

Abstract
The paper presents analysis of our template-based and free docking predictions in the joint CASP12/CAPRI37 round. A new scoring function for template-based docking was developed, benchmarked on the Dockground resource, and applied to the targets. The results showed that the function successfully discriminates the incorrect docking predictions. In correctly predicted targets, the scoring function was complemented by other considerations, such as consistency of the oligomeric states among templates, similarity of the biological functions, biological interface relevance, etc. The scoring function still does not distinguish well biological from crystal packing interfaces, and needs further development for the docking of bundles of α-helices. In the case of the trimeric targets, sequence-based methods did not find common templates, despite similarity of the structures, suggesting complementary use of structure- and sequence-based alignments in comparative docking. The results showed that if a good docking template is found, an accurate model of the interface can be built even from largely inaccurate models of individual subunits. Free docking however is very sensitive to the quality of the individual models. However, our newly developed contact potential detected approximate locations of the binding sites.
Keywords: protein recognition, protein-protein interactions, modeling of protein complexes, structure prediction
INTRODUCTION
Protein docking methodologies can be roughly divided into free docking, where sampling of the binding modes is performed with no regard to the experimentally determined complexes of similar proteins, and template-based or comparative docking, where such complexes determine the docking predictions. Docking usually consist of the global scan/search for an approximate structure of the complex, with computationally inexpensive objective function, followed by scoring/refinement of the putative matches, using more accurate functions.1,2 The scoring function can be derived from the physical principles, or based on known protein structures (knowledge-based or statistical potentials3–6). Statistical potentials have been successfully applied to many problems, such as discrimination of the native structure from decoys,7,8 fold recognition,9 structure prediction,10 protein docking,11–13 protein design,14,15 and prediction of protein stability and affinity.16–18 Recently, we proposed a semi-analytical approach to the development of contact potentials for protein structure prediction based on the Potts model.19
To detect a protein-protein template, a search against a diverse library of protein-protein complexes should be performed according to some measure of sequence/structure similarity of the target and the template. A popular TM-score,20 originally developed for assessing the quality of individual protein structures, has been also successfully used in the template-based docking.21 However, the performance of the TM-score-based scoring deteriorates significantly when target and templates have only moderate structural similarity (TM-score ~ 0.4 – 0.6).22 The TM-score is based on geometry only. Thus, it can be supplemented by terms accounting for other properties of the interacting proteins. In this paper, we introduce a combined function for the scoring of template-based docking predictions, which was systematically benchmarked on the datasets of protein-protein complexes from our Dockground resource (http://dockground.compbio.ku.edu).23 We present a detailed analysis of our successes and failures in modeling of 10 targets offered to the CAPRI participants in CASP12 (CAPRI round 37) with the emphasis on the use of our new combined scoring function and our recently developed contact potential.
METHODS
Datasets
For the training and testing of the scoring function, we used a subset of 572 simulated unbound hetero complexes24 and the template library25 of 4,950 co-crystallized binary complexes from Dockground.
Docking protocols
The template search was performed by HHsuite version 2.0.16. First, the sequence profile (MSA) of the target was built by running three iterations of HHblits26 against UniProt20, a database of non-redundant sequence profiles covering the entire sequence space. The e-value cutoff was set at 10−3 and the default values were used for other alignment-tuning parameters. The resulting HMM profile was run against a database of HMMs for all PDB sequences (PDB70) by HHsearch27 in the local alignment mode using Viterbi algorithm. The oligomeric states of the detected templates were identified using REMARK 350 of the PDB files (author determined biological unit). In case of multiple biological units, the first one was used.
In benchmarking of the scoring function, the template-based docking was performed by full28 and partial structure alignment29 protocols (FSA and PSA, correspondingly). For the CAPRI targets, only FSA docking was utilized as HHSearch detects templates using full protein sequences. Target proteins were structurally aligned to the template monomers by TM-align to generate putative matches, ranked by the smaller of the two TM-scores. The resulting models were rescored by the combined scoring function presented in this paper and by the atom-atom contact potential AACE18.19
The free docking was performed by our FFT (Fast Fourier Transform) program GRAMM30,31 at low resolution, with 3.5 Å grid step and 10° angular interval. Top 100,000 matches with the best shape complementarity were retained for further analysis. The resulting matches were rescored by the AACE18 potential.
Scoring terms
For the scoring we used four template-independent measures: (i) buried interface surface area per chain ΔSASA, (ii) penalty for clashing interface atoms Scl, (iii) desolvation energy Edes, and (iv) interface energy calculated by the semi-empirical atom-atom contact potential AACE18 EAACE18.19 Scl quantifies the number of clashes as the intersection of van der Waals spheres (radii taken from Tsai et al.32) of the two interacting proteins, normalized by ΔSASA (what is average penetration in Ref.33). Edes is the sum of buried atomic interface surface areas weighted by the atomic solvation parameters.34 Rapid calculations of solvent-accessible surfaces were performed by the approximate Le Grand and Merz algorithm35 and the use of k-d trees36 for quick retrieval of spatially adjacent atom pairs; the latter was also used to speed-up detection of interface atom pairs required by AACE18. The scoring of the template-based docking was complemented by: (i) the average TM-score of two target/template alignments STM, (ii) the average sequence identity of the interface residues in these two alignments SseqID, and (iii) fraction of the interface contacts shared by the template and the docking model SFSC.37
The quantities included in the combined scoring function have significantly different ranges. The STM, SseqID and SFSC terms have values from 0 to 1, and were incorporated into the function as such. The average penetration of the docking models are usually within 0 – 0.4Å range, rarely exceeding 1Å.33 Thus, Scl was also included without normalization. On the other hand, ΔSASA, Edes and EAACE18 span several orders of magnitude, without strict bounds (except the lower 0Å2 limit for ΔSASA). Thus, they were normalized as
| (1) |
where γCDF(z; α, β) is the cumulative distribution function of the gamma distribution with shape parameter α and rate β at point z = X0 + sgn × X, , X is the raw value of the parameter, and X0 and sgn are the parameters transforming the raw values into a positive number, since the gamma distribution is defined for the positive arguments only. The gamma distributions were fitted to the raw values of the parameters for all near-native (acceptable or better according to the CAPRI criteria38) FSA/PSA models (Supporting Information Fig. S1). The values of the fitting parameters are in Table S1.
Combined scoring function
The combined scoring function was constructed using linear logistic regression model
| (2) |
where
| (3) |
is the linear combination of the scoring terms Si. The training procedure aimed at finding the optimal weights ωi such that S is assigned the largest negative value (i.e., F(S) → 0) for the incorrect predictions, and the largest positive value (F(S) → 1) for the near-native models. The procedure was carried out using the MASS package39 for the R statistical programing language (glm function) separately for 2,957,706 FSA and 4,341,220 PSA docking models resulting in two sets of the optimal coefficients (Table 1).
Table 1.
Weights ωi in the combined scoring function (Eq. 3). Values in parentheses correspond to the simplified function S = ω0 + ωTMSTM.
| term | FSA | PSA | ||
|---|---|---|---|---|
|
| ||||
| ωi | |z-value| a | ωi | |z-value| a | |
| ω0 | −8.84 (−10.31) | 177.4 | −7.17 (−9.41) | 181.0 |
| Scl | −6.36 b | 80.1 | −7.09 | 124.0 |
| SAACE18 | 3.57 | 55.5 | 3.58 | 72.8 |
| SFSC | 5.44 | 37.0 | 4.93 | 45.7 |
| STM | 5.11 (12.16) | 36.1 | 4.49 (13.72) | 37.3 |
| SΔSASA l | 2.87 | 28.1 | 2.31 | 30.8 |
| Sdesol | 1.69 | 16.5 | 1.28 | 17.8 |
| SseqID | 1.11 | 5.9 | 3.34 | 20.1 |
Z-values of logistic regression coefficient ωi reflect the uncertainty of the estimate (larger values correspond to the smaller errors).
Negative Scl weights are penalty for the clashes.
RESULTS AND DISCUSSIONS
Detection of the near-native matches by the combined scoring function in template-based docking
To compare the performance of the combined scoring function and the original TM-score based algorithm, we analyzed correlation of Fcomb and FTM values from Eq. (2) for the FSA and PSA models with the full (Eq. 3) and the TM-only function S = ω0 + ωTMSTM, correspondingly (the optimal coefficients for latter are in parenthesis in Table 1). At higher FTM scores, FSA docking predictions tend to be near-native with interface RMSDs < 4Å (Fig. 1A) which correlates with the results of the previous studies37,40 that used different datasets. Similar trend was observed for the FSA docking, characterized by the Fcomb score (Fig. 1B). 283 targets, or 49.5% of the dataset, have a near-native model at the top of the prediction lists (top 1 success rate), ranked by the FTM. Ranking by the Fcomb, brings a near-native model to the top of the prediction pool for additional 43 targets and purges the near-native model from the top position for 4 targets only (increase in the top 1 success rate by 6.8%, Fig. 2A). For the PSA docking, top 1 success rate is 40.6 % (233 targets) and 56.5 % (323 targets) when predictions are ranked by the FTM and Fcomb, correspondingly (Fig. 2B).
Figure 1.

Comparison of the combined and TM-only scoring functions. Interface RMSD of 2,957,706 FSA docking predictions for 572 test complexes are plotted against FTM(S) with S = ω0 + ωTMSTM (A) and Fcomb(S) with S from Eq. (3) (B). Data for 4,341,220 PSA models are qualitatively similar and thus not shown. Lower panels display correlations between FTM and Fcomb, separately, for near-native and incorrect FSA (C, D) and PSA (E, F) predictions, correspondingly. Data in panels C and E are plotted for 7,056 FSA and 11,400 PSA near native models, respectively.
Figure 2.
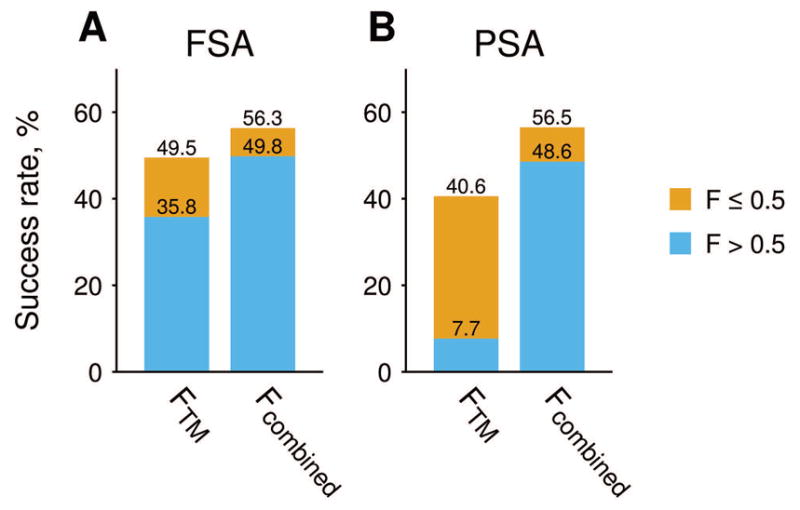
Top 1 FSA (A) and PSA (B) docking success rates. FTM and Fcombined are, respectively, logistic regression models based on TM-score alone and on the combined scoring function from Eq. 2 (see caption to Figure 1 for details). Contributions from the near-native models with high-reliability score (> 0.5) is in blue, and fraction of the models with the low-reliability score (< 0.5) is in yellow.
In a linear regression model, the F(S) = 0.5 value should separate incorrect docking models from the near-native ones. Indeed, most of the incorrect FSA and PSA models for the targets in our dataset have both FTM and Fcomb < 0.5 (Fig 1D, F). On the other hand, a significant part of the near-native FSA and PSA models have the value of the scoring function > 0.5 threshold (Fig 1C, E). Ranking of the models by Fcomb increases this fraction further (upper left quadrants in Fig. 1C, E). This trend is even more pronounced for top 1 models (Fig. 2), where ~28% FSA and 81% PSA of top 1 near-native models have FTM < 0.5, while only 12% FSA and 17% PSA of the near native models have Fcomb < 0.5. Relatively small number of PSA models with FTM < 0.5 is due to generally smaller values of TM-scores for the interface alignments.25 The detailed comparison of the FSA and PSA protocols is provided elsewhere.41 Here we want to point out that the combined scoring function not only increases the overall docking success rate for both FSA and PSA protocols, but also improves discrimination of the incorrect docking predictions. However, a significant part of the near-native models in the prediction pool still have both FTM and Fcomb < 0.5 (low-reliability scores, lower left quadrants in Fig. 1C, E), which points to the need of further development of the scoring function.
Successful targets
For all successful targets, HHpred identified highly reliable templates (HHpred probability ~ 100%) either for both proteins in the target (hetero-n-mers), or templates in oligomeric state that is consistent among the pool of identified templates and matches the oligomeric state of the target (homo-n-mers). In this group, there were one homo-dimer (T119 in CAPRI nomenclature and T0917 in CASP), three homo-trimers (T110/T0860, T111/T0867, and T112/T0881), one homo-octamer (T118/T0881) and one hetero-dimer (T120/T0921-T0922).
Homo-dimer T119/T0917
The CAPRI task for this target was to dock two identical models of 407-residue protein (including 6-His tag at the N-terminal). The monomer was an easy template-based CASP target - most of the server models (155 out of 188) had TM-score > 0.7 to the native structure and the native/model Cα RMSD 1.7 – 3Å. HHpred search yielded 29 templates with probability score 100%, and sequence identities 13 – 33%. Most structures (25 out of 30) came from different bacteria, 3 from eukaryotes (2 from fungi and 1 from kiwi) and only one template (4rfl) originated from the same kingdom of life as the target (archaea). Custom template library for the FSA docking was built from 11 templates, which had the highest TM-score to the CASP server models and form unique interfaces in both asymmetric and biological PDB units. In the case of oligomeric state > 2, all chain combinations in physical contact were considered. The final template library of 17 homodimers was used in the FSA docking of all 187 available CASP server models. The resulting docking models were scored by the combined scoring function and 10 models were selected from the top of the list with maximum diversity of CASP protein models and docking templates (Table S2). Two of these models were of medium and five of acceptable quality, according to the CAPRI criteria. The best model in terms of fraction of predicted native contacts (fnat = 0.6577, acceptable in overall CAPRI evaluation) was built using protein model 2 of the YASARA server42 and chains 1 and 7 from the octameric biounit of 1jq5 structure as the docking template. The best model in terms of the backbone interface RMSD (i-RMSD = 1.98 Å, medium quality) utilized model 5 of the MULTICOM-CONSTRUCT server43 as the monomer structure, and chains A and B from the asymmetric unit of 3iv7 as the docking template. Both template structures are oxidoreductases from Corynebacterium glutamicum bacteria with high structural (TM-score 0.84), but low sequence similarity (18% sequence identity per TM-align), used to generate 4 other near-native models. In addition, one near-native model was generated based on chains A and B from asymmetric unit of 1vlj (oxidoreductase from Thermotoga maritima bacteria). Interestingly, the template from the same kingdom of life (4rfl, sequence ID 23%) yielded incorrect model due to a slightly different protein orientation within the same binding site.
Homo-trimer T110/T0860 (PDB 5fjl)
The CAPRI task was to build a trimeric complex from models of identical subunits of C-terminal domain (residues 327 - 463) of fiber protein (UNIPROT ID F4MI11) from raptor siadenovirus 1. HHpred found only one highly reliable (probability 100% and e-value 1.3×10−49) template (fiber head domain from of the turkey siadenovirus 3) with the sequence identity 20%. This protein had 5 PDB structures (3zpe, 3zpf, 4cw8, 4d62 and 4d63), all monomeric in the asymmetric unit, but in trimeric oligomeric state in the biological unit. The custom template library was, however, assembled only from two PDB entries (3zpe and 4cw8) containing no modified residues (as 4zpf) and only small ligands (PO4) away from the interfaces. 4d62 and 4d63 contain significantly larger ligands at the PO4 site, which we deemed as a possible cause for the interface distortion, and thus excluded these structures from the modeling. Five protein models, selected among 150 CASP stage 2 server models with the lowest values of AACE18 contact potential, were docked by FSA using these two templates. This yielded 7 medium and 3 acceptable quality models (Table S3). All models had consistently high F(S) values, but significantly different AACE18 energies.
Homo-trimer T111/T0867
The CAPRI participants were asked to assemble a trimer from models of C-terminal (residues 228 to 331) of fiber 1 protein from Lizard adenovirus 2 (UNIPROT ID A0A076FUN0). Again, HHpred found only one highly reliable (probability 100% and e-value 1.4×10−70) template (fiber protein from snake adenovirus 1) with the sequence identity 48%. This protein had 5 PDB structures (4d0u, 4d1g, 4d1f, 4d0v and 4umi). All of them are 3-meric in the biological units, however with different oligomeric states in the asymmetric units (monomer in 4umi, 4-mers in 4d0v and 4d0u and 12-mers in 4d1f and 4d1g). Compared to other templates, 4umi has 6 extra residues in the N-terminal, which could affect trimeric interface architecture and thus was excluded from consideration. Custom template library was built from unique pairwise combinations of chains in the asymmetric units of 4d0u and 4d1f. The main model for docking was selected among 20 CASP stage 1 server models with the lowest AACE18 energy (only one model had distinguishably lower AACE18 value). Additional 4 models with lowest AACE18 energies, originating from different groups, were selected among 150 CASP stage 2 models. FSA docking resulted in 4 high- and 3 medium-quality models (Table S4), whereas the remaining three incorrect models were built based on crystallographic interfaces of the templates. If the crystallographic interface was comparable in size with the biological one (models 3 and 5 in Table S4 and Fig. 3), such interfaces were indistinguishable by our procedures. However, the AACE18 value clearly stood out for the smaller crystallographic interface (model 6 in Table S4, Fig. 3).
Figure 3.

Correct (A) and incorrect (B and C) trimeric assemblies for T111/T0867. Structures are models 1, 3 and 6 in the CAPRI submission. Other characteristics of the models are in Table S4.
Homo-trimer T112/T0881
For this target, the trimeric assembly was to be built from the models of C-terminal domain (residues 258 to 461) of goose adenovirus 4 (UNIPROT ID I3PMP2). HHpred detected two distinct high-reliability templates, fiber heads from fowl adenovirus fiber protein 2 (sequence identity 20%, probability 100%, and e-value 1.8×10−59, to 2vtw) and fiber protein 1 (sequence identity 14%, probability 100%, and e-value 6.4×10−56, to 2ium and 2iun). These two templates have very similar structures (TM-score 0.82), low sequence identity 15%, and consistent oligomeric state in both asymmetric and biological units. 2uin contains Ca2+ ions close to the interface, and thus custom template library was constructed from the pairwise combinations of trimeric units of 2vtw and 2ium. For the FSA docking, we selected 10 models from different servers with the lowest AACE18 values. However, model 8 (model 1 from GOAL server44) produced FSA docking models with F(S)< 0.5 only, and thus these results were not included in the final submission. All but two submitted trimers were of acceptable quality (Table S5). The trimeric models 7 and 10 were classified as incorrect, due to i-RMSD and L-RMSD slightly exceeding the CAPRI threshold, caused by poorer loop modeling in the models of the individual protein.
Homo-octamer T118/T0906
The task was to assemble an octamer consisting of models of 363 residue fructose-1,6-bisphosphate aldolase/phosphatase from Thermus thermophilus bacteria (UNIPROT ID Q5SJM8/H9ZRK4). HHpred found two closely homologous proteins, both fructose-1,6-bisphosphate aldolases/phosphatases, originating from different bacteria, Sulfolobus tokodaii (probability 100%, e-value 2.7×10−196, sequence identity 46%, to 3r1m and 1umg) and Pyrobaculum neutrophilum (probability 100%, e-value 7.2×10−195, sequence identity 46%, to 3t2c, 3t2b, 3t2e,3t2f and 3t2g). Both proteins are monomeric in the asymmetric unit and octameric in the biological unit. The PDB structures of the first protein cover smaller portion of the target sequence. Thus, pairwise chain combinations from the 3t2c biounit of the second protein (selected because it has ligands most relevant to the protein function) were used as templates. Model of the targets (from different servers) were selected from all 182 CASP server models by the lowest AACE18 energies, and the highest TM-score to 3t2c. All FSA docking predictions were in the high and the medium quality category for all three distinct interfaces (Table S6 shows characteristics of one such interface). Again, F(S)values were consistently ~1, but the absolute AACE18 interface energies had a wide range.
Hetero-dimer T120/T0921-T0922 (PDB 5aoz for T0921)
This was a classical CAPRI target, where participants were asked to dock models of two different proteins: type 1 cohesin domain (residues 399 – 547) of the cellulosomal scaffolding 2071 residue protein (UNIPROT ID A0AEF4) and dockerin-I domain (residues 569 – 649) of hydrolase GDSL 1056 residue protein (UNIPROT ID UPI0001C3768F) from Ruminococcus flavefaciens bacteria. HHpred identified 8 heterodimeric templates from different bacteria (2ccl, chains A and B; 2vn6AB; 2y3nAB; 3ul4AB; 4dh2AB; 4iu3AB; 4uypAD and 4wkzBA). All these templates perform function similar to the target complex, share ~20% sequence identity to the target subunits, and are similar in structure (TM-score ~ 0.8) and sequence (~30% sequence identity). Because of the small size of the template library, we were able to perform FSA cross-docking for all 187 CASP server models for the first and second proteins. Resulting docking models were scored by F(S) and AACE18. The models with the lowest AACE18 values (the F(S) values were consistently 0.6 – 0.7) were selected for the final submission (Table S7), except model 1, which had an unfavorable AACE18 interface energy, but the highest target/template TM-scores. This model was subjected (although unsuccessfully) to manual adjustment of the ligand (smaller protein) position (model 2 in Table S7). This procedure resulted in 5 acceptable and 3 medium-quality models. Model 9 was incorrect due to displacement of the interface helices in the CASP model of the ligand.
Unsuccessful targets
Homo-dimer T114/T0875
The CAPRI task was to build a homo-dimeric assembly from the models of LV4 2A2 protein, which is a novel domain (residues 823 – 944) of a very large 2254 residue genome polyprotein from Ljungan virus. For the target sequence, HHpred found 4 templates with probability > 99%, sequence identities ~ 20%, and rather moderate e-values ~10−30: (1) human retinoic acid receptor responder protein 3 (2lkt, chain A); (2) human HRAS-like suppressor 3 (4q95, chain A); (3) human group XVI phospholipase A2 (4dot, chain A); and (4) human HRAS-like suppressor 2 (4dpz, chain A). Only 4q95 was hetero-dimeric in the asymmetric unit and tetrameric in the biological unit. All others were monomeric in both asymmetric and biological units. Thus, custom template library was built from the pairwise combinations of chains from the biounit and from PISA-generated crystallographic interfaces of monomeric 4dot and 4dpz (details of the procedure are published elsewhere45). For docking, we selected 11 CASP stage 2 models with the lowest AACE18 energies and the highest TM-scores to the HHpred templates. FSA docking based on the custom template library yielded, however, models of questionable reliability (low F(S) and/or high AACE18 values, models 4 – 6 in Table S8). Thus, we also performed FSA docking using our full-structure template library from Dockground25 and included in the final submission models with the lowest AACE18 values (models 1 – 3 in Table S8). All template-based models had low F(S) values. Thus, we also performed free docking using 11 previously selected models. For the final submission, we selected the models with the lowest AACE18 values (models 7 – 10 in Table S8). All submitted models, however, were incorrect because of the poor quality of the selected individual protein models. For example, our best docking prediction (model 7 in Table S8), which partially captures the correct interface (fnat = 0.27), utilized CASP model with TM-score 0.4 and RMSD 13.5 Å to the native structure. Even the best CASP model for that protein had model-to-native TM-score 0.49 and RMSD 8.6 Å, which is beyond the accuracy needed for docking.37
Homo-dimer T116/T0893 (PDB 5idj)
The task was to assemble homo-dimer from models of mutated (E340Q) ligand-bound histidine kinase domain (residues 304 – 545) of 691 residue protein cell cycle histidine kinase CckA from Caulobacter crescentus bacteria (UNIPROT Q9X688). HHpred found 30 templates with the probability > 99%, e-values ~10−13 - 10−27 and sequence identity 30 - 13 %). Most of the templates originating from different bacteria were homodimers or had a higher oligomeric state in either asymmetric or biological units. Interfaces for these templates had 4-helix bundle architecture and different ligands. The only template from the same bacteria as target (4q20) was crystallized as the ligand-free structure. The second group of templates (with the alignment characteristics indistinguishable from those of the main group) came from human. They had different architecture (interacting β-strands) of the interfaces between chains in homo- and hetero-dimers. For this target, we performed FSA docking using all 150 CASP stage 2 server models and a custom library from all identified templates. Ten resulting models were selected by the F(S) maximum, AACE18 minimum, and diversity of the CASP servers’ models. However, despite the high F(S) and the low AACE values, all submitted models were incorrect (Table S9). Models built on templates from distant organisms (4 and 7 on human and 8 on archaea template) had different interface architecture. Models built on bacterial templates had the overall correct interface architecture, but incorrect length (inherited from the CASP models) and mutual positions of the interface helices (Fig. 4). This spectacular failure correlates with our previous study,41 where we pointed out that 4-helix bundles are prone to erroneous FSA docking. Another reason for the failure was the low overall accuracy of the CASP models (the best CASP models had model-to-native TM-score 0.68 and RMSD 13.2Å, or TM-score 0.53, but lower RMSD 8.9 Å).
Figure 4.

Native structure 5idj (A) and docking models (B and C) of T116/T0893. Predicted complexes are models 1 (B) and 7 (C) in the CAPRI submission. Other characteristics of the models are in Table S9.
Hetero-dimer T113/T0884-T0885
The docking was to be performed using models of two distinct proteins: small C-terminal part (residues 3395 - 3469) of a very large exoprotein (UNIPROT B3R1C1) and a relatively small 116 residue protein CPX19-CDII2 (UNIPROT B3R1C2). Both proteins were from Cupriavidus taiwanensis bacteria. HHpred did not identify any docking template for this target, even for the individual proteins (a small number of low reliability templates). Thus, we performed free docking utilizing 4 CASP models for the receptor (T0885) and 30 models for the ligand (T0884), selected by AACE18 and their proximity to the HHpred templates (3l9t and 2vgl for T0885, and 2b5u for T0884). Resulting models were scored by AACE18, and 10 final models were selected from the top 30 models by visual inspection. All submitted docking models were incorrect (Table S10) due to poor quality of the CASP models for T0884 (model-to-native TM-score ~0.4 and RMSD 5 – 11Å). However, in all docking predictions, location of the binding site was nearly correctly identified on both receptor and ligand (fnat ≠ 0).
Dimer of hetero-dimers T117/T0903-T0904 (PDB 5a7d)
This was the most challenging target, where the task was to assemble a tetramer from models of two Drosophila melanogaster proteins: the receptor (TPR region residues 25-406 of 658 residue PINS protein, UNIPROT ID Q9VB22, CASP target T0903) and the ligand (residues 283 – 623 of 859 residue INSC protein, UNIPROT ID Q24367, domain annotation absent, CASP target T0904). For the target sequences, HHpred detected PDB 4a1s (Fig. 5B), where slightly different PINS sequence portion (residues 1-368) interacts with the 40-residue peptide from the INSC (residues 301-340). This structure contains four chains in the asymmetric unit, although the biological assembly was deemed to be dimeric. No other template appropriate for docking was found. Thus, we decided to dock the rest of the INSC to the PINS. We cut the peptide part of the sequence, plus 5 residues from all 150 CASP stage 2 server T0904 models. Then, we performed free docking of the truncated models to 4a1s with a constraint that the first residue of the truncated ligand should be within 20Å of the C terminal of the peptide chains in 4a1s. Resulting models were scored by the AACE18 potential for each ligand model. Top 1 models for three best ligand models were selected for further processing. The second truncated ligand was symmetrically placed on the free PINS subunit. The missing residues were manually added either connecting truncated ligand to closer peptide (models 1, 4 and 5 in Table S11) or to the most distant peptide (models 2, 3 and 6). We also performed free docking utilizing full ligand models and peptide-free 4a1s (models 7–9). Finally, we performed double docking, where the first of the receptor subunit was docked against the ligand models, and then two best docked configurations were docked to each other (model 10). All submitted models were incorrect. However, models 1,4 and 5 (Fig. 5C) had some native contacts predicted correctly due to the correctly linked peptide portion of the ligand sequence, also present in the native structure (Fig. 5A). The main reason for this target failure was a completely different orientation of the receptor subunits in the native 5a7d and the template 4a1s structures. The native structure contains 12 chains in the asymmetric unit, but the PDB file headers report biologically relevant hetero-dimeric assembly. Thus, most likely, 5 out of 6 interfaces evaluated by CAPRI assessors were crystal packing artifacts.
Figure 5.

Native structure 5a7d (A), template 4a1s (B) and docking model (C) for T117/T0903-T0904. The docking prediction is model 5 in the CAPRI submission. Other characteristics of the model are in Table S11.
CONCLUDING REMARKS
We presented a new scoring function for template-based docking, benchmarked on the non-redundant set of protein-protein complexes from Dockground, and analyzed our performance in the CASP12/CAPRI37 docking round. The results showed that the template-based docking predictions for the CAPRI/CASP targets with the small values of the function (< 0.5) were incorrect. The large values of the function were necessary but not sufficient for the prediction to be correct. In all successfully predicted targets, the large values of the function for the template-based docking were complemented by other considerations, such as consistency of the oligomeric states among templates, similarity of the biological functions, biological interface relevance, etc. For example, docking of T111 showed that our scoring function poorly distinguishes biological from crystal packing interfaces. The scoring function needs further development for the docking of “simple” interfaces such as bundles of helices (see results for T116). In the case of the trimeric targets, sequence-based methods did not find common templates, despite similarity of the structures (TM-score 0.55–0.60). This suggest complementary use of structure- and sequence-based alignments in comparative docking. Another lesson of this joint round is that if a good docking template is found, an accurate model of the interface can be built even from largely inaccurate models of the individual subunits. On the other hand, the free docking is very sensitive to the quality of the individual protein models.37 Thus, we did not make any successful free docking predictions in this round, because models of the individual proteins were not accurate enough. However, our contact potential allowed us to detect approximate locations of the binding sites.
Supplementary Material
Acknowledgments
This study was supported by NIH grant R01GM074255 and NSF grants DBI1262621, DBI1565107 and CNS1337899.
References
- 1.Vakser IA. Protein-protein docking: From interaction to interactome. Biophys J. 2014;107:1785–1793. doi: 10.1016/j.bpj.2014.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moal IH, Moretti R, Baker D, Fernandez-Recio J. Scoring functions for protein–protein interactions. Curr Opin Struct Biol. 2013;23:862–867. doi: 10.1016/j.sbi.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka S, Scheraga HA. Medium- and long-range interaction parameters between amino acids for predicting three-dimensional structures of proteins. Macromolecules. 1976;9(6):945–950. doi: 10.1021/ma60054a013. [DOI] [PubMed] [Google Scholar]
- 4.Miyazawa S, Jernigan RL. Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules. 1985;18(3):534–552. [Google Scholar]
- 5.Sippl MJ. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J Mol Biol. 1990;213(4):859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- 6.Lazaridis T, Karplus M. Effective energy functions for protein structure prediction. Curr Opin Struct Biol. 2000;10(2):139–145. doi: 10.1016/s0959-440x(00)00063-4. [DOI] [PubMed] [Google Scholar]
- 7.Park B, Levitt M. Energy functions that discriminate X-ray and near-native folds from well-constructed decoys. J Mol Biol. 1996;258(2):367–392. doi: 10.1006/jmbi.1996.0256. [DOI] [PubMed] [Google Scholar]
- 8.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Prot Sci. 2002;11(11):2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buchete NV, Straub JE, Thirumalai D. Development of novel statistical potentials for protein fold recognition. Curr Opin Struc Biol. 2004;14(2):225–232. doi: 10.1016/j.sbi.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Skolnick J. In quest of an empirical potential for protein structure prediction. Curr Opin Struct Biol. 2006;16(2):166–171. doi: 10.1016/j.sbi.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Mintseris J, Weng Z. Atomic contact vectors in protein-protein recognition. Proteins. 2003;53(3):629–639. doi: 10.1002/prot.10432. [DOI] [PubMed] [Google Scholar]
- 12.Chuang GY, Kozakov D, Brenke R, Comeau SR, Vajda S. DARS (Decoys As the Reference State) potentials for protein-protein docking. Biophys J. 2008;95(9):4217–4227. doi: 10.1529/biophysj.108.135814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu SY, Vakser IA. DECK: Distance and environment-dependent, coarse-grained, knowledge-based potentials for protein-protein docking. BMC Bioinformatics. 2011;12:280. doi: 10.1186/1471-2105-12-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu C, Li X, Liang J. Developing optimal non-linear scoring function for protein design. Bioinformatics. 2004;20(17):3080–3098. doi: 10.1093/bioinformatics/bth369. [DOI] [PubMed] [Google Scholar]
- 15.Boas FE, Harbury PB. Potential energy functions for protein design. Curr Opin Struct Biology. 2007;17(2):199–204. doi: 10.1016/j.sbi.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320(2):369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 17.Zhang C, Vasmatzis G, Cornette JL, DeLisi C. Determination of atomic desolvation energies from the structures of crystallized proteins. J Mol Biol. 1997;267(3):707–726. doi: 10.1006/jmbi.1996.0859. [DOI] [PubMed] [Google Scholar]
- 18.Bordner AJ, Abagyan RA. Large-scale prediction of protein geometry and stability changes for arbitrary single point mutations. Proteins. 2004;57(2):400–413. doi: 10.1002/prot.20185. [DOI] [PubMed] [Google Scholar]
- 19.Anishchenko I, Kundrotas PJ, Vakser IA. Contact energies in proteins and protein complexes inferred from the Potts model. 2017 doi: 10.1016/j.bpj.2018.07.035. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57(4):702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33(7):2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Negroni J, Mosca R, Aloy P. Assessing the applicability of template-based protein docking in the twilight zone. Structure. 2014;22(9):1356–1362. doi: 10.1016/j.str.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Douguet D, Chen HC, Tovchigrechko A, Vakser IA. DOCKGROUND resource for studying protein-protein interfaces. Bioinformatics. 2006;22:2612–2618. doi: 10.1093/bioinformatics/btl447. [DOI] [PubMed] [Google Scholar]
- 24.Kirys T, Ruvinsky AM, Singla D, Tuzikov AV, Kundrotas PJ, Vakser IA. Simulated unbound structures for benchmarking of protein docking in the DOCKGROUND resource. BMC bioinformatics. 2015;16:243. doi: 10.1186/s12859-015-0672-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anishchenko I, Kundrotas PJ, Tuzikov AV, Vakser IA. Structural templates for comparative protein docking. Proteins. 2015;83:1563–1570. doi: 10.1002/prot.24736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Remmert M, Biegert A, Hauser A, Soding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2012;9(2):173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 27.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21(7):951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 28.Sinha R, Kundrotas PJ, Vakser IA. Docking by structural similarity at protein-protein interfaces. Proteins. 2010;78:3235–3241. doi: 10.1002/prot.22812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinha R, Kundrotas PJ, Vakser IA. Protein Docking by the Interface Structure Similarity: How Much Structure Is Needed? PLoS ONE. 2012;7(2):e31349. doi: 10.1371/journal.pone.0031349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, Vakser IA. Molecular surface recognition: Determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci USA. 1992;89:2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vakser IA. Protein docking for low-resolution structures. Protein Eng. 1995;8:371–377. doi: 10.1093/protein/8.4.371. [DOI] [PubMed] [Google Scholar]
- 32.Tsai J, Taylor R, Chothia C, Gerstein M. The packing density in proteins: Standard radii and volumes. J Mol Biol. 1999;290:253–266. doi: 10.1006/jmbi.1999.2829. [DOI] [PubMed] [Google Scholar]
- 33.Anishchenko I, Kundrotas PJ, Vakser IA. Structural quality of unrefined models in protein docking. Proteins. 2017;85(1):39–45. doi: 10.1002/prot.25188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eisenberg D, McLachlan AD. Solvation energy in protein folding and binding. Nature. 1986;319(6050):199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 35.Le Grand SM, Merz KMJ. Rapid approximation to molecular surface area via the use of Boolean logic and look-up tables. J Comput Chem. 1993;14(3):349–352. [Google Scholar]
- 36.Bentley JL. Multidimensional binary search trees used for associative searching. Commun ACM. 1975;18:509–517. [Google Scholar]
- 37.Anishchenko I, Kundrotas PJ, Vakser IA. Modeling complexes of modeled proteins. Proteins. 2017;85:470–478. doi: 10.1002/prot.25183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mendez R, Leplae R, De Maria L, Wodak SJ. Assessment of blind predictions of protein–protein interactions: Current status of docking methods. Proteins. 2003;52:51–67. doi: 10.1002/prot.10393. [DOI] [PubMed] [Google Scholar]
- 39.Venables WN, Ripley BD, Venables WN. Modern applied statistics with S. New York: Springer; 2002. p. xi.p. 495. [Google Scholar]
- 40.Kundrotas PJ, Zhu Z, Janin J, Vakser IA. Templates are available to model nearly all complexes of structurally characterized proteins. Proc Natl Acad Sci USA. 2012;109:9438–9441. doi: 10.1073/pnas.1200678109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kundrotas PJ, Vakser IA. Global and local structural similarity in protein-protein complexes: Implications for template-based docking. Proteins. 2013;81(12):2137–2142. doi: 10.1002/prot.24392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krieger E, Vriend G. New Ways to Boost Molecular Dynamics Simulations. J Comput Chem. 2015;36(13):996–1007. doi: 10.1002/jcc.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cao RZ, Wang Z, Cheng JL. Designing and evaluating the MULTICOM protein local and global model quality prediction methods in the CASP10 experiment. Bmc Struct Biol. 2014;14:13. doi: 10.1186/1472-6807-14-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joo K, Joung I, Lee SY, Kim JY, Cheng QY, Manavalan B, Joung JY, Heo S, Lee J, Nam M, Lee IH, Lee SJ, Lee J. Template based protein structure modeling by global optimization in CASP11. Proteins. 2016;84:221–232. doi: 10.1002/prot.24917. [DOI] [PubMed] [Google Scholar]
- 45.Kundrotas PJ, Vakser IA, Janin J. Structural templates for modeling homodimers. Prot Sci. 2013;22(11):1655–1663. doi: 10.1002/pro.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.