

ABSTRACT
Oral streptococci are generally considered commensal organisms; however, they are becoming recognized as important associate pathogens during the development of periodontal disease as well as being associated with several systemic diseases, including as a causative agent of infective endocarditis. An important virulence determinant of these bacteria is an ability to evade destruction by phagocytic cells, yet how this subversion occurs is mostly unknown. Using Streptococcus gordonii as a model commensal oral streptococcus that is also associated with disease, we find that resistance to reactive oxygen species (ROS) with an active ability to damage phagosomes allows the bacterium to avoid destruction within macrophages. This ability to survive relies not only on the ROS resistance capabilities of the bacterium but also on ROS production by macrophages, with both being required for maximal survival of internalized bacteria. Importantly, we also show that this dependence on ROS production by macrophages for resistance has functional significance: S. gordonii intracellular survival increases when macrophages are polarized toward an activated (M1) profile, which is known to result in prolonged phagosomal ROS production compared to that of alternatively (M2) polarized macrophages. We additionally find evidence of the bacterium being capable of both delaying the maturation of and damaging phagosomes. Taken together, these results provide essential insights regarding the mechanisms through which normally commensal oral bacteria can contribute to both local and systemic inflammatory disease.
KEYWORDS: macrophages, oral microbiology, phagocytosis
INTRODUCTION
Oral streptococci are generally considered commensal bacteria; however, it is becoming clear that they are not always innocuous. For example, the full development of periodontal disease is not dependent on the presence of a single pathogenic microorganism but also requires an intact commensal oral microbiota, including oral streptococci (1, 2). In addition, oral streptococci have been found to be associated with a number of extraoral diseases, including cardiovascular disease, adverse pregnancy outcomes, and rheumatoid arthritis (3, 4), as well as being major causative agents of infectious endocarditis, a rare disease with a mortality rate near 40% (5–7).
Streptococcus gordonii is a well-studied, ubiquitous, normally commensal oral streptococcus. It is a member of the mitis group of viridans group streptococci (8–10), and the mitis group members are the streptococci most commonly isolated from patients with endocarditis (5–7, 11, 12). In addition to having a role in endocarditis, oral viridans group streptococci in general, and S. gordonii specifically, are becoming recognized as “accessory pathogens” in the development of periodontal disease, through interspecies interactions with overt periodontal pathogens (13–16).
The professional phagocytes are the first cellular line of defense against invading pathogens. However, in chronic inflammatory diseases, including periodontal disease, phagocytes can also make significant contributions to the pathology of disease (17–20). In the oral environment, while neutrophils play an essential role in the prevention of periodontal disease, at least in part due to their constant transiting into the gingival crevicular space and interaction with the oral microbiome (21, 22), macrophages also have critically important roles in the initiation, resolution, and pathology of gingivitis and periodontal disease (23, 24). Phagocytes also play important roles in oral streptococcus-mediated endocarditis development (25–27). Indeed, strains of S. gordonii capable of causing disease in animal models of endocarditis are more resistant to destruction by phagocytes (26). So while evasion of phagocytes is part of the pathogenicity mechanism of streptococci (28, 29), resulting from activation of platelets and subsequent formation of fibrinous endovascular vegetations encapsulating bacteria which can lead to endocarditis (25, 30, 31), an additional determinant for the development of endocarditis is an ability of streptococci to avoid destruction after uptake by a variety of phagocytic cells (26–28, 32–34). Many of the mechanistic details allowing for S. gordonii resistance to destruction by phagocytes remain poorly understood. In addition, phagocytes, including macrophages, alter their phenotypes during inflammation, such as during periodontal disease or endocarditis (24, 25, 35–38). How these changes in phagocyte phenotypes might result in altered interactions with oral streptococci is currently unclear.
In this study, we show that S. gordonii strains with endocarditis pathogenic potential are able to survive within macrophages at a significantly higher rate than strains without pathogenic potential. This ability to survive relies on an interdependence between the phagosomal reactive oxygen species (ROS) production characteristics of macrophages, which are known to change depending on the macrophage activation profile, and the ROS resistance capabilities of S. gordonii, with both being required for maximal survival of the bacterium. Additionally, we found active bacterial subversion of phagosomal maturation, which may allow for avoidance of phagosomal destruction, suggesting a dual-stage requirement for S. gordonii to evade phagocytic destruction.
RESULTS
S. gordonii survives within macrophages in a ROS resistance-dependent manner.
While phagocytes are specially equipped to kill microorganisms, a surprising number of microbes possess defense mechanisms allowing for their survival within these cells (39, 40). S. gordonii is thought to have this ability, though this has not been tested thoroughly. We initially expanded upon the bactericidal assay performed by others (26, 34) and performed a gentamicin protection assay (41). This assay, in which only cells surviving within the phagocyte are recovered, confirmed that S. gordonii strain DL1, which can be causative for endocarditis (5–7, 26), has the ability to survive within lipopolysaccharide (LPS)-activated RAW264.1 macrophages for 2 h postphagocytosis, at a significantly higher rate than that for the nonpathogenic strain SK12 (Fig. 1A), which is unable to cause endocarditis in an animal model (26) despite being able to bind platelets as effectively as DL1 (42).
FIG 1.
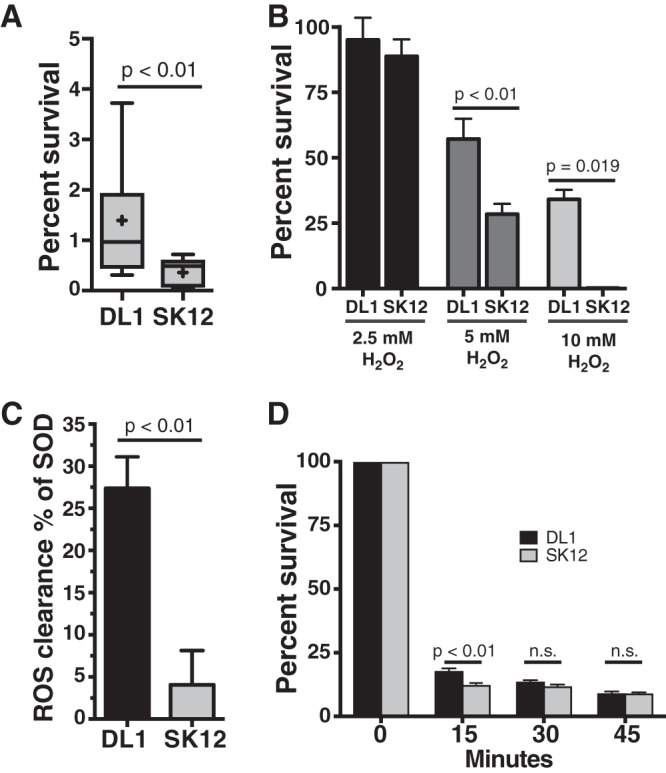
S. gordonii DL1 has increased survival within macrophages compared to that of S. gordonii SK12. (A) Percent survival of S. gordonii strains DL1 and SK12, with and without endocarditis pathogenic potential, respectively, within RAW264.7 macrophages for 2 h postuptake. The box plot with whiskers shows the 10th to 90th percentiles, and “+” indicates the mean for each strain. (B) Survival of S. gordonii strains DL1 and SK12 in the presence of increasing concentrations of hydrogen peroxide for 30 min. Data shown are means and standard errors. DL1 survived at significantly higher rates than those of SK12 in the presence of both 5 mM and 10 mM H2O2. (C) S. gordonii DL1 removes generated superoxide significantly more efficiently than SK12 does, protecting cytochrome c from reduction in the presence of generated superoxide. The graph shows means and standard errors, normalized by setting the positive-control purified superoxide dismutase value at 100%. (D) DL1 and SK12 survival rates after exposure to pH 3 for the indicated times. Data shown are means and standard errors. All data are for a minimum of 4 independent experiments and a minimum of 5 technical replicates per experiment. P values were calculated with unpaired t tests (A and C) or one-way (B) or two-way (D) analysis of variance (ANOVA) with the Sidak multiple-comparison test. n.s., not significant.
Two major tools that macrophages use to kill microbes are creating toxic ROS via the NADPH oxidase (NOX2) complex and highly acidifying the phagosome (39). Since S. gordonii, like all viridans group streptococci, produces its own ROS as a metabolic by-product in the form of H2O2 (43, 44) and has noncatalase ROS resistance mechanisms (45–47), we hypothesized that ROS resistance may be an important mechanism in allowing S. gordonii to survive within phagocytes. Indeed, we found that S. gordonii strain DL1 was better equipped to survive in the presence of H2O2 (Fig. 1B), as well as being more capable of removing generated superoxide from solution (Fig. 1C), than strain SK12. We additionally checked the ability of the strains to survive under low-pH conditions but found no differences in survival at low pH after 15 min (Fig. 1D).
ROS production by macrophages is required for optimum S. gordonii survival.
Because we preactivated the RAW264.7 macrophages prior to performing the killing assays, which increases phagosomal ROS production (48–50), we next tested the requirement for ROS production by macrophages for the survival of S. gordonii. When the macrophages were treated with diphenyleneiodonium (DPI), a NOX2 inhibitor that prevents phagosomal ROS production (51–55), intracellular survival of the pathogenic strain DL1 decreased to levels not significantly different from that of the nonpathogenic strain SK12 (Fig. 2A). While DPI is capable of inhibiting other flavoproteins in addition to the NOX2 complex, including mitochondrial complex I, which can affect the viability of the cells, the inhibition of other flavoproteins often requires DPI concentrations higher than those used here (56). Indeed, the viability of our macrophages was not decreased during the period in which we used the drug (see Fig. S1 in the supplemental material).
FIG 2.
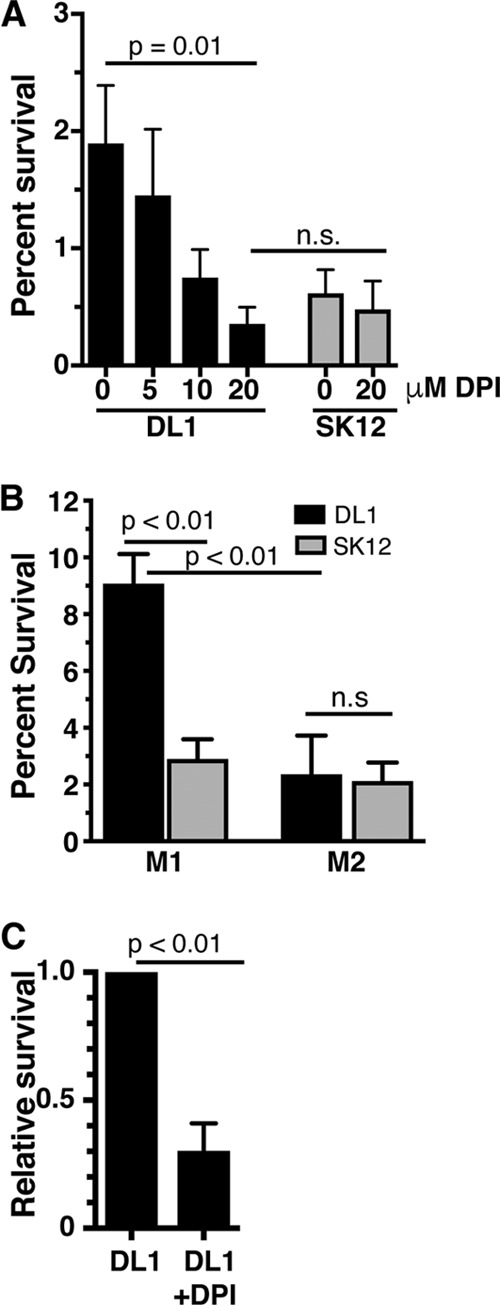
ROS production by macrophages is required for optimal S. gordonii survival. (A) Survival of S. gordonii within RAW264.7 macrophages, with increasing amounts of diphenyleneiodonium (DPI) added to inhibit NOX2 ROS production within the phagosome. Increasing levels of DPI led to significant reductions in survival of DL1, to levels not significantly different from SK12 survival in macrophages with or without DPI added. (B) Survival of S. gordonii within polarized human monocyte-derived macrophages. DL1 survived significantly better in GM-CSF-matured, IFN-γ/LPS-polarized (M1) macrophages than SK12 did, while both strains were equally killed by M-CSF-matured, IL-4-polarized (M2) macrophages. (C) Survival of DL1 within M1 polarized human monocyte-derived macrophages, with 20 μM DPI added to inhibit NOX2 ROS production within the phagosome. The data were normalized by setting DL1 survival at 1. All data are means and standard errors for 4 to 8 independent experiments, with a minimum of 2 technical replicates per experiment. P values were calculated by one-way ANOVA with the Sidak multiple-comparison test (A and B) or by unpaired t test (C).
Macrophages are increasingly being understood to have a variety of phenotypes along a continuum, with the extremes referred to as inflammatory, activated “M1” macrophages and tissue repair, enhanced “M2” macrophages (57). In general, M1 macrophages are regarded as being better equipped to kill pathogens than M2 macrophages (39, 40). This is partly due to phagosomes of macrophages having variable ROS and acidification profiles depending on the activation profile, with M1 macrophages producing more bactericidal phagosomal ROS, with concomitant weaker acidification, than resting or M2 macrophages (50, 54, 58, 59). Because of the dependence of S. gordonii survival on ROS production within the phagosome (Fig. 2A), and since most inflammatory diseases, including endocarditis and periodontitis, result in inflammation and macrophage activation (25, 35, 37, 38), we next examined the ability of S. gordonii to survive within macrophages that have been polarized to either the M1 or M2 phenotype. For these experiments, we used primary monocyte-derived macrophages because they are the predominant in vitro system for studying macrophage polarization (57). We tested S. gordonii survival in polarized human monocyte-derived macrophages with known differences in phagosome maturation, i.e., granulocyte-macrophage colony-stimulating factor (GM-CSF)-matured macrophages activated with gamma interferon (IFN-γ) and LPS (M1 macrophages), which produce phagosomal ROS for longer times and have weaker acidification, and macrophage colony-stimulating factor (M-CSF)-matured macrophages activated with interleukin-4 (IL-4) (M2 macrophages) (50, 54). In M1 macrophages, we found that the rate of S. gordonii DL1 survival was significantly higher than that of SK12, while in M2 macrophages DL1 survival was not significantly different from that of SK12 (Fig. 2B).
As with the RAW cell killing assays, we used a gentamicin protection assay for determinations of S. gordonii survival within monocyte-derived macrophages. A potential issue with the use of gentamicin is that macrophages can be highly pinocytotic, and the membrane-impermeant gentamicin drug can be taken up and enter phagosomes containing bacteria, affecting the bacterial survivability independent of the phagosome bactericidal properties (60, 61). To test for such effects, we varied the amount of time the human monocyte-derived macrophages were incubated with gentamicin post-bacterial uptake, using times of 30 min to 2 h (Fig. S2). We did see differences in the survivability of S. gordonii in RAW macrophages with the different gentamicin incubation times, with the survival rates of S. gordonii within RAW264.7 macrophages obtained with 30 min of gentamicin treatment (Fig. S2) corresponding to those reported for another viridans group streptococcus species, S. parasanguinis (33), as well as to our results (Fig. 1). In contrast, we found no significant difference in the survival of S. gordonii within monocyte-derived macrophages. However, to minimize unintended consequences, we subsequently limited incubation with gentamicin to 30 min.
We also tested human monocyte-derived M1 macrophage ROS production requirements for S. gordonii survival and found that the relative bacterial survival within the macrophages was significantly reduced upon treatment with the inhibitor DPI (Fig. 2C).
Phagosomes containing S. gordonii exhibit delayed maturation.
While ROS resistance can help microorganisms to survive initial phagocytosis, ROS resistance alone is unlikely to allow for extended survival within phagocytes, with long-term survival usually involving bacterium-induced damage or modification to phagosomes (39, 62). As a marker of mature phagolysosomes, the rate of LAMP1 acquisition by maturing phagosomes can be an initial gauge of damage or other bacterium-induced phagosomal modifications (63–65). We therefore examined LAMP1 acquisition by S. gordonii-containing phagosomes in activated RAW macrophages via immunofluorescence assay (Fig. 3A and B). We found that LAMP1 was slower to accumulate on DL1-containing phagosomes than on phagosomes containing the nonpathogenic strain SK12 or heat-killed DL1. In addition, a maximum of approximately 80% of internalized DL1 organisms obtained LAMP1 after 60 min, and this percentage even subsequently appeared to decrease, yet LAMP1 was acquired by over 95% of phagosomes containing SK12 or heat-killed DL1 within 20 to 30 min and remained associated for the full time examined (Fig. 3B).
FIG 3.
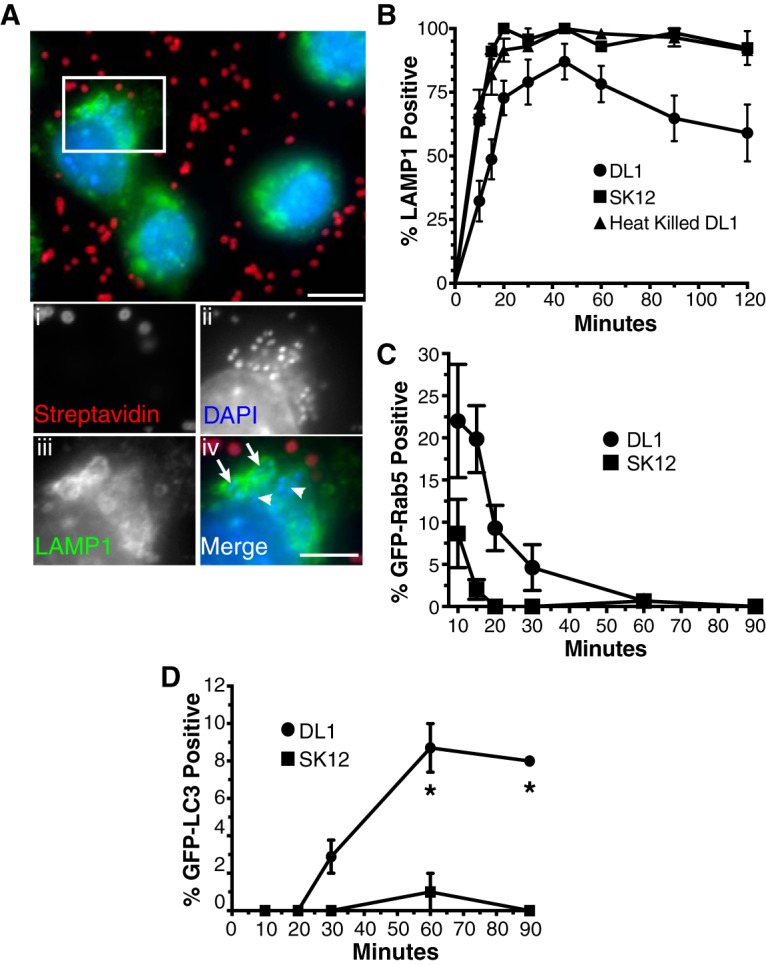
Phagosomes containing S. gordonii DL1 exhibit altered maturation profiles. (A and B) The rates of LAMP1 acquisition by phagosomes containing S. gordonii were determined by immunofluorescence staining and quantification following synchronized phagocytosis. (A) Example immunofluorescence images of LAMP1-positive (arrows in panel iv) and -negative (arrowheads in panel iv) phagosomes containing engulfed S. gordonii (streptavidin negative). Bar = 10 μm (5 μm in panel iv). (B) Percentage of LAMP1-positive S. gordonii-containing phagosomes versus time postengulfment. Heat-killed DL1- and live SK12-containing phagosomes all quickly acquired LAMP1, while, on average, fewer than 80% of phagosomes containing live DL1 obtained the phagolysosomal marker over 120 min. Data shown are means ± standard errors for at least 4 independent experiments per condition, with a minimum of 50 phagosomes per time point per experiment examined. The LAMP1 acquisition curves for DL1 versus SK12 and heat-killed DL1 are significantly different by one-phase association fitting (P < 0.001). (C and D) Synchronized phagocytosis of S. gordonii was performed with RAW264.7 macrophages transfected with GFP-Rab5 (C) or GFP-LC3 (D), and quantification of marker-positive S. gordonii-containing phagosomes over time was performed. Data shown are mean percentages ± standard errors for at least 3 independent experiments, with a minimum of 50 phagosomes per time point per experiment examined. (C) The rates of GFP-Rab5 loss from DL1- and SK12-containing phagosomes are significantly different by one-phase decay fitting (P < 0.001). (D) Asterisks indicate significant differences (P < 0.01) in GFP-LC3 labeling at individual time points by unpaired t test, using the Holm-Sidak method for multiple comparisons.
A number of nonexclusive causes of reduced LAMP1 staining of phagosomes for live but not killed DL1 include (i) S. gordonii-induced damage to the phagosome, (ii) S. gordonii-induced inhibition of phagosome maturation, and (iii) increased phagosomal ROS production by the macrophage in an attempt to overcome bacterial resistance (which will delay phagosomal maturation) (39, 54, 66, 67). When we next tested an earlier stage of macrophage phagosome maturation by examining the rate of clearance of the early endosome/phagosome marker Rab5 from S. gordonii-containing phagosomes, we again saw significant differences in the maturation rate of phagosomes, with extended Rab5 retention on DL1-containing phagosomes compared to that on SK12-containing phagosomes (Fig. 3C). Because we saw that ROS dynamics were important for bacterial survival (Fig. 2), we first hypothesized that there may be bacterium-induced changes in ROS production as opposed to polarization-induced changes by macrophages that could account for the delayed maturation of DL1-containing phagosomes. Indeed, a larger percentage of the DL1- than SK12-containing phagosomes in RAW macrophages acquired the autophagosomal marker LC3 (Fig. 3D). Such acquisition can be indicative of LC3-assisted phagocytosis (LAP), where Toll-like receptor signaling occurring from within undamaged phagosomes can lead to accumulation of LC3 on the phagosomal limiting membrane (68). LAP has been reported to result in various changes in phagosomal maturation, such as increased phagosomal maturation for expedited degradation of the phagosomal contents (68, 69) or increased phagosomal ROS activity and delayed maturation (52, 70). Our results indicated a reduction in phagosomal maturation (Fig. 3B), leading to the possibility of the latter scenario. However, M1 activation of macrophages promotes increased phagosomal ROS activity that is separate from LAP (50, 54). Still, to ensure that additional (potentially LAP-based) changes in ROS production within macrophage phagosomes containing S. gordonii were not occurring, we used luminol-based detection to examine intracellular ROS production by M1-activated macrophages containing DL1 or SK12 (Fig. 4). We did not detect any ROS production differences, suggesting that LAP was likely not a reason for the increased LC3 accumulation on S. gordonii-containing phagosomes.
FIG 4.
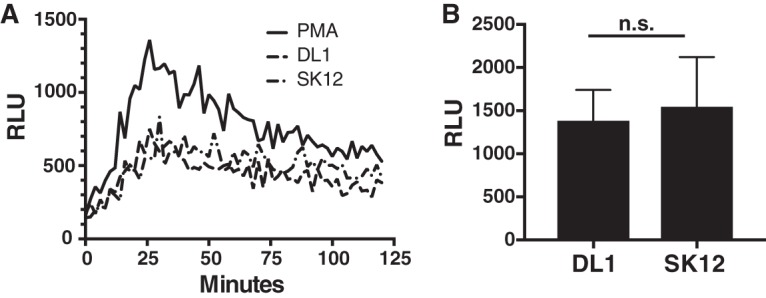
ROS response to S. gordonii by M1 activated macrophages does not vary with the strain. Luminol-based ROS detection was used to quantify the ROS response by M1 activated macrophages during interaction with S. gordonii strains DL1 and SK12. (A) Example time course of luminol luminescence detection, shown in relative luminescence units (RLU). (B) Mean peak RLU (with standard error) detected over 5 independent experiments.
S. gordonii can disrupt phagosome integrity.
Increased LC3 acquisition by phagosomes (Fig. 3D) may also be due to overt bacterium-induced damage to the phagosomal membrane, leading to activation of galectin- or ubiquitin-mediated macroautophagy, giving the macrophage an opportunity to reenvelop and destroy the bacterium (68). As advanced endocarditis lesions show nearly exclusively extracellular bacteria (26, 27), escape from the phagosome is likely in the path to S. gordonii virulence. We therefore next used a lysosomal fluorescein isothiocyanate (FITC)-dextran assay for the sensitive detection of S. gordonii-induced damage to phagolysosomes. FITC-dextran can be used as a pH-sensitive ratiometric fluorophore to measure the pH of the compartment in which it resides (71, 72). When loaded into the lysosome, dextran will enter the phagosome as the fusion of lysosomes occurs during phagosomal maturation, allowing for pH measurements of the maturing phagosomes, and if the phagosomes are damaged FITC-dextran will report the loss of lysosomal acidity. With severe phagosomal damage the FITC-dextran will escape into the cytoplasm, where it is readily detectable even if the bacterium itself is reenveloped (66, 68, 73). We saw a significant increase in the average pH reported by pulse-chased FITC-dextran within M1 activated macrophages that contained S. gordonii compared to that for control M1 macrophages, with the level for those containing S. gordonii DL1 (mean pH = 6.19; standard deviation [SD] = 0.34) being significantly higher than those for both control (mean pH = 5.4; SD = 0.17) and SK12-containing (mean pH = 5.64; SD = 0.15) macrophages (Fig. 5A). When M1 macrophages were treated with DPI, the pH reported by FITC-dextran for those containing S. gordonii DL1 was reduced significantly (mean pH = 5.78; SD = 0.16), as seen with other phagocytic cargo (54, 58), though it was still higher than that for control M1 macrophages not containing bacteria (Fig. 5A). To examine the rates of dextran escape into the cytosol, pH heat maps of FITC-dextran-containing macrophages were generated, and the percentage of cells showing clear cytoplasmic FITC-dextran (reporting pH of >6) was quantified (Fig. 5B and C). We found that S. gordonii DL1 caused significantly more lysosomal/phagolysosomal damage than both control cells and SK12 (Fig. 5C).
FIG 5.

Phagosome damage is detectable upon S. gordonii uptake. Lysosomes of human monocyte-derived M1 macrophages were loaded with FITC-dextran and then allowed to phagocytose S. gordonii or were treated with LLME as a positive control for lysosomal damage. (A) Ratiometric imaging of live cells revealed that the mean pH of FITC-dextran in M1 macrophages with phagocytosed S. gordonii DL1 was significantly higher than that for control cells. Treatment of macrophages with 10 μm DPI during phagocytosis of DL1 (DL1 + DPI) significantly reduced the pH reported by FITC-dextran. The scatterplot shows a minimum of 85 cells per condition quantified over at least 5 independent experiments. Lines show the mean pH ± standard deviation. P values were calculated by one-way ANOVA and Tukey's multiple-comparison test. (B) Example of pH heat map of FITC-dextran in macrophages with or without phagocytosed S. gordonii after ratiometric image calculations. Bar = 10 μm. (C) The percentage of macrophages with visible high-pH dextran, indicating damaged phagolysosomes, was quantified in 4 to 6 independent experiments. A minimum of 100 cells per condition per experiment (overall total of 600 to 1,100 individual cells per condition) were quantified. P values were calculated by one-way ANOVA and Tukey's multiple-comparison test.
DISCUSSION
Oral streptococci are generally considered commensal microorganisms, yet they are becoming recognized as accessory pathogens required for the full development of periodontal disease, are common agents of infective endocarditis, and have been associated with other extraoral systemic diseases (3, 4, 16). The ability to survive in the presence of phagocytic cells is important for oral streptococci to be pathogenic (26, 27), yet the conditions under which oral streptococci can survive encounters with phagocytes are not well defined. In the present study, we found that the oral streptococcus S. gordonii survives to a greater degree within macrophages when they are inflammatory (M1) activated macrophages than when they have been activated toward an alternative (M2) phenotype. This is surprising, as M1 macrophages are generally better equipped to kill pathogens than nonactivated or M2 macrophages (39, 40). However, it is known that macrophages have phagosome maturation profiles, including reactive ROS production and proteolytic capabilities, that vary broadly depending on their polarization state (50, 54, 74, 75). Generally, inflammatory (M1) macrophages have more extensive phagosomal ROS responses and slower acidification than those of regulatory or wound-healing resident macrophages (54, 58, 76).
While the production of ROS by phagocytes is generally microbicidal, successful pathogens often have defenses against ROS-induced damage (39, 45, 77). Oral streptococci produce high levels of H2O2 during their normal metabolism (43, 44), with correspondingly high resistance to damage induced by ROS (45, 46). Indeed, ROS produced by oral streptococci have been suggested to play a bacteriostatic role against other bacteria in oral biofilms (78, 79). Our data show that S. gordonii strains with pathogenic potential have increased ROS resistance capabilities compared to those of a nonpathogenic strain. Importantly, the ROS resistance of S. gordonii appears to be more important than resistance to low phagosomal pH and/or resistance to phagosomal proteolytic enzymes activated at low pH, as S. gordonii resists killing less effectively within M2 macrophages than within M1 activated macrophages, which have a prolonged period of phagosomal ROS production (54). Further, the treatment of M1 macrophages with DPI to inhibit phagosomal ROS production and allow for a lowering of phagosomal pH (Fig. 5A) (54, 58) also decreased the survival of S. gordonii within macrophages (Fig. 3C).
Periodontal disease, like all inflammatory diseases, causes recruitment of significant numbers of inflammatory phagocytes (36, 80, 81), including an increased number of M1 activated macrophages in active lesions (24, 35–38). The resistance we see in streptococcal killing by activated macrophages may therefore be important in allowing for the bacterial contribution to periodontal disease as an accessory pathogen. In addition, the increased resistance to intracellular killing may allow for increased systemic dissemination, as seen with other oral bacteria, such as Porphyromonas gingivalis (82), and/or allow for streptococci to resist phagocytic killing once they have infiltrated the endocardium during the development of endocarditis. Which, if any, of these factors are at play is the subject of future studies.
Our results indicate that maturation of S. gordonii-containing phagosomes, as determined by Rab5 and LAMP1 acquisition, was inhibited. As we also found S. gordonii to be capable of damaging the phagosome, the bacterium-induced phagosomal damage is likely a factor resulting in the observed delay of maturation and increased LC3 acquisition through macroautophagy induction (68, 83). How the phagosome can be actively damaged by S. gordonii is currently unclear, as the alpha-hemolytic streptococci, including S. gordonii, do not produce streptolysin L or O, the pore-forming toxins of the beta-hemolytic streptococci (84). However, there are a number of factors produced by viridans group streptococci that are known or potential endocarditis risk factors, though most are involved in allowing bacterial binding to platelets, which is thought to be an intermediary for invading a damaged endocardium (30, 31). For example, the sialic acid-binding proteins Hsa and GspA (85, 86) are required for optimal infectivity in an experimental model (87). PadA and SspA/SspB are also involved in binding and activating platelets (42, 88). However, other pathogenic factors that may be involved in allowing S. gordonii to survive within phagocytes include PpiA, a surface lipid-anchored protein reported to interfere with phagocytosis (29, 89), and autolysin (AtlA), a virulence factor of Streptococcus pneumoniae that is important in the development of pneumonia (90) and of Streptococcus mutans and Staphylococcus aureus that is involved in the development of endocarditis (28, 91, 92). Future studies will be needed to address the detailed mechanisms allowing for S. gordonii phagosomal escape.
Overall, we find that strains of the normally commensal organism S. gordonii that are also capable of acting as pathogens are significantly better at surviving within macrophages than those that are not and that the bacterium avoids destruction by using a combination of its natural resistance to ROS and an active ability to damage phagosomes. Importantly, we also provide evidence that S. gordonii is better able to withstand destruction within activated (M1) macrophages than within alternatively activated (M2) macrophages, suggesting that shifting of macrophage inflammatory states to combat infection may inadvertently allow a normally commensal bacterium to contribute to disease development and severity.
MATERIALS AND METHODS
Cell culture.
RAW264.7 macrophages were obtained from ATCC and grown in RPMI medium (Lonza) supplemented with 10% fetal calf serum (Caisson) and 2 mM l-glutamine (Corning) at 37°C and 5% CO2. Prior to phagocytosis experiments, the RAW264.7 macrophages were activated for 2 to 24 h with 100 ng/ml lipopolysaccharide (LPS) (from Salmonella enterica serotype Minnesota strain Re595; Sigma-Aldrich). For human monocyte-derived macrophages, blood was obtained from healthy donors in accordance with our institutional review board (IRB)-approved protocol (approval 626714), and peripheral blood mononuclear cells (PBMCs) were isolated by isosmotic density gradient centrifugation using 1-Step polymorphs (Accurate Chemical). The PBMCs were plated on tissue culture plastic or glass coverslips to allow for the purification of monocytes by adherence to the surface, with other components of PBMCs being washed away, as previously described (93). The adherent monocytes were then differentiated for 5 days with 50 ng/ml GM-CSF or 50 ng/ml M-CSF (BioLegend). GM-CSF-matured cells were then activated for 24 h with 10 ng/ml IFN-γ (BioLegend) and for 12 h with 100 ng/ml LPS (Calbiochem), and M-CSF-matured cells were activated for 2 days with 20 ng/ml IL-4 (BioLegend), as previously described (54).
Bacterial culture and phagocyte killing assays.
S. gordonii was grown in brain heart infusion (BHI) medium (BD Biosciences) supplemented with 0.5% yeast extract (IBI Scientific) at 37°C. All experiments, unless otherwise noted, used mid-log-phase bacterial cultures that were briefly sonicated (30 s) to break bacterial chains into individual bacteria. For experiments using heat-killed bacteria, S. gordonii was grown to mid-log phase as described above and then incubated at 60°C for 60 min prior to use.
For determination of survival within macrophages, a modification of previously described protocols was performed (41, 94). Briefly, after adding bacteria to the macrophages (multiplicity of infection [MOI] = 5:1) and an initial centrifugation step to bring bacteria into contact with the macrophages (125 × g for 1 min), the cells were coincubated for 30 min at 37°C. The macrophages were washed with phosphate-buffered saline (PBS) to remove external bacteria and then incubated for 2 h in normal growth medium supplemented with 200 μg/ml gentamicin (Sigma) to ensure that any remaining external bacteria were killed. Alternatively, after 30 min of incubation with gentamicin, the cells were washed and further incubated for 90 min without gentamicin to minimize the uptake of the antibiotic by the macrophages (60). Cells were then lysed with sterile distilled water, serially diluted, and plated onto BHI agar plates. After growth at 37°C for 2 days, the number of CFU was determined and the number of “surviving bacteria” per condition calculated.
To determine the rate of phagocytosis of the bacteria by macrophages, additional wells of macrophages were incubated with bacteria as described above for 30 min, but after extensive washing with PBS, the cells were lysed with water and serially plated on BHI agar plates to determine the initial number of “phagocytosed bacteria.” Alternatively, the bacteria were prelabeled with biotin-hydrazide (Invitrogen) according to the manufacturer's instructions, and after 30 min of incubation under conditions identical to those described above, the macrophages were washed with ice-cold PBS, any remaining external bacteria were stained with streptavidin-Alexa 594 (Thermo Fisher) at 4°C, and then the cells were fixed with 4% paraformaldehyde (PFA), counterstained with DAPI (4′,6-diamidino-2-phenylindole), and mounted for microscopy. The phagocytic index (mean number of internalized bacteria per macrophage) was determined by counting the bacteria within a minimum of 50 macrophages per condition. The phagocytic index was subsequently used to determine the number of phagocytosed bacteria per well by using the known number of macrophages per condition as determined by counting on a hemocytometer. In either case, the percent survival of bacteria was calculated by dividing the number of “surviving bacteria” at 2 h postphagocytosis by the calculated number of “phagocytosed bacteria” (i.e., number of surviving bacteria/number of phagocytosed bacteria × 100). While we saw no significant differences in the phagocytic index between the strains of bacteria, there was a nonsignificant trend for a change dependent on the macrophage polarization state (see Fig. S3 in the supplemental material). However, when the percent survival was calculated by incorporating the phagocytic index for each experiment as described above, any changes in phagocytosis rate between conditions were accounted for.
H2O2 killing assays.
Overnight cultures of S. gordonii were subcultured, grown to mid-log phase (optical density at 600 nm [OD600] of 0.5), added to PBS (with and without H2O2), and incubated for 30 min at room temperature, after which they were serially diluted and plated on BHI plates. Percent survival was calculated based on the number of CFU in PBS with H2O2 compared to that in PBS alone (95).
pH killing assays.
Overnight S. gordonii cultures were harvested and resuspended in 0.1 M glycine at pH 7.0 and then concentrated in 1/10 the original culture volume in 0.1 M glycine buffer, pH 3.0. After 15, 30, and 45 min, samples were taken for serial dilution and plating on BHI plates (96). Percent survival was calculated as described above.
Bacterial superoxide removal.
Using a xanthine oxidase superoxide generation system (97), the ability of S. gordonii to remove superoxide ion from solution before the superoxide was able to reduce cytochrome c (measured as the ΔA550 value) was determined and normalized to that of purified superoxide dismutase (SOD) (Sigma), with the rate of superoxide removal by SOD being set at 100% (98).
Luminol ROS detection assay.
Human monocytes were isolated and matured into M1 macrophages, as described above, in 96-well tissue culture plates. For luminol ROS detection experiments, cells were preincubated with 250 μg/ml horseradish peroxidase (HRP) (Alfa Aesar) for 15 min to allow for fluid-phase uptake into the endosomal system. The cells were then placed in Hanks balanced salt solution (HBSS) (Corning) with 250 μg/ml HRP and 50 μM luminol (Tokyo Chemical Industry). S. gordonii cultures were concentrated and added at an MOI of 50:1. After 1 min of centrifugation (125 × g) followed by bacterial uptake for 5 min, SOD (50 U/ml) and catalase (2,000 U/ml) were added to eliminate external ROS. Luminescence was read on a Synergy temperature-controlled plate reader (Bio-Tek) by reading once per minute, with a 1,000-ms integration time, for 90 min at 37°C (99, 100).
Transfections, immunofluorescence assay, and microscopy.
Transfections of plasmids were performed on RAW 264.7 cells by use of TransIT-X2 (Mirus Bio, WI, USA) according to the manufacturer's directions, using the previously described green fluorescent protein (GFP)-Rab5 (101, 102) or GFP-LC3 (52, 103) construct. For immunostaining, cells were fixed in 4% PFA in PBS for 60 min, permeabilized using 0.1% Triton X-100 for 5 min, and then immunostained using an anti-LAMP primary antibody (clone ID4B [104]; obtained from the Developmental Studies Hybridoma Bank maintained at the University of Iowa) and an Alexa 488-conjugated goat anti-rat secondary antibody (Jackson ImmunoReseach), as previously described (105).
For phagosome time course maturation experiments, bacteria were prelabeled with biotin-hydrazide (Invitrogen) according to the manufacturer's instructions prior to phagocytosis. The bacteria were added to macrophages (MOI = 10:1), centrifuged for 1 min (125 × g) to force contact, coincubated in complete RPMI medium for 10 min at 37°C, washed with ice-cold PBS, and incubated with Alexa 594-streptavidin for 2 min in PBS at 4°C to label external bacteria, which were subsequently excluded from analysis. The cells were then returned to complete RPMI medium at 37°C and incubated for various times before being fixed in 4% PFA and either immunostained and counterstained with DAPI or, if the macrophages were transfected with GFP-Rab5 or GFP-LC3, counterstained with DAPI.
Microscopy was performed on a Nikon Eclipse TE2000-u instrument equipped with a Spot RT740 camera or a Zeiss Axio Imager Z1 Axiophot or Zeiss Axio Observer instrument equipped with an AxioCam MrM monochrome cooled charge-coupled device (CCD) camera. All image analyses were performed with ImageJ v1.46 or higher.
FITC-dextran ratiometric imaging.
Macrophages grown on coverslips were incubated with 150 μg/ml FITC-dextran overnight, followed by a 4-h chase in normal growth medium. The preloading of lysosomes with FITC-dextran allowed for detection of damage to lysosomes/phagosomes by ratiometric imaging as previously described (66, 72, 73, 106). Briefly, ratiometric imaging of FITC-dextran fluorescence in live cells was performed by capturing two fluorescence images with a single emission filter (535/40 nm), but with two different excitation wavelengths: 435 nm (using a 435/20-nm filter), where FITC excitation is pH insensitive, and 492 nm (using a 480/30-nm filter), where FITC excitation is pH sensitive. An in situ calibration was performed by measuring cells with the intracellular pH fixed at 9.0, 7.5, 7.0, 6.5, 6.0, 5.5, 5.0, 4.5, and 4.0 by using the ionophore nigericin (10 μg/ml) in clamping buffer (140 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 5 mM glucose, and appropriate buffer for the pH [15 mM HEPES or 15 mM morpholineethanesulfonic acid {MES}]). Image processing (ImageJ) determined the average ratio of emission by the 492-nm image divided by the emission by the 435-nm image at each pH. The data were fit to a 4-parameter sigmoid model, which was used to convert images of test samples into values representing pH that were then pseudocolored to represent detected pH levels. Treatment of cells with 1 mM l-leucyl-l-leucine methyl ester (LLME) for 2 h was used as a positive control for phagosomal/lysosomal damage (66, 107). For imaging of S. gordonii-containing macrophages, bacteria were added to macrophages after loading with FITC-dextran as described above (MOI = 10:1), centrifuged for 1 min (125 × g), and then incubated for 30 min at 37°C before imaging of macrophages containing bacteria.
Statistical analysis.
All statistical analyses were performed with Prism v7 (GraphPad Software, Inc.). Individual statistical tests used are described in the figure legends, with a significance (α) level of 0.05 used throughout.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by award R03DE025062 from the NIH-NIDCR (to J.G.K.) and by internal University at Buffalo funding.
We thank Stefan Ruhl for providing bacterial strains and for helpful discussions.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00858-17.
REFERENCES
- 1.Irie K, Novince CM, Darveau RP. 2014. Impact of the oral commensal flora on alveolar bone homeostasis. J Dent Res 93:801–806. doi: 10.1177/0022034514540173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han YW, Wang X. 2013. Mobile microbiome: oral bacteria in extra-oral infections and inflammation. J Dent Res 92:485–491. doi: 10.1177/0022034513487559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kesavalu L, Lucas AR, Verma RK, Liu L, Dai E, Sampson E, Progulske-Fox A. 2012. Increased atherogenesis during Streptococcus mutans infection in ApoE-null mice. J Dent Res 91:255–260. doi: 10.1177/0022034511435101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mylonakis E, Calderwood SB. 2001. Infective endocarditis in adults. N Engl J Med 345:1318–1330. doi: 10.1056/NEJMra010082. [DOI] [PubMed] [Google Scholar]
- 6.Murdoch DR, Corey GR, Hoen B, Miró JM, Fowler VG, Bayer AS, Karchmer AW, Olaison L, Pappas PA, Moreillon P, Chambers ST, Chu VH, Falcó V, Holland DJ, Jones P, Klein JL, Raymond NJ, Read KM, Tripodi MF, Utili R, Wang A, Woods CW, Cabell CH, International Collaboration on Endocarditis-Prospective Cohort Study (ICE-PCS) Investigators. 2009. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the International Collaboration on Endocarditis-Prospective Cohort Study. Arch Intern Med 169:463–473. doi: 10.1001/archinternmed.2008.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tleyjeh IM, Steckelberg JM, Murad HS, Anavekar NS, Ghomrawi HMK, Mirzoyev Z, Moustafa SE, Hoskin TL, Mandrekar JN, Wilson WR, Baddour LM. 2005. Temporal trends in infective endocarditis: a population-based study in Olmsted County, Minnesota. JAMA 293:3022–3028. doi: 10.1001/jama.293.24.3022. [DOI] [PubMed] [Google Scholar]
- 8.Marsh PD, Martin MV. 2009. The resident oral microflora, p 24–44. In Oral microbiology, 5th ed. Elsevier, New York, NY. [Google Scholar]
- 9.Teles C, Smith A, Ramage G, Lang S. 2011. Identification of clinically relevant viridans group streptococci by phenotypic and genotypic analysis. Eur J Clin Microbiol Infect Dis 30:243–250. doi: 10.1007/s10096-010-1076-y. [DOI] [PubMed] [Google Scholar]
- 10.Jensen A, Scholz CFP, Kilian M. 2016. Re-evaluation of the taxonomy of the mitis group of the genus Streptococcus based on whole genome phylogenetic analyses, and proposed reclassification of Streptococcus dentisani as Streptococcus oralis subsp. dentisani comb. nov., Streptococcus tigurinus as Streptococcus oralis subsp. tigurinus comb. nov., and Streptococcus oligofermentans as a later synonym of Streptococcus cristatus. Int J Syst Evol Microbiol 66:4803–4820. doi: 10.1099/ijsem.0.001433. [DOI] [PubMed] [Google Scholar]
- 11.Douglas CW, Heath J, Hampton KK, Preston FE. 1993. Identity of viridans streptococci isolated from cases of infective endocarditis. J Med Microbiol 39:179–182. doi: 10.1099/00222615-39-3-179. [DOI] [PubMed] [Google Scholar]
- 12.Naveen Kumar V, van der Linden M, Menon T, Nitsche-Schmitz DP. 2014. Viridans and bovis group streptococci that cause infective endocarditis in two regions with contrasting epidemiology. Int J Med Microbiol 304:262–268. doi: 10.1016/j.ijmm.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Wright CJ, Xue P, Hirano T, Liu C, Whitmore SE, Hackett M, Lamont RJ. 2014. Characterization of a bacterial tyrosine kinase in Porphyromonas gingivalis involved in polymicrobial synergy. Microbiologyopen 3:383–394. doi: 10.1002/mbo3.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitmore SE, Lamont RJ. 2011. The pathogenic persona of community-associated oral streptococci. Mol Microbiol 81:305–314. doi: 10.1111/j.1365-2958.2011.07707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daep CA, Novak EA, Lamont RJ, Demuth DR. 2011. Structural dissection and in vivo effectiveness of a peptide inhibitor of Porphyromonas gingivalis adherence to Streptococcus gordonii. Infect Immun 79:67–74. doi: 10.1128/IAI.00361-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hajishengallis G, Lamont RJ. 2016. Dancing with the stars: how choreographed bacterial interactions dictate nososymbiocity and give rise to keystone pathogens, accessory pathogens, and pathobionts. Trends Microbiol 24:477–489. doi: 10.1016/j.tim.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winterbourn CC, Kettle AJ, Hampton MB. 2016. Reactive oxygen species and neutrophil function. Annu Rev Biochem 85:765–792. doi: 10.1146/annurev-biochem-060815-014442. [DOI] [PubMed] [Google Scholar]
- 18.Bartold PM, Van Dyke TE. 2017. Host modulation: controlling the inflammation to control the infection. Periodontol 2000 75:317–329. doi: 10.1111/prd.12169. [DOI] [PubMed] [Google Scholar]
- 19.Arango Duque G, Descoteaux A. 2014. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol 5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sica A, Mantovani A. 2012. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delima AJ, Van Dyke TE. 2003. Origin and function of the cellular components in gingival crevice fluid. Periodontol 2000 31:55–76. doi: 10.1034/j.1600-0757.2003.03105.x. [DOI] [PubMed] [Google Scholar]
- 22.Ryder MI. 2010. Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontol 2000 53:124–137. doi: 10.1111/j.1600-0757.2009.00327.x. [DOI] [PubMed] [Google Scholar]
- 23.Hasturk H, Kantarci A, Van Dyke TE. 2012. Oral inflammatory diseases and systemic inflammation: role of the macrophage. Front Immunol 3:118. doi: 10.3389/fimmu.2012.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sima C, Glogauer M. 2013. Macrophage subsets and osteoimmunology: tuning of the immunological recognition and effector systems that maintain alveolar bone. Periodontol 2000 63:80–101. doi: 10.1111/prd.12032. [DOI] [PubMed] [Google Scholar]
- 25.Werdan K, Dietz S, Löffler B, Niemann S, Bushnaq H, Silber R-E, Peters G, Müller-Werdan U. 2014. Mechanisms of infective endocarditis: pathogen-host interaction and risk states. Nat Rev Cardiol 11:35–50. doi: 10.1038/nrcardio.2013.174. [DOI] [PubMed] [Google Scholar]
- 26.Young Lee S, Cisar JO, Bryant JL, Eckhaus MA, Sandberg AL. 2006. Resistance of Streptococcus gordonii to polymorphonuclear leukocyte killing is a potential virulence determinant of infective endocarditis. Infect Immun 74:3148–3155. doi: 10.1128/IAI.00087-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durack DT. 1975. Experimental bacterial endocarditis. IV. Structure and evolution of very early lesions. J Pathol 115:81–89. [DOI] [PubMed] [Google Scholar]
- 28.Jung C-J, Zheng Q-H, Shieh Y-H, Lin C-S, Chia J-S. 2009. Streptococcus mutans autolysin AtlA is a fibronectin-binding protein and contributes to bacterial survival in the bloodstream and virulence for infective endocarditis. Mol Microbiol 74:888–902. doi: 10.1111/j.1365-2958.2009.06903.x. [DOI] [PubMed] [Google Scholar]
- 29.Cho K, Arimoto T, Igarashi T, Yamamoto M. 2013. Involvement of lipoprotein PpiA of Streptococcus gordonii in evasion of phagocytosis by macrophages. Mol Oral Microbiol 28:379–391. doi: 10.1111/omi.12031. [DOI] [PubMed] [Google Scholar]
- 30.Fitzgerald JR, Foster TJ, Cox D. 2006. The interaction of bacterial pathogens with platelets. Nat Rev Microbiol 4:445–457. doi: 10.1038/nrmicro1425. [DOI] [PubMed] [Google Scholar]
- 31.Petersen HJ, Keane C, Jenkinson HF, Vickerman MM, Jesionowski A, Waterhouse JC, Cox D, Kerrigan SW. 2010. Human platelets recognize a novel surface protein, PadA, on Streptococcus gordonii through a unique interaction involving fibrinogen receptor GPIIbIIIa. Infect Immun 78:413–422. doi: 10.1128/IAI.00664-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen P-M, Chen H-C, Ho C-T, Jung C-J, Lien H-T, Chen J-Y, Chia J-S. 2008. The two-component system ScnRK of Streptococcus mutans affects hydrogen peroxide resistance and murine macrophage killing. Microbes Infect 10:293–301. doi: 10.1016/j.micinf.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 33.Chen Y-YM, Shieh H-R, Chang Y-C. 2013. The expression of the fim operon is crucial for the survival of Streptococcus parasanguinis FW213 within macrophages but not acid tolerance. PLoS One 8:e66163. doi: 10.1371/journal.pone.0066163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yajima A, Takahashi Y, Shimazu K, Urano-Tashiro Y, Uchikawa Y, Karibe H, Konishi K. 2009. Contribution of phosphoglucosamine mutase to the resistance of Streptococcus gordonii DL1 to polymorphonuclear leukocyte killing. FEMS Microbiol Lett 297:196–202. doi: 10.1111/j.1574-6968.2009.01673.x. [DOI] [PubMed] [Google Scholar]
- 35.Lappin DF, MacLeod CP, Kerr A, Mitchell T, Kinane DF. 2001. Anti-inflammatory cytokine IL-10 and T cell cytokine profile in periodontitis granulation tissue. Clin Exp Immunol 123:294–300. doi: 10.1046/j.1365-2249.2001.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lam RS, O'Brien-Simpson NM, Lenzo JC, Holden JA, Brammar GC, Walsh KA, McNaughtan JE, Rowler DK, van Rooijen N, Reynolds EC. 2014. Macrophage depletion abates Porphyromonas gingivalis-induced alveolar bone resorption in mice. J Immunol 193:2349–2362. doi: 10.4049/jimmunol.1400853. [DOI] [PubMed] [Google Scholar]
- 37.Yu T, Zhao L, Huang X, Ma C, Wang Y, Zhang J, Xuan D. 2016. Enhanced activity of the macrophage M1/M2 phenotypes and phenotypic switch to M1 in periodontal infection. J Periodontol 87:1092–1102. doi: 10.1902/jop.2016.160081. [DOI] [PubMed] [Google Scholar]
- 38.Topoll HH, Zwadlo G, Lange DE, Sorg C. 1989. Phenotypic dynamics of macrophage subpopulations during human experimental gingivitis. J Periodontal Res 24:106–112. doi: 10.1111/j.1600-0765.1989.tb00864.x. [DOI] [PubMed] [Google Scholar]
- 39.Flannagan RS, Cosio G, Grinstein S. 2009. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol 7:355–366. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 40.Price JV, Vance RE. 2014. The macrophage paradox. Immunity 41:685–693. doi: 10.1016/j.immuni.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 41.Vaudaux P, Waldvogel FA. 1979. Gentamicin antibacterial activity in the presence of human polymorphonuclear leukocytes. Antimicrob Agents Chemother 16:743–749. doi: 10.1128/AAC.16.6.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kerrigan SW, Jakubovics NS, Keane C, Maguire P, Wynne K, Jenkinson HF, Cox D. 2007. Role of Streptococcus gordonii surface proteins SspA/SspB and Hsa in platelet function. Infect Immun 75:5740–5747. doi: 10.1128/IAI.00909-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.García-Mendoza A, Liébana J, Castillo AM, de la Higuera A, Piédrola G. 1993. Evaluation of the capacity of oral streptococci to produce hydrogen peroxide. J Med Microbiol 39:434–439. doi: 10.1099/00222615-39-6-434. [DOI] [PubMed] [Google Scholar]
- 44.Barnard JP, Stinson MW. 1996. The alpha-hemolysin of Streptococcus gordonii is hydrogen peroxide. Infect Immun 64:3853–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yesilkaya H, Andisi VF, Andrew PW, Bijlsma JJE. 2013. Streptococcus pneumoniae and reactive oxygen species: an unusual approach to living with radicals. Trends Microbiol 21:187–195. doi: 10.1016/j.tim.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Jakubovics NS, Smith AW, Jenkinson HF. 2002. Oxidative stress tolerance is manganese (Mn(2+)) regulated in Streptococcus gordonii. Microbiology (Reading, Engl) 148:3255–3263. doi: 10.1099/00221287-148-10-3255. [DOI] [PubMed] [Google Scholar]
- 47.Tseng H-J, McEwan AG, Paton JC, Jennings MP. 2002. Virulence of Streptococcus pneumoniae: PsaA mutants are hypersensitive to oxidative stress. Infect Immun 70:1635–1639. doi: 10.1128/IAI.70.3.1635-1639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Newburger PE, Dai Q, Whitney C. 1991. In vitro regulation of human phagocyte cytochrome b heavy and light chain gene expression by bacterial lipopolysaccharide and recombinant human cytokines. J Biol Chem 266:16171–16177. [PubMed] [Google Scholar]
- 49.Cassatella MA, Bazzoni F, Flynn RM, Dusi S, Trinchieri G, Rossi F. 1990. Molecular basis of interferon-gamma and lipopolysaccharide enhancement of phagocyte respiratory burst capability. Studies on the gene expression of several NADPH oxidase components. J Biol Chem 265:20241–20246. [PubMed] [Google Scholar]
- 50.Casbon A-J, Long ME, Dunn KW, Allen L-AH, Dinauer MC. 2012. Effects of IFN-γ on intracellular trafficking and activity of macrophage NADPH oxidase flavocytochrome b558. J Leukoc Biol 92:869–882. doi: 10.1189/jlb.0512244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ellis JA, Mayer SJ, Jones OT. 1988. The effect of the NADPH oxidase inhibitor diphenyleneiodonium on aerobic and anaerobic microbicidal activities of human neutrophils. Biochem J 251:887–891. doi: 10.1042/bj2510887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MAO, Glogauer M, Grinstein S, Brumell JH. 2009. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A 106:6226–6231. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foote JR, Behe P, Frampton M, Levine AP, Segal AW. 2017. An exploration of charge compensating ion channels across the phagocytic vacuole of neutrophils. Front Pharmacol 8:94. doi: 10.3389/fphar.2017.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Canton J, Khezri R, Glogauer M, Grinstein S. 2014. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol Biol Cell 25:3330–3341. doi: 10.1091/mbc.E14-05-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rybicka JM, Balce DR, Chaudhuri S, Allan ERO, Yates RM. 2012. Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner. EMBO J 31:932–944. doi: 10.1038/emboj.2011.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I, Ghigo D. 2008. Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr Drug Metab 9:686–696. doi: 10.2174/138920008786049285. [DOI] [PubMed] [Google Scholar]
- 57.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege J-L, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA. 2014. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mantegazza AR, Savina A, Vermeulen M, Pérez L, Geffner J, Hermine O, Rosenzweig SD, Faure F, Amigorena S. 2008. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 112:4712–4722. doi: 10.1182/blood-2008-01-134791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Duménil A-M, Seabra MC, Raposo G, Amigorena S. 2006. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 60.Drevets DA, Canono BP, Leenen PJ, Campbell PA. 1994. Gentamicin kills intracellular Listeria monocytogenes. Infect Immun 62:2222–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flannagan RS, Heit B, Heinrichs DE. 2016. Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell Microbiol 18:514–535. doi: 10.1111/cmi.12527. [DOI] [PubMed] [Google Scholar]
- 62.Smith LM, May RC. 2013. Mechanisms of microbial escape from phagocyte killing. Biochem Soc Trans 41:475–490. doi: 10.1042/BST20130014. [DOI] [PubMed] [Google Scholar]
- 63.Desjardins M, Huber LA, Parton RG, Griffiths G. 1994. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J Cell Biol 124:677–688. doi: 10.1083/jcb.124.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Botelho RJ, Grinstein S. 2011. Phagocytosis. Curr Biol 21:R533–R538. doi: 10.1016/j.cub.2011.05.053. [DOI] [PubMed] [Google Scholar]
- 65.Flannagan RS, Jaumouillé V, Grinstein S. 2012. The cell biology of phagocytosis. Annu Rev Pathol 7:61–98. doi: 10.1146/annurev-pathol-011811-132445. [DOI] [PubMed] [Google Scholar]
- 66.Davis MJ, Swanson JA. 2010. Technical advance: caspase-1 activation and IL-1β release correlate with the degree of lysosome damage, as illustrated by a novel imaging method to quantify phagolysosome damage. J Leukoc Biol 88:813–822. doi: 10.1189/jlb.0310159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Genestet C, Le Gouellec A, Chaker H, Polack B, Guery B, Toussaint B, Stasia MJ. 2014. Scavenging of reactive oxygen species by tryptophan metabolites helps Pseudomonas aeruginosa escape neutrophil killing. Free Radic Biol Med 73:400–410. doi: 10.1016/j.freeradbiomed.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 68.Randow F, Youle RJ. 2014. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 15:403–411. doi: 10.1016/j.chom.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. 2011. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A 108:17396–17401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Romao S, Gasser N, Becker AC, Guhl B, Bajagic M, Vanoaica D, Ziegler U, Roesler J, Dengjel J, Reichenbach J, Munz C. 2013. Autophagy proteins stabilize pathogen-containing phagosomes for prolonged MHC II antigen processing. J Cell Biol 203:757–766. doi: 10.1083/jcb.201308173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sattler N, Monroy R, Soldati T. 2013. Quantitative analysis of phagocytosis and phagosome maturation. Methods Mol Biol 983:383–402. doi: 10.1007/978-1-62703-302-2_21. [DOI] [PubMed] [Google Scholar]
- 72.Ohkuma S, Poole B. 1978. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci U S A 75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davis MJ, Gregorka B, Gestwicki JE, Swanson JA. 2012. Inducible renitence limits Listeria monocytogenes escape from vacuoles in macrophages. J Immunol 189:4488–4495. doi: 10.4049/jimmunol.1103158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsang AW, Oestergaard K, Myers JT, Swanson JA. 2000. Altered membrane trafficking in activated bone marrow-derived macrophages. J Leukoc Biol 68:487–494. [PubMed] [Google Scholar]
- 75.Balce DR, Li B, Allan ERO, Rybicka JM, Krohn RM, Yates RM. 2011. Alternative activation of macrophages by IL-4 enhances the proteolytic capacity of their phagosomes through synergistic mechanisms. Blood 118:4199–4208. doi: 10.1182/blood-2011-01-328906. [DOI] [PubMed] [Google Scholar]
- 76.Rybicka JM, Balce DR, Khan MF, Krohn RM, Yates RM. 2010. NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes. Proc Natl Acad Sci U S A 107:10496–10501. doi: 10.1073/pnas.0914867107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nathan C, Shiloh MU. 2000. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A 97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kreth J, Merritt J, Shi W, Qi F. 2005. Competition and coexistence between Streptococcus mutans and Streptococcus sanguinis in the dental biofilm. J Bacteriol 187:7193–7203. doi: 10.1128/JB.187.21.7193-7203.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jakubovics NS, Gill SR, Vickerman MM, Kolenbrander PE. 2008. Role of hydrogen peroxide in competition and cooperation between Streptococcus gordonii and Actinomyces naeslundii. FEMS Microbiol Ecol 66:637–644. doi: 10.1111/j.1574-6941.2008.00585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jotwani R, Palucka AK, Al-Quotub M, Nouri-Shirazi M, Kim J, Bell D, Banchereau J, Cutler CW. 2001. Mature dendritic cells infiltrate the T cell-rich region of oral mucosa in chronic periodontitis: in situ, in vivo, and in vitro studies. J Immunol 167:4693–4700. doi: 10.4049/jimmunol.167.8.4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fine N, Hassanpour S, Borenstein A, Sima C, Oveisi M, Scholey J, Cherney D, Glogauer M. 2016. Distinct oral neutrophil subsets define health and periodontal disease states. J Dent Res 95:931–938. doi: 10.1177/0022034516645564. [DOI] [PubMed] [Google Scholar]
- 82.Carrion J, Scisci E, Miles B, Sabino GJ, Zeituni AE, Gu Y, Bear A, Genco CA, Brown DL, Cutler CW. 2012. Microbial carriage state of peripheral blood dendritic cells (DCs) in chronic periodontitis influences DC differentiation, atherogenic potential. J Immunol 189:3178–3187. doi: 10.4049/jimmunol.1201053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kreibich S, Emmenlauer M, Fredlund J, Rämö P, Munz C, Dehio C, Enninga J, Hardt W-D. 2015. Autophagy proteins promote repair of endosomal membranes damaged by the Salmonella type three secretion system 1. Cell Host Microbe 18:527–537. doi: 10.1016/j.chom.2015.10.015. [DOI] [PubMed] [Google Scholar]
- 84.Sierig G, Cywes C, Wessels MR, Ashbaugh CD. 2003. Cytotoxic effects of streptolysin O and streptolysin S enhance the virulence of poorly encapsulated group A streptococci. Infect Immun 71:446–455. doi: 10.1128/IAI.71.1.446-455.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bensing BA, Sullam PM. 2002. An accessory sec locus of Streptococcus gordonii is required for export of the surface protein GspB and for normal levels of binding to human platelets. Mol Microbiol 44:1081–1094. doi: 10.1046/j.1365-2958.2002.02949.x. [DOI] [PubMed] [Google Scholar]
- 86.Takahashi Y, Sandberg AL, Ruhl S, Muller J, Cisar JO. 1997. A specific cell surface antigen of Streptococcus gordonii is associated with bacterial hemagglutination and adhesion to alpha2-3-linked sialic acid-containing receptors. Infect Immun 65:5042–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Takahashi Y, Takashima E, Shimazu K, Yagishita H, Aoba T, Konishi K. 2006. Contribution of sialic acid-binding adhesin to pathogenesis of experimental endocarditis caused by Streptococcus gordonii DL1. Infect Immun 74:740–743. doi: 10.1128/IAI.74.1.740-743.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Keane C, Petersen HJ, Tilley DO, Haworth J, Cox D, Jenkinson HF, Kerrigan SW. 2013. Multiple sites on Streptococcus gordonii surface protein PadA bind to platelet GPIIbIIIa. Thromb Haemost 110:1278–1287. doi: 10.1160/TH13-07-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mukouhara T, Arimoto T, Cho K, Yamamoto M, Igarashi T. 2011. Surface lipoprotein PpiA of Streptococcus mutans suppresses scavenger receptor MARCO-dependent phagocytosis by macrophages. Infect Immun 79:4933–4940. doi: 10.1128/IAI.05693-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Berry AM, Lock RA, Hansman D, Paton JC. 1989. Contribution of autolysin to virulence of Streptococcus pneumoniae. Infect Immun 57:2324–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mani N, Baddour LM, Offutt DQ, Vijaranakul U, Nadakavukaren MJ, Jayaswal RK. 1994. Autolysis-defective mutant of Staphylococcus aureus: pathological considerations, genetic mapping, and electron microscopic studies. Infect Immun 62:1406–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shibata Y, Kawada M, Nakano Y, Toyoshima K, Yamashita Y. 2005. Identification and characterization of an autolysin-encoding gene of Streptococcus mutans. Infect Immun 73:3512–3520. doi: 10.1128/IAI.73.6.3512-3520.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Davies JQ, Gordon S. 2005. Isolation and culture of human macrophages. Methods Mol Biol 290:105–116. [DOI] [PubMed] [Google Scholar]
- 94.Honma K, Mishima E, Sharma A. 2011. Role of Tannerella forsythia NanH sialidase in epithelial cell attachment. Infect Immun 79:393–401. doi: 10.1128/IAI.00629-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Honma K, Mishima E, Inagaki S, Sharma A. 2009. The OxyR homologue in Tannerella forsythia regulates expression of oxidative stress responses and biofilm formation. Microbiology (Reading, Engl) 155:1912–1922. doi: 10.1099/mic.0.027920-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nikitkova AE, Haase EM, Vickerman MM, Gill SR, Scannapieco FA. 2012. Response of fatty acid synthesis genes to the binding of human salivary amylase by Streptococcus gordonii. Appl Environ Microbiol 78:1865–1875. doi: 10.1128/AEM.07071-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McCord JM, Fridovich I. 1969. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 244:6049–6055. [PubMed] [Google Scholar]
- 98.Lynch MC, Kuramitsu HK. 1999. Role of superoxide dismutase activity in the physiology of Porphyromonas gingivalis. Infect Immun 67:3367–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dahlgren C, Karlsson A, Bylund J. 2007. Measurement of respiratory burst products generated by professional phagocytes. Methods Mol Biol 412:349–363. doi: 10.1007/978-1-59745-467-4_23. [DOI] [PubMed] [Google Scholar]
- 100.Lock R, Johansson A, Orselius K, Dahlgren C. 1988. Analysis of horseradish peroxidase-amplified chemiluminescence produced by human neutrophils reveals a role for the superoxide anion in the light emitting reaction. Anal Biochem 173:450–455. doi: 10.1016/0003-2697(88)90213-8. [DOI] [PubMed] [Google Scholar]
- 101.Roberts RL, Barbieri MA, Pryse KM, Chua M, Morisaki JH, Stahl PD. 1999. Endosome fusion in living cells overexpressing GFP-rab5. J Cell Sci 112:3667–3675. [DOI] [PubMed] [Google Scholar]
- 102.Vieira OV, Bucci C, Harrison RE, Trimble WS, Lanzetti L, Gruenberg J, Schreiber AD, Stahl PD, Grinstein S. 2003. Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol Cell Biol 23:2501–2514. doi: 10.1128/MCB.23.7.2501-2514.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen JW, Murphy TL, Willingham MC, Pastan I, August JT. 1985. Identification of two lysosomal membrane glycoproteins. J Cell Biol 101:85–95. doi: 10.1083/jcb.101.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kay JG, Murray RZ, Pagan JK, Stow JL. 2006. Cytokine secretion via cholesterol-rich lipid raft-associated SNAREs at the phagocytic cup. J Biol Chem 281:11949–11954. doi: 10.1074/jbc.M600857200. [DOI] [PubMed] [Google Scholar]
- 106.Canton J, Grinstein S. 2015. Measuring lysosomal pH by fluorescence microscopy. Methods Cell Biol 126:85–99. doi: 10.1016/bs.mcb.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 107.Aits S, Kricker J, Liu B, Ellegaard A-M, Hämälistö S, Tvingsholm S, Corcelle-Termeau E, Høgh S, Farkas T, Holm Jonassen A, Gromova I, Mortensen M, Jäättelä M. 2015. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 11:1408–1424. doi: 10.1080/15548627.2015.1063871. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.