

ABSTRACT
Streptococcus pneumoniae is a frequent colonizer of the upper respiratory tract and a leading cause of bacterial pneumonia. The innate immune system senses pneumococcal cell wall components, toxin, and nucleic acids, which leads to production of inflammatory mediators to initiate and control antibacterial defense. Here, we show that the cGAS (cyclic GMP-AMP [cGAMP] synthase)-STING pathway mediates detection of pneumococcal DNA in mouse macrophages to primarily stimulate type I interferon (IFN) responses. Cells of human individuals carrying HAQ TMEM173, which encodes a common hypomorphic variant of STING, were largely or partly defective in inducing type I IFNs and proinflammatory cytokines upon infection. Subsequent analyses, however, revealed that STING was dispensable for restricting S. pneumoniae during acute pneumonia in mice. Moreover, explorative analyses did not find differences in the allele frequency of HAQ TMEM173 in nonvaccinated pneumococcal pneumonia patients and healthy controls or an association of HAQ TMEM173 carriage with disease severity. Together, our results indicate that the cGAS/STING pathway senses S. pneumoniae but plays no major role in antipneumococcal immunity in mice and humans.
KEYWORDS: innate immunity, cGAS, STING, Streptococcus pneumoniae, pneumonia, type I interferons, single-nucleotide polymorphism, human
INTRODUCTION
Pneumonia kills more children under 5 years of age than any other disease worldwide, causing approximately 2 million deaths annually (1), and probably even more in elderly as well as immunocompromised individuals. Streptococcus pneumoniae is a leading cause of community-acquired pneumonia (CAP) (2, 3). This bacterium alone caused an estimated 826,000 deaths and 14.5 million cases of serious illness globally in 2000 (1). A better understanding of the bacterial interaction with the host, the antibacterial immune response, and individual risk factors might help in the development of novel preventive and/or therapeutic approaches to lower the high morbidity and mortality associated with S. pneumoniae.
The interaction of S. pneumoniae with the host usually starts with the asymptomatic colonization of the upper respiratory tract. Pneumonia only develops when pneumococci get access to the lower respiratory tract by aspiration and resist prompt elimination by the immune system (2, 3). The innate immune system relies on so-called pattern recognition receptors (PRRs) expressed by macrophages, dendritic cells, and other pulmonary cells to sense infections (4, 5). These PRRs include Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), C-type lectin receptors (CLRs), and cytosolic DNA sensors (4, 6, 7). Among those, TLR2, TLR9, NOD2, NLRP3, AIM2, and Mincle have been identified to mediate inflammatory responses and antibacterial defense during pneumococcal pneumonia (8–18). In addition, we recently demonstrated that a STING-dependent cytosolic DNA-sensing pathway activates type I interferon (IFN) responses to S. pneumoniae infection in murine macrophages (19).
STING is both an adapter molecule mediating cytosolic DNA recognition and a direct receptor for the bacterial cyclic dinucleotides (CDNs) cyclic-di-AMP (c-di-AMP) and cyclic-di-GMP (c-di-GMP) (20–23). In the STING-dependent cytosolic DNA sensing pathway, cyclic GMP-AMP (cGAMP) synthase (cGAS) has been identified as the primary receptor of microbial and endogenous DNA (24). cGAS directly binds to double-stranded DNA (dsDNA) and subsequently catalyzes the production of the second messenger 2′3′-cGAMP, which in turn activates STING (25).
Recent studies found that TMEM173, the gene encoding STING, is polymorphic (26, 27). The most common allele besides the wild-type (WT) allele R232 is HAQ TMEM173, which contains a haplotype comprised of three nonsynonymous single-nucleotide polymorphisms (SNPs), R71H-G230A-R293Q. Ectopically expressed HAQ STING exhibited diminished capability to activate type I IFN production in response to endogenous and bacterial CDNs or Listeria monocytogenes infection compared to that of WT STING (26, 27). In contrast, the frequent R232H SNP impairs activation by bacterial CDNs but not by endogenous 2′3′-cGAMP (27, 28). In addition, rare gain-of-function mutations in TMEM173 have been associated with autoinflammatory syndromes (29, 30).
Here, we demonstrate that the cGAS/STING axis detects S. pneumoniae infection, and that this sensing is attenuated in cells of individuals carrying HAQ TMEM173 but not R232H TMEM173. Experiments in a mouse model of pneumococcal pneumonia as well as analyses aimed at elucidating a potential association between HAQ TMEM173 or R232H TMEM173 and pneumococcal pneumonia indicate, however, that the cGAS/STING pathway is largely dispensable for antipneumococcal defense in mice and humans.
RESULTS
Type I IFN production during pneumococcal infection is dependent on the cGAS-STING pathway.
In order to assess the relevance of STING for the induction of type I IFNs upon S. pneumoniae infection, bone marrow-derived macrophages (BMDMs) derived from WT and Tmem173−/− mice were infected with S. pneumoniae or a capsule-deficient isogenic mutant or were stimulated with pneumococcal DNA. In confirmation of previously published gene-silencing experiments from our group (19), type I IFN induction in response to S. pneumoniae or bacterial DNA was impaired in STING-deficient cells (Fig. 1A and B). Moreover, expression of the IFN-stimulated gene Ccl5 was likewise dependent on STING (Fig. 1C). We next investigated the involvement of cGAS in S. pneumoniae-induced type I IFN responses. We found that short interfering RNA (siRNA)-mediated silencing of cGAS reduced type I IFN responses to pneumococcal infection or stimulation with S. pneumoniae DNA (see Fig. S1 in the supplemental material). Furthermore, subsequent experiments using BMDMs from cGas−/− mice showed that cGAS deficiency leads to a strongly reduced type I IFN induction upon S. pneumoniae infection or DNA stimulation (Fig. 1D and E). As expected, however, cGas−/− cells were not attenuated in responding to cGAMP. Moreover, expression of Ccl5 was also reduced in cGAS-deficient cells after pneumococcal infection (Fig. 1F). To elucidate the relevance of the STING-dependent pathway for activation of type I IFN production during pneumococcal pneumonia in vivo, we intranasally infected WT and STING-deficient mice with S. pneumoniae. Expression of Ifnb and Ccl5 was almost completely or partly inhibited in lungs of mice lacking STING compared to organs from WT animals (Fig. 1G and H). Altogether, these results demonstrate that type I IFN responses induced by S. pneumoniae in mice are dependent on the cGAS-STING pathway.
FIG 1.
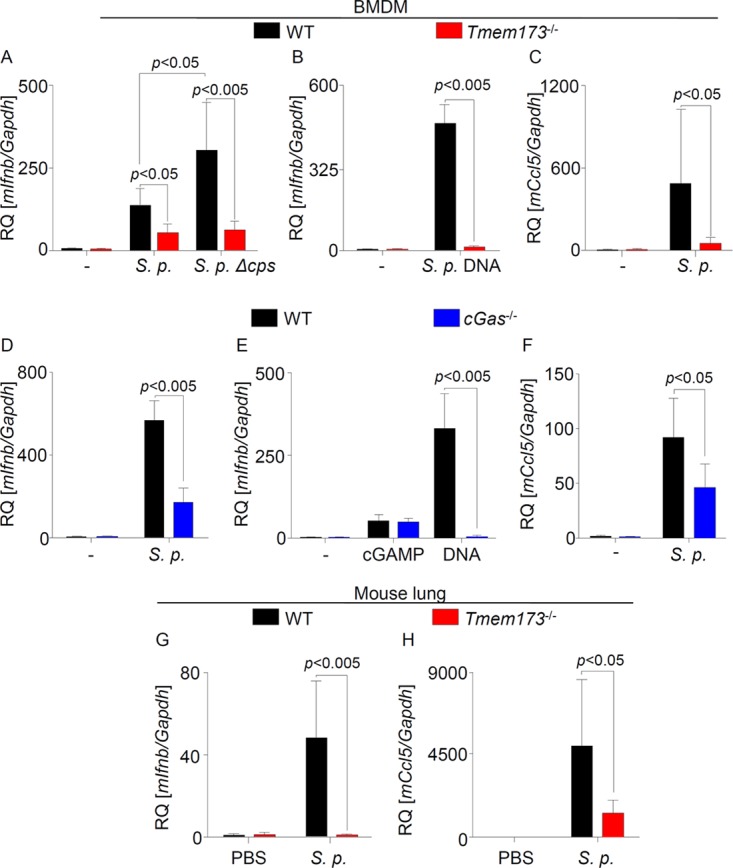
cGAS and STING mediate type I IFN responses to S. pneumoniae infection. (A and B) WT and Tmem173−/− mouse BMDMs were left untreated, were infected with S. pneumoniae (S. p.) or its Δcps isogenic mutant strain, or were stimulated with pneumococcal DNA for 6 h. Expression of Ifnb was quantified by qRT-PCR. (C) WT and Tmem173−/− BMDMs were left untreated or were infected with S. pneumoniae, and expression of Ccl5 was quantified by qRT-PCR. (D to F) BMDMs from cGas−/− mice were infected with S. pneumoniae or stimulated with synthetic DNA or cGAMP, and expression of Ifnb (D and E) and Ccl5 (F) was quantified by qRT-PCR. (G and H) WT and Tmem173−/− mice were intranasally infected with S. pneumoniae, and the expression of Ifnb and Ccl5 in the lungs was measured 48 h postinfection by qRT-PCR. Data from in vitro analyses are shown as means ± standard deviations (SD) from three independent experiments, measured in technical duplicates. Mouse data represent analyses of 7 mice. Statistical analyses were performed with the Mann-Whitney U test. Comparisons with a P value of <0.05 were considered significant. RQ, relative quantification.
The cGAS-STING pathway has little effect on production of proinflammatory cytokines during pneumococcal infection of mice.
We next investigated whether proinflammatory cytokine production was influenced by the cGAS/STING pathway as well. To this aim, we measured proinflammatory cytokine production in S. pneumoniae-infected BMDMs as well as in lungs of mice infected with S. pneumoniae. Although the levels of interleukin-1β (IL-1β) and IL-6 in the supernatant of STING-deficient cells infected with S. pneumoniae were slightly reduced compared with those of their WT counterparts (Fig. 2A to C and Fig. S2), cells from cGas−/− mice produced levels of proinflammatory cytokines similar to those of WT cells (Fig. 2D to F). Moreover, production of these proinflammatory cytokines in the lungs of S. pneumoniae-infected mice was not affected by STING (Fig. 2G to I). Overall, these results indicate that the cGAS/STING signaling pathway has little effect on proinflammatory cytokine production in infections with S. pneumoniae.
FIG 2.

Proinflammatory cytokine production is partly dependent on STING during S. pneumoniae infection in vitro but not in vivo. (A to C) WT and Tmem173−/− BMDMs were infected with S. pneumoniae for 16 to 18 h, and proinflammatory cytokine production was determined by sandwich ELISA. (D to F) Cytokine protein concentration in the supernatant of cGas−/− and WT cells infected for 16 to 18 h with S. pneumoniae was measured by sandwich ELISA. (G to I) Cytokine production in whole lung homogenates of S. pneumoniae-infected mice was quantified by ELISA. Data are shown as means ± SD. Data are representative of 4 (A to C) or 3 (D to F) independent experiments carried out in duplicates. (G to I) Data represent analyses of 7 mice. Data were analyzed through the Mann-Whitney U test. Comparisons with a P value of <0.05 were considered significant.
Endogenous HAQ STING is impaired in mediating type I IFN responses to S. pneumoniae infection or stimulation with pneumococcal DNA.
Previous studies showed that HAQ STING was strongly impaired in mediating responses to bacterial CDNs and DNA (26, 27, 31). To assess whether endogenous HAQ STING affects type I IFN and proinflammatory cytokine responses to S. pneumoniae and pneumococcal DNA, we screened 564 individuals for the presence of HAQ TMEM173, isolated peripheral blood mononuclear cells (PBMCs) from 4 individuals identified as homozygotic HAQ TMEM173 carriers, cultured the cells for 7 days before infection/stimulation to allow differentiation into a macrophage-like state, and compared those cells to cells expressing WT STING. All donors were WT for amino acid position 232 (R232). Intriguingly, cells from HAQ TMEM173 carriers showed impaired type I IFN expression and production of the IFN-stimulated cytokine IP-10 following S. pneumoniae infection or pneumococcal DNA stimulation (Fig. 3A and Fig. S3A). Furthermore, S. pneumoniae-stimulated IL-1β production was also partly reduced in cells expressing HAQ STING (Fig. 3B and Fig. S3B), whereas release of tumor necrosis factor alpha (TNF-α) and IL-6 was not or was only insignificantly affected (Fig. 3C and D and Fig. S3C and D).
FIG 3.
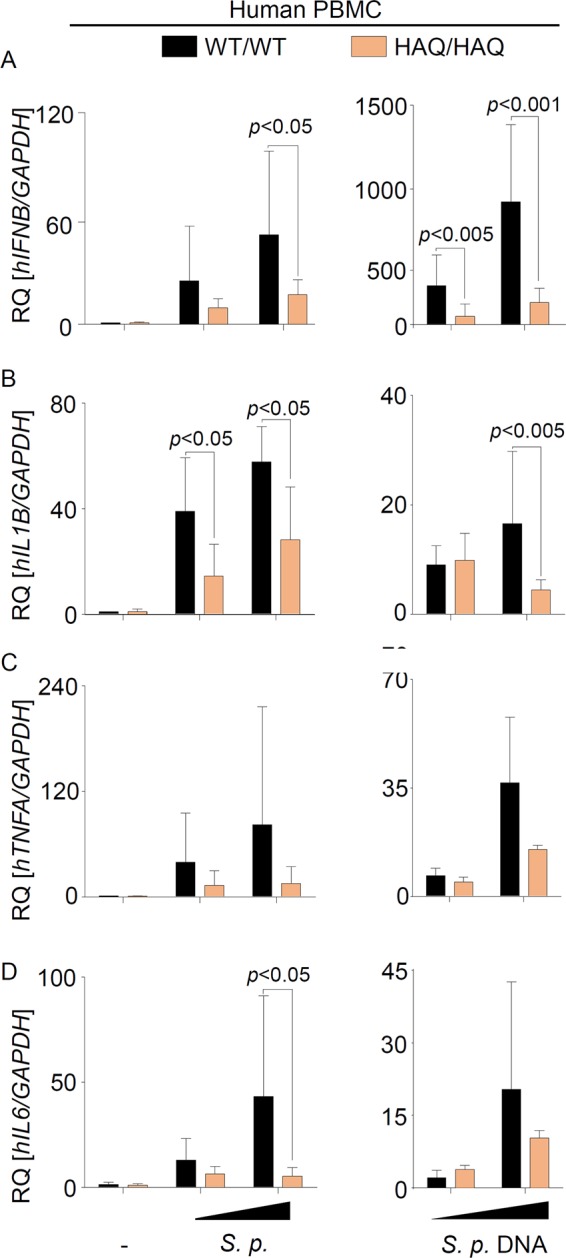
Endogenous HAQ STING is impaired in mediating type I IFN and proinflammatory cytokine responses to S. pneumoniae infection. PBMCs from healthy volunteers were isolated, cultivated for 7 days, and subsequently infected with S. pneumoniae at a multiplicity of infection of 2.5 or 10 or stimulated with 0.2 or 1 μg/ml of pneumococcal DNA for 6 h. The expression of IFNB (A), IL1B (B), TNFA (C), and IL-6 (D) was assessed by qRT-PCR. Data are represented as means ± SD from 4 independent experiments carried out in triplicates. Comparisons with a P value of <0.05 were considered significant.
Additional experiments using the human monocytic cell line THP-1, which has previously been found to express HAQ STING (28, 32) and which we confirmed to carry HAQ TMEM173 in homozygosity, showed only a weak type I IFN induction in response to DNA, cGAMP, or S. pneumoniae challenge (Fig. S4). Indeed, while WT STING-expressing PBMCs showed a 50- to 1,000-fold IFNB induction upon S. pneumoniae or DNA stimulation, respectively (Fig. 3A), the bacterium induced only a 9-fold increase in IFNB expression in THP-1 cells. Notably, CRISPR/Cas9-mediated deletion of cGAS (33) further weakened type I IFN induction in THP-1 cells (Fig. S4). Altogether, these data reveal that endogenous HAQ STING is a hypomorphic variant that is strongly impaired (but not deficient) in mediating innate immune responses to S. pneumoniae.
The STING isoform R232H is functional in mediating immune responses to pneumococcal infection.
The R232H variant of STING occurs in approximately 13.7% of the human population. While R232H STING has been shown to respond normally to DNA, it was found to be defective in inducing immune responses to bacterial CDN stimulation (27, 28). To evaluate the capacity of endogenous R232H STING in inducing immune responses against S. pneumoniae infection or pneumococcal DNA stimulation, we screened healthy volunteers for the presence of this allele, isolated cells from 3 individuals carrying the R232H allele in homozygosity, and compared their response to that of cells from volunteers carrying WT TMEM173. In agreement with previous studies (27, 28), cells expressing R232H STING exhibited reduced responses to exogenous CDNs (unpublished data). The presence of the R232H STING, however, did not influence the type I IFN or proinflammatory cytokine response against pneumococcal infection or DNA stimulation (Fig. 4A to D and Fig. S5). These data indicate that recognition of pneumococcal CDNs does not play a major role in mediating immune responses during pneumococcal infection.
FIG 4.
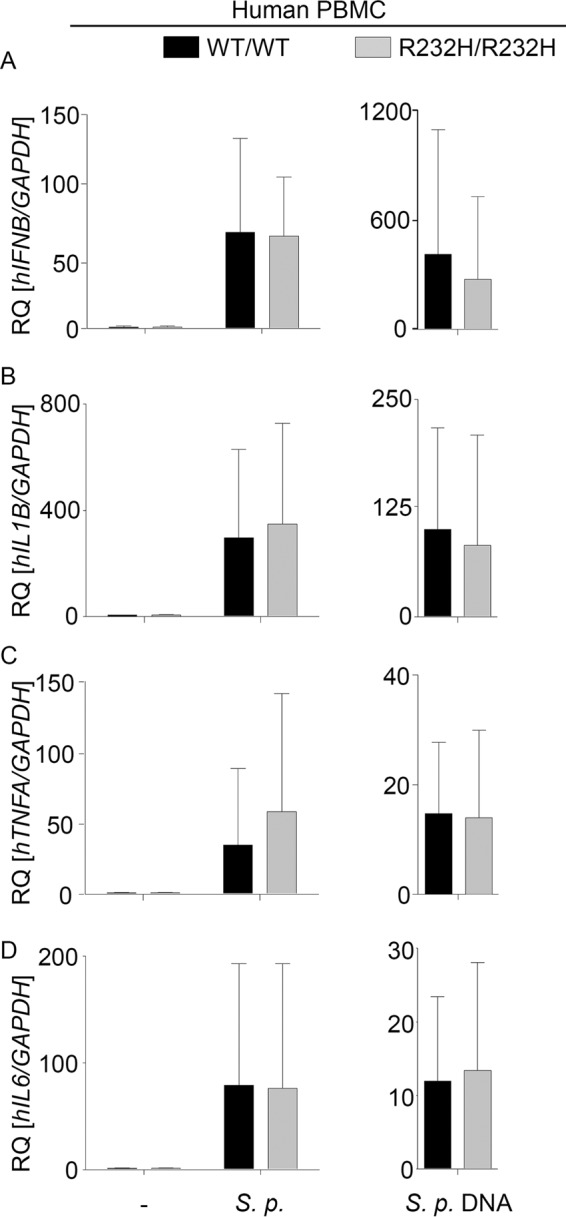
R232H variant of STING responds normally to S. pneumoniae infection or stimulation with pneumococcal DNA. PBMCs from healthy volunteers were isolated by density gradient centrifugation. Seven days after isolation, cells were challenged for 6 h with S. pneumoniae at a multiplicity of infection of 2.5 or stimulated for the same period with 1 μg/ml pneumococcal DNA. RNA was isolated, and the expression of IFNB (A), IL1B (B), TNFA (C), and IL-6 (D) was determined by qRT-PCR. Data are shown as the RQ of specified mRNAs. Data represent the means ± SD from 3 independent experiments carried out in triplicates. Differences were assessed with the Mann-Whitney U test.
STING is dispensable for the antibacterial defense against S. pneumoniae in mice.
In order to evaluate the relevance of STING in the antibacterial defense against S. pneumoniae in mice, we intranasally infected WT and Tmem173−/− animals with a high infection dose (5 × 106 CFU/mice) and a low infection dose (5 × 105 CFU/mice) and measured bacterial burdens in the lungs, spleens, and blood. We found no differences between bacterial burdens in WT and Tmem173−/− animals (Fig. 5A to F). These results indicate that STING is not required for antibacterial immunity against S. pneumoniae in mice.
FIG 5.
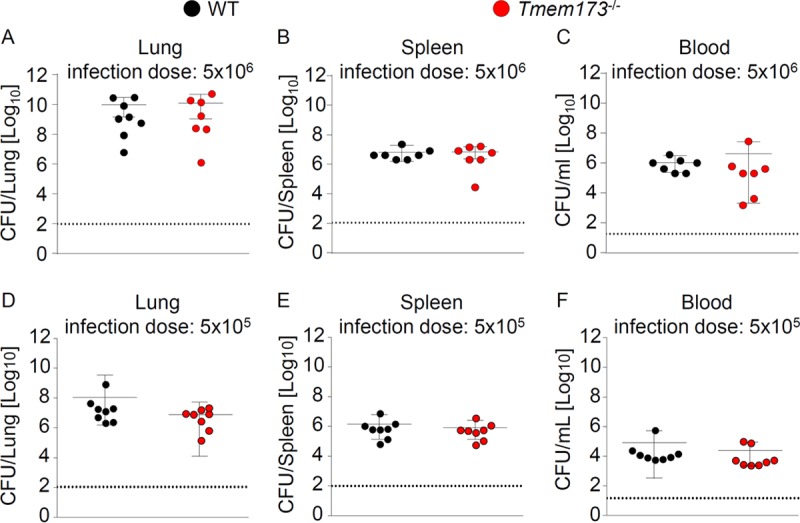
STING does not contribute to antipneumococcal defense in mice. WT and STING-deficient mice were intranasally infected with 5 × 106 CFU (A to C) or 5 × 105 CFU (D to F) S. pneumoniae, and the bacterial burdens in the lungs (A and D), spleen (B and E), and blood (C and F) were measured 48 h postinfection. Data represent means ± SD from 7 or 8 mice per group. Comparisons were performed with the Mann-Whitney U test.
Expression of common STING variants seems to have no major impact on susceptibility of humans to pneumococcal pneumonia.
In an attempt to assess a potential association between carriage of the HAQ TMEM173 allele and an increased susceptibility to pneumococcal pneumonia, we next compared the frequencies of the HAQ TMEM173 allele in nonvaccinated patients suffering from pneumococcal pneumonia and healthy controls (Table 1). None of the groups analyzed deviated significantly from Hardy-Weinberg equilibrium. The analysis revealed no difference in HAQ allele frequency between our patient and control cohorts and did not provide evidence for substantially altered odds of pneumococcal disease in HAQ TMEM173 carriers. Similarly, the frequency of the R232H allele in our pneumococcal pneumonia patient group was not different from that of our healthy control cohorts. We then conducted an ordered logistic regression analysis in order to test whether carriage of HAQ or R232H TMEM173 is associated with severity of pneumonia. Patients were grouped in three different levels of increasing severity according to the CURB-65 scores (34). As expected, age, but not gender, was significantly associated with increased severity of the disease. On the contrary, carriage of neither HAQ nor R232 TMEM173 showed a significant effect on pneumonia severity (Table 2). Collectively, our explorative analyses strongly suggest that carriage of HAQ or R232H TMEM173 is not a major risk factor for acute pneumococcal pneumonia in nonvaccinated individuals. However, due to limited sample size, the possibility of small effects actually being present cannot be excluded.
TABLE 1.
Distribution of TMEM173 HAQ and TMEM173 R232H in patients and healthy controls
| Parameter | Value(s) for: |
||
|---|---|---|---|
| Patients | Control group | Replication control groupa | |
| No. | 87 | 92 | 564 |
| Proportion female [% (n)] | 48 (42) | 53.2 (49) | 63.7* (359) |
| Age [yr; median (range)] | 66 (36–94) | 72 (35–93) | 32.0* (22–74) |
| TMEM173 HAQ [% (n)] | |||
| Heterozygous | 24 (21) | 20.6 (19) | 24.6 (139) |
| Homozygous | 1 (1) | 0 (0) | 1.4 (8) |
| Haplotype carrier | 25 (22) | 20.6 (19) | 26.1 (147) |
| Allele frequency | 0.132 | 0.103 | 0.137 |
| TMEM173 R232H [% (n)] | |||
| Heterozygous | 23.0 (20) | 29.3 (27) | 23.8 (134) |
| Homozygous | 0 (0) | 2.2 (2) | 1.8 (10) |
| Haplotype carrier | 23.0 (20) | 31.5 (29) | 25.5 (144) |
| Allele frequency | 0.115 | 0.168 | 0.137 |
*, P < 0.05 compared to the patient group.
TABLE 2.
Distribution of TMEM173 HAQ and TMEM173 R232H in patients grouped according to severity of pneumonia
| Parameter | Severitya |
P valueb | ||
|---|---|---|---|---|
| Low (CURB-65 = 0–1) | Middle (CURB-65 = 2) | High (CURB-65 = 3–5) | ||
| Proportion female [% (n)] | 42.6 (23) | 45.4 (10) | 83.3 (5) | 0.37 |
| Age [yr; median (range)] | 57.5 (37–94) | 74 (36–94) | 73 (72–81) | 0.004 |
| TMEM173 HAQ [% (n)] | ||||
| Haplotype carrier | 27.8 (15) | 13.6 (3) | 50 (3) | 0.778 |
| TMEM173 R232H [% (n)] | ||||
| Haplotype carrier | 18.5 (10) | 36.4 (8) | 33.3 (2) | 0.256 |
Severity was defined by following the guidelines of the British Thoracic Society (34). From 87 patients initially included in the study, 5 were not considered for this analysis, as information regarding pneumonia severity was not available.
Significance was evaluated through an ordered logistic regression model.
DISCUSSION
Type I IFNs were discovered more than half a century ago as key antiviral mediators (35) whose activity against viruses largely depends on the transcriptional induction of IFN-stimulated genes with direct antiviral activities (36). In addition, increasing evidence obtained in recent years indicates that type I IFNs also play important roles in bacterial infections. They have been shown to induce the expression of proteins with antibacterial activity (36, 37) and to regulate interleukin (38) as well as chemokine (39, 40) production. According to this pleiotropic function of type I IFNs, they have been demonstrated to be either beneficial or harmful for the host, probably depending on the pathogen and the mode of infection. In models of lung infections, for example, type I IFNs enhance host resistance to Legionella pneumophila (37, 41–43), Chlamydia pneumoniae (44), and Pseudomonas aeruginosa (45) but increase susceptibility to Mycobacterium tuberculosis (46) and Staphylococcus aureus (47). Several studies also reported that mice lacking receptors for type I IFNs exhibit impaired antibacterial defense against S. pneumoniae in models of nasal carriage, pneumonia, and sepsis (48–51), although other studies examining invasive serotype 1 strain (52) or postinfluenza pneumococcal infections indicated that this cytokine family can also have detrimental effects for the host (39, 40).
In this report, we show that production of type I IFNs during S. pneumoniae infection depends on recognition of bacterial DNA by the cGAS/STING pathway, and that this pathway is dispensable for antibacterial defense during acute pneumococcal pneumonia in mice. Moreover, we show that expression of the hypomorphic HAQ variant of STING dramatically reduces S. pneumoniae-induced type I IFN responses, but that it appears to have no major influence on the susceptibility of nonvaccinated individuals toward pneumococcal pneumonia or on disease severity. We speculate that the divergent effect of type I IFN versus STING deficiencies on pneumococcal infections can be explained by the capacity of cGAS/STING to also regulate the production of other mediators in addition to type I IFNs (53, 54). Indeed, STING-dependent signaling has been shown to activate various transcription factors, such as IRF3, NF-κB, and STAT6 (21, 53). STING deficiency might lead to a reduced production of unknown factors that impair antipneumococcal immunity, and this effect might compensate for the defect in type I IFN and IL-1β production. Moreover, human macrophages expressing HAQ STING were still able to produce small amounts of type I IFNs, which might be sufficient for mediating potentially protective effects on antibacterial immunity.
We did not find an association between carriage of TMEM173 variants with pneumococcal pneumonia or severity of the disease in nonvaccinated patients. However, this might be different in vaccinated individuals, since STING has recently been indicated to affect B cell responses in mice (55), and expression of a mouse equivalent of the HAQ allele reduced the efficacy of pneumococcal vaccination using Pneumovax 23 in mice (31).
In L. monocytogenes infection, sensing of bacterial c-di-AMP by STING has recently been shown to trigger type I IFN production (56). Similar to Listeria spp. and most other Gram-positive bacteria, S. pneumoniae is able to produce c-di-AMP (57). However, our data demonstrating that DNA sensing by cGAS is required for pneumococcus-induced type I IFN production suggest that recognition of c-di-AMP is dispensable for innate immune sensing of S. pneumoniae. In line with this conclusion, cGAS-deficient cells exhibited decreased IFN induction in response to S. pneumoniae infection. Additionally, cells carrying the R232H allele, which has been reported to affect bacterial CDN recognition (28), were fully capable of responding to S. pneumoniae infection. Furthermore, distribution of the R232H SNP was equal in the pneumococcal pneumonia patients and healthy controls analyzed. However, while STING-deficient BMDMs showed a slightly diminished production of IL-1β and IL-6 upon S. pneumoniae infection, cGas−/− cells exhibited no defect in proinflammatory cytokine production. Thus, we cannot exclude a minor contribution of a STING-dependent but cGAS-independent sensing mechanism in stimulating proinflammatory responses in vitro.
Our analyses indicate that deficiency in the STING-dependent pathway had stronger effects on the production of proinflammatory cytokines in human PBMC-derived macrophages than in murine BMDMs. This difference might be explained by the differential expression of other PRRs known to detect pneumococci between murine and human macrophages, which might compensate for the lack of cGAS/STING-dependent sensing.
In summary, we show that cGAS/STING senses pneumococcal DNA during S. pneumoniae infection to primarily stimulate production of type I IFNs and to contribute to proinflammatory cytokine production in human cells. Analyses of mice lacking STING as well as individuals expressing common STING variants, however, suggest that this pathway plays no major role in antipneumococcal host defense.
MATERIALS AND METHODS
Ethics statement.
Healthy volunteers from whom PBMCs were isolated provided written informed consent, and the study procedures were in agreement with the local ethics committee guidelines (Charité–Universitätsmedizin Berlin). Samples from patients with pneumococcal pneumonia were kindly provided by the CAPNETZ study group. This prospective multicenter study (German Clinical Trials Register DRKS00005274) was approved by the ethical review board of each participating clinical center (reference number of the leading ethics committee of Medical Faculty of Otto-von-Guericke-University in Magdeburg, 104/01; reference number for Medical School Hannover, 301/2008; see www.capnetz.de for participating centers) and was performed in accordance with the Declaration of Helsinki. All patients provided written informed consent prior to enrollment in the study. All animal experiments were approved by institutional (Charité–Universitätsmedizin Berlin) and governmental animal welfare committees (LAGeSo Berlin; approval ID G0440/12).
Bacterial strains.
The S. pneumoniae D39 strain, belonging to serotype 2, as well as the Δply and Δcps isogenic mutant strains (58), were used for in vitro infection assays. S. pneumoniae strain serotype 3 (NCTC 7978) was used to induce pneumonia in mice. Bacteria were cultured on Columbia blood agar plus 5% sheep blood for 12 h at 37°C. Culture of the mutant strains also required the addition of 40 μl of the following antibiotics on the agar plate: D39Δcps strain, 50 mg/ml kanamycin; D39Δply strain, 1 mg/ml erythromycin.
BMDMs.
Bone marrow-derived macrophages (BMDMs) were isolated from femurs and tibiae of WT, Tmem173−/−, and cGas−/− female mice on a C57BL/6 background. Before infection, cells were grown in RPMI 1640 containing 30% L929 cell supernatant and 20% fetal calf serum (FCS) for 10 days.
Cell transfection and infection.
Mouse BMDMs were infected with the above-mentioned strains of S. pneumoniae at a multiplicity of infection of 1, centrifuged at 200 × g for 5 min, and then incubated for 6 h at 37°C. Bacterial DNA or synthetic dsDNA (ISD, or interferon stimulatory DNA; Invivogen) was transfected at a concentration of 1 μg/ml into the cells using Lipofectamine 2000 (Invitrogen). For siRNA-mediated cGas downregulation, BMDMs were transfected 48 h prior to infection with control nonsilencing siRNA or with a specific siRNA targeting cGas using HiPerfect (Qiagen).
Murine model of pneumococcal pneumonia.
Female WT and Tmem173−/− mice (23, 59), 8 to 14 weeks old on a C57BL/6 background, were housed in individually ventilated cages, with food and water provided ad libitum. Mice were anesthetized by intraperitoneal injection of ketamine (1.6 mg) and xylazine (0.5 mg) and intranasally infected with 5 × 105 or 5 × 106 CFU S. pneumoniae serotype 3 (NCTC 7978) in 20 μl of phosphate-buffered saline (PBS). Body weight and temperature were monitored every 12 h. Two days after infection, mice were anesthetized, heparinized, and euthanized through final blood withdrawal. Lungs were then flushed via the pulmonary artery and removed for subsequent analyses. Bacterial counts were assessed by plating serial dilutions of lung or spleen homogenates or blood on Columbia blood agar plates.
Quantitative reverse transcription-PCR (qRT-PCR).
Total RNA was isolated from cultured cells or mouse lung homogenates using the PerfectPure RNA purification system (5 Prime) or TRIzol (Life Technologies), respectively. cDNA was synthesized from total RNA using a high-capacity reverse transcription kit (Applied Biosystems), and quantitative PCR was performed using TaqMan assays (Life Technologies) or self-designed primer sets on an ABI 7300 instrument (Applied Biosystems). The input was normalized to the average expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in untreated cells or PBS-treated mice.
ELISA.
Protein concentrations of IP-10, IL-6, TNF-α, and IL-1β were quantified by commercially available sandwich enzyme-linked immunosorbent assay (ELISA) kits (eBioscience). Protein concentrations were quantified in a FilterMax F5 multimode microplate reader (Molecular Devices) and read at 450 nm.
Subjects.
Peripheral blood was collected from 14 healthy volunteers carrying HAQ, R232H, or WT TMEM173 and belonging to a cohort group (n = 564) assembled at the Institute of Microbiology, Charité University Medical Center Berlin. The association study between the HAQ haplotype and pneumococcal pneumonia was performed using DNA samples obtained from 87 patients with S. pneumoniae-induced community-acquired pneumonia (CAP) without antipneumococcal vaccination. Samples were provided by the CAPNETZ study group, a German multicenter prospective cohort study for CAP. A detailed description of the CAPNETZ methodology has been provided before (60). The control groups were composed of a subgroup of healthy individuals of similar age and sex distribution (n = 92) from the PolSenior program, an interdisciplinary project, designed to evaluate health and socioeconomic status of Polish Caucasians aged ≥65 years (61) and the cohort of the Institute of Microbiology, Charité University Medical Center Berlin.
TMEM173 genotyping.
Genomic DNA was collected in buccal swabs and isolated by employing the Gentra Puregene buccal cell kit from Qiagen by following the manufacturer's instructions. Genotyping of TMEM173 R71H (rs11554776), G230A (rs78233829), R293Q (rs7380824), and the TMEM173 R232H SNP (rs1131769) was performed by PCR using fluorescence-labeled hybridization fluorescence resonance energy transfer probes (TIB Molbiol) accompanied by melting curve analysis in a LightCycler 480 (Roche Diagnostics). Primers and probes used were the following (where f-primer indicates forward primer, r-primer indicates reverse primer, and uppercase letters within the sequences [e.g., “XI”] indicate a label within the oligonucleotide probe, with “I” indicating a base analogue and “X” the modification): rs11554776 f-primer, ggagtgacacacgttgg; r-primer, gcctagctgaggagctg; simple probe, LC640-ctggagtggaXItgtggcgcag-PH; rs78233829 f-primer, gggtctcactcctgaatcaggt; r-primer, ccgatccttgatgcaagca; anchor probe, LC640-cagtttatccaggaagcgaatgttggg-PH; sensor probe, ggtcagcggtctgctgg-FL; rs7380824 f-primer, accctggtaggcaatga; r-primer, gcttagtctggtcttcctcttac; anchor probe, LC640-ggcctgctcaagcctatcctcccgg-PH; sensor probe, cctcaagtgtccggcagaagagtt-FL; rs1131769 f-primer, cccactcccctgcacactt; r-primer, tggataaactgcccaagcagac; anchor probe, LC640-aggatcgggtttacagcaacagca-PH; sensor probe, ggtgaccatgctggcatc-FL.
Infection and stimulation of human PBMCs.
Fifty-milliliter aliquots of whole peripheral blood were drawn from healthy volunteers, and PBMCs were isolated by density gradient centrifugation (62, 63). Briefly, whole blood was diluted 1:1 with PBS and layered onto 20 ml Histopaque-1077 (Sigma-Aldrich). The gradient then was centrifuged at 800 × g for 25 min at room temperature, and the PBMCs were collected from the interface. PBMCs were washed twice with PBS and resuspended in RPMI medium supplemented with 10% FCS and 1% l-glutamine. Half of the cell medium was replaced every 2 days, and the cells were kept on culture for 7 days to allow differentiation into a macrophage-like state before infection or stimulation. Infection with S. pneumoniae and stimulation with bacterial DNA were performed as described before for mouse BMDMs.
Human monocytic THP-1 cells.
cGAS−/− (33) and control THP-1 human cells were maintained under standard culture conditions. Cell differentiation into a macrophage-like state was induced by resuspension of cells in culture medium containing 80 nM phorbol myristate acetate (PMA) for 48 h.
Statistics.
Data analysis was carried out using Prism software (GraphPad Software). Groups were contrasted using a two-tailed Mann-Whitney U test. The association study was performed employing a chi-square test for association and calculation of odds ratios. A Fisher's exact test, together with calculation of exact confidence intervals, was used if applicable. The analysis of association between allele carriage and disease severity was conducted through an ordered logistic regression using SPSS (IBM Corporation). Differences with a P value of <0.05 were regarded as significant.
Supplementary Material
ACKNOWLEDGMENTS
We are thankful to the healthy volunteer for donating blood, the CAPNETZ foundation for providing samples from pneumococcal pneumonia patients, the investigators in the local clinical centers, and all practitioners, physicians, and respiratory specialists cooperating within the network. We thank Frank P. Mockenhaupt (Charité–Universitätsmedizin Berlin) for help with the statistical analyses, as well as Sven Hammerschmidt (Interfaculty Institute for Genetics and Functional Genomics, Ernst-Moritz-Arndt Universität Greifswald, Greifswald, Germany) and Tim Mitchell (College of Medical and Dental Sciences, University of Birmingham, Birmingham, United Kingdom) for sharing bacterial strains. Moreover, we are grateful to Veit Hornung (Gene Center Munich, Germany) for providing cGAS-deficient THP-1 cells, Herbert “Skip” Virgin (Washington University School of Medicine, St. Louis, MO) for his kind permission to use cGas−/− mice, and Anca Dorhoi (Max Planck Institute for Infection Biology, Berlin, Germany) for providing those animals.
This work was supported by the Deutsche Forschungsgemeinschaft (GRK1673/B5 to J.S.M.R. and B.O., GRK1673/A5 to R.R.S., SFB/TR84 project A1/A5 to B.O., projects C3 and C6 to M.W., and project B1 to N.S.), the German Ministry for Education and Research (CAPSyS; grant TP4 to M.W.), the Ministry of Science and Higher Education (MEIN), Poland (PBZ-MEiN-9/2/2006–K143/P01/2007/1 to M.P.K.), and the National Institutes of Health (grants R21AI099346 and T32AI022295 to J.C.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00849-17.
REFERENCES
- 1.Nature Reviews Microbiology. 2009. Closing the GAPP on pneumonia. Nat Rev Microbiol 7:838. doi: 10.1038/nrmicro2273. [DOI] [PubMed] [Google Scholar]
- 2.van der Poll T, Opal SM. 2009. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374:1543–1556. doi: 10.1016/S0140-6736(09)61114-4. [DOI] [PubMed] [Google Scholar]
- 3.Henriques-Normark B, Tuomanen EI. 2013. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harb Perspect Med 3:a010215. doi: 10.1101/cshperspect.a010215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Opitz B, van Laak V, Eitel J, Suttorp N. 2010. Innate immune recognition in infectious and noninfectious diseases of the lung. Am J Resp Crit Care Med 181:1294–1309. doi: 10.1164/rccm.200909-1427SO. [DOI] [PubMed] [Google Scholar]
- 5.Chaput C, Sander LE, Suttorp N, Opitz B. 2013. NOD-like receptors in lung diseases. Front Immunol 4:393. doi: 10.3389/fimmu.2013.00393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schroder K, Tschopp J. 2010. The inflammasomes. Cell 140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 7.Wu J, Chen ZJ. 2014. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol 32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 8.Albiger B, Dahlberg S, Sandgren A, Wartha F, Beiter K, Katsuragi H, Akira S, Normark S, Henriques-Normark B. 2007. Toll-like receptor 9 acts at an early stage in host defence against pneumococcal infection. Cell Microbiol 9:633–644. doi: 10.1111/j.1462-5822.2006.00814.x. [DOI] [PubMed] [Google Scholar]
- 9.Davis KM, Nakamura S, Weiser JN. 2011. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J Clin Investig 121:3666–3676. doi: 10.1172/JCI57761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang R, Tsuchiya K, Kawamura I, Shen Y, Hara H, Sakai S, Yamamoto T, Fernandes-Alnemri T, Yang R, Hernandez-Cuellar E, Dewamitta SR, Xu Y, Qu H, Alnemri ES, Mitsuyama M. 2011. Critical roles of ASC inflammasomes in caspase-1 activation and host innate resistance to Streptococcus pneumoniae infection. J Immunol 187:4890–4899. doi: 10.4049/jimmunol.1100381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koppe U, Suttorp N, Opitz B. 2012. Recognition of Streptococcus pneumoniae by the innate immune system. Cell Microbiol 14:460–466. doi: 10.1111/j.1462-5822.2011.01746.x. [DOI] [PubMed] [Google Scholar]
- 12.McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, Smeaton S, El-Rachkidy R, McLoughlin RM, Mori A, Moran B, Fitzgerald KA, Tschopp J, Petrilli V, Andrew PW, Kadioglu A, Lavelle EC. 2010. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog 6:e1001191. doi: 10.1371/journal.ppat.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Opitz B, Puschel A, Schmeck B, Hocke AC, Rosseau S, Hammerschmidt S, Schumann RR, Suttorp N, Hippenstiel S. 2004. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem 279:36426–36432. doi: 10.1074/jbc.M403861200. [DOI] [PubMed] [Google Scholar]
- 14.Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, Reppe K, Meixenberger K, Dorhoi A, Ma J, Holmes A, Trendelenburg G, Heimesaat MM, Bereswill S, van der Linden M, Tschopp J, Mitchell TJ, Suttorp N, Opitz B. 2011. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol 187:434–440. doi: 10.4049/jimmunol.1003143. [DOI] [PubMed] [Google Scholar]
- 15.Fatykhova D, Rabes A, Machnik C, Guruprasad K, Pache F, Berg J, Toennies M, Bauer TT, Schneider P, Schimek M, Eggeling S, Mitchell TJ, Mitchell AM, Hilker R, Hain T, Suttorp N, Hippenstiel S, Hocke AC, Opitz B. 2015. Serotype 1 and 8 pneumococci evade sensing by inflammasomes in human lung tissue. PLoS One 10:e0137108. doi: 10.1371/journal.pone.0137108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kostadinova E, Chaput C, Gutbier B, Lippmann J, Sander LE, Mitchell TJ, Suttorp N, Witzenrath M, Opitz B. 2016. NLRP3 protects alveolar barrier integrity by an inflammasome-independent increase of epithelial cell adherence. Sci Rep 6:30943. doi: 10.1038/srep30943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Behler-Janbeck F, Takano T, Maus R, Stolper J, Jonigk D, Tort Tarres M, Fuehner T, Prasse A, Welte T, Timmer MS, Stocker BL, Nakanishi Y, Miyamoto T, Yamasaki S, Maus UA. 2016. C-type lectin Mincle recognizes glucosyl-diacylglycerol of Streptococcus pneumoniae and plays a protective role in pneumococcal pneumonia. PLoS Pathog 12:e1006038. doi: 10.1371/journal.ppat.1006038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rabes A, Zimmermann S, Reppe K, Lang R, Seeberger PH, Suttorp N, Witzenrath M, Lepenies B, Opitz B. 2015. The C-type lectin receptor Mincle binds to Streptococcus pneumoniae but plays a limited role in the anti-pneumococcal innate immune response. PLoS One 10:e0117022. doi: 10.1371/journal.pone.0117022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koppe U, Hogner K, Doehn JM, Muller HC, Witzenrath M, Gutbier B, Bauer S, Pribyl T, Hammerschmidt S, Lohmeyer J, Suttorp N, Herold S, Opitz B. 2012. Streptococcus pneumoniae stimulates a STING- and IFN regulatory factor 3-dependent type I IFN production in macrophages, which regulates RANTES production in macrophages, cocultured alveolar epithelial cells, and mouse lungs. J Immunol 188:811–817. doi: 10.4049/jimmunol.1004143. [DOI] [PubMed] [Google Scholar]
- 20.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. 2011. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin L, Hill KK, Filak H, Mogan J, Knowles H, Zhang B, Perraud AL, Cambier JC, Lenz LL. 2011. MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J Immunol 187:2595–2601. doi: 10.4049/jimmunol.1100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. 2013. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin L, Xu LG, Yang IV, Davidson EJ, Schwartz DA, Wurfel MM, Cambier JC. 2011. Identification and characterization of a loss-of-function human MPYS variant. Genes Immun 12:263–269. doi: 10.1038/gene.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yi G, Brendel VP, Shu C, Li P, Palanathan S, Cheng Kao C. 2013. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS One 8:e77846. doi: 10.1371/journal.pone.0077846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. 2013. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep 3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, Goudin N, Fremond ML, Nitschke P, Molina TJ, Blanche S, Picard C, Rice GI, Crow YJ, Manel N, Fischer A, Bader-Meunier B, Rieux-Laucat F. 2014. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Investig 124:5516–5520. doi: 10.1172/JCI79100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, Lee CC, DiMattia MA, Cowen EW, Gonzalez B, Palmer I, DiGiovanna JJ, Biancotto A, Kim H, Tsai WL, Trier AM, Huang Y, Stone DL, Hill S, Kim HJ, St Hilaire C, Gurprasad S, Plass N, Chapelle D, Horkayne-Szakaly I, Foell D, Barysenka A, Candotti F, Holland SM, Hughes JD, Mehmet H, Issekutz AC, Raffeld M, McElwee J, Fontana JR, Minniti CP, Moir S, Kastner DL, Gadina M, Steven AC, Wingfield PT, Brooks SR, Rosenzweig SD, Fleisher TA, Deng Z, Boehm M, Paller AS, Goldbach-Mansky R. 2014. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371:507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel S, Blaauboer SM, Tucker HR, Mansouri S, Ruiz-Moreno JS, Hamann L, Schumann RR, Opitz B, Jin L. 2017. The common R71H-G230A-R293Q human TMEM173 is a null allele. J Immunol 198:776–787. doi: 10.4049/jimmunol.1601585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, Sivick KE, Zeng Q, Soares KC, Zheng L, Portnoy DA, Woodward JJ, Pardoll DM, Dubensky TW Jr, Kim Y. 2015. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med 7:283ra252. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mankan AK, Schmidt T, Chauhan D, Goldeck M, Honing K, Gaidt M, Kubarenko AV, Andreeva L, Hopfner KP, Hornung V. 2014. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J 33:2937–2946. doi: 10.15252/embj.201488726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim WS, Baudouin SV, George RC, Hill AT, Jamieson C, Le Jeune I, Macfarlane JT, Read RC, Roberts HJ, Levy ML, Wani M, Woodhead MA, Pneumonia Guidelines Committee of the BTS Standards of Care Committee. 2009. BTS guidelines for the management of community acquired pneumonia in adults: update 2009. Thorax 64(Suppl 3):iii1–iii55. doi: 10.1136/thx.2009.121434. [DOI] [PubMed] [Google Scholar]
- 35.Isaacs A, Lindenmann J. 1957. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 147:258–267. doi: 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- 36.MacMicking JD. 2012. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol 12:367–382. doi: 10.1038/nri3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, Kempa S, Peter A, Mollenkopf HJ, Dorhoi A, Kershaw O, Gruber AD, Sander LE, Witzenrath M, Herold S, Nerlich A, Hocke AC, van Driel I, Suttorp N, Bedoui S, Hilbi H, Trost M, Opitz B. 2016. IFNs modify the proteome of Legionella-containing vacuoles and restrict infection via IRG1-derived itaconic acid. PLoS Pathog 12:e1005408. doi: 10.1371/journal.ppat.1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castiglia V, Piersigilli A, Ebner F, Janos M, Goldmann O, Dambock U, Kroger A, Weiss S, Knapp S, Jamieson AM, Kirschning C, Kalinke U, Strobl B, Muller M, Stoiber D, Lienenklaus S, Kovarik P. 2016. Type I interferon signaling prevents IL-1beta-driven lethal systemic hyperinflammation during invasive bacterial infection of soft tissue. Cell Host Microbe 19:375–387. doi: 10.1016/j.chom.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, Belperio JA, Cheng G, Deng JC. 2009. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Investig 119:1910–1920. doi: 10.1172/JCI35412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamura S, Davis KM, Weiser JN. 2011. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Investig 121:3657–3665. doi: 10.1172/JCI57762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lippmann J, Muller HC, Naujoks J, Tabeling C, Shin S, Witzenrath M, Hellwig K, Kirschning CJ, Taylor GA, Barchet W, Bauer S, Suttorp N, Roy CR, Opitz B. 2011. Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell Microbiol 13:1668–1682. doi: 10.1111/j.1462-5822.2011.01646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schiavoni G, Mauri C, Carlei D, Belardelli F, Pastoris MC, Proietti E. 2004. Type I IFN protects permissive macrophages from Legionella pneumophila infection through an IFN-gamma-independent pathway. J Immunol 173:1266–1275. doi: 10.4049/jimmunol.173.2.1266. [DOI] [PubMed] [Google Scholar]
- 43.Sporri R, Joller N, Albers U, Hilbi H, Oxenius A. 2006. MyD88-dependent IFN-gamma production by NK cells is key for control of Legionella pneumophila infection. J Immunol 176:6162–6171. doi: 10.4049/jimmunol.176.10.6162. [DOI] [PubMed] [Google Scholar]
- 44.Rothfuchs AG, Trumstedt C, Mattei F, Schiavoni G, Hidmark A, Wigzell H, Rottenberg ME. 2006. STAT1 regulates IFN-alpha beta- and IFN-gamma-dependent control of infection with Chlamydia pneumoniae by nonhemopoietic cells. J Immunol 176:6982–6990. doi: 10.4049/jimmunol.176.11.6982. [DOI] [PubMed] [Google Scholar]
- 45.Carrigan SO, Junkins R, Yang YJ, Macneil A, Richardson C, Johnston B, Lin TJ. 2010. IFN regulatory factor 3 contributes to the host response during Pseudomonas aeruginosa lung infection in mice. J Immunol 185:3602–3609. doi: 10.4049/jimmunol.0903429. [DOI] [PubMed] [Google Scholar]
- 46.Stanley SA, Johndrow JE, Manzanillo P, Cox JS. 2007. The type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol 178:3143–3152. doi: 10.4049/jimmunol.178.5.3143. [DOI] [PubMed] [Google Scholar]
- 47.Martin FJ, Gomez MI, Wetzel DM, Memmi G, O'Seaghdha M, Soong G, Schindler C, Prince A. 2009. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J Clin Investig 119:1931–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maier BB, Hladik A, Lakovits K, Korosec A, Martins R, Kral JB, Mesteri I, Strobl B, Muller M, Kalinke U, Merad M, Knapp S. 2016. Type I interferon promotes alveolar epithelial type II cell survival during pulmonary Streptococcus pneumoniae infection and sterile lung injury in mice. Eur J Immunol 46:2175–2186. doi: 10.1002/eji.201546201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parker D, Martin FJ, Soong G, Harfenist BS, Aguilar JL, Ratner AJ, Fitzgerald KA, Schindler C, Prince A. 2011. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. mBio 2:e00016-. doi: 10.1128/mBio.00016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macri G, Ruggeri A, Leanderson T, Teti G. 2007. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol 178:3126–3133. doi: 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- 51.LeMessurier KS, Hacker H, Chi L, Tuomanen E, Redecke V. 2013. Type I interferon protects against pneumococcal invasive disease by inhibiting bacterial transmigration across the lung. PLoS Pathog 9:e1003727. doi: 10.1371/journal.ppat.1003727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hughes CE, Harvey RM, Plumptre CD, Paton JC. 2014. Development of primary invasive pneumococcal disease caused by serotype 1 pneumococci is driven by early increased type I interferon response in the lung. Infect Immun 82:3919–3926. doi: 10.1128/IAI.02067-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen H, Sun H, You F, Sun W, Zhou X, Chen L, Yang J, Wang Y, Tang H, Guan Y, Xia W, Gu J, Ishikawa H, Gutman D, Barber G, Qin Z, Jiang Z. 2011. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 147:436–446. doi: 10.1016/j.cell.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 54.Poli C, Augusto JF, Dauve J, Adam C, Preisser L, Larochette V, Pignon P, Savina A, Blanchard S, Subra JF, Chevailler A, Procaccio V, Croue A, Creminon C, Morel A, Delneste Y, Fickenscher H, Jeannin P. 2017. IL-26 confers proinflammatory properties to extracellular DNA. J Immunol 198:3650–3661. doi: 10.4049/jimmunol.1600594. [DOI] [PubMed] [Google Scholar]
- 55.Zeng M, Hu Z, Shi X, Li X, Zhan X, Li XD, Wang J, Choi JH, Wang KW, Purrington T, Tang M, Fina M, DeBerardinis RJ, Moresco EM, Pedersen G, McInerney GM, Karlsson Hedestam GB, Chen ZJ, Beutler B. 2014. MAVS, cGAS, and endogenous retroviruses in T-independent B cell responses. Science 346:1486–1492. doi: 10.1126/science.346.6216.1486. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Woodward JJ, Iavarone AT, Portnoy DA. 2010. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bai Y, Yang J, Eisele LE, Underwood AJ, Koestler BJ, Waters CM, Metzger DW, Bai G. 2013. Two DHH subfamily 1 proteins in Streptococcus pneumoniae possess cyclic di-AMP phosphodiesterase activity and affect bacterial growth and virulence. J Bacteriol 195:5123–5132. doi: 10.1128/JB.00769-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Noske N, Kammerer U, Rohde M, Hammerschmidt S. 2009. Pneumococcal interaction with human dendritic cells: phagocytosis, survival, and induced adaptive immune response are manipulated by PavA. J Immunol 183:1952–1963. doi: 10.4049/jimmunol.0804383. [DOI] [PubMed] [Google Scholar]
- 59.Jin L, Getahun A, Knowles HM, Mogan J, Akerlund LJ, Packard TA, Perraud AL, Cambier JC. 2013. STING/MPYS mediates host defense against Listeria monocytogenes infection by regulating Ly6C(hi) monocyte migration. J Immunol 190:2835–2843. doi: 10.4049/jimmunol.1201788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Welte T, Suttorp N, Marre R. 2004. CAPNETZ-community-acquired pneumonia competence network. Infection 32:234–238. doi: 10.1007/s15010-004-3107-z. [DOI] [PubMed] [Google Scholar]
- 61.Bledowski P, Mossakowska M, Chudek J, Grodzicki T, Milewicz A, Szybalska A, Wieczorowska-Tobis K, Wiecek A, Bartoszek A, Dabrowski A, Zdrojewski T. 2011. Medical, psychological and socioeconomic aspects of aging in Poland: assumptions and objectives of the PolSenior project. Exp Gerontol 46:1003–1009. doi: 10.1016/j.exger.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 62.Eitel J, Krull M, Hocke AC, N′Guessan PD, Zahlten J, Schmeck B, Slevogt H, Hippenstiel S, Suttorp N, Opitz B. 2008. Beta-PIX and Rac1 GTPase mediate trafficking and negative regulation of NOD2. J Immunol 181:2664–2671. doi: 10.4049/jimmunol.181.4.2664. [DOI] [PubMed] [Google Scholar]
- 63.Eitel J, Meixenberger K, van Laak C, Orlovski C, Hocke A, Schmeck B, Hippenstiel S, N′Guessan PD, Suttorp N, Opitz B. 2012. Rac1 regulates the NLRP3 inflammasome which mediates IL-1beta production in Chlamydophila pneumoniae infected human mononuclear cells. PLoS One 7:e30379. doi: 10.1371/journal.pone.0030379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.