote the contact predictions.
3.4 Relationship between number of effective sequences and precision of contact prediction
Study of the relationship between the number of effective sequences (Meff) in the alignment and the precision of the predicted contacts can provide useful insights on estimating the accuracy of the predicted contacts. A direct comparison between Meff and precision is less meaningful if Meff is calculated for the whole target sequence and the contact precision are evaluated at the domain level. Hence, we also calculated Meff using our Meff calculation method at the domain level. Figure 2 plots the precisions of top L/5 contact predictions of the domains in CASP12 dataset against the logarithm of their number of sequences (N) in the alignments generated for the whole targets and the logarithm of the number of effective sequences (Meff) at the domain level, respectively. The Pearson’s correlation between the precision and log(N) is 0.47, lower than 0.66 between the precision and log(Meff) at the domain level. According to the plot between contact prediction precision and Meff in Figure 2, it can be inferred that multiple sequence alignments with at least around 100 effective sequences at domain level has a good chance to produce 50% precise contact predictions, whereas, when the Meff is more than 1000, the precision has a high chance to reach above 70 to 80%, for L/5 long-range contacts.
Figure 2.
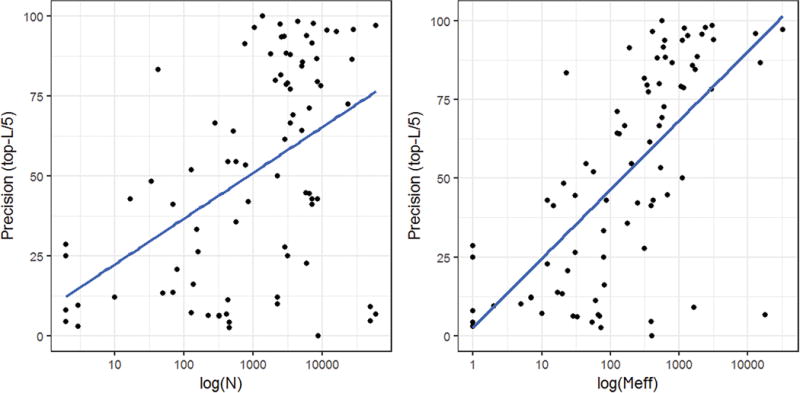
The precision of top L/5 long-range contacts predicted by MULITCOM-CONSTRUCT is plotted against the logarithm of number of sequences (N) in the alignments generated for the whole targets (left) and the logarithm of number of effective sequences (Meff) calculated for the domains (right) on the CASP12 dataset. The Pearson’s correlation coefficients of the precision with log(N) and log(Meff) are 0.47 and 0.66, respectively.
However, there are some exceptional cases where the contact prediction precision is very low even though the number of sequences in the multiple sequence alignment is high. MULTICOM-CONSTRUCT’s contact precision for top L/5 long-range contacts is only 4.6% for the domain T0898-D1, whose number of sequences in the alignment is very high (~50K) and the number of effective sequences is 389. When checking the quality of coevolution-based features of this domain, we observed that all individual coevolution-based features also had low-quality contact predictions. However, in general, 389 effective sequences should be sufficient to produce coevolution-based features and final contact predictions of decent quality. After checking the sequence alignment of this domain, we find that most of the sequences have gaps for the first domain (i.e. T0898-D1) and cover only the second domain of the target, such that the Meff for the second domain is much higher, 1648. Moreover, although the multiple sequence alignment has many sequences, most sequences are extremely short, having only around 30 valid residues (non-gaps), and are not useful for predicting long-range contacts with sequence separation >= 24. To verify our observation through Meff calculations, we modified our program to calculate Meff so that aligned gaps were also considered as a match (gap was considered as 21st amino acid) and calculated new Meff. For this domain, such a gap-considered Meff is just 2, suggesting that the poor coverage is the cause of the poor contact prediction. Another exceptional case is MULTICOM-CONSTRUCT’s precision for top L/5 long-range contacts is only 7% for the domain T0893-D1, although the multiple sequence alignment generated with 75% coverage threshold has 63,308 sequences with Meff of 17,939. For this domain, all standard coevolution-based features also have poor predictions. We suspect one reason for the low contact precision is the unusual shape of the domain as its tertiary structure consists of just two long helices side by side, whereas the other domain (T0893-D2) of regular shape in the same target has a much higher Meff resulting in long-range contact predictions of 97% precision. These exceptions suggest that, sometimes, coevolution-based contact prediction methods can fail to produce accurate contacts even in the presence of a large number of sequences in the alignments, possibly because many of the sequences in the alignment are false positive homologous sequences or do not align well with target domains. Therefore, in addition to alignment depth as measured by number of (effective) sequences, alignment quality needs to be considered for assessing the accuracy of co-evolution-based contact prediction.
3.5 Impact of alignment parameters on the quality and depth of multiple sequence alignments
Our alignment generation algorithm gradually switches to pick lower quality multiple sequence alignments when high-coverage and highly homologous sequences cannot be found. For deciding when to use a lower quality alignment, we set a threshold of minimum 2.5L sequences in the alignment. We run HHblits with three pre-specified coverage options and JackHMMER with six different e-value thresholds. For example, when HHblits search with 75% coverage option produces an alignment having less than 2.5L sequences, we check the output of the search with 68% coverage, and so on. To analyze if these parameters were well tuned, we studied two subsets - (a) all the targets where we used the results of HHblits search with 75% coverage, and (b) all the targets where we used JackHMMER with e-value threshold of 1E−40. For these two sets of targets, to study how the various parameters influence the quality of the multiple sequence alignment (and ultimately the quality of contact prediction), we generated multiple sequence alignment with all kinds of parameter settings. In other words, for the first subset where we had chosen HHblits alignments with 75% coverage in CASP12 experiment, we regenerated the alignments with all three coverage options (60%, 68%, and 75%) and predicted contacts using the coevolution-based method CCMpred, respectively. For this set, surprisingly, the precision of contacts predicted using the alignments generated with coverage parameter of 60% is slightly higher, on average, than the ones predicted using the coverage parameter of 75%. The average precisions of top L/2 long-range contacts for the three coverage thresholds (60%, 68%, and 75%) are 61.8%, 60.7%, and 58.1% respectively (see Table S3). This is true for both multi-domain and single-domain targets in the dataset, suggesting that only one HHblits search with coverage option of 60% is generally sufficient to generate good results. Similarly, for the second set of targets where we had used JackHMMER with e-value threshold of 1E−40, we regenerated the alignments with all six e-value thresholds (1, 1E−4, 1E−10, 1E−20, 1E−30, and 1E−40) and predicted contacts using the coevolution-based method CCMpred. On this dataset, the best precision is obtained when alignments are selected with less stringent criteria of 1E−10 or 1E−20 e-value threshold. While the mean precision for these domains is 61.8% and 61.7% at e-value threshold of 1E−30 and 1E−40, the precision increases to 63.5% at the threshold of 1E−10 and 1E−20 (see Table S4). These results suggest that JackHMMER searches with e-value threshold of 1E−30 and 1E−40 need not to be run. In addition to these analyses on the contact predictions of CCMpred, we also predicted contacts using FreeContact method and observed similar results confirming our conclusion.
3.6 Impact of the convergence of coevolution methods on contact prediction
During our experiment, the coevolution-based tool PSICOV sometime could not converge within several hours, either because there were too few sequences or too many sequences in the alignment or because the input sequence was long. Hence, we ran three PSICOV jobs with different parameters in parallel and picked the one that finished within the waiting time limit, based on a preferred order. The preferred order for selecting PSICOV predictions was ‘d = 0.03’ followed by ‘r = 0.001’ and ‘r = 0.01’. To verify if this preference order was effective, from the dataset of all the targets for which native structures were available for us, we selected the targets for which a multiple sequence alignment with at least 5 sequences could be generated and for which all three PSICOV jobs converged without any time limit constraint, resulting in a dataset of 60 domains. On this dataset, the mean precision of top L/5 long-range contacts for the options ‘d=0.03’, ‘r=0.001’, and ‘r=0.01’ are 35.4%, 33.3%, and 18.1%, respectively (see Table S5). The relatively higher precision of the option ‘d=0.03’ and much lower precision of the option ‘r=0.01’ validates that our preference order is fine.
Further, to check how much accuracy was lost due to the five-hour time limit, from the above set of 60 domains, we selected the domains for which we could not select the first PSICOV job (with d=0.03 option) because of the time limit and had instead selected the second PSICOV job (with r = 0.001 option). This resulted in a set of 10 domains for which the mean precision of top L/5 and L/2 long-range contacts were 57.5% and 41% when the contacts were predicted with the ‘r=0.001’ option. However, had we waited for long enough to let the first set of jobs finish for these targets, the mean precision would have increased to 64.9% and 46.9% for top L/5 and L/2 contacts respectively.
Overall, the experiments show that generating reliable multiple sequence alignments is not a straightforward process. The definition of ‘a useful alignment’ also depends upon the coevolution-based method used to predict contacts from the alignment. While some of these methods are resource expensive and take longer to run, other methods are relatively fast and are almost independent of the alignment size and length of the protein sequence. Hardware resources and the waiting time limit available for co-evolution feature generation can influence the decision to generate and pick the best alignments. In general, coevolution-based methods take longer to run if the size of alignment (number of sequences in alignment) is big. In some case, CCMpred can run on CPUs for more than a day and PSICOV can run for days. If the hardware resources are limited, it is appropriate to attempt to obtain a reasonable, but less extensive alignment before running these tools. For instance, if HHBlits coverage option of 75% produces 90K sequences, it may be appropriate to increase the coverage threshold to a higher value like 80% to obtain an alignment of smaller size for which the coevolution-based methods can make predictions within a time limit.
3.7 Three-dimensional model reconstruction using the predicted contacts
The primary objective of predicting contacts is to use them for three-dimensional structure prediction. In this context, with the contacts predicted by MULTICOM-CONSTRUCT, we built three-dimensional models using our fragment-free ab initio folding tool CONFOLD 1.0 [14] to study the usefulness of the predicted contacts. CONFOLD is guided by predicted contacts and secondary structures only, and hence is a good method to build models to study the independent value of the predicted contacts. Using CONFOLD, we built five models for each target in the CASP12 dataset with five sets of contacts - top 0.8L, 1.0L, 2.0L, 3.0L, and 4.0L contacts, without removing short-range or medium-range contacts. To be consistent with other similar works, we built models for the whole target sequence first, without using any knowledge of domains, and then evaluated the predicted models against structural domains. Furthermore, since the number of contacts selected to build models greatly influences the quality of the reconstructed models, we selected ‘best of five’ models for our analysis. Our reconstruction results (summarized in Table S6), shows that in general, predicted contacts and secondary structures alone could recover the folds of 15 out of the 87 domains, i.e. with TM-score [23] greater than 0.5. We investigated structural domains for which the accuracy of the models was low, and found that many of them are from multi-domain proteins, which are hard for all ab initio methods to fold as whole. This suggests that dividing multi-domain proteins into individual domains before folding them with predicted contacts is desirable. For each of the structural domains, we also studied the relationship between the best reconstructed models and the quality of the contact sets selected for the reconstruction. The Pearson’s correlation coefficient between the TM-score of the reconstructed models and precision of long-range, medium-range, and short-range contacts are 0.60, 0.42, and 0.34, respectively, indicating long-range contacts are most useful for tertiary structure modeling. We also find that the proportion of the number of long-range, medium-range, and short-range contacts in the native structures is more similar to the proportion of the contacts that were used to build the best models, suggesting that contact-selection i.e. the number of short-range, medium-range, and long-range contacts to select for building models, is important for accurate reconstruction.
As an example, we discuss the reconstruction of a free-modeling domain T0900-D1. T0900-D1 consisting of 102 residues is a complicated beta-sheet domain having 194 long-range, 31 medium-range, and 27 short-range contacts. Of the five sets of contacts selected for reconstruction (0.8L, 1L, 2L, 3L, and 4L), the second set of top 1L contacts generated best models for this domain. This top 1L set of 60 long-range, 30 medium-range, and 13 short-range contacts generated the top model with 0.43 TM-score, almost recovering the fold of the protein. Despite predicted contacts being very precise (i.e. top L/5 precision of 95% and top L precision of 60%) for this domain, the less accurate reconstruction can be attributed to the poor distribution of predicted contacts used to build the models (see Figure3 (A) and (B)). The correctly predicted contacts only cover a portion of the structure of this domain. In a different experiment, we reconstructed this domain using all true contacts and obtained a model with 0.9 TM-score and 1.4 Å RMSD, which is near native. These examples suggest that the gap between the reconstruction accuracy of using true contacts and that of using only predicted contacts alone (i.e. without using other information like structural templates or fragments), is still wide and the contact-based protein folding requires more research.
Figure 3.
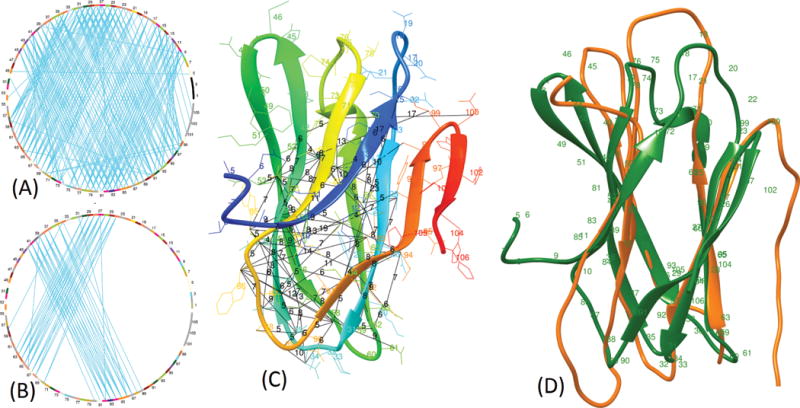
Visualization of the top L contacts predicted using MULTICOM-CONSTRUCT and reconstructed model for the domain T0900-D1. Chord diagram for the long-range contacts in the native structure are shown in (A) and the top L contacts predicted by MULTICOM-CONSTRUCT shown in (B). MULTICOM-CONSTRUCT predicted contacts are highlighted in the native structure with actual distances between the residues shown in black (C) and the reconstructed structure (in orange) superimposed with the native structure (in green) is shown in (D).
Supplementary Material
Acknowledgments
The work was partially supported by a NIH grant (R01GM093123) to JC. We thank CASP12 organizers and assessor to make contact prediction data available. Particularly, we would like to devote this work to Dr. Anna Tramontano who co-organized CASP12 before she passed away and had made great contributions to the CASP community for many years.
Footnotes
4. Conflict of Interest
The authors declare there is no conflict of interest.
References
- 1.Zhang W, Yang J, He B, et al. Integration of QUARK and I-TASSER for Ab Initio Protein Structure Prediction in CASP11. Proteins Struct Funct Bioinforma. 2016;84:76–86. doi: 10.1002/prot.24930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ovchinnikov S, Kim DE, Wang RY-RR, et al. Improved de novo structure prediction in CASP11 by incorporating coevolution information into Rosetta. 2016;84:67–75. doi: 10.1002/prot.24974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duarte JM, Sathyapriya R, Stehr H, et al. Optimal contact definition for reconstruction of contact maps. BMC Bioinformatics. 2010;11:283. doi: 10.1186/1471-2105-11-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vassura M, Margara L, Di lena P, et al. FT-COMAR: Fault tolerant three-dimensional structure reconstruction from protein contact maps. Bioinformatics. 2008;24:1313–1315. doi: 10.1093/bioinformatics/btn115. [DOI] [PubMed] [Google Scholar]
- 5.Eickholt J, Cheng J. Predicting protein residue-residue contacts using deep networks and boosting. Bioinformatics. 2012;28:3066–72. doi: 10.1093/bioinformatics/bts598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng J, Baldi P. Improved residue contact prediction using support vector machines and a large feature set. BMC Bioinformatics. 2007;8:113. doi: 10.1186/1471-2105-8-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di lena P, Nagata K, Baldi P. Deep architectures for protein contact map prediction. Bioinformatics. 2012;28:2449–2457. doi: 10.1093/bioinformatics/bts475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones DT, Singh T, Kosciolek T, Tetchner S. MetaPSICOV: Combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics. 2014;31:btu791. doi: 10.1093/bioinformatics/btu791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skwark MJ, Raimondi D, Michel M, Elofsson A. Improved Contact Predictions Using the Recognition of Protein Like Contact Patterns. PLoS Comput Biol. 2014 doi: 10.1371/journal.pcbi.1003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang S, Sun S, Li Z, et al. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. PLoS Comput Biol. 2017 doi: 10.1371/journal.pcbi.1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seemayer S, Gruber M, Söding J. CCMpred - Fast and precise prediction of protein residue-residue contacts from correlated mutations. Bioinformatics. 2014;30:3128–3130. doi: 10.1093/bioinformatics/btu500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones DT, Buchan DWA, Cozzetto D, Pontil M. PSICOV: Precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics. 2012;28:184–90. doi: 10.1093/bioinformatics/btr638. [DOI] [PubMed] [Google Scholar]
- 13.Kaján L, Hopf TA, Kalaš M, et al. FreeContact: fast and free software for protein contact prediction from residue co-evolution. BMC Bioinformatics. 2014;15:85. doi: 10.1186/1471-2105-15-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adhikari B, Bhattacharya D, Cao R, Cheng J. CONFOLD: Residue-residue contact-guided ab initio protein folding. Proteins. 2015;83:1436–49. doi: 10.1002/prot.24829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Remmert M, Biegert A, Hauser A, Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2011;9:173–5. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 16.Johnson LS, Eddy SR, Portugaly E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics. 2010;11:431. doi: 10.1186/1471-2105-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eickholt J, Cheng J. A study and benchmark of DNcon: a method for protein residue-residue contact prediction using deep networks. BMC Bioinformatics. 2013;14(Suppl 1):S12. doi: 10.1186/1471-2105-14-S14-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freund Y, Schapire RE. A Decision-Theoretic Generalization of On-Line Learning and an Application to Boosting. J Comput Syst Sci. 1997;55:119–139. doi: 10.1006/jcss.1997.1504. [DOI] [Google Scholar]
- 19.Adhikari B, Nowotny J, Bhattacharya D, et al. ConEVA: a toolbox for comprehensive assessment of protein contacts. BMC Bioinformatics. 2016;17:517. doi: 10.1186/s12859-016-1404-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Xu J. Predicting protein contact map using evolutionary and physical constraints by integer programming. Bioinformatics. 2013;29:i266–73. doi: 10.1093/bioinformatics/btt211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinch LN, Li W, Monastyrskyy B, et al. Evaluation of free modeling targets in CASP11 and ROLL. Proteins Struct Funct Bioinforma. 2016 doi: 10.1002/prot.24973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kosciolek T, Jones DT. Accurate contact predictions using covariation techniques and machine learning. Proteins Struct Funct Bioinforma. 2015 doi: 10.1002/prot.24863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.