

Abstract
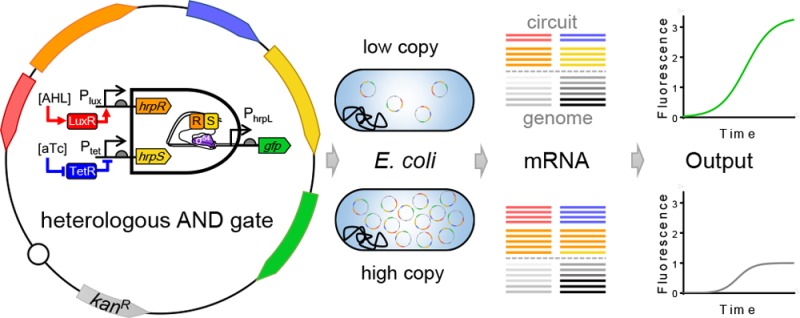
Synthetic biology approaches commonly introduce heterologous gene networks into a host to predictably program cells, with the expectation of the synthetic network being orthogonal to the host background. However, introduced circuits may interfere with the host’s physiology, either indirectly by posing a metabolic burden and/or through unintended direct interactions between parts of the circuit with those of the host, affecting functionality. Here we used RNA-Seq transcriptome analysis to quantify the interactions between a representative heterologous AND gate circuit and the host Escherichia coli under various conditions including circuit designs and plasmid copy numbers. We show that the circuit plasmid copy number outweighs circuit composition for their effect on host gene expression with medium-copy number plasmid showing more prominent interference than its low-copy number counterpart. In contrast, the circuits have a stronger influence on the host growth with a metabolic load increasing with the copy number of the circuits. Notably, we show that variation of copy number, an increase from low to medium copy, caused different types of change observed in the behavior of components in the AND gate circuit leading to the unbalance of the two gate-inputs and thus counterintuitive output attenuation. The study demonstrates the circuit plasmid copy number is a key factor that can dramatically affect the orthogonality, burden and functionality of the heterologous circuits in the host chassis. The results provide important guidance for future efforts to design orthogonal and robust gene circuits with minimal unwanted interaction and burden to their host.
Keywords: gene circuit, host circuit interaction, orthogonality, metabolic burden, RNA-Seq, synthetic biology
Synthetic biology holds great potential for cell engineering by introducing synthetic gene regulatory circuits to the host chassis with the goal to generate predictable behavior. Typically heterologous gene networks are designed and assumed to be orthogonal (no direct genetic crosstalk interaction) to the host cell genetic background.1−3 The hypothesis is largely based on the fact that the heterologous components do not naturally exist or have no homologues in the host chassis, and hence are less likely to produce unintended regulatory interaction with the endogenous genetic elements.4,5 Such orthogonality assumption is expected to lead to no or minimal interference on the host cell gene expression and physiology and thus allow to increase the functional predictability and compatibility of the introduced circuits. On the other hand, wholly orthogonal circuits would be unlikely to exist since the imported circuits will use some shared cellular resources such as metabolites, energy equivalents as well as replication, transcription and translation machineries. However, the design of the circuits themselves may provide some space to mitigate crosstalk and resource competition to minimize their physiological interference on the host chassis.6−8
To date, a number of orthogonal genetic devices and circuits have been constructed to perform various functions and have demonstrated the great potential of using orthogonal components to generate robust host cell behavior.1−3,9−12 For example, a previously engineered orthogonal AND gate circuit has been shown to work reliably among nearly all seven typically used E. coli strains, whereas the same circuit using an alternative endogenous promoter as one input (i.e., Plux replaced by Plac) failed to function in six out of seven these host strains.1 This reported circuit–host compatibility assay indicate that the use of orthogonal gene elements for a circuit help to eliminate potential unintended interactions between the circuit and the host genetic programs. However, most of the presumed orthogonal components and circuits have been designed based on prior literature knowledge and bioinformatics analysis, and have not been experimentally tested for their effects on the host cell genetic machinery. To a large extent, this has been limited by the lack of routinely available yet widely affordable methods to perform genomic wide profiling of gene expression. Ideally, a genetic device should be as orthogonal as possible to their host chassis to facilitate its reuse and reliability in different cellular contexts, i.e., having minimal interruption on the host gene expression and imposing low metabolic load on the host growth.
Here we used RNA sequencing (RNA-Seq)13 to quantify the entire transcriptome to study the interactions between a representative heterologous AND gate circuit and the host Escherichia coli under various conditions including different circuit designs and plasmid copy numbers. We envision that such genome-wide gene expression profiling will enable a quantitative measure of the orthogonality and effect of the various imported circuits on their host, which in turn could provide important insights and guidance for future efforts to design more orthogonal and robust gene circuits with minimal unwanted interaction and burden to their host. We show that the heterologous circuits themselves have little effect on the host gene expression profile, whereas the circuit plasmid copy number matters more with medium copy number plasmid having more prominent effect on the host transcriptome than its low copy number counterpart. In contrast, the circuits have stronger influence on the host cell growth with a metabolic load proportional to the circuit copy number. Moreover, we show that variation of copy number, an increase from low to medium copy, caused different types of change observed in the behavior of components in the AND gate circuit leading to an unbalance and distortion of the two gate-inputs and thus the attenuation of the gate output. Taken together, we demonstrate that the circuit plasmid copy number is a key factor that can dramatically affect the orthogonality, load and functionality of the introduced heterologous gene circuits in the host chassis,14,15 and that RNA-Seq is a powerful method for characterizing and debugging circuits that goes beyond the limitation of traditionally used fluorescent reporters.16,17
Results
Orthogonal AND Gate Circuit Design and RNA-Seq Assays
We first chose a previously reported modular and orthogonal AND gate that has been built and characterized in Escherichia coli,1 as the candidate heterologous gene circuit to study its interaction with the host genetic machinery. The AND gate circuit is designed to comprise an orthogonal σ54-dependent hrpR/hrpS heteroregulation module from the hrp (hypersensitive response and pathogenicity) system of Type III secretion in Pseudomonas syringae.18−20 The AND gate (Figure 1A) comprises two coactivating genes hrpR and hrpS and one σ54-dependent hrpL promoter, and can integrate two interchangeable signal inputs to generate one output. The output hrpL promoter is activated only when both the codependent HrpR and HrpS enhancer-binding proteins are expressed and form a heteromeric complex.
Figure 1.

Experimental design for studying the interactions between the heterologous AND gate gene circuits and the E. coli host transcriptome. (A) Schematics showing the composition of the different gene circuits and their potential interaction with the genetic background of the host E. coli K-12 MG1655 cell. The host genome is shown on the left, whereas the different plasmids carrying circuits are shown on the right and the bottom. In total three different circuits are carried in two types of plasmid of different copy number, i.e., one medium copy (pSB3K3, p15A ori) and one low copy (pSB4K5, pSC101 ori). The composition of each circuit is illustrated inside the cognate plasmid backbone. (B) The dynamic output fluorescence responses of the AND gate circuit under four input inductions. The red curves and data points are the scenario when the circuit is on the medium copy plasmid (pSB3K3), whereas the black curves and data points are for circuits on the low copy plasmid (pSB4K5). The four input inductions used are 100 nM AHL plus 20 ng/mL aTc, 100 nM AHL only, 20 ng/mL aTc only, and no inducers. Cells were grown in M9-glycerol media at 37 °C. Error bars, s.d. (n = 3). a.u., arbitrary units.
The circuit core elements, hrpR and hrpS and the hrpL promoter, are imported from the Pseudomonas syringae. Using online BLAST software to align their genetic sequences against the genomic sequences of Escherichia coli MG1655, no significant sequence similarity was found between them, indicating low homology of these heterologous genetic elements to the E. coli host. Due to the requirement of modularity, both the inputs and output of the AND gate were designed to be promoters, enabling the inputs to be wired to any input promoters and the output to be connected to any gene modules downstream to drive various cellular responses.21−23 Here we used the exogenous aTc inducible Ptet and AHL inducible Plux promoters as the two inputs. Both promoters and their cognate receptor genes tetR and luxR are exogenous to the E. coli genome. Similarly, the BLAST results of the two input promoter sequences also showed no significant similarity. Hence, we assume these heterologous genetic elements do not tend to interact with the endogenous ones in the host, i.e., the rational for orthogonality.
To compare conditions of different circuit compositions, we also built constructs that comprise only the two input promoters of the AND gate with gfp reporters (Figure 1A). Thus, with the condition of empty plasmid alone as the control, we analyzed three types of gene circuits, namely the AND-gate, Inputs-gfp and empty plasmid. Since the circuit copy number could be another influencing factor, we considered two conditions of plasmid copy number here, i.e., one medium copy number (pSB3K3) and one low copy number (pSB4K5). The copy number of a plasmid in the host is determined by their origin of replication. The plasmid pSB3K3 with p15A ori produces medium copy number (∼15–20 copies per cell) of plasmids in host cells, while the plasmid pSB4K5 with pSC101 ori produces low copy numbers (∼5 copies per cell).24 Both plasmids have the same kanamycin resistance (kanR) to minimize differences (Figure S1E,F). Figure 1B shows the dynamic output fluorescence response of the AND gate circuit under four logic input inductions when hosted on the two plasmids with different copy numbers.
In total, we have 6 different conditional combinations from the above three genetic circuit compositions and two plasmid copy numbers. Accordingly, we have generated 7 RNA-Seq samples in total (Table 1), among which Sample 1 and Sample 2 are biological replicates of the same condition, i.e., the AND-gate in pSB3K3 (Figure S1A). This duplicate was used to validate the high quality and repeatability of the RNA-Seq performed and at the same time to control the total sequencing cost. The correlation of gene expression (Figure S4) between the two replicate samples (S1 and S2) is significantly high (R2 = 0.9788), indicating excellent reproducibility of the RNA-Seq data. This is also reflected in the uniform mapped sequencing read profiles of the plasmid hosted genes from the duplicate samples (Figure S5A,B). Table 3 shows the different paired comparisons between the 7 RNA-Seq samples. In total there are ten paired comparisons among the seven samples: i.e., C1 is for studying the RNA-Seq repeatability between biological replicates (S1 vs S2); C2, C3, C4 are grouped for studying different circuit loads in the medium copy plasmid (pSB3K3, p15A ori) and C8, C9, C10 are grouped for studying different circuit loads in the low copy number plasmid (pSB4K5, pSC101 ori); C5, C6, and C7 are grouped for studying the effect caused by the change of copy number of the plasmid hosting the same circuit.
Table 1. Summary of the RNA-Seq Samples in This Study.
| sample # | circuit insert | hosting plasmid | copy number |
|---|---|---|---|
| S1 | AND-gate | pSB3K3 | medium |
| S2 | AND-gate | pSB3K3 | medium |
| S3 | Inputs-gfp | pSB3K3 | medium |
| S4 | none | pSB3K3 | medium |
| S5 | AND-gate | pSB4K5 | low |
| S6 | Inputs-gfp | pSB4K5 | low |
| S7 | none | pSB4K5 | low |
Table 3. Ten Paired Comparisons between RNA-Seq Samples and Identified Number of DEGsa.
| paired comparison | DEGs (χ2-test) | DEGs (edgeR) | DEGs overlapped |
|---|---|---|---|
| C1 (S1 vs S2) | 63 | 13 | 13 |
| C2 (S1/2 vs S3) | 50 | 25 | 25 |
| C3 (S3 vs S4) | 111 | 47 | 46 |
| C4 (S1/S2 vs S4) | 67 | 41 | 41 |
| C8 (S5 vs S6) | 14 | 8 | 8 |
| C9 (S6 vs S7) | 201 | 64 | 62 |
| C10 (S5 vs S7) | 137 | 42 | 42 |
| C5 (S1/2 vs S5) | 356 | 168 | 129 |
| C6 (S3 vs S6) | 481 | 387 | 273 |
| C7 (S4 vs S7) | 1265 | 941 | 627 |
This table shows the number of DEGs in each paired comparison highlighting the conditions (C5, C6, C7) attributing to the effect of changes in circuit plasmid copy number. S1/2 represents means of the gene expression values of the two replicate Samples 1 and 2. Ten paired comparisons performed among the 7 samples: C1 is for studying RNA-Seq repeatability between biological replicates (S1 vs S2); C2, C3, C4 are grouped for studying different circuit loads in the medium copy plasmid (pSB3K3); and C8, C9, C10 are grouped for studying different circuit loads in the low copy plasmid (pSB4K5); C5, C6, and C7 are grouped for studying the effect caused by changes in plasmid copy number of the same circuit.
Table 2 summarizes the RNA-Seq sequencing data set obtained and the mapping of the reads to the E. coli host genome and the cognate circuit plasmids. It shows that around 70–90% total reads were successfully mapped to the host genome and about 2–30% total reads were mapped to the plasmid across all samples. To obtain the expression level for each gene, we counted the number of reads mapped to each gene according to their location in the chromosome or plasmid. The reads were then normalized according to the gene length to obtain the relative expression level for each gene (RPKM value). The distribution of the expression levels of all host genes across all seven samples follows an expected approximately normal distribution (Figure S3).
Table 2. Summary of the RNA-Seq Dataset with Mapping to the E. coli Host Genome and Circuit Plasmid.
| features | S1 | S2 | S3 | S4 | S5 | S6 | S7 |
|---|---|---|---|---|---|---|---|
| total reads | 19 625 015 | 19 304 441 | 18 016 312 | 15 732 889 | 12 522 316 | 26 114 723 | 21 623 806 |
| GC content | 45% | 45% | 44% | 46% | 46% | 46% | 46% |
| genome mapped | 13 777 962 | 12 805 437 | 11 722 783 | 13 170 363 | 10 465 952 | 21 954 375 | 19 153 164 |
| host genes mapped | 8 161 736 | 7 624 657 | 6 737 923 | 7 604 894 | 6 345 500 | 12 926 900 | 11 963 227 |
| plasmid mapped | 3 458 262 | 3 963 547 | 3 823 566 | 357 583 | 514 191 | 816 200 | 65 752 |
| plasmid genes mapped | 3 371 909 | 3 886 264 | 3 766 222 | 309 625 | 477 403 | 747 665 | 38 594 |
Circuit Metabolic Load Increases with Its Copy Number in the Host
To probe the metabolic load of gene circuits imposed on their hosts, we monitored cell growth by measuring cell density periodically for all sample conditions. Figure 2A shows the cell growth curves for each sample culture. It can be seen that cells containing the empty plasmid alone (Samples 4 and 7) have the fastest growth among all conditions, whereas cells containing circuits on the low copy number plasmid (pSB4K5) had faster growth rates than their counterparts on the medium copy number plasmid (pSB3K3). This indicates the imported gene circuits have affected the host cell growth with a projected load increasing with their copy numbers in the host. We view the observed metabolic load could be linked to the competitive usage of shared cellular resources between the host endogenous machineries and the inserted synthetic circuit as indicated previously.6,8,25
Figure 2.

Host cell metabolic load imposed by gene circuit of varying compositions. (A) Cell growth curves of the seven samples used in this study. The vertical dashed line indicates the snapshot time point when the cells were sampled for RNA-Seq. Error bars, s.d. (n = 3). (B) Fitted growth model parameter values of each sample. μm, the growth rate of the cells at exponential growth phase; λ, the lag time before cells entering exponential growth phase A; the maximum growth density achieved. (C) Sample growth rates (μm) derived from the fitted cell growth model at log phase. The grow rate for AND-gate in pSB3K3 is the mean of that of Samples 1 and 2. (D) Metabolic load of the circuit imposed on the host. The metabolic loads are calculated from their cognate sample cell growth rates by setting the least affected sample (i.e., S7 carrying the low copy number plasmid pSB4K5 only), as the reference (zero load).
To obtain exact cell growth rate, we fit the cell growth data to the modified Gompertz model for cell growth.26Figure 2B lists the fitted growth model parameter values for each sample condition with Figure S2 displaying the model fitting performance. It shows that the growth rate (μm) for each sample ranks in the descending order as Sample 7 > Sample 4 > Sample 5 > Sample 6 > Sample 2 > Sample 1 ⩾ Sample 3. Notably cells with gene circuits hosted on the low copy number plasmid have higher growth rates than those with the same circuits hosted on the medium copy number plasmid (Figure 2C).
We next calculated the metabolic load for each circuit by measuring the relative reduction in growth rate in comparison to a reference condition.7 We used the fastest growing sample (S7), i.e., host carrying the low copy number plasmid pSB4K5 alone, as the reference (zero load) to obtain the metabolic load for all other sample constructs following the equation detailed in the Materials and Methods section.
Figure 2D shows that the metabolic load induced from the same gene circuit are significantly lower when it was hosted on the low copy number plasmid, in particular for the conditions with a complete AND gate circuit. The empty plasmid pSB3K3 showed a negligible load difference compared to the reference empty pSB4K5, suggesting that plasmid replication from low to medium number bears only a small fitness cost. The more pronounced metabolic load associated with the AND gate and Inputs-gfp circuits implies that the expression of circuit parts represents a higher fitness penalty than plasmid replication. In addition, the AND gate circuit imposed a lower load on the host compared to the Inputs-gfp circuit in both plasmids. This is likely due to that the later circuit produced significantly higher GFP proteins from its two gfp reporters which are highly stable and hence readily accumulate compared to its counterpart transcription factors in the AND gate circuit. Taken together, these data demonstrate that the copy number of a gene circuit has a pivotal role for its metabolic load imposed on the host, whereas its hosting plasmid only has a minor impact. Typically the circuit metabolic load is increasing with its copy number in the host cell.
Plasmid Copy Number Outweighs Circuit Composition for Effect on Host Gene Expression
To explore genome-wide interaction between gene circuit and the host, we applied hierarchical clustering of host gene expression derived from the RNA-Seq data set for all samples. The result (Figure 3) showed that the samples containing the same copy number plasmids clustered together, despite the apparent lower metabolic load associated with copy numbers per se, and indicating the plasmid copy number outweighs the gene circuit composition. Overall the host genes can be divided into 4 expression clusters. Clusters 1 and 2 comprise genes whose expression levels in the presence of the low copy hosting plasmid are higher than those in the presence of the medium copy number hosting plasmid, whereas Clusters 3 and 4 show the opposite. Notably host genes within Clusters 1 and 4 display a more consistent expression pattern.
Figure 3.

Genome-wide host gene expression clustering showing prominent effect caused by changes in circuit plasmid copy number. The expression of 3084 host genes in the six sample conditions are hierarchically clustered (the additional 1431 genes are excluded according to the clustering criteria due to their atypical absolute gene expression values). The result shows that the samples containing the plasmid of same copy number clustered together (Samples 1–4 containing medium copy number plasmid pSB3K3, Samples 5–7 containing low copy number plasmid pSB4K5). Overall the host genes can be divided into four expression clusters. The DEGs from the paired comparisons of C5, C6 and C7 tend to be located in the Clusters 1 and 4, which are related to the impact caused by the change in circuit plasmid copy number. S1/2 are the means of the gene expression values of the two replicate Samples 1 and 2. The color bar indicates the scale for gene expression (normalized across the seven samples for each gene).
We next identified differentially expressed genes (DEGs) across all compared comparisons of C1–C10 (Table 3) using two statistical methods, i.e., χ2-test and edgeR, to cross-validate by minimizing potential false positives. Table 3 summarizes identified DEGs including the overlapped DEGs cross-validated by the aforementioned two methods. It shows that the three paired comparisons of C5, C6 and C7 resulted in the highest numbers of identified DEGs, highlighting the prominent effect corresponding to the variation of plasmid copy number. The DEGs from C5–C7 tend to be located in Clusters 1 and 4, which possess highly consistent gene expression patterns (Figure 3). In contrast, paired comparisons of C2–C4 and C8–C10 for studying the effect of different circuit loads in the same type of plasmids only produced moderate numbers of DEGs. Taken together, these results indicate that the plasmid copy number outweighs circuit composition among contributing factors that affect host gene expression, although copy number only marginally affected the apparent metabolic load and growth rates.
To further investigate what host cellular processes may have been affected by the heterologous gene circuits, we studied the change of expression levels of genes required for protein biosynthesis and regulation, including genes encoding for the transcription and translation machineries, transcription regulation genes, housekeeping genes and essential genes (Table S5–S9). Figure 4A shows that the circuits have little effect on the host transcription process including DNA polymerase, RNA polymerase, transcription termination factor and various transcription-related genes across all samples. The circuits affected the translation process mainly on tRNA related genes (Figure 4C) but with little effect on ribosome and ribosome related genes (Figure 4B). There is minor effect on the host transcription factor genes (Figure 4D) though largely owing to the copy number increase of the circuit plasmid (in C6 and C7). The circuits did not show any obvious interference on the 39 host housekeep genes (Figure 4E). Figure 4F shows the C5–C7 paired comparisons contain the highest numbers of DEGs across the 703 host essential genes,27 corroborating the aforementioned prominent effect caused by copy number increase of the circuit plasmid. Overall, the results demonstrate that the heterologous gene circuits only had minor effect on cells biosynthesis machinery.
Figure 4.

Host gene expression profile comparison across all RNA-Seq samples. The blue curve represents the mean expression value of each gene in the seven samples. The red curve represents the upper bound of 1.44 times the mean expression value, whereas the green curve represents the lower bound of 1/1.44 times the mean value. Hence, if the gene expression value is significantly outside the region between the red and green curves, it is likely that the gene will be a candidate of differentially expressed genes. The gene expression profiles of DNA polymerases, RNA polymerases, transcriptional termination factors and other transcription related genes (A), ribosomes and related genes (B), tRNA and tRNA related genes (C), transcription factors (D), housekeeping genes (E) and essential genes (F) are shown, respectively. The differentially expressed genes resulted from each paired comparison are listed in the upper right-hand corner of each figure panel. In all panels, genes on the horizontal axis are ranked in ascending order by their mean values of gene expression (RPKM).
Next, we performed functional enrichment analysis among the identified overlapped DEGs using the online tool DAVID.28,29Table S3 compares cellular processes by the change of copy number of plasmid hosting otherwise same constructs, broadly showing a wide range of similarly affected metabolic processes and including those involved in the cell’s major biosynthesis and energy production pathways (carbon metabolism, nitrogen metabolism, respiration, transport). We conclude that the introduced plasmid copy numbers affect the overall cellular expression profiles, but that these changes per se lead only to small growth differences and indicating that cells can adapt well to costs associated with replicating low and medium copy number plasmids.
The significant metabolic burden observed between the AND gate and Inputs-gfp circuit plasmids compared with empty vectors suggested that the expressed circuit parts explain growth penalties, either through their specific interference with host functionality (crosstalk) or through costs associated with their expression (luxR, tetR, hrpR, hrpS, gfp; Figure 1A). Table S4 shows the functional annotations of DEGs in pairwise comparisons between different circuit compositions with empty plasmids. The most predominant differences in gene expression are associated with GO processes involved in amino acid biosynthesis (general amino acid, tryptophan aromatic amino acid and nitrogen compound biosynthesis) and specific KEGG pathways required for alanine, aspartate and glutamate biosynthesis, as well as numerous ABC transporters, including several amino acid transporters for lysine/arginine/ornithine (argT), glutamine (glnH), glutamate/aspartate (gltL), arginine (artJ) and branched amino acids (livK). These findings strongly suggest that protein production of the introduced synthetic components place an amino acid burden on the host cell that could to a large degree account for the metabolic burden observed. The higher copy number plasmids expressing circuit parts clearly impose a high metabolic burden compared with the low copy number versions, correlating with more DEGs involved in amino acid synthesis, assuming that higher plasmid copy numbers result in overall higher expression rates. This assumption is supported by the higher transcription rates of the antibiotic resistance and origin of replication control genes in pSB3K3 compared to pSB4K5 (Figure S6).
We did not observe any striking or specific differences in expression patterns between constructs harboring AND gate or Inputs-gfp that could indicate any specific cross talk between host and synthetic parts, lending support to the notion that the heterologous synthetic circuits introduced no obvious genetic cross talk.
Copy Number Variation Caused Contrary Changes in Circuit Components
Notably we found the AND gate circuit behaved differently in the two plasmids of different copy numbers. Figure 1B shows that the output fluorescence of AND gate hosted in medium copy number plasmid pSB3K3 is about half that when hosted in low copy number plasmid pSB4K5. This is counterintuitive since generally the expression level of a gene is expected to proportional to its copy number in the host cell.
To investigate potential underlying cause, we examined the mRNA levels of all genes in the two circuits (i.e., AND-gate and Inputs-gfp) from their transcription profiles30 (gene transcription activity) on the two types of hosting plasmids (Figure 5). It reveals that the transcript levels of the constitutively expressed luxR and tetR genes (Figure 5A and 5B) are in proportion to their plasmid copy number, which is also reflected by the expression levels of the antibiotic resistance and origin of replication control genes (Figure S6) in the two circuit plasmids. However, we note the regulated hrpR and hrpS genes under the two inducible promoters expressed quite differently on the two plasmids. Whereas both hrpR and hrpS were transcribed at similar levels when hosted in the low copy number plasmid pSB4K5, the hrpR transcription level is consistently much higher than that of hrpS when hosted in the medium copy number plasmid pSB3K3 (Figure 5A). This is also consistent with the significantly differential expression levels of the two gfp reporter genes downstream the two inducible promoters of Ptet and Plux in the Inputs-gfp circuit when hosted on the medium copy number plasmid (Figure 5B). Strikingly, hrpR transcription was increased significantly while hrpS transcription was decreased drastically when the circuit moved from the low copy number plasmid to the medium copy number one. Because HrpR and HrpS need to form a hetero hexamer to activate its target hrpL output promoter, the total activator complex available in the host would be determined by the lower level of the two component molecules, i.e., displaying the short-board-effect and explaining the aforementioned counterintuitive output attenuation of the AND gate circuit present in higher copy number.
Figure 5.

Transcription profiles of the genes in the circuits hosted on the two plasmids of different copy numbers. (A) The transcript read counts of the genes in the AND gate circuit that are carried on the two plasmids of different copy number (i.e., medium copy pSB3K3 and low copy pSB4K5, S1/2 (mean) vs S5). (B) The transcript read counts of the genes in the Inputs-gfp circuit that are carried on the two plasmids of different copy number (i.e., medium copy pSB3K3 and low copy pSB4K5, S3 vs S6). (C) The transcriptional profiles of the AND gate circuit in S1, S2 (pSB3K3) and S5 (pSB4K5). (D) The transcriptional profiles of the Inputs-gfp circuit in S3 (pSB3K3) and S6 (pSB4K5). Note that the read counts of the two gfp transcripts can be distinguished by their different 5′ UTRs (RBSs) downstream their inducible promoters (i.e., gfp-Plux and gfp-Ptet). See also Figure S5 and S6.
Cleary the data shows that an increase in copy number has caused contrary changes observed in the behavior of components in the AND gate circuit leading to the unbalance and distortion of the two gate inputs and thus the output attenuation. We view that such contrast in behavior change is due to the difference in mode-of-action of the two inducible promoters of which the Plux is activator receptor (LuxR) regulated and Ptet is repressor receptor (TetR) regulated, and discuss this further in the Discussion section.
Discussion
In this study we applied RNA whole transcriptome sequencing to probe the interactions between imported heterologous gene circuits and the host E. coli using various circuit compositions and different copy number plasmids. The method provides genome-wide gene expression profiling that enables a quantitative measure of the orthogonality and effect of the imported circuits on their host. Though the circuits have utilized many host resources including DNA polymerases, RNA polymerases, transcription factors, ribosomes and other translation related factors, it is striking that the circuits present in low copy number did not significantly affect the gene expression of these resource related factors and neither the transcription regulation in the host. This provides evidence that the heterologous AND circuit studied is highly “orthogonal” to their host genetic background, and the orthogonality design between an imported circuit and the host could be vital to help reduce any potential genome-wide interactions.1,4
However, we found that the circuit plasmid copy number significantly impacts on the metabolic load,31 orthogonality and functionality14 of the introduced heterologous gene circuits in the host chassis. The gene circuits imposed notable metabolic load on the host, whereas empty plasmids did not, resulting in cell growth reduction that is generally in proportion to their copy numbers in the host, suggesting that expression of the synthetic genes are responsible. The analysis of the number of differentially expressed genes in the host transcriptome and their clustering collectively shows that an increase in the circuit plasmid copy number has led to more prominent increase in the interference between the circuits and the host genetic background in contrast to the change with circuit compositions alone. Our results reveal that the plasmid copy number that is concomitant with higher gene expression, outweighs circuit composition for their effect on differential host gene expression. Our data revealing a large number of genes involved in nitrogen metabolism, amino acid biosynthesis and transport affected in the host when expressing synthetic circuit parts from high copy number plasmids suggests resource competition between host and synthetic circuit at the level of translation. Thereby as a rule of thumb, we propose that it will be beneficial to design and implement functional gene circuits in low copy number, and predict low expressional levels to be similarly advantageous, in the host if possible. In return this will help increase the orthogonality and robustness of the underlying circuits with reduced or minimized host physiological interference, in particular for large scale circuits comprising many parts.32 That said, it is recognized that low copy number of molecules may increase the noise within a biological system.33,34 Hence, attention should be paid to designing the exact copy number and the expression levels of relevant genes within a circuit with an aim to achieve a desired balanced system behavior. In some cases, it could be worthwhile and necessary to use multiple compatible plasmids with different copy numbers to address the resource allocation, robustness and modularity requirements pertaining to the design of a particular circuit.1,32 In addition, the amounts of available host cellular resources (e.g., proteases, ribosomes, amino acids, sigma factors) may vary depending on the strains used which could have significant impact on circuit behavior.1,7,35
We show that the change in circuit plasmid copy number could cause contrary changes to be observed in the behavior of different components within the circuits, which can lead to the imbalance of the predesigned/tested stoichiometry among the underlying circuit blocks. This has been evidenced by the disproportionate transcription of the two input promoters and the subsequent drastic output attenuation of the AND gate circuit when the circuit migrating from a low copy number plasmid to a medium copy number one. Our previous characterization of receptor-mediated small molecule inducible promoters have revealed that a low concentration of the repressor receptor (e.g., TetR) in the cell can significantly increase the sensitivity and dynamic range, whereas a high activator receptor (e.g., LuxR) concentration will achieve the same outcome.34 A copy number increase would be equivalent to the effect of an increased concentration of both the constitutively expressed receptors (TetR and LuxR) in the cytoplasm, leading to contrary changes of the output behavior of the repressor and activator receptor-mediated promoters. This is also echoed by the evidence that output fluorescence from the GFP reporters under the two inducible promoters in the Inputs-gfp circuit exhibited contrary change when migrating from the low copy plasmid to the medium copy one (Figure S7). Thus, we view such contrast in copy number-induced behavior change are owing to the difference in mode-of-action of the two inducible promoters of which the Plux is activator receptor (LuxR) mediated, whereas the Ptet is repressor receptor (TetR) mediated.
The study exemplifies that RNA-Seq represents a new powerful method for characterizing and debugging circuits that goes beyond the limitation of traditionally used fluorescent reporters. RNA-Seq uses next-generation sequencing to reveal the presence and quantity of all RNAs in a biological sample at a given moment, including for genes of both the circuit and host. Thereby it produces a global snapshot of the internal workings of the intact gene circuits in real action, which provides unprecedented detailed information to assist identifying any imbalanced or failed circuit nodes or components such as the two disproportionate AND-gate inputs disclosed in this work. That being said, the method presently has its own limitation that it would only provide the mRNA levels but not the protein levels at a given moment. We think that this may be complemented by new emerging technology such as the genome-wide ribosome profiling (Ribo-Seq)36 or selected reaction monitoring-based mass spectrometry proteomics37 that could help quantify the relative levels of translated proteins corresponding to all transcripts in a host. Combining these methods can serve as powerful tools for more accurately diagnosing gene circuits and probing their interactions with the host genomic background. Moreover, the high cost of RNA-Seq could be reduced with newly adapted versions in the field such as the RNAtag-Seq,38,39 which uses DNA barcodes to uniquely “tag” RNAs from each sample, allowing multiple samples to be pooled early before RNA library preparation in a single reaction and being sequenced together. Such multiplexed approach simplifies library preparation and significantly reduces library preparation costs, resulting in lower time and cost per sample.
Materials and Methods
Plasmid Circuit Construction
Plasmid construction and DNA manipulations were performed following standard molecular biology techniques. The hrpR, hrpS genes, hrpL promoter, the aTc (anhydrotetracycline, rbs30-tetR-B0015-Ptet2) and AHL (3OC6HSL, rbs30-luxR-B0015-Plux2) inducible promoters were synthesized by GENEART following the BioBrick standard (http://biobricks.org),24 by eliminating the four restriction sites (EcoRI, XbaI, SpeI and PstI) for the BioBrick standard via synonymous codon exchange and flanking with prefix and suffix sequences containing the appropriate restriction sites and RBS (ribosome binding site) sequences. The double terminator BBa_B0015 (http://partsregistry.org) was used to terminate gene transcription in all cases. pSB3K3 (p15A ori, kanR) and pSB4K5 (pSC101, kanR)24 was used to clone and characterize all the genetic constructs in this study. The GFP (gfpmut3b, BBa_E0840) reporter was from the Registry of Standard Biological Parts (http://partsregistry.org). The various RBS sequences (Table S10) for each gene construct were introduced by PCR amplification (using PfuTurbo DNA polymerase from Stratagene and an Eppendorf Mastercycler gradient thermal cycler) with primers containing the corresponding RBS sequences and appropriate restriction sites. The constitutive promoters used were assembled from two annealed single stranded primers flanked with appropriate restriction sites. All circuit constructs were assembled following the BioBrick DNA assembly method and verified by DNA sequencing (Beckman Coulter Genomics) prior to their use. Primers were synthesized by Sigma-Aldrich. Further information can be found in Figure S1 (plasmid maps) and Table S10 (part genetic sequences) describing the circuit constructs used. All plasmids used are available upon request and selected plasmids may be obtained from the Addgene repository (https://www.addgene.org/Baojun_Wang/).
Strains, Media and Growth Conditions
Plasmid cloning work was performed in E. coli TOP10 strain, whereas all circuit construct characterization were all performed in E. coli K-12 NCM3722 strain. Cells were cultured in M9 minimal media (11.28 g/L M9 salts, 1 mM thiamine hydrochloride, 0.2% (w/v) casamino acids, 2 mM MgSO4, 0.1 mM CaCl2, 0.4% (v/v) glycerol). The kanamycin used was 25 μg/mL. Cells inoculated from single colonies on freshly streaked LB plates were grown overnight in 5 mL M9 in sterile 30 mL universal tubes at 37 °C with shaking (200 rpm). Overnight cultures were diluted into prewarmed M9 media at OD600 = 0.02 for the day cultures (100 mL in 500 mL flasks), which were all induced by 100 nM AHL plus 20 ng/mL aTc and grown for 4 h at 37 °C prior to be harvested for RNA-Seq sample preparation. For fluorescence assay by fluorometry, diluted cultures were also loaded into a 96-well microplate (Bio-Greiner, chimney black, flat clear bottom) and induced with 5 μL (for single input induction) or 10 μL (for double input induction) inducers of varying concentrations to a final volume of 200 μL per well by a multichannel pipet. The microplate was covered by a UV transparent lid to counteract evaporation and incubated in the fluorometer (BMG FLUOstar) with continuous shaking (200 rpm, linear mode, 37 °C) between each cycle of repetitive measurements. Chemical reagents and inducers used were analytical grade from Sigma-Aldrich. For cell growth curve assay, diluted cultures were cultured separately in 200 mL flasks at a volume of 50 mL and cell absorbance (OD600) were measured by a spectrophotometer (Jenway Genova Plus) around every 30 min by sampling half ml culture into 1 mL cuvettes that have been preloaded half mL M9 media.
Assay of Gene Expression
Fluorescence levels of gene expression were assayed by fluorometry at the cell population level. Cells grown in 96-well plates were monitored and assayed using a BMG FLUOstar fluorometer for repeated absorbance (OD600) and fluorescence (485 nm for excitation, 520 ± 10 nm for emission, Gain = 1000) readings (20 min/cycle). The fluorometry data of gene expression were first processed in BMG Omega Data Analysis Software (v1.10) and were analyzed in Matlab after being exported. The medium backgrounds of absorbance and fluorescence were determined from blank wells loaded with M9 media and were subtracted from the readings of other wells. The fluorescence/OD600 (Fluo./OD600) at a specific time for a sample culture was determined after subtracting its triplicate-averaged counterpart of the negative control cultures (GFP-free) at the same time.
RNA-Seq Sample Preparation and Sequencing
E. coli NCM3722 cultures were grown and growth stopped 4 h post day dilution by adding 1/10 volume of 5% phenol 95% ethanol (v/v). Cells were harvested by centrifugation (4500g for 30 min). Supernatants were discarded and pellets drained by gravity flow for 5 min. Pellets wet weights were measured by subtracting the weights of cognate empty tubes. There are 7 samples in total containing 6 different plasmid constructs in the E. coli host. Samples 1 and 2 are biological replicates of the same construct (AND gate in pSB3K3), Sample 3 (Inputs-gfp in pSB3K3), Sample 4 (empty pSB3K3), Sample 5 (AND gate in pSB4K5), Sample 6 (Inputs-gfp in pSB4K5), Sample 7 (empty pSB4K5). The pellet cell samples were frozen at −80 °C before sent out on dry ice to vertis Biotechnologie AG for RNA-Seq In brief, the cell pellets were incubated with lysozyme for 15 min at room temperature. The total RNA was then isolated using the mirVana RNA isolation kit (Invitrogen) including DNase treatment. Primary transcript enrichment was achieved by rRNA depletion and treatment with Terminator exonuclease (Epicenter) to remove other processed RNAs. RNA was fragmented using RNaseIII and cDNA libraries were built including PCR amplification with barcoded sequencing adaptors. Samples were pooled in approximately equimolar amounts to form one cDNA pool. The cDNA pool was sequenced on an Illumina HiSeq 2000 machine. The short sequence alignment software “Bowtie”40 was used to map RNA-Seq reads (about 20 million each sample) on the E. coli MG1655 annotated genome (NCBI accession number NC_000913) and the cognate plasmid circuit sequences of each sample. The number of mapped reads for each gene was determined according to their annotated location features (NCBI gff format). The expression levels of genes were subsequently determined using the normalized measure of RPKM (Reads Per Kilobase of transcript per Million mapped reads).41 Read mapping were visualized using the Integrative Genomics Viewer tool (IGV).42 To increase accuracy, under the assumption of normal distribution, we treated genes with the expression values that are out of the typical range of μ ± 3σ as exceptions and thus did not take them into account for subsequent statistical comparison analysis. Here, we filtered out those genes (Table S2) due to their expression levels are either too high or too low following this criteria. The obtained RNA-Seq data set can be openly accessed and downloaded from the Edinburgh DataShare Repository with the DOI: http://dx.doi.org/10.7488/ds/2119.
Cell Growth Rate Modeling and Metabolic Load Calculation
The cell growth curve for each sample, as described by the measured cell density (OD600), were fitted using the Gompertz model,26 an S-shaped function as shown below.
where μm stands for bacterial growth rate at exponential growth phase; A is the maximum cell density that the culture would be achieved; λ is the lag time before cells entering exponential growth phase. The nonlinear least-squares fitting function (cftool) in Matlab (MathWorks R2014a) was applied to fit the experimental data of cell growth to parametrize the growth model (Figure 2B and Figure S2).
Metabolic load is calculated following the method defined previously,7i.e., the relative growth rate reduction against a reference sample. Here we used the fastest growing Sample 7, i.e., host carrying the empty low copy number plasmid pSB4K5, as the reference to calculate the metabolic load for all other sample constructs following the equation
where μm and μmc are the cell growth rates of a sample and the selected reference sample, respectively.
Gene Expression Clustering and Differential Expression Analysis
The hierarchical clustering function (clustergram) in Matlab was used to cluster gene expression levels in all sequenced transcriptomes with the exception that the mean gene expression levels of the two biological repeats (Samples 1 and 2) were treated as one condition. Hierarchical clustering was performed twice, on both directions, row (gene) wise and column (sample/condition) wise to obtain the heat map with dendrograms as shown in Figure 3.
To minimize potential false positives, two parallel methods were used to find differentially expressed genes between compared conditions. The first method used is the combined 2-fold expression change detection and χ2-test. Differentially expressed genes were determined when both the expression levels between compared conditions having more than 2-fold difference and the false discovery rate-adjusted p-value <0.005 from the χ2-test. For the second method, the software edgeR43 was used. Since duplicate is available for one circuit condition, as suggested by edgeR, we used the duplicate samples (S1 and S2) to calculate the dispersion value in the experiment (0.025) which was subsequently adopted for all other paired comparison analysis in this study. The p-values and FDRs associated with the DEGs were provided in the Supporting Information of gene expression analysis. The online tool DAVID28,29 was used for the functional enrichment analysis among identified differentially expressed genes. Gene functions were retrieved from the Gene Ontology biological processes44 and KEGG pathway databases.45
Acknowledgments
The work was supported by UK BBSRC project grant [BB/N007212/1], Leverhulme Trust research grant [RPG-2015-445] and Wellcome Trust-UoE Institutional Strategic Support Fund. XW acknowledges funding support from the Scottish Universities Life Sciences Alliance and China Scholarship Council.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.7b00328.
Author Contributions
BW and JS conceived the project and designed the experiment. QL and BW performed the experiment and data analysis. All the authors took part in the interpretation of results and preparation of materials for the manuscript. BW led the study and wrote the manuscript.
The authors declare no competing financial interest.
Notes
The obtained RNA-Seq data set can be openly accessed and downloaded from the Edinburgh DataShare Repository with the DOI: http://dx.doi.org/10.7488/ds/2119. All plasmids used are available upon request and selected plasmids may be obtained from the Addgene repository https://www.addgene.org/Baojun_Wang/.
Supplementary Material
References
- Wang B.; Kitney R. I.; Joly N.; Buck M. (2011) Engineering modular and orthogonal genetic logic gates for robust digital-like synthetic biology. Nat. Commun. 2, 508. 10.1038/ncomms1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon T. S.; Lou C.; Tamsir A.; Stanton B. C.; Voigt C. A. (2012) Genetic programs constructed from layered logic gates in single cells. Nature 491, 249–253. 10.1038/nature11516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodius V. A.; Segall-Shapiro T. H.; Sharon B. D.; Ghodasara A.; Orlova E.; Tabakh H.; Burkhardt D. H.; Clancy K.; Peterson T. C.; Gross C. A.; Voigt C. A. (2013) Design of orthogonal genetic switches based on a crosstalk map of σs, anti-σs, and promoters. Mol. Syst. Biol. 9, 702. 10.1038/msb.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley R. W.; Buck M.; Wang B. (2016) Tools and principles for microbial gene circuit engineering. J. Mol. Biol. 428, 862–888. 10.1016/j.jmb.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Brophy J. A. N.; Voigt C. A. (2014) Principles of genetic circuit design. Nat. Methods 11, 508–520. 10.1038/nmeth.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceroni F.; Algar R.; Stan G.-B.; Ellis T. (2015) Quantifying cellular capacity identifies gene expression designs with reduced burden. Nat. Methods 12, 415–418. 10.1038/nmeth.3339. [DOI] [PubMed] [Google Scholar]
- Cardinale S.; Joachimiak M. P.; Arkin A. P. (2013) Effects of genetic variation on the E. coli host-circuit interface. Cell Rep. 4, 231–237. 10.1016/j.celrep.2013.06.023. [DOI] [PubMed] [Google Scholar]
- Qian Y.; Huang H.-H.; Jiménez J. I.; Del Vecchio D. (2017) Resource competition shapes the response of genetic circuits. ACS Synth. Biol. 6, 1263. 10.1021/acssynbio.6b00361. [DOI] [PubMed] [Google Scholar]
- Stanton B. C.; Nielsen A. A. K.; Tamsir A.; Clancy K.; Peterson T.; Voigt C. A. (2014) Genomic mining of prokaryotic repressors for orthogonal logic gates. Nat. Chem. Biol. 10, 99–105. 10.1038/nchembio.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An W.; Chin J. W. (2009) Synthesis of orthogonal transcription-translation networks. Proc. Natl. Acad. Sci. U. S. A. 106, 8477–8482. 10.1073/pnas.0900267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brödel A. K.; Jaramillo A.; Isalan M. (2016) Engineering orthogonal dual transcription factors for multi-input synthetic promoters. Nat. Commun. 7, 13858. 10.1038/ncomms13858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha M.; Salis H. M. (2015) A portable expression resource for engineering cross-species genetic circuits and pathways. Nat. Commun. 6, 7832. 10.1038/ncomms8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Gerstein M.; Snyder M. (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileyko Y.; Joh R. I.; Weitz J. S. (2008) Small-scale copy number variation and large-scale changes in gene expression. Proc. Natl. Acad. Sci. U. S. A. 105, 16659–16664. 10.1073/pnas.0806239105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. W.; Gyorgy A.; Cameron D. E.; Pyenson N.; Choi K. R.; Way J. C.; Silver P. A.; Del Vecchio D.; Collins J. J. (2016) Creating single-copy genetic circuits. Mol. Cell 63, 329–336. 10.1016/j.molcel.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T. K.; Khalil A. S.; Collins J. J. (2009) Next-generation synthetic gene networks. Nat. Biotechnol. 27, 1139–1150. 10.1038/nbt.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Buck M. (2012) Customizing cell signaling using engineered genetic logic circuits. Trends Microbiol. 20, 376–384. 10.1016/j.tim.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Hutcheson S. W.; Bretz J.; Sussan T.; Jin S.; Pak K. (2001) Enhancer-binding proteins HrpR and HrpS interact to regulate hrp-encoded type III protein secretion in Pseudomonas syringae strains. J. Bacteriol. 183, 5589–5598. 10.1128/JB.183.19.5589-5598.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Barahona M.; Buck M. (2014) Engineering modular and tunable genetic amplifiers for scaling transcriptional signals in cascaded gene networks. Nucleic Acids Res. 42, 9484–9492. 10.1093/nar/gku593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic M.; James E. H.; Burrows P. C.; Rego F. G. M.; Buck M.; Schumacher J. (2011) Regulation of the co-evolved HrpR and HrpS AAA+ proteins required for Pseudomonas syringae pathogenicity. Nat. Commun. 2, 177. 10.1038/ncomms1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Barahona M.; Buck M. (2013) A modular cell-based biosensor using engineered genetic logic circuits to detect and integrate multiple environmental signals. Biosens. Bioelectron. 40, 368–376. 10.1016/j.bios.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Buck M. (2014) Rapid engineering of versatile molecular logic gates using heterologous genetic transcriptional modules. Chem. Commun. 50, 11642–11644. 10.1039/C4CC05264A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley R. W.; Buck M.; Wang B. (2016) Recognizing and engineering digital-like logic gates and switches in gene regulatory networks. Curr. Opin. Microbiol. 33, 74–82. 10.1016/j.mib.2016.07.004. [DOI] [PubMed] [Google Scholar]
- Shetty R.; Endy D.; Knight T. (2008) Engineering BioBrick vectors from BioBrick parts. J. Biol. Eng. 2, 5. 10.1186/1754-1611-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorochowski T. E.; Avcilar-Kucukgoze I.; Bovenberg R. A. L.; Roubos J. A.; Ignatova Z. (2016) A minimal model of ribosome allocation dynamics captures trade-offs in expression between endogenous and synthetic Genes. ACS Synth. Biol. 5, 710–720. 10.1021/acssynbio.6b00040. [DOI] [PubMed] [Google Scholar]
- Zwietering M. H.; Jongenburger I.; Rombouts F. M.; van’t Riet K. (1990) Modeling of the bacterial growth curve. Appl. Environ. Microbiol. 56, 1875–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H.; Lin Y.; Gao F.; Zhang C.-T.; Zhang R. (2014) DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 42, D574–D580. 10.1093/nar/gkt1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G.; Sherman B. T.; Hosack D. A.; Yang J.; Gao W.; Lane H. C.; Lempicki R. A. (2003) DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4, R60. 10.1186/gb-2003-4-9-r60. [DOI] [PubMed] [Google Scholar]
- Huang D. W.; Sherman B. T.; Lempicki R. A. (2008) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jiang K.; Zhang S.; Lee S.; Tsai G.; Kim K.; Huang H.; Chilcott C.; Zhu T.; Feldman L. J. (2006) Transcription profile analyses identify genes and pathways central to root cap functions in maize. Plant Mol. Biol. 60, 343–363. 10.1007/s11103-005-4209-4. [DOI] [PubMed] [Google Scholar]
- Jones K. L.; Kim S.-W.; Keasling J. D. (2000) Low-copy plasmids can perform as well as or better than high-copy plasmids for metabolic engineering of bacteria. Metab. Eng. 2, 328–338. 10.1006/mben.2000.0161. [DOI] [PubMed] [Google Scholar]
- Nielsen A. A. K.; Der B. S.; Shin J.; Vaidyanathan P.; Paralanov V.; Strychalski E. A.; Ross D.; Densmore D.; Voigt C. A. (2016) Genetic circuit design automation. Science 352, aac7341. 10.1126/science.aac7341. [DOI] [PubMed] [Google Scholar]
- Elowitz M. B.; Levine A. J.; Siggia E. D.; Swain P. S. (2002) Stochastic gene expression in a single cell. Science 297, 1183–1186. 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Wang B.; Barahona M.; Buck M. (2015) Amplification of small molecule-inducible gene expression via tuning of intracellular receptor densities. Nucleic Acids Res. 43, 1955–1964. 10.1093/nar/gku1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley R. W.; Wang B. (2015) Designer cell signal processing circuits for biotechnology. New Biotechnol. 32, 635–643. 10.1016/j.nbt.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.-W.; Burkhardt D.; Gross C.; Weissman J. S. (2014) Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157, 624–635. 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P.; Aebersold R. (2012) Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Methods 9, 555–566. 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- Shishkin A. A.; Giannoukos G.; Kucukural A.; Ciulla D.; Busby M.; Surka C.; Chen J.; Bhattacharyya R. P.; Rudy R. F.; Patel M. M.; Novod N.; Hung D. T.; Gnirke A.; Garber M.; Guttman M.; Livny J. (2015) Simultaneous generation of many RNA-seq libraries in a single reaction. Nat. Methods 12, 323–325. 10.1038/nmeth.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorochowski T. E.; Espah Borujeni A.; Park Y.; Nielsen A. A.; Zhang J.; Der B. S.; Gordon D. B.; Voigt C. A. (2017) Genetic circuit characterization and debugging using RNA-seq. Mol. Syst. Biol. 13, 952. 10.15252/msb.20167461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B.; Trapnell C.; Pop M.; Salzberg S. L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25–R25. 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A.; Williams B. A.; McCue K.; Schaeffer L.; Wold B. (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Robinson J. T.; Thorvaldsdóttir H.; Winckler W.; Guttman M.; Lander E. S.; Getz G.; Mesirov J. P. (2011) Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. D.; McCarthy D. J.; Smyth G. K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M. D.; Wakefield M. J.; Smyth G. K.; Oshlack A. (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14. 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M.; Goto S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.