

Abstract
The functional and biological significance of the selected CASP12 targets are described by the authors of the structures. The crystallographers discuss the most interesting structural features of the target proteins and assess whether these features were correctly reproduced in the predictions submitted to the CASP12 experiment.
Keywords: X-ray Crystallography, NMR, CASP, Protein Structure Prediction
Introduction
Integrity of the CASP experiment rests on the blind prediction principle requesting models to be built on proteins of unknown structures. To get a supply of modeling targets, the CASP organization relies on the help of the experimental structural biology community. In the latest seven experiments (2002–2014), the vast majority (>80%) of CASP targets came from structural genomics centers participating in the Protein Structure Initiative (PSI) program. With the end of the PSI in 2015, CASP faced a challenging task of replenishing the target supply normally provided by the PSI Centers. Dealing with this problem required diversification of target sources and going beyond the existing network of the recurring CASP target providers. Soliciting for targets, the organizers directly approached a wider set of structure determination groups, and also worked out a better protocol for obtaining and analyzing information about the structures placed on hold with the PDB. These efforts bore fruits, and 82 targets were secured for the CASP12 experiment. This number is quite impressive (considering that targets were collected in a short 3-month span of time) and is only somewhat smaller than the number of targets in a typical PSI-era CASP experiment (cf. 100 targets in the most recent CASP11 experiment). It is also worth mentioning that CASP12 targets came from 33 different protein crystallography groups stationed in 17 countries worldwide. Because of this variety, CASP12 targets exhibited wide diversity of sizes (from 75 to 670 residues), difficulties (from high accuracy modeling targets to new folds), quaternary structure composition (from single-domain targets to hetero-complexes), organisms (from rare extremophilic archaea from the depths of the Red Sea to Homo sapiens), and protein types (from globular to viral and membrane). Such diversity is vital for comprehensive testing of prediction methods. CASP organizers, who are co-authors of this paper, want to thank every experimentalist who contributed to CASP12 and thereby helped promote the development of more effective protein structure prediction methods. The list of all crystallographers who contributed targets for the CASP12 experiment is provided in Table 1 of the Supplementary material.
This manuscript is the fourth in a series of CASP target highlight papers1–3. The chapters of the paper reflect the views of the contributing authors on twelve CASP12 targets: 1) the flagellar cap protein from Pseudomonas aeruginosa – T0886; 2) bacteriophage AP205 coat protein – T0859; 3) toxin-immunity protein complex from the contact-dependent growth inhibition system of Cupriavidus taiwanensis – T0884/T0885; 4) sorbitol dehydrogenase from Bradyrhizobium japonicum – T0889; 5) C-terminal domain of human gasdermin-B – T0948; 6) receptor-binding domain of the whitewater arroyo virus glycoprotein – T0877; 7) glycoside hydrolase family 141 founding member BT1002 – T0912; 8) a DNA-binding protein from Aedes aegypti – T0890; 9) snake adenovirus-1 LH3 hexon-interlacing protein – T0909; 10) an ice-binding protein from Antarctica – T0883; 11) a domain of UDP-glucose glycoprotein glucosyltransferase from Chaetomium thermophilum – T0892; and 12) a cohesin from Ruminococcus flavefaciens scaffoldin protein complexed with a dockerin – T0921/T0922. The results of the comprehensive numerical evaluation of CASP12 models are available at the Prediction Center website (http://www.predictioncenter.org). The detailed assessment of the models by the assessors is provided elsewhere in this issue.
1. FliD, the flagellar cap protein from Pseudomonas aeruginosa PAO1 (CASP: T0886, Ts886, PDB: 5FHY) – provided by Sandra Postel and Eric J. Sundberg
Bacterial flagella are long helical cell appendages that are important for bacterial motility and pathogenicity 4. These extracellular hollow filaments are formed by thousands of copies of FliC (flagellin) molecules and connected via a hook to the flagellar rotary motor anchored in the bacterial membrane 5. The motor drives the propeller-like motion of the filament, which confers swimming motility to the bacteria 6. An important structural and functional component of bacterial flagella is the flagellar capping protein, FliD, that is located at the distal end of the flagellar filament 7. Unfolded FliC molecules are translocated from the cell cytoplasm through the hollow filament pore to the tip of the growing flagellum where FliD regulates flagellar assembly by chaperoning and sorting FliC proteins. An absence of FliD leads to improperly constructed filaments and, consequently, impaired bacterial motility and infectivity 8. In the most commonly studied organism for flagella, Salmonella serovar Typhimurium, FliD is known to form a homopentameric complex on the tip of the flagellum, as shown in a low-resolution cryo-EM structure 7,9,10. Until recently, these data provided the only available structural insight to FliD. Our crystal structure of a large fragment of FliD, FliD78–405, from Pseudomonas aeruginosa PAO1 was the first high-resolution structure of any FliD from any bacterium, providing novel details concerning FliD function 11.
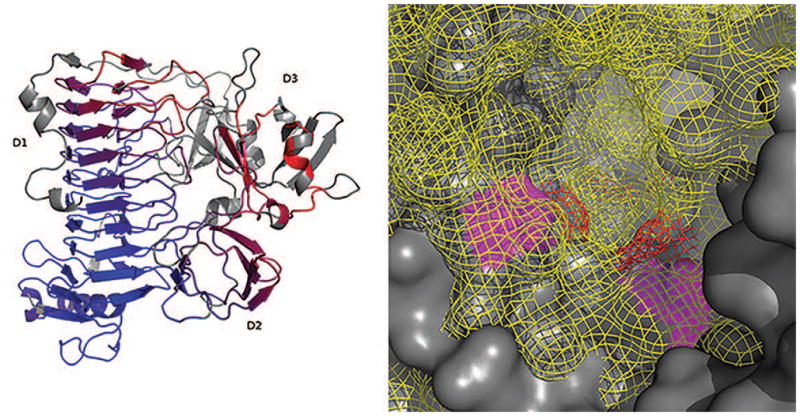
In our crystal structure 11, the Pseudomonas FliD78-405 monomer exhibits an L-shaped structure (Figure 1A), which can be divided into two globular domains and a helical region. Domain D3 is a loop insertion into domain D2, and both domains have structural similarity to other flagellar proteins. Residues 309 to 405 of FliD78-405 are highly flexible as revealed by hydrogen/deuterium exchange (HDX) and we were also unable to model those residues in our structure. Full-length Pseudomonas FliD1-474 encodes predicted N- (residues 1 to 77) and C-terminal (residue 406 to 474) coiled coil domains that prohibited crystallization in our hands.
Figure 1. (A) Crystal structure of the Pseudomonas FliD78-405 monomer subunit.

in which the domain D3 (CASP domain D2, green), domain D2 (CASP domain D1, blue) and the helical region (red), which belongs to domain D1 (not evaluated in CASP), are indicated. (B) Side view (top panel) and top view (bottom panel) showing cartoon representations of the hexameric FliD78-405 crystal structure. Each monomer subunit is colored distinctly. (C) SAXS-generated molecular envelope of the monomeric FliD1-474 with the CASP prediction model T0886TS036_1 (cyan). (D) Superposition of CASP prediction models T0886TS247_1_D1 (orange) and T0886TS247_1_D2 (orange) with D2 (CASP domain D1, blue) and D3 (CASP domain D2, green) of the FliD78-405 monomer crystal structure. (E) Superposition of CASP prediction model T0886TS247_1 (orange) with the FliD78-405 monomer crystal structure (domain coloring as in Panel A). (F) Superposition of CASP prediction model T0886TS247_1 (orange) with the E. coli FliD43-416 crystal structure 5H5V (magenta). (G) Superposition of CASP prediction models T0886TS247_1 (orange), T0886TS011_1 (cyan), T0886TS064_1_1 (light blue), T0886TS411_1 (yellow) with the FliD78-405 monomer crystal structure (domain coloring as in Panel A). (H) Superposition of CASP prediction models T0886TS247_1-D2 (orange), T0886TS064_1_1-D2 (light blue), T0886TS011_1-D2 (cyan), T0886TS411_1-D2 (yellow), T0886TS456_1-D2 (dark grey), T0886TS173_1_1-D2 (red) with D3 of the FliD78-405 monomer crystal structure (green).
In contrast to the Salmonella FliD, which forms a pentamer, Pseudomonas FliD adopts a hexameric oligomeric state in the crystal structure (Figure 1B), as well as in solution and functions as a hexamer in vivo 11. The number of protofilaments that comprise the flagellar filament upon which FliD oligomers reside varies among bacteria 12, suggesting that FliD stoichiometries also vary between bacteria, which is supported by our results. More recently, the crystal structure of FliD from E. coli, which includes all residues except the N- and C-terminal coiled coils showed that this FliD protein also forms a hexamer 13.
Pseudomonas FliD was included in CASP12 as a regular target T0866 and small-angle X-ray scattering (SAXS)-assisted target Ts886. SAXS data of the monomeric full-length protein, FliD1-474, for which no crystal structure yet exists, were collected and the data provided to the modelers to aid the structure prediction process of the shorter construct that we had crystallized. All the SAXS-assisted target models exhibit low similarity to the FliD crystal structure as shown in an overlay of the best model Ts886TS036_1 with our crystal structure in Figure 1C, but do fit well into the SAXS envelope (Figure 1C).
The models obtained during the regular prediction round without using the SAXS envelopes to assist model-building vary greatly. The highest ranked model T0886TS247_1 closely resembles the crystal structure of Pseudomonas FliD78-405 on the individual domain level (Figure 1D). However, the connection between domain D2 (CASP domain D1) and domain D3 (CASP domain D2) diverges resulting in a relative positioning of these two domains that is different than in the crystal structure (Figure 1E). The low resolution of the SAXS molecular envelope of FliD1-474 is potentially compatible with multiple, various domain arrangements and may have made it difficult to predict the exact positioning of the individual domains (Figure 1C). Residues 309 to 405 of FliD78-405, which we could not model in the crystal structure due to poor or missing electron density, were in general modeled as helical bundles in T0886TS247_1. A superposition with the recently solved crystal structure of E. coli FliD43-416 (PDB 5H5V 13), which covers a larger fragment of FliD, shows the correct prediction of helical bundles in those regions (Figure 1F). However, the bundles are placed in a different orientation relative to the D2 and D3 domains, and do show a differences in the placement of individual helices. These discrepancies between the model and the experimental structure may be due to the high flexibility in the linker region and in the helical regions that we detected by HDX11.
Compared to T0886TS247_1, all of the other models exhibit substantially less similarity to the FliD78-405 crystal structure (Figure 1G). Models of domain D3 (CASP domain D2) alone, however, exhibited greater likenesses to the crystal structure, with secondary structural elements generally predicted properly (Figure 1H). This might be related to the lower flexibility (as shown by HDX) of domain D3 in comparison to the rest of the FliD molecule. Overall, FliD seemed to be a difficult target to model, despite the SAXS data provided, and only domain D3 appeared to yield models by multiple modeling groups that resembled the actual crystal structure very well.
2. Structure of bacteriophage AP205 coat protein (CASP: T0859; PDB: 5FS4, 5JZR, 5LQP) - provided by Kaspars Tars, Roman I. Koning and Guido Pintacuda
ssRNA phages like MS2, Qβ and AP205 infect various gram-negative bacteria and are among the simplest known viruses used for decades as models to study various problems in molecular biology. Lately, ssRNA phages and their components have found several applications, notably in vaccine development 14. Capsid of ssRNA phages contains 178 copies of coat protein (CP) and a single copy of maturation protein, responsible for attachment of phage particles to bacterial receptor 15. When produced in bacteria, recombinant CP of ssRNA phages spontaneously assembles in virus-like particles (VLPs), containing 180 copies of CP. Due to strong interactions between two adjacent CP monomers, VLPs can be regarded as built from 90 CP dimers.
In general, VLPs are empty, non-infectious shells of viruses, devoid of genomic nucleic acid, but morphologically similar to the corresponding viruses. VLPs have several applications, the best known of which is vaccine development. For example, VLPs of Hepatitis B virus have been used as successful vaccines for a few decades16. VLPs can be used not only as vaccines against the disease caused by the virus of VLP origin, but also as scaffolds to induce strong immune response against virtually any antigen17. In this case, multiple copies of the antigen of interest should be attached to the surface of VLP. The immune system recognizes patterns of regularly repeating antigens on VLP surface as a potential threat to the organism, inducing highly elevated titres of antibodies and stronger T-cell responses compared to free antigen 18. To avoid pre-existing immune responses, pathogens that do not target humans are preferable as carriers of antigens. For this purpose, VLPs of ssRNA phages like MS2, Qβ and AP205 have been widely used 14.
For creation of vaccine candidate, the antigen of choice can be efficiently attached to VLPs by genetic fusion of CP and antigen genes. Since antigens must be presented on the surface of VLPs, the knowledge of the exact three-dimensional structure of VLP provides useful information about suitable sites of insertion of antigens in coat protein sequences. Due to folding problems, large insertions are often tolerated only at either N- or C-termini of CP, but this is possible only if the terminal end of CP is well exposed on the VLP surface. However, in VLPs of ssRNA phages studied so far, like MS2 19, Qβ 20, GA 21, PP7 22, PRR1 23 and Cb5 24 both terminal ends are poorly exposed on the surface. Additionally, three N- and three C- terminal ends of neighbouring CP dimers on the VLP surface are clustered together, resulting in steric clashes among any N- or C-terminal insertions. Instead, a so-called AB loop is well exposed and well separated from AB loops of neighbouring CP subunits, but only relatively short amino acid sequences can be inserted in it without compromising the VLP stability. In contrast, AP205 VLPs have been known before to tolerate significantly longer insertions at both C- and N- termini25, but the structural reason for this remained unknown. Since we failed to obtain high resolution crystals of recombinant AP205 VLPs, we constructed and crystallized an assembly-deficient AP205 CP mutant, capable to form dimers, but not VLPs. The obtained crystal structure was further fitted into a medium resolution cryo-EM map of native recombinant AP205 VLPs. Additionally, a solid-state NMR structure of AP205 coat protein was obtained from labelled AP205 VLPs. The obtained results revealed that compared to related ssRNA phages, the structure of AP205 CP is circularly permuted 26, meaning that about 20 N-terminal residues including the first β-strand are found at the C-terminal part instead. This feature is made possible due to the close proximity of N- and C-terminal parts of two monomers within the dimer (Figure2AB). The result is that in AP205 VLPs both N- and C- termini are found in the same position as AB loops in other phages (Figure 2CD). This provides a structural basis for construction of vaccine candidates using AP205 VLPs.
Figure 2. Structural features of bacteriophage AP205 coat protein.

Coat protein in AP205 and related phages, such as MS2, builds very stable dimers. Two monomers are shown in different shades of grey (panels A and B). Notice the close proximity of N- (blue) and C- (red) termini in dimers. 90 dimers further assemble into VLPs (panels C and D). In MS2, AB loop (green) is the most exposed structure on the surface of VLPs. Compared to MS2, in AP205 the first β-strand (yellow) is shifted to the C-terminus, although it remains in the same position in 3D. As a result, in AP205, C-and N- termini are the most exposed features on VLPs. In panel (E), crystal structure of AP205 monomer (green) is superimposed with the modeled structure (blue and red). The overall fold of model is approximately correct, except that it lacks C-terminal β-strand. Residues 1-39 (blue) are correctly placed in respect to the sequence, corresponding to the first four β-strands. For the rest of model (red) residues are placed incorrectly according to the sequence and out-of-register errors occur. Notice also that position of N-terminus is relatively well predicted, while C-terminus is in a very different position.
Out of 499 models submitted on CASP12 target T0859, only one had a reasonably accurate overall structure (Figure 2E, red and blue). Model T0859TS001, made by researchers at Francis Crick institute, included almost all of the actual secondary structure elements apart from the C-terminal β-strand, which is unique for AP205, compared to other similar phages. About one third of the protein, comprising approximately 40 N-terminal residues was placed fairly accurately in respect to sequence, as compared to the crystal structure. This means that researchers correctly deduced that the first β-strand is missing in AP205. After residue 40, progressively increasing out-of-register errors occur in the model. At the C-terminal part the register shift is about 20 residues. Due to this shift, the C-terminal residues are modeled as α-helix although in crystal structure they form the extra (C-terminal) β-strand, not observed in similar phages. Therefore, the C-terminal part is not modeled correctly and does not suggest the placement of C-termini on the surface of VLP, close to AB loops in related phages. Even though the overall precision of the model is somewhat limited, the model correctly suggests that N-terminal part is indeed well-exposed on the surface of VLP and occupies the position of AB loops in related phages. If experimental data had not been available, the model T0859TS001 would have provided significant biologically relevant information for construction of VLP based vaccines.
3. Structure of the toxin-immunity protein complex from the contact-dependent growth inhibition system of Cupriavidus taiwanensis (CASP: T0884/T0885, PDB: 5T87) – provided by Karolina Michalska, Christopher S. Hayes, Celia W. Goulding and Andrzej Joachimiak
Contact-dependent growth inhibition (CDI) is an important mechanism of inter-cellular competition found in Gram-negative bacteria. Bacteria utilizing the CDI system (CDI+) use diverse CdiB-CdiA two-partner secretion systems to deliver protein toxins directly into neighboring bacteria 27,28. CdiB is an outer membrane transport protein exporting the CdiA effector onto the cell surface. CdiA recognizes specific receptors on susceptible bacteria and translocates its C-terminal toxin domain (CdiA-CT) into the target cell 29–31. The variable CdiA-CT toxin region is usually demarcated by a conserved peptide motif, such as the VENN sequence found in enterobacterial CdiAs 32. Different CdiA-CTs can be fused to heterologous CdiA proteins at the VENN motif to generate novel chimeric effectors 28,32,33. CdiA proteins carry a variety of toxin domains, most commonly exhibiting nuclease or pore-forming activities 32–35. To protect against self-inhibition, CDI+ bacteria produce CdiI immunity proteins, which bind and neutralize cognate CdiA-CT toxins.
We have selected the CdiA-CT/CdiI complex from Cupriavidus taiwanensis LMG 19424 for structural analysis. PSI-BLAST searches for CdiA-CT homologs recover several predicted S-type pyocins from Pseudomonas species and MafB toxins from Neisseria species 36 (50–70% sequence identity). Other hits include CdiA-CT domains from Rhizobium leguminosarum and Achromobacter strains, and Rhs peptide-repeat proteins from Streptomyces species. All of these homologs are predicted to mediate inter-bacterial competition 37,38, though none have been validated experimentally. An HHpred-based search identified the C-terminal domain of 16S rRNA-cleaving colicin E3 39,40 as a possible structural homolog having 9% sequence identity to CdiA-CT. The CdiI immunity protein is less conserved than CdiA-CT, with homologs sharing ~30–40% sequence identity. An HHpred analysis recovered proteins with α-helical hairpin repeats, with the armadillo-like γ-COP coatomer (13% sequence identity with CdiI) being the closest match.
The 2.40 Å resolution crystal structure of the CdiA-CT/CdiI complex (Figure 3A) shows that the toxin putative catalytic domain (75 residues) consists of a central four-stranded antiparallel β-sheet, sandwiched by two N- and C-terminal α-helices and one 310 helix. The immunity protein (116 residues) is composed of three consecutive α-hairpins creating an armadillo-like structure. The N-terminal β-strand of CdiI protrudes from the helical body to complement the CdiA-CT β-sheet, potentially influencing toxin conformation. This arrangement also suggests that the N-terminal segment of CdiI is likely disordered in the free CdiI. A Dali server search for CdiA-CT structural homologs identified only low-similarity matches: inorganic triphosphatase (Z-score 3.7, RMSD 3.3 Å, PDB:3TYP) (Figure 3B) and WW domain of human transcription elongation regulator 1 (Z-score 3.5, RMSD 2.9 Å, PDB:2DK7). More distant hits include E. coli ParE toxin (Z-score 3.0, RMSD 2.4 Å, PDB:3KXE) (Figure 3C), which belongs to the barnase/EndoU/colicin E5-D/RelE (BECR) family (PMID:22731697). Although structurally related, these toxins display different activities: ParE family poison DNA gyrase 41, RelE is a ribosome-dependent mRNase 42, and colicins D/E5 cleave the anticodon loops of specific tRNAs 43. Therefore, the exact biochemical function of CdiA-CT cannot be predicted easily and may include RNase or DNase activity. The CdiI fold is well-represented in the PDB and is a popular scaffold for designer proteins. The closest match corresponds to human deoxyhypusine hydroxylase (Z-score 12.3, RMSD 2.0 Å, PDB:4D4Z), followed by protein phosphatase 2 (Z-score 12.3, RMSD 2.5 Å, PDB:2IE3) and other proteins with virtually no sequence similarity to CdiI. Though many of the homologs engage in protein-protein interactions, none are annotated as an immunity protein.
Figure 3. The CdiA-CT/CdiICtai complex.

(A) Experimental structure with the most conserved residues and their interactions shown in stick representation. The CdiA-CT toxin domain is shown in teal and the CdiI immunity protein in pink. Hydrogen bonds are depicted as red broken lines. Superposition of CdiA-CT with (B) the closest PDB homolog, inorganic triphosphatase (coral, PDB:3TYP), (C) with ParE toxin from E. coli (yellow, PDB:3KXE) and (D) with model T0884TS183_1 (purple) and refined model TR884TS118_1 (blue). The strand β1 from CdiI is shown for reference. (E) Superposition of CdiI with model T0885TS005_2 (cyan) and refined model TR885TS247_1 (blue).
Antitoxin proteins often bind over nuclease toxin active sites to prevent substrate access. Typically, nuclease toxins are highly electropositive and the cognate immunity proteins carry complementary acidic residues to promote electrostatic interactions. CdiA-CT contains several basic residues, including conserved His212, His214 and Arg216 (Figure 3A), which may be key catalytic residues. CdiI is more electrostatically neutral than previously characterized immunity proteins. It directly interacts with the toxin’s putative active site using the conserved His72, Arg75 and Asp108 residues, which form a hydrogen bond, stacking interaction and salt-bridge, respectively. As mentioned above, β1 of CdiI complements the toxin fold.
For the CASP12 competition, CdiA-CT and CdiI were modeled as monomers and as a hetero-complex.
For CdiA-CT (T0884), the best model (out of 185 total monomeric predictions) was generated by QUARK (T0884TS183_1), which uses ab initio algorithms with no global template information. This model scored 66 GDT_TS points, 10 points higher than the next model, T0884TS236_1 generated by MULTICOM-construct. The highest-scoring regular prediction model T0884TS183_1 was subsequently released for refinement, where it was further improved to GDT_TS of 76 by the PKUSZ_Wu_group (TR884TS118_1). Model T0884TS183_1-D1 closely resembles the crystal structure, though helix α1 is misoriented and the β3-β4 hairpin is distorted (Figure 3D). However, we note that toxin helix α1 is constrained by the immunity protein in the CdiA-CT/CdiI complex. Therefore, it is possible that the free toxin domain adopts the conformation predicted by the computational model. Toxin residues that interact with the immunity protein are generally located in proper positions, though a more accurate spatial prediction of β4 would bring the conserved His212 and His214 to better agreement with the crystal structure.
CdiI (T0885) is a more straightforward structure prediction target with fewer discrepancies among the 190 predicted models. The best five models for this target were generated by the BAKER-ROSETTAserver group, with the top model T0885TS005_2 scoring 88 (out of 100) GDT_TS points (Figure 3E). The next model in the accuracy ranking was generated by the MULTICOM-novel group scoring 15 GDT_TS points below the best. As we found with CdiA-CT, the major misalignments were observed for peripheral elements (β1 and the C-terminus of helix α6) involved in protein-protein interactions. Similarly to the CdiA-CT, the best server model for CdiI, T0885TS005_2, was released for the refinement (without the 11 N-term residues trimmed by the assessors) and was further improved to 95 GDT_TS points (TR885TS247_1).
These examples show that computational prediction can yield models with correct folds, and when combined with sequence conservation analysis, can inform rational mutagenesis and biochemical analyses.
Even though the monomeric subunits of the CdiA-CT/CdiI (T0884/T0885) hetero-complex were predicted quite well, the full complex was modeled poorly. Although some of the multimeric models reached reasonable global accuracy scores (e.g., LDDT of 0.73 for TS239_1), the visual inspection showed that all models left the putative active site of toxin fully exposed and failed to properly predict the correct protein-protein interface. Accuracy of interface contacts in the submitted predictions is rather poor, with the highest recall of 23.4% achieved in the prediction TS203_3, where subunit molecules partly overlap. Thus, for the CdiA-CT/CdiI complex, in silico approaches did not provide useful information to confidently determine complex organization important for understanding function and catalysis.
4. Sorbitol dehydrogenase (BjSDH) from Bradyrhizobium japonicum (CASP: T0889; PDB: 5JO9) - provided by Leila Lo Leggio, Folmer Fredslund and Gert-Wieland Kohring
Rare sugars are defined as monosaccharides and their derivatives which are rare in nature, and these sugars have attracted interest for potential medical and food applications 44. Consequently, enzymes able to produce and interconvert rare sugars have also attracted attention. One such enzyme is the Zn-independent short chain dehydrogenase from Bradyrhizobium japonicum (BjSDH) which uses NAD+/NADH as a non-covalently bound cofactor. We initiated structural studies of BjSDH (CASP ID T0889) as part of a collaborative EU project devoted to the development of an electro-enzymatic flow-cell device for the production of rare sugars 45. BjSDH was selected for structure determination due to some favorable properties. First of all, while BjSDH preferentially catalyses the oxidation of D-glucitol (a synonym for D-sorbitol) to D-fructose, it can also catalyse the oxidation of L-glucitol to the rare sugar L-sorbose with enzymatic cofactor regeneration and high D-sorbose yield 46 (Figure 4A). Sorbitol dehydrogenases are additionally of particular interest in biosensor technology, since D-sorbitol is a marker for onset of diabetes as well as a food ingredient 47. Furthermore, it is a thermostable enzyme with Tm of 62 °C 46, which is a desirable property for potential industrial use and biosensor technology, as thermostability often correlates with general stability.
Figure 4.

(A) Products of reaction catalyzed by BjSDH with D-glucitol and L-glucitol as substrates; (B) Structure based sequence alignment of region around loop 193-203 covering the active site of BjSDH. Sequences of GatDH, RsSDH and top 5 DALI hits searching with the BjSDH structure are shown; (C) BjSDH structure shown as cartoon (gold) and symmetry related molecule packing against is (grey). Ligands in the structure are shown as sticks, while loop 193-203 in top 5 models from CASP12 are shown as lines; (D) Continuous β-sheet between two monomers in BjSDH crystal structure, and same region in the RsSDH crystal structure.
Structure determination 48 was not straightforward due to limited resolution, which was estimated to be at 2.9Å according to CC1/2 of about 50% in the outer resolution shell 49, but closer to 3.2Å with more conventional evaluation of resolution limit at I/σ(I) around 2. The Molecular Replacement model (PDB code 4NBU 50) had only 29 % sequence identity to the target after structure-based alignment. As all short chain dehydrogenases, BjSDH adopts a Rossman fold 51 and has a catalytic tetrad (Asn112, Ser140, Tyr153 and Lys157). BjSDH was co-crystallized with NAD+ and D-glucitol. D-glucitol could be modeled in the electron density map and phosphate is clearly bound, mimicking part of the cofactor, however a full co-factor molecule could not be modeled. This is probably due to the presence of 1.4 M NaH2PO4/K2HPO4 in the crystallization conditions, competing with the cofactor. Although there is only one molecule in the asymmetric unit, the enzyme forms a tetramer in the crystal structure due to crystallographic symmetry, and this is also assumed to be the predominant form in solution 48.
All the closest structural relatives identified with DALI after structure determination (reported in Fredslund et al 48), have only around 30% sequence identity, and while most are dehydrogenases, none are denoted as sorbitol dehydrogenases. When compared to the DALI results, the most structurally diverse part of the structure is a helix-turn-helix motif or “lid” loop, residues 189–205 in BjSDH, partly responsible for ligand binding. This loop is different in length, sequence and conformation (Figure 4B), compared to enzymes with relatively similar specificity like R. sphaeroides sorbitol dehydrogenase RsSDH 52. The analysis of the DALI results also confirmed that the catalytic tetrad is highly conserved structurally in BjSDH compared to similar dehydrogenases. All the top DALI hits also form tetramers with similar symmetry.
To see if structural features of target T0889 were correctly predicted in CASP12 models, we analyzed the top 5 monomeric models (based on the GDT_TS score) and the top oligomeric model (based on the recall score for interface contacts).
The monomeric models were based solely or in part on the structure of clavulanic acid dehydrogenase from Streptomyces clavuligerus 53 (PDB entry 2JAH or 2JAP), which was also the top DALI hit. Unsurprisingly, the models predict correctly the positioning of the catalytic tetrad and overall predict the structure of BjSDH in a satisfactory manner. However, the helix-turn-helix loop is different in the 5 top scoring models as compared to the crystal structure and the model used for molecular replacement. Since the resolution of the crystal structure is limited, and this loop in particular was difficult to trace in the electron density, there might be errors in the crystallographic model, but the conformation of the loop from several CASP12 models is definitely incompatible with crystal packing (Figure 4C) and cannot accurately represent the conformation it assumes in the crystal. On the other hand, crystal packing could have affected the conformation and furthermore, the loop is involved in ligand binding, which would not be taken into account explicitly by the modeling programs and could also affect its conformation.
One of the most important features of BjSDH was its thermostability46, as the knowledge of its structural determinants may help stabilize related enzymes by protein engineering. In particular, we compared the structure to the sorbitol dehydrogenase RsSDH, for which the melting temperature by CD spectroscopy was also measured and found to be much lower than for BjSDH under similar conditions (Tm of 47 °C vs 62°C). One of the striking features in BjSDH is a much higher proline/glycine ratio compared to RsSDH, a feature which is obvious from the sequence and does not require knowledge of the 3D structure. An additional feature which is likely to affect stability becomes obvious only through analysis of the quaternary structure. As previously mentioned BjSDH is a tetramer in the structure and in solution, as are many members of the short chain dehydrogenase family, and probably also RsSDH52. In BjSDH, two monomers of the tetramer have a large interaction surface via a continuous β-sheet formed between the two monomers, while this is not the case in RsSDH, indicating a less stable tetramer in the latter (Figure 4D). As the top CASP12 models for BjSDH were all based on the clavulanic acid dehydrogenase structure, which is also a tetramer and includes the continuous β-sheet between subunits, the top monomeric models are all compatible with an intersubunit β-sheet formation.
Among the oligomeric models, model TS188_4 from the chuo-u group was the best as judged by the interface contact recall (http://predictioncenter.org/casp12/multimer_results.cgi?target=T0889o). The model represents the same homo-tetrameric assembly as the target structure T0889 (BjSDH) and the PDB structure 2JAH, which was used as a template. The tetramer interfaces are modeled reasonably well, with 72% of the native interface contacts being correctly reproduced, while the constituting monomers lack some details, which may affect the analysis of the protein stability. It should be noted, though, that the top model does not have much added value compared to the 2JAH template, as their superposition yields a Cα RMSD of only 0.7 Å.
In conclusion, the top CASP12 models reproduce correctly some but not all biologically and biotechnologically interesting features of SDH, specifically they cannot predict the lid loop conformation, which is part of the substrate binding pocket, or subtle details of the interactions in the tetramer.
5. Crystal Structure of the C-terminal Domain of Human Gasdermin-B (CASP: T0948; PDB: 5TJ4, 5TJ2, 5TIB) - provided by Kinlin L. Chao and Osnat Herzberg
Biological Significance of Gasdermin-B
The human genome encodes four gasdermins (GSDMA-D) that are expressed in epithelial cells of the gastrointestinal tract and skin, regulating the maintenance of the epithelial cell barrier, cell proliferation, differentiation and programmed cell-death processes 54,55. Based on the different protein levels in cancers, human GSDMA, GSDMC and GSDMD are considered tumor suppressors and GSDMB (CASP12 target T0948), a tumor promoter. GSDMB amplification and GSDMB overexpression lead to poor response to HER2-targeted therapy in HER2-positive breast cancer 56. The N-terminal domain of gasdermins possesses membrane-binding activity, whereas the C-terminal domain autoregulates the lipid binding function. Multiple genome-wide association studies (GWAS) revealed a correlation between single nucleotide polymorphisms (SNPs) in the protein coding and transcriptional regulatory regions of the neighboring GSDMA, GSDMB and ORDML3 genes with susceptibility to asthma 57, type 1 diabetes 58,59, Crohn’s disease, ulcerative colitis 59,60 and rheumatoid arteritis 59,61. Pal and Moult identified 2 GSDMB SNPs (dbSNP:rs2305479 and dbSNP:rs2305480) in linkage disequilibrium with a marker of disease risk 59. They correspond to a Gly299 → Arg299 change (rs230549), and a Pro306 → Ser306 change (rs2305480) in the C-terminal domain of GSDMB (GSDMB_C) (numbering scheme according to Uniprot isoform Q8TAX9-1). Analyses of the 1000 Genomes Project Consortium data 62 showed co-occurrence of the 2 SNPs (Gly299:Pro306 or Arg299:Ser306) with ~50% occurrence of each combination in the general population (Pal and Moult, unpublished). Unlike monogenic diseases which are caused by high penetrance SNPs in single genes, complex-trait diseases are associated with multiple low penetrance SNPs in multiple genes. Most of the SNPs present in a genome are actually not disease causative. However, because of linkage disequilibrium within the genome SNPs the challenge for the large-scale genome sequencing is to reveal the disease causative SNPs. The structural studies of GSDMB_C were undertaken to provide insights into possible mechanisms that the SNPs may contribute to disease risk 63.
Key features of Gasdermin-B C-terminal domain
GSDMB amino acid sequence is homologous to the sequence of Gsdma3, the mouse homolog of GSDMA. The structure of Gsdma3 (PDB 5B5R) revealed 2 domains connected by a long flexible linker. The N-terminal lipid-binding domain folds into an α+β structure, and the C-terminal inhibitory domain adopts an α-helical fold comprising 8 helices 64. The 7-helix bundle topology of GSDMB_C (α5-α11 in our paper 63 describing the crystal structure, PDB 5TJ4, 5TJ2, 5TIB ) is the same as that of Gsdma3, except that it lacks a Gsdma3 subdomain comprising an α-helix and a 3-stranded β-sheet between the last two α-helices (Fig 5A–C).
Figure 5.

(A) Structure-based sequence alignment of the GSDMB (T0948 comprises GSDMB’s C-terminal domain) and mouse Gsdma3 C-terminal domains with secondary structure elements shown above or below the respective sequences. Identical and conservatively replaced residues are colored in red and blue. The alignment was performed using the programs Clustal Omega 118 and ESPript 3 (espript.ibcp.fr/Espript/). (B) Ribbon diagram of the GSDMB_C fold (PDB 5TIB). The α7–α8 GSDMB loop containing the polymorphism residues is colored in red. (C) Superposition of the experimental GSDMB_C structure (colored yellow) and the corresponding Gsdma3 domain that served as a modeling template (blue, 5B5R), (D) Superposition of the experimental GSDMB_C structure (colored yellow) and the best GTD_TS CASP12 scored model of group 251 (green). (E) Superposition of the polymorphism loop of the experimental structure (colored gray with α′ highlighted in orange) with the corresponding loop assessed as the closest (Group 330) based on the position specific criterion (colored cyan with α′ highlighted in magenta).
We determined three crystal structures of the GSDMB_C containing (1) the Arg299:Ser306 pair corresponding to individuals with increased disease risk, (2) the Gly299:Pro306 present in healthy individuals, and (3) the Gly299:Ser306 combination, one from each allele. The second possible combination, Arg299:Pro306, did not yield well diffracting crystals 63. The SNP residues at positions 299 and 306 are located on a loop connecting the α7 and α8 helices of GSDMB (Figure 5AB). Three GSDMB_C structures provide 16 independently determined molecules in their asymmetric units: 6 with Ser at position 306 and 10 molecules with Pro at that position. All 16 versions of this loop contain a 5-residue α-helix (α′, Pro309-Ser313) (Figure 5AB). However, the loops with Ser306 adopt an additional well-ordered 4-residue helical turn (Met303-Ser306) between the α7 and α′ helices (Figure 5B). By contrast, the loops with a Pro306 do not form this helical turn and each loop version assumes different backbone conformations 63. In addition, a Gly299→ Arg299 alters the charge distribution on the protein surface. Examination of the structures shows that, unlike a more flexible Ser306 side chain, Pro306 cannot be accommodated at the end of the helical turn because its side chain would clash with main chain carbonyl atoms of the preceding residues. One or both of these changes may contribute to the susceptibility of individuals to develop diseases by possibly modulating the selectivity and binding affinity of its N-terminal domain to lipids or the association with partner proteins, for example HSP90β or fatty acid synthase 65.
CASP12 predictions for the functionally important regions of GSDMB_C
The 166-residue GSDMB_C CASP12 target sequence (T0948) contained the Arg299:Ser306 pair found in individuals with increased disease risk (PDB 5TIB). The publication of the full-length Gsdma3 structure shortly prior to the CASP12 prediction deadline provided a homologous template for T0948 (PDB 5B5R 64). T0948 and the 198-residue Gsdma3 C-terminal domain share 34.5% sequence identity, and superpositioning yields a RMSD of 2.3 Å for 113 shared Cα positions (Figure 5C). However, a 33 amino acid Gsdma3 subdomain between α10 and the last helix (Gsdma3 α12 or GSDMB α11) corresponds to a disordered loop in GSDMB that is too short to form an analogous subdomain (Met366–Tyr382) 63, and therefore could not be predicted. This Gsdma3 region is functionally important because it interacts with a segment on the N-terminal domain that is involved in membrane disruption 64.
A total of 422 predictions for T0948 were deposited in CASP12, and 150 of them had GDT_TS scores > 70. The Gsdma3-based models for T0948 were quite accurate for the well-aligned core 7-helix bundle region, but not for the functionally important polymorphism loop. The superposed structures of GSDMB_C and the highest GDT_TS scored model, from group 251 (myprotein-me server, Skwark and colleagues) illustrate the similarity within the core 7-helix bundle (Figure 5D). However, the predictions for the polymorphism loop conformation (i.e. residues Arg299–Val322 of GSDMB corresponding to Arg54-Val77 in T0948) were poor, presumably because the GSDMB loop is 8 residues longer than that of Gsdma3 and lacks significant sequence homology 63 (Figure 5A). Encouragingly, many top models (although not TS251_1-D1, Figure 5D) predicted the α′ helix (Pro309-Ser313) in the polymorphism loop. However, its length was overestimated and its orientation was wrong in all cases. Examination of the CASP analyses tables including position-specific alignment shows that large differences exist even for the polymorphism loop closest to the crystal structure (e.g., group 330, Laufer_seed, Perez and colleagues - Figure 5E). No group reproduced in their prediction the 4-residue helical turn preceding Ser306, a key structural difference that distinguishes the GSDMB produced by Crohn’s, ulcerative colitis, and asthma patients from that of healthy individuals. Thus, the GSDMB example shows that prediction of the conformations of large loops that deviate substantially from their template structures has not yet achieved the level of accuracy required for drawing conclusions about structure-function relationships.
6. Receptor-binding domain of the Whitewater Arroyo Virus glycoprotein: studying pathogenicity from a structural point of view (CASP: T0877; PDB: 5NSJ) – provided by Amir Shimon and Ron Diskin
Some enveloped RNA viruses from the Arenaviridae family attach to Transferrin Receptor 1 (TfR1) and use it as a cellular receptor for cell entry. For binding to TfR1, they utilize the receptor-binding domain (GP1) that is part of their class-I trimeric spike complex. Several arenaviruses can infect humans and cause acute disease due to their ability to bind the human-TfR1 (hTfR1) in addition to TfR1 from rodents and bats that naturally serve as hosts for these viruses.
Since both pathogenic and non-pathogenic arenaviruses use similar rodent-TfR1 receptors but only the pathogenic viruses can utilize hTfR1, we wanted to understand what the structural barriers are that prevent non-pathogenic viruses from doing so. This information is important if we want to understand the molecular mechanisms that may allow non-pathogenic viruses to emerge into the human population as novel pathogens. To compare non-pathogenic and pathogenic arenaviruses, we crystallized the GP1 domain from the non-pathogenic Whitewater Arroyo virus (WWAV) 66,67 and compared its structure with the GP1 from the pathogenic Machupo arenavirus determined in complex with hTfR1 by the Harrison group68.
This structural information allowed us to analyze a putative interaction of WWAV-GP1 with hTfR1 (Figure 6A). We found several structural features that preclude hTfR1 usage 69, including electrostatic incompatibility between WWAV-GP1 and hTfR1 (Figure 6B). Interestingly, similar incompatibilities equally affect the pathogenic viruses. These pathogenic viruses can nevertheless use hTfR1 due to more elaborated sets of weak interactions throughout their binding sites that allow them to energetically overcome the structural incompatibilities 69. Thus, viruses within this family make different interactions with TfR1, giving rise to a range of affinities toward TfR1, which ultimately determine their potential to utilize hTfR1 despite the structural barriers 69.
Figure 6. The structure of WWAV-GP1 compared to the top three models.

(A): Ribbon diagrams of the WWAV-GP1 colored in rainbow and shown in a putative complex with hTfR1 (surface representation) (PDB ID: 3KAS). (B): A potential charge-repulsion between two negatively charged groups on WWAV and hTfR1 that was identified using this analysis. (C): Comparison of the top three models from ‘MULTICOM-construct’, ‘MULTICOM-novel’, and ‘GOAL’ (designated S236, S345, and S220, respectively) with WWAV-GP1. (D): A close-up view comparing the loops of WWAV-GP1 that interact with hTfR1 to the top model. Structures were rendered using PyMOL (www.pymol.org).
This study required an accurate structure of WWAV-GP1. Sequence conservation of viral glycoproteins like the GP1 domains from TfR1-tropic viruses is generally very low, due to rapid evolution under strong immunological pressure (i.e. 24 % identity between the GP1s of Machupo and Whitewater Arroyo viruses). Thus, a modeling approach may not fully reveal the fine details that are needed for such an analysis. In CASP12, the GP1 domain from WWAV was designated as a target for automated servers (T0877). Most of the predictors were able to provide models that faithfully represent the overall structure of this domain with GDT_TS > 50. We compared the top three models to the crystal structure of WWAV-GP1 (Figure 6C). ‘MULTICOM-construct’, ‘MULTICOM-novel’, and ‘GOAL’ achieved the best overall ranking with GDT_TS of 67.8, 68.7, and 70.3, respectively. The central β-sheet and the α-helices were modeled correctly along the primary structure but slightly deviate from their real positions in space. Interestingly, a disulfide bond that WWAV has but is not shared by GP1 domains for which structural information was previously available, was not modeled although the cysteine residues were placed in their correct orientations. Since this bond influences the local geometry of a near-by loop, the modelers were unable to accurately model its conformation. In general, the conformations of the loops from the various predictors cluster together, but deviate from the real structure of WWAV-GP1. Considering the goal of our study, this is a major drawback since some of the important contacts that GP1 makes with TfR1 are mediated through these loops (Figure 6D). Thus, modeling loops is a challenging task and since loops are often involved in protein-protein interactions, bona fide structural information would be preferred for the type of analysis that we have performed.
7. Structure features and biological significance of a new glycoside hydrolase family 141 founding member BT1002 (CASP: T0912; PDB: 5MPQ) - provided by Didier Ndeh, Arnaud Baslé and Harry J. Gilbert
Rhamnogalacturonan II (RG-II) is a primary cell wall pectin of plants present in fruits, vegetables, wine and chocolate. It is the most complex carbohydrate known and despite its remarkable structural complexity, it is highly conserved across the plant kingdom 70,71. RG-II is a complex 10 kDa acidic polysaccharide 70,72. To elucidate how the human gut microbiota (HGM) has evolved to utilise complex glycans we investigated the RG-II degradome of the prominent gut microbe Bacteroides thetaiotaomicron. The organism is capable of metabolising RG-II in in-vitro growth experiments, and combined transcriptomic and biochemical data revealed that at least 23 enzymes induced in culture conditions with RG-I as the sole carbon source are directly involved in its metabolism 72,73. The organism is capable of cleaving 20 out of the 21 unique glycosidic linkages in RG-II and biochemical evidence suggests that the CASP12 target T0912 (BT1002) is one of 7 novel enzymes recruited by B. thetaiotaomicron to achieve this purpose72.
BT1002 is a novel α-L-fucosidase and founding member of the new glycoside hydrolase family 141 (GH141) 74. BT1002 targets the complex tetrasaccharide structure mXFRA found in RG-II. The importance of BT1002 in RG-II metabolism is exemplified by the fact that genetic mutants lacking the enzyme are unable to metabolise mXFRA during in-vitro growth on RG-II, leading to accumulation of mXFRA in the growth medium. This implies that the enzyme is unique and indispensable for the breakdown of its target in RG-II.
We solved the BT1002 phase problem using selenomethionine single-wavelength anomalous diffraction. The crystallized construct diffracted to a resolution of 2 Å. It comprises 624 amino acids of which 605 were modeled (PDB ID 5MQP). BT1002 contains 12 α-helices and 50 β-strands forming 6 sheets. The catalytic domain is made of the C-terminal and N-terminal ends of the protein (residues 19-113 and 300-618 respectively), which fold into a β-helix. An extended loop of the catalytic domain comprising residues 323 to 370 mediates contacts between the β-helix and the β-sandwich domains (D1 and D2) made of residues 114 to 299. Domain D3 is flanked by two α-helices (Figure 7, panel A). While efforts to identify specific active site interactions between BT1002 and its tetrasaccharide target are ongoing, we identified two aspartates (Asp523 and Asp564) as potential catalytic residues through site directed mutagenesis72. The residues are 6.1 Å apart in a pocket suggesting an acid-base assisted double displacement mechanism. The closest structural homolog we found using a DALI search with the catalytic domain was a GH-120 β-xylosidase (PDB code 3VSU) with a root mean square deviation of 2.7 Å. While the active site pockets are conserved their primary sequence (20% identity), their catalytic centers and their specificities are very different.
Figure 7. (A): Cartoon representation of BT1002.
(5MPQ, chain A) aligned with T0912TS349_1 in pymol (sequence alignment followed by structural superposition with Cα atoms only). Residues are colored by a RMSD gradient (dark blue is a good alignment and red are higher deviations). Residues not used are colored grey. The domain are labelled D1 to D3. (B): Binding pocket surface representation. The predicted model (T0912TS303_1) surface is represented in solid dark grey and the PDB model surface in yellow mesh. The putative catalytic residues in the predicted model are colored magenta and red in the PDB model.
The BT1002 protein was included in CASP as target T0912 and was evaluated in the full-length and domain-based modes (domain D1: residues 24-113 and 299-622; D2: 114-154 and 258-299; D3: 155-257). Out of the 456 models submitted on the target, 175 models scored 40 GDT_TS or higher. Considering large size of the target and its multi-domain composition, such prediction can be considered as successful. The best top ranked model (i.e. the best model among models assigned as #1 by each of the groups) was submitted by the wfMESHI-TIGRESS group (T0912TS303_1, GDT_TS=48.2). To illustrate how well different regions of the protein are predicted, we aligned the BT1002 crystal structure with a mid-range model (T0912TS349_1, HHPred1, GDT_TS=40.8). The result is presented in Figure 7 (panel A) where colder colors indicate a close match and hotter colors a higher RMSD (residues in grey were not used). The backbone of the catalytic domain D1 was very well predicted with the 11 parallel β-strand stacks of the β-helix correctly identified (194 models scored above GDT_TS=50 with the best model’s GDT_TS=66.4). This is not surprising as such a domain is well described with multiple examples in the PDB data bank. Side chain positioning is more distant to the crystal protein structure. For instance, the catalytic residues Asp564 and Asp523 are separated by about 9 Å in the best D1 model rather than 6.1 Å in the crystal structure. The domain D2 was also correctly modeled overall (85 models scored above GDT_TS=50 with the best model’s GDT_TS=77.7). The third domain was poorly predicted, with the best model scoring only GDT_TS=42.0. Nevertheless, this model (T0912TS247_1-D3) correctly predicted the β-strands and the β-sandwich, though with a register error. As a consequence, the flanking α-helices were missed. The overall fold prediction accuracy is essential for this target. Indeed, the binding pocket important for ligand recognition and binding, is not only constituted by the surface of the catalytic domain D1 and its extended loop but also the surface of domain D3. Therefore we had to consider only the full target predictions. Figure 7 (panel B) shows an overlay of the best predicted model (T0912TS303_1) and the experimental model (5MQP). The PDB model surface represented as a yellow mesh is clearly smaller than the predicted model surface in dark grey. Additionally, the putative catalytic residues are more distant in the predicted model (magenta surface) than in the PDB model (red mesh).
In summary, the BT1002 structure prediction results are very encouraging but show the challenges facing the community in order to elucidate complex biological functions.
8. A cryptic DNA-binding protein from Aedes aegypti (CASP: T0890; PDB: N/A) - provided by Reinhard Albrecht and Marcus D. Hartmann
During their development, pupating insects (holometabola) may accumulate uracil in the DNA of larval tissues. The protein UDE has been implicated in the development of holometabola in the late larval stages as a uracil-DNA degrading factor. At the time of its experimental identification in Drosophila larval extracts, homologs were only found in holometabola75. Its sequence revealed a domain organization with a tandem sequence repeat in the N-terminal half, and several conserved motifs in the C-terminal half of the protein. In some holometabola, only one copy of the N-terminal tandem repeat is found, and it was shown for UDE from Drosophila melanogaster (DmUDE), that the first copy of the tandem repeat may be functionally dispensable76. Now, however, with more genomes sequenced, sequence searches result in a more diverse picture, including UDE proteins with a more complex domain arrangement in holometabola and homologs in plant-pathogenic fungi.
With its developmental implications and narrow phylogenetic distribution, UDE posed an attractive target for the development of insecticides specific to holometabola, or fungicides specific to certain plant pathogens. Initially, UDE caught our attention as we just had identified a novel uracil-binding mode in the protein cereblon, which we thought could be linked to the recognition of uracil in DNA, and which can be mimicked by the binding of the drug thalidomide 77,78. Inspired by the topicality of the Zika virus at that time, we decided to tackle the UDE protein from the yellow fever mosquito Aedes aegypti (AaUDE; AAEL003864), a major virus vector.
AaUDE is a canonical UDE protein with the N-terminal tandem repeat and a length of 306 residues; In vitro, it showed DNA binding properties similar to DmUDE. While full-length AaUDE withstood crystallization attempts, a recombinant protein corresponding to a proteolytic fragment encompassing residues 87-277, thus omitting the first copy of the tandem repeat and the potentially flexible C-terminal end, yielded well-diffracting crystals. The structure, which we solved via SAD phasing using a platinum derivative (CASP target T0890), shows an all-helical two-domain protein. The N-terminal domain corresponds to the second copy of the tandem repeat and forms a three-helix bundle, while the C-terminal half is folded into a compact domain consisting of six helices; the interfacial surface area between the two domains amounts to about 500 Å2 (Figure 8A).
Figure 8. The crystal structure of AaUDE(87-277) in comparison to the best DALI matches and CASP predictions.

(A) The full crystal structure in cartoon representation. (B) The crystal structure (red) superimposed with the best DALI matches for the N-terminal (PDB: 3UN9; DALI Z-score 7.5) and the C-terminal domain (PDB: 3TD7; DALI Z-score 10.1). (C) The two best CASP predictions for the N-terminal domain (D1), models T0890TS236_1 (MULTICOM-construct) and T0890TS486_1 (TASSER), yielded a GDT_TS of 68.0 and 67.7 for D1 and of 30.0 and 31.8 for the whole structure. (D) The best CASP predictions for the C-terminal domain (D2). T0890TS250_1 (Seok-server) yielded a GDT_TS of 74.8 for D2 and 44.7 for the whole structure. T0890TS119_1 represents the three almost identical models T0890TS119_1 (HHPred0), T0890TS349_1 (HHPred1) and T0890TS313_1 (HHGG), which yielded a GDT_TS of 69.8, 69.8 and 70.5 for D2 and of 40.8, 40.8 and 41.0 for the whole structure. T0890TS464_1 (tsspred2) yielded a GDT_TS of 59.2 for D2 and 33.4 for the whole structure.
A DALI search with the full structure returned many hits for the N-terminal domain, but only one hit for the C-terminal domain. For the N-terminal domain, the hits yielded Z-scores of up to 7.5. It had previously been predicted to be a three-helix bundle and had been implicated in DNA binding76. This notion is supported by our crystal structure, as this domain presents extended stretches of positively charged residues along its helices. The highest-scoring DALI hit was, however, the single hit for the C-terminal domain. With a Z-score of 10.1 it matches a non-conserved additional C-terminal domain of the mimivirus sulfhydryl oxidase R596, which had previously been described as an ORFan domain of novel fold, and which is functionally not understood79 (Figure 8B).
For the CASP predictors, AaUDE posed a tough but not intractable target. There were many good predictions for the simpler N-terminal domain (T0890-D1), and a few good predictions for the C-terminal domain (T0890-D2). Curiously, none of the groups could predict both domains. The five best overall models, ranging between a GDT_TS of 44.7 and 33.4 (submitted by the Seok-server, HHGG, HHPred1, HHPred0 and tsspred2) owe their accuracy to the correctly identified similarity of the C-terminal domain to the aforementioned mimivirus ORFan domain. They fail, however, to reasonably predict the N-terminal domain. The overall models from rank six on mostly contain fair-to-good predictions of the N-terminal but not the C-terminal domain, as they miss the link to the mimivirus protein. The best-matching predictions for the individual domains are depicted in Figure 8C and 8D. Despite the good predictions for the individual domains, the inter-domain interface and thus the relevant biological assembly could not be predicted.
9. The snake adenovirus 1 LH3 hexon-interlacing protein (CASP: T0909; PDB: 5G5N and 5G5O) – provided by Thanh H. Nguyen, Abhimanyu K. Singh, and Mark J van Raaij
Adenoviruses are non-enveloped double-stranded DNA viruses with a diameter of around 100 nm 80. At the vertices of the icosahedral adenovirus particles, a pentameric penton base protein is located, while the faces are covered with trimeric hexon proteins. Fiber proteins protrude from the penton bases and are responsible for primary host cell recognition 81. Internalization of human adenoviruses is known to be mediated by the penton base protein interacting with cell surface integrins, but some other adenoviruses lack known integrin-binding motifs in their penton base sequence. Five genera of adenoviruses are known, one of which is the Atadenovirus genus. Atadenoviruses infect birds, snakes, lizards, ruminants or possums. The LH3 gene is a genus-specific atadenovirus gene found at the left end of the genome. The LH3 gene product is believed to be involved in stabilization of the viral capsid 82,83. The LH3 protein forms trimeric protrusions on the faces of the atadenovirus particle 83. In total, four LH3 trimers are present on each of the faces, and 80 in the entire atadenovirus particle.
The Snake Atadenovirus 1 LH3 protein (CASP target T0909) was expressed in E. coli, crystallized, the structure was solved using SAD from a mercury derivative crystal and refined using native data of a different crystal form at 2.0 Å resolution84. Evidence of proteolysis was observed and is consistent with the first 25 residues missing from the experimentally determined structure (Figure 9). The structure revealed a compact, knob-like trimer of right-handed β-helices, as predicted by the BetaWrap server 85. The missing part was evident when fitting the structure into an 11 Å cryo-EM map of SnAdV-1 84.
Figure 9. Crystal structure of SnAdV-1 LH3 in comparison with the best CASP12 model.

Superposition of one of the best predicted regular (monomeric) models (T0909TS303_1, magenta) onto a monomer (left; side view) and the trimer (middle; top view, C-termini closest to the reader) of the experimentally determined structure (cyan). On the right, one of the best predicted trimeric models (T0909TS247_1o, orange) is shown viewed from the bottom, N-termini closest to the reader. Chain termini are indicated where possible and a loop that is disordered in two monomers of the trimer in the crystal structure is highlighted by asterisks.
Each LH3 monomer contains eleven β-helical rungs stacked on top of each other. Each β-helical rung consists of three β-strands that form long parallel β-sheets with their counterparts from the other rungs. The β-sheets are named PB1, PB2 and PB3, following the nomenclature proposed by Mayans et al 86. Turns between β-strands are named T1 (between PB1 and PB2), T2 (between PB2 and PB3), and T3 (between PB3 and PB1). PB1 connects to PB2 mainly by short β-turns, at the trimer interface, while PB2 connects to PB3 and PB3 to PB1 by longer loops.
Amino acid ladders are observed in the structure of the LH3 protein, as is common for β-helical structures 86,87. Asparagine-, isoleucine- and phenylalanine- ladders are found in the core of each monomer, stabilizing the basic β-helical architecture of the monomer. The asparagine ladder (residues 193, 214, 248 and 291) is located right at the T1 turn, while the isoleucine (residues 68, 98, 134, 167, 311, 357) and phenylalanine (residues 103, 139, 172, 195) ladders are found in the PB1 and PB2 sheets, respectively. A ladder containing isoleucines and a leucine (Ile84, Ile147, Ile179 and Leu125) is present in the PB3 sheet. It is possible that the hydrogen bonds in the asparagine ladder help avoid out-of-register interactions when the β-helix folds.
A structural homology search using the DALI server88 showed the best matches for tailspikes from Bacillus phage phi29 89, Shigella phage Sf6 90 and Salmonella phage P22 91. Structure superposition between SnAdV-1 LH3 and Sf6 TSP with its ligands revealed a strikingly similar β-helix topology, despite the low sequence identity (13%). It should be noted that the Shigella phage SF6 tailspike has endorhamnosidase activity. At the binding site, loops from T2 and T3 turns were found to be involved in the interaction with the lipopolysaccharide substrate. Superposition of the two structures do not show conservation of the loop conformations, however, it is possible to form a potential ligand binding groove in the structure of SnAdV-1 LH3 either between two subunits or on the surface of a single monomer (like in the phage P22 tailspike 91). Evidence for non-conserved binding sites among bacteriophage tailspike proteins was discussed previously 92. The structural similarity with bacteriophage tailspikes and its location on the viral cell surface suggested the LH3 protein may be involved in binding a (carbohydrate) ligand. However, we have not been able to demonstrate this or a role for the LH3 protein in host interaction.
Structural superposition of the crystal structure and the best CASP12 models showed they share a similar β-helical fold. The β-helix motif was predicted correctly. The best model, with a DALI z-score of 30.4, suggested a structure comprising three anti-parallel β-sheets PB1, PB2 and PB3 connected by β-turns T1, T2 and T3, as observed in the experimentally determined structure. The length and orientation of β-strands are represented quite accurately, although there are some mismatches. Surface loop conformations are, as expected, predicted much less reliably. Structural superposition of the other CASP12 models also showed that the main β-helix is generally predicted accurately, but loop conformations are different. Most of the β-strands in the models have correct length and location, which is impressive given the low sequence identity (less than 15%) of the SnAdV-1 LH3 protein to known structures. The N-terminal α-helix is identified and, for the most part, the asparagine and hydrophobic amino acid ladders are predicted correctly. It is noteworthy that the N-terminal, virus-facing part of the protein, appears to be somewhat better predicted than the C-terminal, virus-distal part.
It should be kept in mind that SnAdV-1 LH3 protein is a homo-trimer. The standard predictions did not use this given feature. However, some of the predictions that took the homo-trimeric state into account correctly predicted the trimerization interface and reproduced almost 40% of the native interface contacts. This, in turn, might have assisted us in solving the structure by molecular replacement without having to resort to a heavy atom derivative (searching for independent monomers is also possible, but more difficult than searching for correctly assembled trimers). The availability of a SAXS envelope might also have helped to derive an accurate trimeric model computationally, even without prior knowledge of the oligomeric state (see SAXS paper, this issue).
10. Crystal structure of an ice binding protein from an Antarctic Biological Consortium (CASP:T0883; PDB:6EIO) – provided by Valentina Nardone, Marco Mangiagalli and Marco Nardini)
Organisms exposed to permanent subzero temperatures or seasonal temperature dropping are protected from freezing damage by producing Ice Binding Proteins (IBPs) which adsorb to the ice surface and stop ice crystal growth in a non-colligative manner93. A measurable effect of ice binding is that IBPs decrease the water freezing temperature, thereby creating a thermal hysteresis (TH) gap between the melting and the freezing temperature94. TH has been explained by the fact that IBP induces a micro curvature on the ice surface. In this way, ice growth is restricted in between the adsorbed IBP and the curved surface. This makes the association of other water molecules thermodynamically unfavorable, causing the decrease of water freezing temperature. The second activity of IBPs is the ice recrystallization inhibition (IRI), which prevents the growth of large ice crystals at the expenses of smaller ones. Growth of these large crystals causes dehydration and cellular damage 95. Because of these properties, in recent years the potential application of IBPs has been recognized in several different fields in which materials and substances have to be preserved from freezing, including food processing, cryopreservation, cryosurgery, fishery and agricultural industries, and anti-icing materials development93,96.
IBPs have been isolated in different species, including fishes, insects, plants, algae, fungi, yeasts and bacteria. Proteins from different sources share the ability to bind ice crystals, but they can exhibit very diverse 3D structures, including small globular proteins, single α-helices, four helix bundles, polyproline type II helix bundles and β-solenoids. This structural diversity suggests that ice binding activity arose independently multiple times in evolution 93.
As a result, it is very difficult to determine the structural features important for ice binding. Structural studies may provide useful information on the ice-binding sites and on their mechanism of action. For instance, structural comparison of IBPs with different folds may highlight common general features, such as the presence of single/multiple flat surfaces and their hydrophobic/hydrophilic residue distribution, in order to grant an efficient ice binding. Furthermore, many IBPs contain threonine-rich repeats, such as Thr-X-Thr or Thr-X-Asx, usually located on the protein surface. The comparison of position/conformation of these repeats in structurally diverse IBPs, coupled with site-directed mutagenesis studies, could help recognize their role in ice binding.
We focused our attention on EfcIBP, a bacterial IBP identified by metagenomic analysis of the Antarctic ciliate Euplotes focardii and the associated bacterial consortium. Tested for its effects on ice, recombinant EfcIBP shows atypical combination of TH and IRI activities not reported in other bacterial IBPs. Its TH activity was only 0.53 °C at 50 μM, but it had one of the highest IRI activities described to date, with an effective concentration in the nanomolar range. As a result, EfcIBP effectively protected purified proteins and bacterial cells from ice damages. Furthermore, the presence in the EfcIBP sequence of a secretion signal seems to indicate that EfcIBP might be either concentrated around cells or anchored at the cell surface, permitting the entire consortium to thrive/survive at challenging temperatures 97. To shed light on the antifreeze properties of EfcIBP at the molecular level it is crucial to elucidate its ice-binding mechanism through a combination of structural and molecular biology studies. Therefore, we solved the EfcIBP structure by means of X-ray crystallography.
EfcIBP crystals diffracted to atomic resolution (up to 0.84 Å), and the EfcIBP structure was solved by molecular replacement with the crystal structure of the IBP from the antarctic bacteria Colwellia sp. (PDB-code 3WP9; DALI Z-score of 32.3, residue identity of 38%) as a search model 98. The overall structure of EfcIBP consists of a right-handed β-helix with a triangular cross-section formed by three faces made by parallel β-sheets, and by an additional single 5-turn α-helix, aligned along the axis of the β-helix. The first face of the β-helix (9 β-strands) is screened from the solvent region by the long α-helix and by the N-terminal region. This protein surface is, therefore, not suited for the interaction with ice crystals. The second face (8 β-strands) is flat and regular, while the third (8 β-strands) is only partly flat, with two β-strands which markedly diverge towards the exterior of the protein body. The latter two faces are fully exposed to the solvent region and, therefore, potentially suited for the interaction with ice crystals. Interestingly, both faces host multiple threonine-rich repeats, a feature not found so far in IBPs with fold similar to EfcIBP.
Overall, the CASP12 results on target T0883 indicate that right-handed β-helix can be predicted extremely well. All β-strands of the three faces of the EfcIBP structure are correctly positioned as well as the 5-turn α-helix, aligned along the β-helix axis. It should be noted, however, that the β-strand located immediately after the α-helix is correctly placed within the β-helix fold in the model but is shifted by two residues, such that the preceding loop is two residues longer and the following loop two residues shorter than in the experimental structure.
The top ten ranked models (CASP GDT_TS score >89.0) are characterized by an RMSD of ~1.4 Å for the core of the protein (181 Cα pairs over 207 residues). The structure of the first 9 N-terminal residues is not predicted correctly partly because this region is shorter in the homologous proteins used as templates, partly because its conformation might be selected by crystal contacts and, therefore, difficult to predict. The CASP12 models contain a deletion, correctly identified at the top of the right-handed β-helix, where a small cap subdomain of about 12 residues is present in homologous proteins. In this region, however, the Gly-Pro-Pro sequence at the closure of the deletion does not superimpose well with the corresponding EfcIBP crystal structure.
Finally, it is worth noting that the overall quality of the CASP12 prediction does not seem to improve significantly when multiple protein templates are used for modeling instead of a single template. This is probably due to the high structural conservation and rigidity of the β-helix scaffold which tolerates insertion/deletion of several residues without any significant perturbation of the core structure and which is reproduced similarly in all protein templates.
11. The TRXL1 domain of Chaetomium thermophilum UGGT (CASP: T0892; PDB: 5MU1, 5MZO, 5N2J and 5NV4) – provided by Pietro Roversi, Alessandro T. Caputo, Johan C. Hill and Nicole Zitzmann
One of the last unsolved mysteries of the eukaryotic endoplasmic reticulum glycoprotein folding quality control (ERQC) machinery is its single checkpoint enzyme, the ER UDP-glucose glycoprotein glucosyltransferase (UGGT). Once monoglucosylated by this enzyme, glycoproteins are retained in the ER bound to the lectins calnexin and/or calreticulin and the associated chaperones and foldases that assist their folding99. The mechanism by which UGGT recognizes and glucosylates a large variety of misfolded glycoprotein substrates remains unknown.
The N-terminal ~1200 residues of UGGT harbor the enzyme’s misfold sensing activity 100,101. The lack of any obvious sequence homology of this portion of UGGT with proteins of known fold led to the creation of a UGGT-specific protein fold family (Pfam family PF06427) which gathers all known eukaryotic UGGT N-terminal sequences. The most recent secondary structure and domain boundary predictions for UGGT detected three thioredoxin-like (TRXL) domains in this region102,103. The canonical TRXL fold (Pfam family PF13848) comprises a thioredoxin fold (a four-stranded β sheet sandwiched between three α-helices, TRX=βαβ–αββα Pfam family PF00085, red in Figure 10), modified by the insertion of a 4-helix subdomain (TRXL=βαβ–αααα–αββα blue in Figure 10)104,105.
Figure 10. The TRXL1 domain of CtUGGT.

(A) In blue, the CtUGGT TRXL1 N-terminal α-helical subdomain (residues 43-110). In red, the TRXL1 thioredoxin subdomain (residues 111-216). The disulphide bridge C138-C150 is represented as spheres. (B) The structure of the closest structural homologue to CtUGGT TRXL1, Staphylococcus aureus DsbA, with the α-helical insertion subdomain (residues 63-129) in blue and the thioredoxin subdomain (residues 14-62 and 130-177) in red. In (A) and (B) N- and C-termini are denoted by the letters “N” and “C”, respectively. (C) The superposition of the top ten CASP12 T0892 models, overlayed on the CtUGGT TRXL1 crystal structure in the region of the N-terminal helical subdomain and the first helix of the thioredoxin subdomain. The CtUGGT TRXL1 crystal structure is colored and represented as in panel A. The top ten CASP12 T0892 models are in ribbon representation and colored as follows: T0892TS011_1:green; T0892TS011_2: cyan; T0892TS017_1: magenta; T0892TS017_2: yellow; T0892TS017_5: grey; T0892TS411_2; T0892TS017_3: salmon pink; T0892TS079_5: violet; T0892TS479_3: steel blue; T0892TS320_4: orange. A black star marks the hinge between the helical subdomain and the thioredoxin subdomain. A dotted circle marks the first helix in the thioredoxin subdomain. (D) The superposition of the top two CASP12 T0892 models (T0892TS011_1 and T0892TS011_2, in green and cyan respectively, in ribbon representation), overlayed on the CtUGGT TRXL1 crystal structure in the region of the C-terminal thioredoxin subdomain, without its first α-helix. The CtUGGT TRXL1 crystal structure is colored and represented as in panel A. The wrongly predicted first two strands of the thioredoxin subdomain are circled, and an asterisk marks the incorrectly predicted α-helix for the stretch of residues 151-164 of CtUGGT TRXL1.
To aid our understanding of UGGT structure and function, we determined four distinct crystal structures of Chaetomium thermophilum UGGT, aka CtUGGT 106. An unexpected structural feature of the UGGT molecule is the unusual subdomain structure of the first thioredoxin-like domain (TRXL1), encoded by residues 43-216 in CtUGGT. The published sequence–based secondary structure predictions in this region was rather accurate, with most helices and sheets correctly predicted from sequence – but the UGGT TRXL1 domain boundaries were not well predicted 104,105.
Indeed, the UGGT TRXL1 domain folds with sequential pairing of a helical subdomain with a thioredoxin subdomain (blue and red in Figure 10), while all other known TRXL domains present a helical subdomain as an insertion within the thioredoxin subdomain (see for example in Figure 10B the closest structural homologue of CtUGGT TRXL1, Staphylococcus aureus DsbA, PDB ID 3BD2). The CtUGGT crystal structures also reveal that the CtUGGT TRXL1 domain harbors a disulfide bridge between Cys138 and Cys150 (represented as spheres in Figure 10A).
We submitted the CtUGGT TRXL1 sequence to CASP12 (target T0892) in order to test prediction methods for their ability to model i) its non-canonical subdomain structure, in which an N-terminal α–helical subdomain is followed by a C-terminal thioredoxin subdomain and ii) the presence of a disulfide bridge between CtUGGT TRXL1 C138 and C150.
We compare here the top 10 CASP12 T0892 models (as ranked by the GDT_TS score on the CASP12 results server) to the coordinates of the TRXL1 domain in the 2.8 Å CtUGGT crystal structure (PDB ID 5NV4), residues 43-216. The overall RMSDCα across the ensemble of the top ten T0892 models is 10.7 Å over 174 Cαs107. All these CASP12 T0892 models predict an N-terminal 4-helix subdomain followed by a C-terminal subdomain which resembles to various degrees a TRX fold. None of the top T0892 CASP12 models predicts the CtUGGT TRXL1 C138-C150 disulfide bond.
If one restricts the analysis to the CtUGGT TRXL1 N-terminal, helical subdomain (residues 43-110) and the first α-helix (residues 111-126) of the C-terminal, thioredoxin subdomain, the top ten T0892 models align rather well with each other and with the crystal structure. The overall RMSDCα for the ten structures over these 84 Cαs is 1.7 Å. The major differences between the CASP12 T0892 models in the 43-126 portion arise at the hinge (CtUGGT residues 108-111, denoted by a black star in Figure 10C) between the helical subdomain and the first α-helix of the thioredoxin subdomain. The two top-ranked CASP12 models (T0892TS011_1 and T0892TS011_2, green and cyan in Figures 10C#x02013;D) show a different hinge region from the rest. As a result of these differences, in the same top-ranking two models, the relative angle between the N-terminal helical subdomain and the first helix of the thioredoxin subdomain also differs from the crystal structure and the rest of the T0892 CASP12 ensemble of models. The CtUGGT 111-126 α-helix is marked by a dotted circle in Figure 10C.
In the C-terminal thioredoxin subdomain (residues 111-216), the top ten CASP12 T0892 models align poorly with each other and with the crystal structure of the target. The overall RMSDCα for the ten models over these 84 Cαs is 9.5 Å107. Only the two top ranking CASP12 T0892 models (T0892TS011_1 and T0892TS011_2, green and cyan in Figures 10C–D) correctly contain a 4-stranded β-sheet at the center of the TRXL1 thioredoxin subdomain. Even restricting attention to these two models only, across residues 127-216 the RMSDCα between the models and the crystal structure is still as high as 6.5 Å over 90 Cαs107 (see Figure 10D). In particular, the first two β-strands of the thioredoxin subdomain β-sheet in the models do not superimpose well on the same β-strands in the crystal structure (circled in Figure 10D). Moreover, in both models, the stretch of sequence 151-164 – which immediately follows those strands - is wrongly predicted to fold as an α-helix (marked by an asterisk in Figure 10D) which is not present in the crystal structure.
Overall, none of the models predict the CtUGGT TRXL1 C138-C150 disulfide bond, and the 128-181 region between the first TRX helix and the third TRX strand is not well defined in any of the models. On the other hand, the best CASP12 T0892 models are successful in predicting the structure of the N-terminal 4-helix subdomain, and the two top-scoring ones also manage to correctly predict that the domain is a linear fusion of an N-terminal 4-helix subdomain and a C-terminal subdomain of TRX fold. In summary, as far as this target was concerned, the CASP12 predictors did well, but did not put us out of our job just yet.
12. Structural characterization of the third cohesin from Ruminococcus flavefaciens scaffoldin protein, ScaB (RfCohScaB3) complexed with a group 1a dockerin (RfDoc1a) (CASP: T0921/T0922; PDB: 5AOZ (RfCohScaB3), 5M2O (RfCohScaB3/Doc1a complex) – provided by Pedro Bule, Ana Luisa Carvalho, Carlos M.G.A. Fontes and Shabir Najmudin
The plant cell wall represents a major untapped global source of carbon and energy. Herbivores, in particular ruminants, are able to utilize this energy source thanks to the presence of cellulolytic bacteria in their gastrointestinal tract. Ruminococcus flavefaciens, a Gram-positive Firmicute, is a major symbiont in the rumen. R. flavefaciens possesses a highly intricate multi-enzyme complex, termed the cellulosome, which comprises a range of cellulases and hemicellulases that degrade the structural polysaccharides in a highly efficient and concerted way. The assembly of cellulosomes occurs via highly ordered protein–protein interactions between cohesins (Cohs), which are located in multi-modular macromolecular scaffolds (scaffoldins), and dockerin molecules (Docs), which are found in the enzymes or on the scaffoldins themselves 108,109. Strain FD-1 of R. flavefaciens produces one of the most intricate and potentially versatile cellulosomes described to date. The genome of R. flavefaciens FD-1 encodes 223 dockerin-bearing proteins, which are predominantly enzymes displaying catalytic activity that modifies carbohydrates 110. In this highly elaborate cellulosome, scaffoldin B (ScaB) acts as the backbone to which other components attach. ScaB comprises 9 cohesins of 2 distinct types. Cohesins 1 to 4 are similar to the two cohesins of a second, smaller scaffoldin ScaA, whose dockerin binds to ScaB cohesins 5 to 9 through a different protein-protein specificity. ScaB contains a C-terminal dockerin that binds to the cohesin of cell-surface ScaE providing a mechanism to anchor the entire complex to the bacterial cell. A distinct scaffoldin, ScaC, acts as an adaptor that binds predominantly hemicellulases while connecting to the first type ScaB cohesins, thus serving to increase the repertoire of proteins that can be integrated into the complex. In Clostridium species studied so far, enzyme-borne Docs interact with their cognate Cohs through a dual-binding mode 109. Internal dockerin symmetry allows them to bind to cohesins in either of two orientations resulting in two different Coh-Doc conformations that are related by ~180° rotation. This dual-binding mode results from the characteristic internal symmetry of the Doc primary sequence and is believed to add flexibility to the cellulosomal macromolecular organization. Based on primary sequence similarity, R. flavefaciens dockerins are classified in different groups. Recent studies have shown that groups 3 and 6 R. flavefaciens Docs display a single-binding mode for their target Cohs, i.e. binding occurs in one orientation only 111. Intriguingly, Group 1 Docs also do not seem to possess the internal sequence symmetry required to support the dual-binding mode. Thus, modeling studies are required to predict the correct binding mode between various types of Coh-Doc complexes and to predict which amino acid residues act as molecular specificity determinants.
X-ray crystal structures of the third R. flavefaciens cohesin from ScaB (RfCohScaB3) and group 1 Doc (RfDoc1a) in complex with RfCohScaB3 (Figure 11A) were recently solved, and characterized by comprehensive biochemical analyses 112. RfCohScaB3 forms an elongated nine-stranded β-sandwich in a classical jelly-roll topology. The overall RfCohScaB3 structure is similar to other enzyme-borne Doc-binding Cohs (RMSD of less than 3.0 Å between at least 130 Cα atom pairs) despite the very low sequence similarity (4–12%). The major structural differences are in the Doc-binding interface formed by β-strands 8, 3, 6, 5. In turn, the overall tertiary structure of RfDoc1a is very similar to other enzyme-borne Docs (RMSD of less than 2.0 Å between at least 60 Cα atoms; sequence identity 20–32%). The structure contains two Ca2+ ions coordinated by several amino-acid residues, similar to the canonical EF-hand loop motif described in all other Docs 109. The whole of helix-1 makes predominantly hydrophobic interactions with the Coh, while helix-3 interacts mainly through its C-terminus. Ile-39 and Val-43 on helix-1 of the RfDoc1a and Ala-38 and Leu-79 on the binding platform of RfCohScaB3 were shown to be the key specificity determinants.
Figure 11. Structure of the RfCohScaB3-Doc1a complex.
(A) Structure of RfCohScaB3-Doc1a complex with the dockerin in red and the cohesin in blue. The dockerin N- and C- terminus and the α-helices are labeled, and a transparent gray molecular surface of the cohesin is shown. (B) Superposition of CASP12 prediction models T0921TS220_2_D1 (light blue) and T0921TS166_1_D1 (light green) with RfCohScaB3 crystal structure (black). (C) Superposition of CASP12 prediction models T0922TS005_3_D1 (light blue) and T0922TS077_4_D1 (light green) with the RfDoc1a crystal structure (black). Ca2+ ions are depicted as green spheres.
How do the modeling studies of CASP12 compare with the experimental structural studies of RfCohScaB3 (T0921) and RfDoc1a (T0922) and the complex between them? Predictions for both, the RfCohScaB3 and RfDoc1a were very successful, with 147 models for the former and 143 for the latter (out of 186 total for each of the subunits) having GDT_TS scores greater than 50. The top model for each target and a slightly poorer model scoring ~10 GDT_TS below the top model were chosen for comparative purposes. For RfCohScaB3, these were models T0921TS220 from the GOAL group (GDT_TS of 70.7) and T0921TS452 from the Zhou-Sparks-X group (GDT_TS of 60.7). Superpositions of these models using SSM onto the X-ray structure gave RMSD of 2.1 Å for 127 Cα atoms and 2.4 Å for 120 Cα atoms, respectively (Figure 11B). Though the core structure matches really well, there are major differences in the β6-7 and β8-9 loops and in the β8 strand on the dockerin binding interface. Ala 38 is generally in the correct position, but there is considerable variation in the Leu 79 position. For RfDoc1a, we chose T0922TS005 from the BAKER-ROSETTAserver group (the top scorer with GDT_TS of 83.8) and T0922TS077 from the Falcon_Topo group (GDT_TS of 73.7). Superpositions of these models using SSM onto the X-ray structure gave RMSD of 1.4 Å for 69 Cα atoms and 1.6 Å for 63 Cα atoms, respectively (Figure 11C). Generally, the α-helices 1 and 3 are well modeled and consequently so are the key specificity residues, like Ile 39 and Val 43, with differences mainly in the loop regions and N- and C-termini, which are not involved in Coh recognition. However, the modeling of the RfCohScaB3/Doc1a heterocomplex was less successful, with only three models out of 325 (TS203_4 from the Seok group, TS188_1 from the Chuo_U group and TS208_3 from the SVMQA group) correctly modeling half or more of the intermolecular surface contacts compared to the crystal structure. One reason for this could be incorrect modeling of the loops in the binding surface of the cohesins. In these three predicted complexes the cohesins have similar or less prominent loops between β-strands 6 & 7, and 8 & 9 compared to the crystal structure (cf. Figure 11B), thus avoiding steric clashes when complexing with the cognate dockerin models in the single-binding mode.
In summary, the monomeric subunits of the RfCohScaB3/Doc1a complex (T0921/T0922) were modeled very successfully despite relatively low sequence similarity to available homologues, while the whole complex was not. A more advanced approach is needed to predict whether the cohesin-dockerin interaction can operate through a single-binding mode, where only one binding orientation is possible (mainly through helix-1 or through helix-3 of the dockerin) or through a dual binding mode, where the binding can be in one of two orientations (by either helix-1 or helix-3 to the cohesin binding surface).
Discussion
The paper provides insights into structural and functional details of twelve selected CASP12 targets and analyzes to what extent the most interesting features of the targets are reproduced in the predictions from the standpoint of the authors of the structures. Since specific features of the targets are difficult for CASP assessors to address on a large scale, the authors’ insights represent a critical piece of information for both understanding the utility of models and developing protein structure prediction methodologies and assessment strategies.
The examples presented in the article highlight a series of reoccurring themes that challenge current modeling methods and that therefore deserve attention from method developers.
Oligomers. The structural integrity and biological function of proteins often depends on their quaternary structure and the ability to form specific macromolecular complexes. However, protein oligomerization is not always taken into account in modeling. To address this issue, CASP introduced a separate ‘Assembly modeling’ category in CASP12 113, and will continue to encourage modelers to develop methods for predicting hetero- and homo-oligomeric structures. When modelers do predict oligomers, they are more successful in modeling the subunits than full complexes (e.g., T0884/T0885, T0921/T0922). That is not surprising as prediction of complexes oftentimes involves more than just direct docking of the initial subunit models. One of the complications is conformational changes of protein fragments upon complex formation, as in the T0884/T0885 case. In that complex, the N-terminal segment of the immunity protein T0885 is disordered in the predicted free form, and possibly undergoes disorder-to-order transition upon binding to the toxin putative catalytic domain T0884. This transition is important for the physiological function of the complex. In general, advanced modeling techniques capable of accounting for such scenarios are needed. The authors of homo-multimers (e.g., T0909, T0889) suggest that building multimeric models is beneficial for functional annotation of the proteins and that using information about the oligomeric state can help generate better monomeric models. Sometimes higher-order structures are not only desirable, but necessary to maintain the stability of a protein, as exemplified by some of the CASP12 viral protein targets (e.g., T0880, T0909).
Multi-domain proteins. The majority of proteins exist as multi-domain entities 114, and a sizeable portion of targets in each CASP (1/3 in CASP12) is multi-domain proteins. Constituent protein domains can either have independent functions or contribute to the function of a multi-domain protein in cooperation with other domains. In the latter case, the relative orientation of domains may be important for protein activity. For example, surfaces of two structural domains of target T0912 interact to form a pocket responsible for ligand recognition and binding. Therefore, accurate prediction of the full target was necessary in this case. CASP evaluates multi-domain targets as both per-domain and whole-structure models, however only the domain-based results are usually accounted in the assessors’ reports. A more comprehensive evaluation of multi-domain targets may require additional analysis of the biological relevance of inter-domain architecture, and a separate approach for assessment.
Loops. The specific structure of individual loops is often a key for the understanding protein function. Unfortunately, prediction of loop conformations in general has not achieved the level of accuracy required to confidently establish their role in interactions with small molecules or partner proteins (e.g., T0877, T0889, T0948). The problem is more pronounced for long loops that deviate substantially from their homologues. Taking into account the importance of the problem, future assessors might consider a more careful scrutiny of loop modeling accuracy. This might include assessment of the (a) accuracy of the loop main chain in isolation, (b) relationship of the loop to rest of the structure and (c) errors in protein-ligand interactions. The local-structure evaluation measures (e.g., CAD 115, LDDT 116 or SphereGrinder 117) and interface accuracy measures (Interface Contact Score and Interface Patch Distance 113), which were recently introduced in CASP, can be used for this purpose. It also important to evaluate whether a loop conformation is robustly determined experimentally, and not influenced by the crystal environment.
Conserved residues. When faced with a specific biological system, different sources of information should be checked to yield more accurate models. For example, sequence analysis of the protein family may highlight conserved surface residues potentially involved in complex formation. For example, in CDI systems it is known that immunity proteins typically block access to the active site of the toxins. For the CdiA-CT/CdiI complex (T0884/T0885 target), the three highly conserved residues of the CdiI immunity protein (H73, R76 and D109) interact directly with three highly conserved and presumably catalytic residues of CdiA-CT toxin (H181, H183 and R185) identifying part of the surface in contact. Including this information in predictions would strongly constrain possible solutions.
Disulfides. It is well known that closely spaced cysteines tend to form disulfide bridges in extracellular proteins. However, these were not properly modeled in at least two CASP12 targets (T0877, T0892). Since disulfide bonds play an important role in the stability of some proteins, their proper modeling seems to be an easy and obvious way of improving models.
Alignment. In spite of enormous progress, correct sequence alignment remains a challenge in structure modeling and improved methods are likely to enhance modeling accuracy (T0859, T0883). For example, for target T0859, an alignment register shift resulted in an incorrect secondary structure assignment, which in turn hindered surface exposure of functionally important residues.
Purification tags and signal peptides. A number of CASP sequences included purification tags or signal peptides. If not identified and removed before the modeling, these structural extensions of protein domains might complicate modeling routine. Even though it is usually easy to identify the tags and there are several programs to predict the presence of signal peptide sequences, many structure prediction methods still do not make use of them and attempt to build models of these regions (e.g., T0886, T0922).
Low resolution data. Data from low resolution structure determination experiments are expected to help build atomic-resolution models of proteins. However, the data-assisted component of CASP12 showed that utilizing SAXS or cross-linking data had only marginal effect on the atomic-level structure modeling (T0886, T0909). This outcome shows that either the additional information is too coarse-grained to assist current methods or that the computational community has not been able to fully utilize the potential hidden in the data.
We hope that these general conclusions will guide future CASP assessments and encourage methods developers to address the issues.
Supplementary Material
Acknowledgments
Names of the authors contributing to specific sections are provided in the sections’ titles; concept, abstract, introduction, discussion, editing and coordination - by AK, KF, JM and TS.
CASP experiment and open access fees for this manuscript are supported by the US National Institute of General Medical Sciences (NIGMS/NIH), grant number GM100482.
T0859: Grant sponsor: the Latvian Council of Sciences, grant number: 12.094; Grant sponsor: the European Regional Development Fund, grant number: 2010/0314/2DP/2.1.1.1.0/ 10/APIA/VIAA/052); Grant sponsor: Biostruct-X and the Latvian-French cooperation program Osmosis, grant number: 7869.
T0884/T0885: Grant sponsor: National Institutes of Health, grant number: GM102318 (CWG, CSH & subcontract to AJ); Grant sponsor: National Institutes of Health, grant number: GM094585 (to AJ); Grant sponsor: National Institutes of Health, grant number: GM115586 (to AJ); Grant sponsor: U. S. Department of Energy, Office of Biological and Environmental Research, contract number: DE-AC02-06CH11357 (to AJ)
T0889: Initial funding for structure determination was from the European Community’s Seventh Framework Programme (FP7/2007–2013) under grant agreement No. NMP3-SL-2008-213487. Thanks to Harm Otten and Jens-Christian N. Poulsen for their contributions to structure determination of BjSDH.
T0948: Grant sponsor: National Institutes of Health (NIH), grant number: R01GM102810 (to OH and JM).
T0877: Grant sponsor: Israel Science Foundation (ISF), grant number 682/16 to RD.
T0892: ATC and JCH were funded by Wellcome Trust 4-year Studentships 097300/Z/11/Z and 106272/Z/14/Z, respectively; NZ is a Fellow of Merton College, Oxford.
T0909: Grant sponsor: Spanish Ministry of Economy, Industry and Competitiveness, grant number BFU2014-53425-P (to MJvR).
T0921/T0922: Grant sponsor: Fundaçao para a Ciência e a Tecnologia (Lisbon, Portugal), grant numbers PTDC/BIA-MIC/5947/2014 and RECI/BBB-BEP/0124/2012, and SFRH/BD/86821/2012 to PB.
Abbreviations
- CASP
community wide experiment on the Critical Assessment of Techniques for Protein Structure Prediction
- VLP
virus-like particle
- TfR1
Transferrin Receptor 1
- WWAV
Whitewater Arroyo Virus
- GP1
glycoprotein 1
- RG-II
Rhamnogalacturonan-II
- HGM
Human gut microbiota
- GH
Glycoside hydrolases (GH)
- IBP
ice binding protein
- TH
thermal hysteresis
- IRI
ice recrystallization inhibition
Contributor Information
Andriy Kryshtafovych, Genome Center, University of California, Davis, 451 Health Sciences Drive, Davis, California 95616, USA.
Reinhard Albrecht, Department of Protein Evolution, Max Planck Institute for Developmental Biology, Spemannstraße 35, 72076 Tübingen, Germany.
Arnaud Baslé, Institute for Cell and Molecular Biosciences, University of Newcastle, Newcastle upon Tyne NE2 4HH, UK.
Pedro Bule, CIISA - Faculdade de Medicina Veterinária, Universidade de Lisboa, Avenida da Universidade Técnica, 1300-477 Lisboa, Portugal.
Alessandro T. Caputo, Oxford Glycobiology Institute, Department of Biochemistry, University of Oxford, South Parks Road, Oxford OX1 3QU, England, United Kingdom
Ana Luisa Carvalho, UCIBIO, REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516 Caparica, Portugal.
Kinlin L. Chao, Institute for Bioscience and Biotechnology Research, University of Maryland, Rockville, MD 20850
Ron Diskin, Department of Structural Biology, Weizmann Institute of Science, Rehovot, Israel.
Krzysztof Fidelis, Genome Center, University of California, Davis, 451 Health Sciences Drive, Davis, California 95616, USA.
Carlos M.G.A. Fontes, CIISA - Faculdade de Medicina Veterinária, Universidade de Lisboa, Avenida da Universidade Técnica, 1300-477 Lisboa, Portugal
Folmer Fredslund, Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark.
Harry J. Gilbert, Institute for Cell and Molecular Biosciences, University of Newcastle, Newcastle upon Tyne NE2 4HH, UK
Celia W. Goulding, Department of Molecular Biology & Biochemistry /Pharmaceutical Sciences, University of California Irvine, Irvine, CA 92697, USA
Marcus D. Hartmann, Department of Protein Evolution, Max Planck Institute for Developmental Biology, 72076 Tübingen, Germany
Christopher S. Hayes, Department of Molecular, Cellular and Developmental Biology /Biomolecular Science and Engineering Program, University of California, Santa Barbara, Santa Barbara, CA 93106, USA
Osnat Herzberg, Institute for Bioscience and Biotechnology Research, University of Maryland, Rockville, MD 20850; Department of Chemistry and Biochemistry, University of Maryland, College Park, MD 20742.
Johan C. Hill, Oxford Glycobiology Institute, Department of Biochemistry, University of Oxford, South Parks Road, Oxford OX1 3QU, England, United Kingdom
Andrzej Joachimiak, Midwest Center for Structural Genomics /Structural Biology Center, Biosciences Division, Argonne National Laboratory, USA; Department of Biochemistry and Molecular Biology, University of Chicago, Chicago, IL 60637, USA.
Gert-Wieland Kohring, Microbiology, Saarland University, Campus Building A1.5, Saarbrücken, D-66123 Saarland, Germany.
Roman I. Koning, Netherlands Centre for Electron Nanoscopy, Institute of Biology Leiden, Leiden University Einsteinweg 55, 2333 CC Leiden, the Netherlands; Department of Molecular Cell Biology, Leiden University Medical Center, P.O.Box 9600, 2300 RC, Leiden, The Netherlands
Leila Lo Leggio, Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark.
Marco Mangiagalli, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126, Milano, Italy.
Karolina Michalska, Midwest Center for Structural Genomics /Structural Biology Center, Biosciences Division, Argonne National Laboratory, USA.
John Moult, Institute for Bioscience and Biotechnology Research, Department of Cell Biology and Molecular genetics, University of Maryland, 9600 Gudelsky Drive, Rockville, MD 20850, USA.
Shabir Najmudin, CIISA - Faculdade de Medicina Veterinária, Universidade de Lisboa, Avenida da Universidade Técnica, 1300-477 Lisboa, Portugal.
Marco Nardini, Department of Biosciences, University of Milano, Via Celoria 26, 20133 Milano, Italy.
Valentina Nardone, Department of Biosciences, University of Milano, Via Celoria 26, 20133 Milano, Italy.
Didier Ndeh, Institute for Cell and Molecular Biosciences, University of Newcastle, Newcastle upon Tyne NE2 4HH, UK.
Thanh H. Nguyen, Department of Macromolecular Structures, Centro Nacional de Biotecnologia (CSIC), calle Darwin 3, 28049 Madrid, Spain
Guido Pintacuda, Université de Lyon, Centre de RMN à Très Hauts Champs, Institut des Sciences Analytiques (UMR 5280 - CNRS, ENS Lyon, UCB Lyon 1), 69100 Villeurbanne, France.
Sandra Postel, Institute of Human Virology, University of Maryland School of Medicine, Baltimore, MD 21201, USA.
Mark J. van Raaij, Department of Macromolecular Structures, Centro Nacional de Biotecnologia (CNB-CSIC), calle Darwin 3, 28049 Madrid, Spain
Pietro Roversi, Oxford Glycobiology Institute, Department of Biochemistry, University of Oxford, South Parks Road, Oxford OX1 3QU, England, United Kingdom.
Amir Shimon, Department of Structural Biology, Weizmann Institute of Science, Rehovot, Israel.
Abhimanyu K. Singh, School of Biosciences, University of Kent, Canterbury, Kent, CT2 7NJ, United Kingdom
Eric J. Sundberg, Institute of Human Virology, Department of Medicine and Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, MD 21201, USA
Kaspars Tars, Latvian Biomedical Research and Study Center, Rātsupītes 1, LV1067, Riga, Latvia; Faculty of Biology, Department of Molecular Biology, University of Latvia, Jelgavas 1, LV-1004 Riga, Latvia.
Nicole Zitzmann, Oxford Glycobiology Institute, Department of Biochemistry, University of Oxford, South Parks Road, Oxford OX1 3QU, England, United Kingdom.
Torsten Schwede, Biozentrum /SIB Swiss Institute of Bioinformatics, Klingelbergstrasse 50, 4056 Basel, Switzerland.
References
- 1.Kryshtafovych A, Moult J, Bartual SG, Bazan JF, Berman H, Casteel DE, Christodoulou E, Everett JK, Hausmann J, Heidebrecht T, Hills T, Hui R, Hunt JF, Seetharaman J, Joachimiak A, Kennedy MA, Kim C, Lingel A, Michalska K, Montelione GT, Otero JM, Perrakis A, Pizarro JC, van Raaij MJ, Ramelot TA, Rousseau F, Tong L, Wernimont AK, Young J, Schwede T. Target highlights in CASP9: Experimental target structures for the critical assessment of techniques for protein structure prediction. Proteins. 2011;79(Suppl 10):6–20. doi: 10.1002/prot.23196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kryshtafovych A, Moult J, Bales P, Bazan JF, Biasini M, Burgin A, Chen C, Cochran FV, Craig TK, Das R, Fass D, Garcia-Doval C, Herzberg O, Lorimer D, Luecke H, Ma X, Nelson DC, van Raaij MJ, Rohwer F, Segall A, Seguritan V, Zeth K, Schwede T. Challenging the state of the art in protein structure prediction: Highlights of experimental target structures for the 10th Critical Assessment of Techniques for Protein Structure Prediction Experiment CASP10. Proteins. 2014;82(Suppl 2):26–42. doi: 10.1002/prot.24489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kryshtafovych A, Moult J, Basle A, Burgin A, Craig TK, Edwards RA, Fass D, Hartmann MD, Korycinski M, Lewis RJ, Lorimer D, Lupas AN, Newman J, Peat TS, Piepenbrink KH, Prahlad J, van Raaij MJ, Rohwer F, Segall AM, Seguritan V, Sundberg EJ, Singh AK, Wilson MA, Schwede T. Some of the most interesting CASP11 targets through the eyes of their authors. Proteins. 2016;84(Suppl 1):34–50. doi: 10.1002/prot.24942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duan Q, Zhou M, Zhu L, Zhu G. Flagella and bacterial pathogenicity. J Basic Microbiol. 2013;53(1):1–8. doi: 10.1002/jobm.201100335. [DOI] [PubMed] [Google Scholar]
- 5.Arora SK, Ritchings BW, Almira EC, Lory S, Ramphal R. The Pseudomonas aeruginosa flagellar cap protein, FliD, is responsible for mucin adhesion. Infect Immun. 1998;66(3):1000–1007. doi: 10.1128/iai.66.3.1000-1007.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg HC. The rotary motor of bacterial flagella. Annu Rev Biochem. 2003;72:19–54. doi: 10.1146/annurev.biochem.72.121801.161737. [DOI] [PubMed] [Google Scholar]
- 7.Yonekura K, Maki S, Morgan DG, DeRosier DJ, Vonderviszt F, Imada K, Namba K. The bacterial flagellar cap as the rotary promoter of flagellin self-assembly. Science. 2000;290(5499):2148–2152. doi: 10.1126/science.290.5499.2148. [DOI] [PubMed] [Google Scholar]
- 8.Kim JS, Chang JH, Chung SI, Yum JS. Molecular cloning and characterization of the Helicobacter pylori fliD gene, an essential factor in flagellar structure and motility. J Bacteriol. 1999;181(22):6969–6976. doi: 10.1128/jb.181.22.6969-6976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maki-Yonekura S, Yonekura K, Namba K. Domain movements of HAP2 in the cap-filament complex formation and growth process of the bacterial flagellum. Proc Natl Acad Sci U S A. 2003;100(26):15528–15533. doi: 10.1073/pnas.2534343100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yonekura K, Maki-Yonekura S, Namba K. Complete atomic model of the bacterial flagellar filament by electron cryomicroscopy. Nature. 2003;424(6949):643–650. doi: 10.1038/nature01830. [DOI] [PubMed] [Google Scholar]
- 11.Postel S, Deredge D, Bonsor DA, Yu X, Diederichs K, Helmsing S, Vromen A, Friedler A, Hust M, Egelman EH, Beckett D, Wintrode PL, Sundberg EJ. Bacterial flagellar capping proteins adopt diverse oligomeric states. Elife. 2016:5. doi: 10.7554/eLife.18857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galkin VE, Yu X, Bielnicki J, Heuser J, Ewing CP, Guerry P, Egelman EH. Divergence of quaternary structures among bacterial flagellar filaments. Science. 2008;320(5874):382–385. doi: 10.1126/science.1155307. [DOI] [PubMed] [Google Scholar]
- 13.Song WS, Cho SY, Hong HJ, Park SC, Yoon SI. Self-Oligomerizing Structure of the Flagellar Cap Protein FliD and Its Implication in Filament Assembly. J Mol Biol. 2017;429(6):847–857. doi: 10.1016/j.jmb.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Pumpens P, Renhofa R, Dishlers A, Kozlovska T, Ose V, Pushko P, Tars K, Grens E, Bachmann MF. The True Story and Advantages of RNA Phage Capsids as Nanotools. Intervirology. 2016;59(2):74–110. doi: 10.1159/000449503. [DOI] [PubMed] [Google Scholar]
- 15.Koning RI, Gomez-Blanco J, Akopjana I, Vargas J, Kazaks A, Tars K, Carazo JM, Koster AJ. Asymmetric cryo-EM reconstruction of phage MS2 reveals genome structure in situ. Nat Commun. 2016;7:12524. doi: 10.1038/ncomms12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hepatitis B vaccines: WHO position paper--recommendations. Vaccine. 2010;28(3):589–590. doi: 10.1016/j.vaccine.2009.10.110. [DOI] [PubMed] [Google Scholar]
- 17.Jennings GT, Bachmann MF. The coming of age of virus-like particle vaccines. Biol Chem. 2008;389(5):521–536. doi: 10.1515/bc.2008.064. [DOI] [PubMed] [Google Scholar]
- 18.Bachmann MF, Rohrer UH, Kundig TM, Burki K, Hengartner H, Zinkernagel RM. The influence of antigen organization on B cell responsiveness. Science. 1993;262(5138):1448–1451. doi: 10.1126/science.8248784. [DOI] [PubMed] [Google Scholar]
- 19.Valegard K, Liljas L, Fridborg K, Unge T. The three-dimensional structure of the bacterial virus MS2. Nature. 1990;345(6270):36–41. doi: 10.1038/345036a0. [DOI] [PubMed] [Google Scholar]
- 20.Golmohammadi R, Fridborg K, Bundule M, Valegard K, Liljas L. The crystal structure of bacteriophage Q beta at 3. 5 A resolution. Structure. 1996;4(5):543–554. doi: 10.1016/s0969-2126(96)00060-3. [DOI] [PubMed] [Google Scholar]
- 21.Tars K, Bundule M, Fridborg K, Liljas L. The crystal structure of bacteriophage GA and a comparison of bacteriophages belonging to the major groups of Escherichia coli leviviruses. J Mol Biol. 1997;271(5):759–773. doi: 10.1006/jmbi.1997.1214. [DOI] [PubMed] [Google Scholar]
- 22.Tars K, Fridborg K, Bundule M, Liljas L. The three-dimensional structure of bacteriophage PP7 from Pseudomonas aeruginosa at 3. 7-A resolution. Virology. 2000;272(2):331–337. doi: 10.1006/viro.2000.0373. [DOI] [PubMed] [Google Scholar]
- 23.Persson M, Tars K, Liljas L. PRR1 coat protein binding to its RNA translational operator. Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 3):367–372. doi: 10.1107/S0907444912047464. [DOI] [PubMed] [Google Scholar]
- 24.Plevka P, Kazaks A, Voronkova T, Kotelovica S, Dishlers A, Liljas L, Tars K. The structure of bacteriophage phiCb5 reveals a role of the RNA genome and metal ions in particle stability and assembly. J Mol Biol. 2009;391(3):635–647. doi: 10.1016/j.jmb.2009.06.047. [DOI] [PubMed] [Google Scholar]
- 25.Tissot AC, Renhofa R, Schmitz N, Cielens I, Meijerink E, Ose V, Jennings GT, Saudan P, Pumpens P, Bachmann MF. Versatile virus-like particle carrier for epitope based vaccines. PLoS One. 2010;5(3):e9809. doi: 10.1371/journal.pone.0009809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shishovs M, Rumnieks J, Diebolder C, Jaudzems K, Andreas LB, Stanek J, Kazaks A, Kotelovica S, Akopjana I, Pintacuda G, Koning RI, Tars K. Structure of AP205 Coat Protein Reveals Circular Permutation in ssRNA Bacteriophages. J Mol Biol. 2016;428(21):4267–4279. doi: 10.1016/j.jmb.2016.08.025. [DOI] [PubMed] [Google Scholar]
- 27.Ruhe ZC, Low DA, Hayes CS. Bacterial contact-dependent growth inhibition. Trends Microbiol. 2013;21(5):230–237. doi: 10.1016/j.tim.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willett JL, Ruhe ZC, Goulding CW, Low DA, Hayes CS. Contact-Dependent Growth Inhibition (CDI) and CdiB/CdiA Two-Partner Secretion Proteins. J Mol Biol. 2015;427(23):3754–3765. doi: 10.1016/j.jmb.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aoki SK, Malinverni JC, Jacoby K, Thomas B, Pamma R, Trinh BN, Remers S, Webb J, Braaten BA, Silhavy TJ, Low DA. Contact-dependent growth inhibition requires the essential outer membrane protein BamA (YaeT) as the receptor and the inner membrane transport protein AcrB. Mol Microbiol. 2008;70(2):323–340. doi: 10.1111/j.1365-2958.2008.06404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruhe ZC, Nguyen JY, Xiong J, Koskiniemi S, Beck CM, Perkins BR, Low DA, Hayes CS. CdiA Effectors Use Modular Receptor-Binding Domains To Recognize Target Bacteria. MBio. 2017;8(2) doi: 10.1128/mBio.00290-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruhe ZC, Wallace AB, Low DA, Hayes CS. Receptor polymorphism restricts contact-dependent growth inhibition to members of the same species. MBio. 2013;4(4) doi: 10.1128/mBio.00480-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aoki SK, Diner EJ, de Roodenbeke CT, Burgess BR, Poole SJ, Braaten BA, Jones AM, Webb JS, Hayes CS, Cotter PA, Low DA. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature. 2010;468(7322):439–442. doi: 10.1038/nature09490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nikolakakis K, Amber S, Wilbur JS, Diner EJ, Aoki SK, Poole SJ, Tuanyok A, Keim PS, Peacock S, Hayes CS, Low DA. The toxin/immunity network of Burkholderia pseudomallei contact-dependent growth inhibition (CDI) systems. Mol Microbiol. 2012;84(3):516–529. doi: 10.1111/j.1365-2958.2012.08039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morse RP, Nikolakakis KC, Willett JL, Gerrick E, Low DA, Hayes CS, Goulding CW. Structural basis of toxicity and immunity in contact-dependent growth inhibition (CDI) systems. Proc Natl Acad Sci U S A. 2012;109(52):21480–21485. doi: 10.1073/pnas.1216238110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aoki SK, Webb JS, Braaten BA, Low DA. Contact-dependent growth inhibition causes reversible metabolic downregulation in Escherichia coli. J Bacteriol. 2009;191(6):1777–1786. doi: 10.1128/JB.01437-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jamet A, Jousset AB, Euphrasie D, Mukorako P, Boucharlat A, Ducousso A, Charbit A, Nassif X. A new family of secreted toxins in pathogenic Neisseria species. PLoS Pathog. 2015;11(1):e1004592. doi: 10.1371/journal.ppat.1004592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. Polymorphic toxin systems: Comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct. 2012;7:18. doi: 10.1186/1745-6150-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang D, Iyer LM, Aravind L. A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res. 2011;39(11):4532–4552. doi: 10.1093/nar/gkr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carr S, Walker D, James R, Kleanthous C, Hemmings AM. Inhibition of a ribosome-inactivating ribonuclease: the crystal structure of the cytotoxic domain of colicin E3 in complex with its immunity protein. Structure. 2000;8(9):949–960. doi: 10.1016/s0969-2126(00)00186-6. [DOI] [PubMed] [Google Scholar]
- 40.Ng CL, Lang K, Meenan NA, Sharma A, Kelley AC, Kleanthous C, Ramakrishnan V. Structural basis for 16S ribosomal RNA cleavage by the cytotoxic domain of colicin E3. Nat Struct Mol Biol. 2010;17(10):1241–1246. doi: 10.1038/nsmb.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Y, Pogliano J, Helinski DR, Konieczny I. ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol Microbiol. 2002;44(4):971–979. doi: 10.1046/j.1365-2958.2002.02921.x. [DOI] [PubMed] [Google Scholar]
- 42.Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, Ehrenberg M. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell. 2003;112(1):131–140. doi: 10.1016/s0092-8674(02)01248-5. [DOI] [PubMed] [Google Scholar]
- 43.Masaki H, Ogawa T. The modes of action of colicins E5 and D, and related cytotoxic tRNases. Biochimie. 2002;84(5–6):433–438. doi: 10.1016/s0300-9084(02)01425-6. [DOI] [PubMed] [Google Scholar]
- 44.Li Z, Gao Y, Nakanishi H, Gao X, Cai L. Biosynthesis of rare hexoses using microorganisms and related enzymes. Beilstein J Org Chem. 2013;9:2434–2445. doi: 10.3762/bjoc.9.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Z, Etienne M, Quiles F, Kohring GW, Walcarius A. Durable cofactor immobilization in sol-gel bio-composite thin films for reagentless biosensors and bioreactors using dehydrogenases. Biosens Bioelectron. 2012;32(1):111–117. doi: 10.1016/j.bios.2011.11.043. [DOI] [PubMed] [Google Scholar]
- 46.Gauer S, Wang Z, Otten H, Etienne M, Bjerrum MJ, Lo Leggio L, Walcarius A, Giffhorn F, Kohring GW. An L-glucitol oxidizing dehydrogenase from Bradyrhizobium japonicum USDA 110 for production of D-sorbose with enzymatic or electrochemical cofactor regeneration. Appl Microbiol Biotechnol. 2014;98(7):3023–3032. doi: 10.1007/s00253-013-5180-7. [DOI] [PubMed] [Google Scholar]
- 47.Kant R, Tabassum R, Gupta BD. A highly sensitive and distinctly selective D-sorbitol biosensor using SDH enzyme entrapped Ta2O5 nanoflowers assembly coupled with fiber optic SPR. Sensor Actuat B-Chem. 2017;242:810–817. [Google Scholar]
- 48.Fredslund F, Otten H, Gemperlein S, Poulsen JC, Carius Y, Kohring GW, Lo Leggio L. Structural characterization of the thermostable Bradyrhizobium japonicumD-sorbitol dehydrogenase. Acta Crystallogr F Struct Biol Commun. 2016;72(Pt 11):846–852. doi: 10.1107/S2053230X16016927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336(6084):1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Javidpour P, Pereira JH, Goh EB, McAndrew RP, Ma SM, Friedland GD, Keasling JD, Chhabra SR, Adams PD, Beller HR. Biochemical and structural studies of NADH-dependent FabG used to increase the bacterial production of fatty acids under anaerobic conditions. Appl Environ Microbiol. 2014;80(2):497–505. doi: 10.1128/AEM.03194-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rao ST, Rossmann MG. Comparison of super-secondary structures in proteins. J Mol Biol. 1973;76(2):241–256. doi: 10.1016/0022-2836(73)90388-4. [DOI] [PubMed] [Google Scholar]
- 52.Philippsen A, Schirmer T, Stein MA, Giffhorn F, Stetefeld J. Structure of zinc-independent sorbitol dehydrogenase from Rhodobacter sphaeroides at 2. 4 A resolution. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 4):374–379. doi: 10.1107/S0907444904034390. [DOI] [PubMed] [Google Scholar]
- 53.MacKenzie AK, Kershaw NJ, Hernandez H, Robinson CV, Schofield CJ, Andersson I. Clavulanic acid dehydrogenase: structural and biochemical analysis of the final step in the biosynthesis of the beta-lactamase inhibitor clavulanic acid. Biochemistry. 2007;46(6):1523–1533. doi: 10.1021/bi061978x. [DOI] [PubMed] [Google Scholar]
- 54.Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, Sumiyama K, Sagai T, Shiroishi T. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics. 2007;89(5):618–629. doi: 10.1016/j.ygeno.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Carl-McGrath S, Schneider-Stock R, Ebert M, Rocken C. Differential expression and localisation of gasdermin-like (GSDML), a novel member of the cancer-associated GSDMDC protein family, in neoplastic and non-neoplastic gastric, hepatic, and colon tissues. Pathology. 2008;40(1):13–24. doi: 10.1080/00313020701716250. [DOI] [PubMed] [Google Scholar]
- 56.Hergueta-Redondo M, Sarrio D, Molina-Crespo A, Vicario R, Bernado-Morales C, Martinez L, Rojo-Sebastian A, Serra-Musach J, Mota A, Martinez-Ramirez A, Castilla MA, Gonzalez-Martin A, Pernas S, Cano A, Cortes J, Nuciforo PG, Peg V, Palacios J, Pujana MA, Arribas J, Moreno-Bueno G. Gasdermin B expression predicts poor clinical outcome in HER2-positive breast cancer. Oncotarget. 2016;7(35):56295–56308. doi: 10.18632/oncotarget.10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, Depner M, von Berg A, Bufe A, Rietschel E, Heinzmann A, Simma B, Frischer T, Willis-Owen SA, Wong KC, Illig T, Vogelberg C, Weiland SK, von Mutius E, Abecasis GR, Farrall M, Gut IG, Lathrop GM, Cookson WO. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448(7152):470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 58.Saleh NM, Raj SM, Smyth DJ, Wallace C, Howson JM, Bell L, Walker NM, Stevens HE, Todd JA. Genetic association analyses of atopic illness and proinflammatory cytokine genes with type 1 diabetes. Diabetes Metab Res Rev. 2011;27(8):838–843. doi: 10.1002/dmrr.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pal LR, Moult J. Genetic Basis of Common Human Disease: Insight into the Role of Missense SNPs from Genome-Wide Association Studies. J Mol Biol. 2015;427(13):2271–2289. doi: 10.1016/j.jmb.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, D’Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H, Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC, Cho JH International IBDGC. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FA, Zhernakova A, Hinks A, Guiducci C, Chen R, Alfredsson L, Amos CI, Ardlie KG, Barton A, Bowes J, Brouwer E, Burtt NP, Catanese JJ, Coblyn J, Coenen MJ, Costenbader KH, Criswell LA, Crusius JB, Cui J, de Bakker PI, De Jager PL, Ding B, Emery P, Flynn E, Harrison P, Hocking LJ, Huizinga TW, Kastner DL, Ke X, Lee AT, Liu X, Martin P, Morgan AW, Padyukov L, Posthumus MD, Radstake TR, Reid DM, Seielstad M, Seldin MF, Shadick NA, Steer S, Tak PP, Thomson W, van der Helm-van Mil AH, van der Horst-Bruinsma IE, van der Schoot CE, van Riel PL, Weinblatt ME, Wilson AG, Wolbink GJ, Wordsworth BP, Wijmenga C, Karlson EW, Toes RE, de Vries N, Begovich AB, Worthington J, Siminovitch KA, Gregersen PK, Klareskog L, Plenge RM Consortium Y, Consortium B. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42(6):508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Genomes Project C. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chao KL, Kulakova L, Herzberg O. Gene polymorphism linked to increased asthma and IBD risk alters gasdermin-B structure, a sulfatide and phosphoinositide binding protein. Proc Natl Acad Sci U S A. 2017;114(7):E1128–E1137. doi: 10.1073/pnas.1616783114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 65.Hergueta-Redondo M, Sarrio D, Molina-Crespo A, Megias D, Mota A, Rojo-Sebastian A, Garcia-Sanz P, Morales S, Abril S, Cano A, Peinado H, Moreno-Bueno G. Gasdermin-B promotes invasion and metastasis in breast cancer cells. PLoS One. 2014;9(3):e90099. doi: 10.1371/journal.pone.0090099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zong M, Fofana I, Choe H. Human and host species transferrin receptor 1 use by North American arenaviruses. J Virol. 2014;88(16):9418–9428. doi: 10.1128/JVI.01112-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fulhorst CF, Bowen MD, Ksiazek TG, Rollin PE, Nichol ST, Kosoy MY, Peters CJ. Isolation and characterization of Whitewater Arroyo virus, a novel North American arenavirus. Virology. 1996;224(1):114–120. doi: 10.1006/viro.1996.0512. [DOI] [PubMed] [Google Scholar]
- 68.Abraham J, Corbett KD, Farzan M, Choe H, Harrison SC. Structural basis for receptor recognition by New World hemorrhagic fever arenaviruses. Nat Struct Mol Biol. 2010;17(4):438–444. doi: 10.1038/nsmb.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shimon A, Shani O, Diskin R. Structural Basis for Receptor Selectivity by the Whitewater Arroyo Mammarenavirus. J Mol Biol. 2017;429(18):2825–2839. doi: 10.1016/j.jmb.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 70.O’Neill MA, Ishii T, Albersheim P, Darvill AG. Rhamnogalacturonan II: structure and function of a borate cross-linked cell wall pectic polysaccharide. Annu Rev Plant Biol. 2004;55:109–139. doi: 10.1146/annurev.arplant.55.031903.141750. [DOI] [PubMed] [Google Scholar]
- 71.Matsunaga T, Ishii T, Matsumoto S, Higuchi M, Darvill A, Albersheim P, O’Neill MA. Occurrence of the primary cell wall polysaccharide rhamnogalacturonan II in pteridophytes, lycophytes, and bryophytes. Implications for the evolution of vascular plants. Plant Physiol. 2004;134(1):339–351. doi: 10.1104/pp.103.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ndeh D, Rogowski A, Cartmell A, Luis AS, Basle A, Gray J, Venditto I, Briggs J, Zhang X, Labourel A, Terrapon N, Buffetto F, Nepogodiev S, Xiao Y, Field RA, Zhu Y, O’Neill MA, Urbanowicz BR, York WS, Davies GJ, Abbott DW, Ralet MC, Martens EC, Henrissat B, Gilbert HJ. Complex pectin metabolism by gut bacteria reveals novel catalytic functions. Nature. 2017;544(7648):65–70. doi: 10.1038/nature21725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP, Abbott DW, Henrissat B, Gilbert HJ, Bolam DN, Gordon JI. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 2011;9(12):e1001221. doi: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(Database issue):D490–495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bekesi A, Pukancsik M, Muha V, Zagyva I, Leveles I, Hunyadi-Gulyas E, Klement E, Medzihradszky KF, Kele Z, Erdei A, Felfoldi F, Konya E, Vertessy BG. A novel fruitfly protein under developmental control degrades uracil-DNA. Biochem Biophys Res Commun. 2007;355(3):643–648. doi: 10.1016/j.bbrc.2007.01.196. [DOI] [PubMed] [Google Scholar]
- 76.Pukancsik M, Bekesi A, Klement E, Hunyadi-Gulyas E, Medzihradszky KF, Kosinski J, Bujnicki JM, Alfonso C, Rivas G, Vertessy BG. Physiological truncation and domain organization of a novel uracil-DNA-degrading factor. Febs J. 2010;277(5):1245–1259. doi: 10.1111/j.1742-4658.2009.07556.x. [DOI] [PubMed] [Google Scholar]
- 77.Hartmann MD, Boichenko I, Coles M, Zanini F, Lupas AN, Hernandez Alvarez B. Thalidomide mimics uridine binding to an aromatic cage in cereblon. J Struct Biol. 2014;188(3):225–232. doi: 10.1016/j.jsb.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 78.Hartmann MD, Boichenko I, Coles M, Lupas AN, Hernandez Alvarez B. Structural dynamics of the cereblon ligand binding domain. PLoS ONE. 2015;10(5):e0128342. doi: 10.1371/journal.pone.0128342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hakim M, Ezerina D, Alon A, Vonshak O, Fass D. Exploring ORFan domains in giant viruses: structure of mimivirus sulfhydryl oxidase R596. PLoS ONE. 2012;7(11):e50649. doi: 10.1371/journal.pone.0050649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.San Martin C. Latest insights on adenovirus structure and assembly. Viruses. 2012;4(5):847–877. doi: 10.3390/v4050847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Singh AK, Menendez-Conejero R, San Martin C, van Raaij MJ. Crystal structure of the fibre head domain of the Atadenovirus Snake Adenovirus 1. PLoS ONE. 2014;9(12):e114373. doi: 10.1371/journal.pone.0114373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gorman JJ, Wallis TP, Whelan DA, Shaw J, Both GW. LH3, a “homologue” of the mastadenoviral E1B 55-kDa protein is a structural protein of atadenoviruses. Virology. 2005;342(1):159–166. doi: 10.1016/j.virol.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 83.Pantelic RS, Lockett LJ, Rothnagel R, Hankamer B, Both GW. Cryoelectron microscopy map of Atadenovirus reveals cross-genus structural differences from human adenovirus. J Virol. 2008;82(15):7346–7356. doi: 10.1128/JVI.00764-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Menendez-Conejero R, Nguyen TH, Singh AK, Condezo GN, Marschang R, van Raaij MJ, San Martin C. Structure of a reptilian adenovirus reveals a phage tailspike fold stabilizing a vertebrate virus capsid. Structure. 2017 doi: 10.1016/j.str.2017.08.007. In production. [DOI] [PubMed] [Google Scholar]
- 85.Bradley P, Cowen L, Menke M, King J, Berger B. BETAWRAP: successful prediction of parallel beta -helices from primary sequence reveals an association with many microbial pathogens. Proc Natl Acad Sci U S A. 2001;98(26):14819–14824. doi: 10.1073/pnas.251267298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mayans O, Scott M, Connerton I, Gravesen T, Benen J, Visser J, Pickersgill R, Jenkins J. Two crystal structures of pectin lyase A from Aspergillus reveal a pH driven conformational change and striking divergence in the substrate-binding clefts of pectin and pectate lyases. Structure. 1997;5(5):677–689. doi: 10.1016/s0969-2126(97)00222-0. [DOI] [PubMed] [Google Scholar]
- 87.Garnham CP, Campbell RL, Walker VK, Davies PL. Novel dimeric beta-helical model of an ice nucleation protein with bridged active sites. BMC Struct Biol. 2011;11:36. doi: 10.1186/1472-6807-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38(Web Server issue):W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xiang Y, Leiman PG, Li L, Grimes S, Anderson DL, Rossmann MG. Crystallographic insights into the autocatalytic assembly mechanism of a bacteriophage tail spike. Mol Cell. 2009;34(3):375–386. doi: 10.1016/j.molcel.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Muller JJ, Barbirz S, Heinle K, Freiberg A, Seckler R, Heinemann U. An intersubunit active site between supercoiled parallel beta helices in the trimeric tailspike endorhamnosidase of Shigella flexneri Phage Sf6. Structure. 2008;16(5):766–775. doi: 10.1016/j.str.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 91.Steinbacher S, Miller S, Baxa U, Budisa N, Weintraub A, Seckler R, Huber R. Phage P22 tailspike protein: crystal structure of the head-binding domain at 2.3 A, fully refined structure of the endorhamnosidase at 1. 56 A resolution, and the molecular basis of O-antigen recognition and cleavage. J Mol Biol. 1997;267(4):865–880. doi: 10.1006/jmbi.1997.0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leiman PG, Molineux IJ. Evolution of a new enzyme activity from the same motif fold. Mol Microbiol. 2008;69(2):287–290. doi: 10.1111/j.1365-2958.2008.06241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bar Dolev M, Braslavsky I, Davies PL. Ice-Binding Proteins and Their Function. Annu Rev Biochem. 2016;85:515–542. doi: 10.1146/annurev-biochem-060815-014546. [DOI] [PubMed] [Google Scholar]
- 94.Raymond JA, DeVries AL. Adsorption inhibition as a mechanism of freezing resistance in polar fishes. Proc Natl Acad Sci U S A. 1977;74(6):2589–2593. doi: 10.1073/pnas.74.6.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yu SO, Brown A, Middleton AJ, Tomczak MM, Walker VK, Davies PL. Ice restructuring inhibition activities in antifreeze proteins with distinct differences in thermal hysteresis. Cryobiology. 2010;61(3):327–334. doi: 10.1016/j.cryobiol.2010.10.158. [DOI] [PubMed] [Google Scholar]
- 96.Cid FP, Rilling JI, Graether SP, Bravo LA, Mora M, de L, Jorquera MA. Properties and biotechnological applications of ice-binding proteins in bacteria. FEMS Microbiol Lett. 2016;363(11) doi: 10.1093/femsle/fnw099. [DOI] [PubMed] [Google Scholar]
- 97.Mangiagalli M, Bar-Dolev M, Tedesco P, Natalello A, Kaleda A, Brocca S, de Pascale D, Pucciarelli S, Miceli C, Bravslavsky I, Lotti M. Cryo-protective effect of an ice-binding protein derived from Antarctic bacteria. Febs J. 2017;284(1):163–177. doi: 10.1111/febs.13965. [DOI] [PubMed] [Google Scholar]
- 98.Hanada Y, Nishimiya Y, Miura A, Tsuda S, Kondo H. Hyperactive antifreeze protein from an Antarctic sea ice bacterium Colwellia sp. has a compound ice-binding site without repetitive sequences. Febs J. 2014;281(16):3576–3590. doi: 10.1111/febs.12878. [DOI] [PubMed] [Google Scholar]
- 99.Michalak M, Corbett EF, Mesaeli N, Nakamura K, Opas M. Calreticulin: one protein, one gene, many functions. Biochem J. 1999;344(Pt 2):281–292. [PMC free article] [PubMed] [Google Scholar]
- 100.Arnold SM, Kaufman RJ. The noncatalytic portion of human UDP-glucose: glycoprotein glucosyltransferase I confers UDP-glucose binding and transferase function to the catalytic domain. J Biol Chem. 2003;278(44):43320–43328. doi: 10.1074/jbc.M305800200. [DOI] [PubMed] [Google Scholar]
- 101.Guerin M, Parodi AJ. The UDP-glucose:glycoprotein glucosyltransferase is organized in at least two tightly bound domains from yeast to mammals. J Biol Chem. 2003;278(23):20540–20546. doi: 10.1074/jbc.M300891200. [DOI] [PubMed] [Google Scholar]
- 102.Zhu T, Satoh T, Kato K. Structural insight into substrate recognition by the endoplasmic reticulum folding-sensor enzyme: crystal structure of third thioredoxin-like domain of UDP-glucose:glycoprotein glucosyltransferase. Sci Rep. 2014;4:7322. doi: 10.1038/srep07322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Calles-Garcia D, Yang M, Soya N, Melero R, Menade M, Ito Y, Vargas J, Lukacs GL, Kollman JM, Kozlov G, Gehring K. Single-particle electron microscopy structure of UDP-glucose:glycoprotein glucosyltransferase suggests a selectivity mechanism for misfolded proteins. J Biol Chem. 2017 doi: 10.1074/jbc.M117.789495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ferrari DM, Soling HD. The protein disulphide-isomerase family: unravelling a string of folds. Biochem J. 1999;339(Pt 1):1–10. [PMC free article] [PubMed] [Google Scholar]
- 105.Kozlov G, Maattanen P, Thomas DY, Gehring K. A structural overview of the PDI family of proteins. Febs J. 2010;277(19):3924–3936. doi: 10.1111/j.1742-4658.2010.07793.x. [DOI] [PubMed] [Google Scholar]
- 106.Roversi P, Marti L, Caputo AT, Alonzi DS, Hill JC, Dent KC, Kumar A, Levasseur MD, Lia A, Waksman T, Basu S, Soto Albrecht Y, Qian K, McIvor JP, Lipp CB, Siliqi D, Vasiljevic S, Mohammed S, Lukacik P, Walsh MA, Santino A, Zitzmann N. Interdomain conformational flexibility underpins the activity of UGGT, the eukaryotic glycoprotein secretion checkpoint. Proc Natl Acad Sci U S A. 2017;114(32):8544–8549. doi: 10.1073/pnas.1703682114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Theobald DL, Steindel PA. Optimal simultaneous superpositioning of multiple structures with missing data. Bioinformatics. 2012;28(15):1972–1979. doi: 10.1093/bioinformatics/bts243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bayer EA, Belaich JP, Shoham Y, Lamed R. The cellulosomes: multienzyme machines for degradation of plant cell wall polysaccharides. Annu Rev Microbiol. 2004;58:521–554. doi: 10.1146/annurev.micro.57.030502.091022. [DOI] [PubMed] [Google Scholar]
- 109.Fontes CM, Gilbert HJ. Cellulosomes: highly efficient nanomachines designed to deconstruct plant cell wall complex carbohydrates. Annu Rev Biochem. 2010;79:655–681. doi: 10.1146/annurev-biochem-091208-085603. [DOI] [PubMed] [Google Scholar]
- 110.Dassa B, Borovok I, Ruimy-Israeli V, Lamed R, Flint HJ, Duncan SH, Henrissat B, Coutinho P, Morrison M, Mosoni P, Yeoman CJ, White BA, Bayer EA. Rumen cellulosomics: divergent fiber-degrading strategies revealed by comparative genome-wide analysis of six ruminococcal strains. PLoS ONE. 2014;9(7):e99221. doi: 10.1371/journal.pone.0099221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bule P, Alves VD, Leitao A, Ferreira LM, Bayer EA, Smith SP, Gilbert HJ, Najmudin S, Fontes CM. Single Binding Mode Integration of Hemicellulose-degrading Enzymes via Adaptor Scaffoldins in Ruminococcus flavefaciens Cellulosome. J Biol Chem. 2016;291(52):26658–26669. doi: 10.1074/jbc.M116.761643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bule P, Alves VD, Israeli-Ruimy V, Carvalho AL, Ferreira LM, Smith SP, Gilbert HJ, Najmudin S, Bayer EA, Fontes CM. Assembly of Ruminococcus flavefaciens cellulosome revealed by structures of two cohesin-dockerin complexes. Sci Rep. 2017;7(1):759. doi: 10.1038/s41598-017-00919-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lafita A, Bliven S, Kryshtafovych A, Bertoni M, Monastyrskyy B, Duarte JM, Schwede T, Capitani G. Assessment of protein assembly prediction in CASP12. Proteins. 2018 doi: 10.1002/prot.25408. CASP12 Special issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vogel C, Bashton M, Kerrison ND, Chothia C, Teichmann SA. Structure, function and evolution of multidomain proteins. Curr Opin Struct Biol. 2004;14(2):208–216. doi: 10.1016/j.sbi.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 115.Olechnovic K, Kulberkyte E, Venclovas C. CAD-score: a new contact area difference-based function for evaluation of protein structural models. Proteins. 2013;81(1):149–162. doi: 10.1002/prot.24172. [DOI] [PubMed] [Google Scholar]
- 116.Mariani V, Biasini M, Barbato A, Schwede T. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics. 2013;29(21):2722–2728. doi: 10.1093/bioinformatics/btt473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kryshtafovych A, Monastyrskyy B, Fidelis K. CASP prediction center infrastructure and evaluation measures in CASP10 and CASP ROLL. Proteins. 2014;82(Suppl 2):7–13. doi: 10.1002/prot.24399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302(1):205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.