

ABSTRACT
Staphylococcus aureus is an important human pathogen, but studies of the organism have suffered from the lack of a robust tool set for its genetic and genomic manipulation. Here we report the development of a system for the facile and high-throughput genomic engineering of S. aureus using single-stranded DNA (ssDNA) oligonucleotide recombineering coupled with clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9-mediated counterselection. We identify recombinase EF2132, derived from Enterococcus faecalis, as being capable of integrating single-stranded DNA oligonucleotides into the S. aureus genome. We found that EF2132 can readily mediate recombineering across multiple characterized strains (3 of 3 tested) and primary clinical isolates (6 of 6 tested), typically yielding thousands of recombinants per transformation. Surprisingly, we also found that some S. aureus strains are naturally recombinogenic at measurable frequencies when oligonucleotides are introduced by electroporation, even without exogenous recombinase expression. We construct a temperature-sensitive, two-vector system which enables conditional recombineering and CRISPR/Cas9-mediated counterselection in S. aureus without permanently introducing exogenous genetic material or unintended genetic lesions. We demonstrate the ability of this system to efficiently and precisely engineer point mutations and large single-gene deletions in the S. aureus genome and to yield highly enriched populations of engineered recombinants even in the absence of an externally selectable phenotype. By virtue of utilizing inexpensive, commercially synthesized synthetic DNA oligonucleotides as substrates for recombineering and counterselection, this system provides a scalable, versatile, precise, inexpensive, and generally useful tool for producing isogenic strains in S. aureus which will enable the high-throughput functional assessment of genome variation and gene function across multiple strain backgrounds.
KEYWORDS: CRISPR, Cas9, Staphylococcus aureus, genetic engineering, genome editing, mutS, recombineering
IMPORTANCE
Engineering genetic changes in bacteria is critical to understanding the function of particular genes or mutations but is currently a laborious and technically challenging process to perform for the important human pathogen Staphylococcus aureus. In an effort to develop methods which are rapid, easy, scalable, versatile, and inexpensive, here we describe a system for incorporating synthetic, mutagenic DNA molecules into the S. aureus genome and for eliminating cells that lack the engineered mutation. This method allows efficient, precise, and high-throughput genetic engineering of S. aureus strains and will facilitate studies seeking to address a variety of issues about the function of particular genes and specific mutations.
INTRODUCTION
Staphylococcus aureus is a common and highly successful opportunistic pathogen that is responsible for a high burden of patient morbidity and mortality nationwide and worldwide (1). S. aureus underlies diverse clinical diseases, ranging from relatively benign to life-threatening diseases, involving many different organ systems (2–4). Moreover, S. aureus infections, especially noncutaneous infections, are often chronic or relapsing (5–8) and are difficult to eradicate permanently. Despite its medical importance, knowledge of key factors which promote virulence, chronicity, and pathogenicity in S. aureus remains incomplete (9).
Genes contributing to clinically relevant phenotypes in S. aureus are believed to be numerous (10–12) and are frequently regulated in large, multicomponent networks (13, 14). Yet, S. aureus is difficult to manipulate genetically, making it challenging to experimentally test the effects of specific genes or mutations through the construction of isogenic strains. A major advance in the field was heralded by the development of methods to bypass common S. aureus restriction systems, which serve as strong barriers to the introduction of exogenous DNA. The construction of a transgenic cytosine methylase-negative Escherichia coli strain first enabled escape of the S. aureus type IV restriction system (15), and the strain’s subsequent modification to actively mimic methylation profiles of major S. aureus clonal complexes (CCs) further improved efficiencies by avoiding the type III restriction systems (16). Whereas previously only strains carrying spontaneous or induced mutations in their restriction systems could be manipulated, these technologies have now enabled high-efficiency transformation of engineered plasmids into most laboratory and clinical S. aureus strains.
Most established methods for modifying S. aureus genomes rely on rare homologous recombination events with large donor fragments encoding the desired change, with antibiotic-mediated selection for successful allelic exchange (17–19). Although useful, these techniques are relatively inefficient and introduce exogenous genetic material into the host genome along with the targeted mutation (18, 20). Modified approaches utilizing lethal, counterselectable markers, including antisense-secY expression (21), toxic metabolites (19), homing endonucleases (19), and, most recently, clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 (22, 23), have increased enrichment for genetically modified strains to various degrees or have eliminated the need for the introduction of chromosomally integrated markers. Nevertheless, all available genome editing strategies for S. aureus remain laborious and involve the individual cloning of each ~1-kb-to-2-kb homologous repair template, with accompanying protocol optimization (17, 18, 20–22, 24); this strategy is especially difficult to implement for engineering gene deletions, which must be manufactured using splicing by overhang extension (SOE) PCR (15, 23).
To address the methodological need for a facile, versatile, and scalable system for S. aureus precision genomic engineering, here we have developed conditional systems for recombineering (25) and CRISPR/Cas9-mediated counterselection (26) in that organism. First pioneered in E. coli (27), this powerful strategy has subsequently been adapted to other species, including Gram-positive organisms (28–32), although the range of bacteria for which such tools exist remains limited (32). Recombineering, which incorporates mutagenic oligonucleotides into a host genome through the action of bacteriophage-derived single-stranded DNA (ssDNA) recombinases, allows point mutations, variable-length deletions, and small insertions to be precisely engineered using short, commercially synthesized oligonucleotides (25, 31, 33, 34). Following recombineering, CRISPR/Cas9-mediated endonuclease cleavage targeted to the wild-type allele provides counterselection for the engineered change, even in the absence of an externally selectable phenotype, by introducing lethal double-stranded DNA breaks into the genome of unedited cells (27). Because recombineering and counterselection vector construction can be performed using inexpensive, commercially manufactured ssDNA fragments, the system is inherently scalable and amenable to high-throughput applications.
RESULTS
Identification of a single-stranded oligonucleotide recombineering protein with activity in S. aureus.
We initially tested the activity of various known and predicted recombinases in S. aureus by evaluating their ability to mediate ssDNA recombineering of the rpoB H481Y mutation, which confers rifampin resistance (35). We evaluated six different recombinases exogenously expressed from cassette-based shuttle vector pCN50 (36): bet, the recombinase gene utilized in the E. coli λ Red recombineering system (25); EF2132 and orfC, derived from Enterococcus faecalis and Legionella pneumophilia, respectively, both being genes that have been previously shown to have cross-species activity in E. coli (25); gp20, a recombinase originating from S. aureus with weak activity in E. coli (25); and two putative S. aureus recombinases (which we termed recTS2 and recTS3) which we identified on the basis of protein homology to these known recombinases. All proteins were codon optimized for expression in S. aureus. Each expression construct was separately introduced into S. aureus type strain ATCC 29213 (Rosenbach [37]), electrocompetent cells were prepared, and mutagenic oligonucleotide was subsequently introduced by electroporation. Recombineering activity was evaluated by comparing the number of rifampin-resistant colonies produced using the mutagenic oligonucleotide to the number seen after mock electroporation in the absence of the oligonucleotide (Fig. 1A).
FIG 1 .

Recombineering in S. aureus. (A) Activity of different recombinases when expressed exogenously in S. aureus compared to paired, mock-transformation controls lacking mutagenic oligonucleotide. Recombineering utilized mutagenic oligonucleotides encoding rifampin resistance. Recombination frequencies significantly higher than those observed in paired controls (≤P = 0.002 [two-tailed t test]) are indicated by an asterisk. TE, Tris-EDTA. (B) Sequence confirmation of recombineered isolates. Residues 477 through 485 of S. aureus rpoB are shown, with the H481Y missense mutation indicated as a white letter in a purple field. Mutations introduced by the recombineering oligonucleotide are highlighted between wild-type and engineered strains. (C) Recombineering efficiencies across laboratory and clinical strains. Results are shown for wild-type strains and strains exogenously expressing recombinase EF2132 compared to paired mock-transformation controls. Recombination frequencies significantly higher than those observed in controls (≤P = 0.008 [two-tailed t test]) are indicated by a red asterisk for wild-type strains and a purple asterisk for strains expressing recombinase. Sequence type (ST) and clonal complex (CC) data are indicated in brackets next to each strain name. (D) Effect of various oligonucleotide lengths, mismatch repair-evading silent substitutions, and phosphorothioate modifications on recombination efficiency. Recombination frequencies significantly higher than those observed in the mock transformation control (≤P = 0.002 [two-tailed t test]) are indicated by an asterisk. Error bars in all panels indicate standard errors of the means.
Only EF2132 achieved recombineering frequencies significantly (P = 3.2 × 10−5 [two-tailed t test]) greater than the rate of spontaneous rifampin resistance occurring in mock-transformation and recombinase-negative controls, ~2 × 10−5 per cell (Fig. 1A). Confirmatory sequencing of 10 rifampin-resistant recombinants showed that each carried all six mutations encoded by the mutagenic oligonucleotide (two substitutions encoding H481Y plus four silent changes to escape reversion by mismatch repair [MMR] [38]) (Fig. 1B), indicating that the phenotype resulted from incorporation of the synthetic oligonucleotide into the genome rather than from spontaneous, resistance-conferring point mutations. Surprisingly, rifampin-resistant colonies were obtained at approximately twice the spontaneous background mutation rate (P = 0.002 [two-tailed t test]) when recombineering was performed in the absence of exogenous recombinase expression, suggesting that strain ATCC 29213 is naturally recombinogenic at low frequencies.
To assess the generality of EF2132-mediated recombineering function, we repeated the assay in laboratory strain Newman (39), previously characterized clinical isolate N315 (40), and six randomly selected and otherwise uncharacterized primary clinical S. aureus isolates which were obtained from unrelated patient specimens (arbitrarily numbered 1 to 6; Fig. 1C).
Strains ATCC 29213, Newman, and N315 were each transformable with recombineering plasmid passaged through a transgenic E. coli strain that modifies plasmid DNA to confer the adenine methylation profile of S. aureus CC8 (16), consistent with the compatible restriction groups of these isolates’ clonal complexes (CC5 and CC8; see Table S1 in the supplemental material). Recombineering in S. aureus laboratory strain Newman produced statistically significant numbers of recombinants only in the presence of exogenous recombinase expression and achieved a recombineering rate approximately half that seen for ATCC 29213 (7.32 × 10−6 recombinants per cell; P = 1.20 × 10−4 [two-tailed t test]). In contrast, strain N315 achieved recombineering efficiencies (2.5 × 10−3 recombinants per cell; P = 8.06 × 10−6 [two-tailed t test]) that were nearly 2 orders of magnitude greater than those seen with ATCC 29213 and which similarly yielded statistically significant numbers of recombinants in the absence of exogenous recombinase expression (1.02 × 10−6 per cell; P = 1.66 × 10−8 [two-tailed t test]).
Summary of strains and plasmids used. Download TABLE S1, XLSX file, 0.02 MB (20.6KB, xlsx) .
Copyright © 2018 Penewit et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Five of the clinical strains (strains 1, 2, 3, 5, and 6) were transformable with recombineering vector after plasmid artificial modification to actively mimic CC8 adenine methylation (16); using whole-genome analysis, these strains were subsequently determined to belong to CC8 and restriction-compatible groups CC1, CC5, CC15, and CC97 (Table S1). The remaining isolate (strain 4) was transformable only with vector lacking cytosine methylation (15) and was later ascertained to be a member of CC121, explaining its incompatibility with CC8-modified plasmid. All six of the clinical strains carrying recombineering vector and transformed with mutagenic oligonucleotide generated rifampin-resistant colonies at statistically significantly higher frequencies than mock transformations lacking the recombineering oligonucleotide (P = 7.6 × 10−3 to 4.2 × 10−5 [two-tailed t test]), with efficiencies varying by 2 orders of magnitude (range, 3.66 × 10−6 to 1.4 × 10−4 recombinants per cell). Clinical strain 5 alone yielded statistically significant numbers of recombinants in the absence of exogenous recombinase expression (5.0 × 10−6 per cell; P = 1.6 × 10−4 [two-tailed t test]).
Collectively, these data both indicate that expression of EF2123 is able to generally catalyze recombineering across different S. aureus strain backgrounds, regardless of clonal complex or sequence type, albeit at various efficiencies, and reinforce the idea that some isolates are naturally recombinogenic without the expression of exogenous recombinase.
The N315 genome includes an endogenous bacteriophage, ϕN315 (41), and the draft assembly of ATCC 29213 similarly contains evidence of a lysogenic phage sequence (42). We reasoned that the ability of some strains to natively recombineer with ssDNA oligonucleotides could reflect contributions from bacteriophage-carried genes present in their genomes. We therefore evaluated the recombineering capacity of an N315 background from which ϕN315 had been deleted (43). Compared to the parental strain, no reduction in recombineering frequency was observed. This result suggests that currently undefined host-encoded factors, rather than endogenous bacteriophage proteins, influence recombineering efficiencies in S. aureus.
Optimization of recombineering oligonucleotides.
Various properties and modifications of oligonucleotides have been reported to influence recombineering efficiency in E. coli, including length (38), configuration of phosphorothioate bonds which inhibit exonuclease digestion (44), and inclusion of mismatches to escape reversion of changes engineered by the mismatch repair (MMR) system (38). To explore how each of these considerations influences recombineering in S. aureus, we evaluated oligonucleotides which differed in one or more of these features (Fig. 1D).
Optimal recombineering frequencies were obtained with oligonucleotides of 90 bp in length, significantly exceeding the length previously reported to be optimal for E. coli (38). Recombineering efficiency was reduced approximately 2-fold when using 100-bp oligonucleotides, 10-fold for templates of 70 bp, and was not significantly different from background rifampin resistance rates when using 50-bp oligonucleotides. Incorporating phosphorothioate bonds into the oligonucleotide 5′ end yielded the greatest number of recombinants in S. aureus, in comparison to inclusion of that modification on both ends, which is optimal for E. coli (44). Exclusion of additional, silent mutations intended to bypass MMR correction of engineered changes (38) resulted in recombineering efficiencies that were indistinguishable from background mutation levels, indicating that the MMR system represents a major barrier to genome engineering in S. aureus.
In summary, we found that 90-bp oligonucleotides carrying 5′ phosphorothioate bonds and modifications to escape the MMR system promote recombineering most efficiently in S. aureus.
Developing a two-plasmid system for conditional recombineering and counterselection.
To enable recovery of recombineered isolates without a selectable phenotype, we next developed a temperature-sensitive, two-vector system to carry out conditional recombineering and CRISPR/Cas9-mediated counterselection in S. aureus (Fig. 2). The pCN-EF2132tet recombineering vector expresses EF2132 recombinase on a chloramphenicol selectable backbone. The pCas9counter counterselection vector confers erythromycin resistance and expresses a self-sufficient small guide RNA (sgRNA) (45) and Cas9. Targeting of Cas9 to a locus of interest is achieved by cloning a 20-bp DNA oligonucleotide (protospacer element) into the sgRNA backbone which matches the specified locus and is immediately upstream of a Cas9 recognition site (protospacer adjacent motif [PAM] 5′-NGG-3′). Both constructs are built on shuttle vectors and therefore can be propagated and genetically manipulated in E. coli. Genome engineering is accomplished by first transforming pCN-EF2132tet into a given S. aureus strain to make a recombineering-competent lineage and then subsequently introducing the recombineering oligonucleotide concurrently with the counterselection vector. Only cells undergoing successful recombineering are immune to lethal, double-stranded DNA breaks induced by the counterselection plasmid such that erythromycin resistance serves as a selectable proxy for the recombineered change. The growth temperature of engineered cells can be subsequently increased, resulting in loss of the vector system.
FIG 2 .

Overview of recombineering and Cas9-mediated counterselection for S. aureus genome engineering. (A) Temperature-sensitive recombineering vector pCN-EF2132tet, encoding recombinase (green circles), is transformed into the strain to be edited. Key elements of the vector are diagrammed and include recombinase EF2132 followed by a transcriptional terminator; high-copy-number temperature-sensitive S. aureus replicon T181cop-634ts; a chloramphenicol resistance gene for selection in S. aureus; and the E. coli ColE1 origin of replication with an ampicillin resistance selectable marker for maintenance in E. coli. (B) Temperature-sensitive counterselection plasmid pCAS9counter is introduced at the time that recombineering is performed. Key elements of the vector are diagrammed and include a synthetic guide RNA (sgRNA) targeted to the genomic site being modified; Cas9 followed by a transcriptional terminator; low-copy-number temperature-sensitive S. aureus replicon E194ts (which is compatible with T181cop-634ts); an erythromycin resistance gene for selection in S. aureus; and the E. coli ColE1 origin of replication with an ampicillin resistance selectable marker for maintenance in E. coli. (C and D) Recombineering is performed using a mutagenic oligonucleotide (blue curved lines) encoding the desired change (yellow diamond). Transformation with these elements leads to two possible outcomes: integration of the mutagenic oligonucleotide is successful and Cas9 (yellow Pac-Man symbol) is unable to cleave the genome (C), or integration of the mutagenic oligonucleotide does not occur and Cas9 introduces a double-stranded break into the host genome, killing unedited cells (D). (E) Brief growth of bacteria at elevated temperatures which are nonpermissive for the plasmid system results in its loss, leaving isogenic, edited cells.
We tested the efficacy of our Cas9-mediated counterselection strategy in ATCC 29213, a type strain which exhibited representative recombineering efficiencies for S. aureus (Fig. 1C). Recombineering utilized an oligonucleotide encoding both rpoB H481Y and a noncoding mutation which eliminated a nearby PAM, with counterselection targeted to that ablated site. Given this design, successful recombineering with the oligonucleotide provides both rifampin resistance and immunity to Cas9-mediated counterselection, allowing the effects of recombineering and Cas9 activity to be separately investigated.
Consistent with our earlier experiments (Fig. 1C), limited numbers of rifampin-resistant colonies were obtained when S. aureus carrying the recombineering vector was transformed with counterselection plasmids targeted either to an irrelevant biological sequence (GFP) or the rpoB PAM site (Fig. 3A), indicating a low background frequency of spontaneous rifampin resistance. Transformation with the rpoB counterselection vector, however, yielded less than 1% of the erythromycin-resistant colonies obtained from the GFP-targeted control (P = 0.04 [two-tailed t test]), demonstrating that the vast majority of cells taking up active counterselection plasmid were killed by it. When recombineering was performed during cotransformation of the GFP-targeted counterselection vector, colony counts on rifampin-containing medium were also consistent with prior recombineering efficiencies, with slightly higher counts of erythromycin-resistant colonies suggesting higher efficiencies of plasmid transformation than recombineering. When recombineering was performed during cotransformation of the counterselection vector targeted to rpoB, the number of recombinants that were recovered on rifampin media were equivalent to those seen when the GFP-targeted construct was used. Although the numbers of erythromycin-resistant colonies were markedly reduced compared to the numbers seen with the GFP-targeted control (P = 3.0 × 10−5 [two-tailed t test]), those counts were significantly elevated compared to the viable colonies seen with transformation of the rpoB counterselection vector alone (P = 1.1 × 10−4 [two-tailed t test]), evidencing increased survival in the context of recombineering.
FIG 3 .

Recombineering with concurrent Cas9-mediated counterselection. (A) Frequency of colonies recovered using different combinations of counterselection vectors, rifampin resistance recombineering oligonucleotide, and selective media when recombineering oligonucleotides and counterselection vectors are introduced concurrently. (B) Fraction of colonies carrying counterselection vectors which display resistance to rifampin after recombineering with cotransformation of counterselection vectors. Error bars in both panels indicate standard errors of the means.
In contrast to the GFP-targeted control, replica plating of recombineered, rpoB-counterselected cells onto rifampin-containing media indicated that the population was significantly enriched (P = 3.2 × 10−3 [two-tailed t test]) for rifampin-resistant recombinants (Fig. 3B). However, only a fraction of erythromycin-resistant colonies were resistant to rifampin, and we sequenced counterselected isolates at the site of recombineering to further investigate. Of 12 colonies that were resistant to rifampin and erythromycin, 10 carried both the mutations conferring rifampin resistance and those ablating the PAM, consistent with the intended recombineering product, while the remaining two were wild type, suggesting separate escape mutations which inactivated the counterselection plasmid and conferred rifampin resistance. A total of 9 of 10 erythromycin-resistant colonies which were sensitive to rifampin were similarly wild type, while 1 harbored only the ablated PAM mutation. Collectively, these results indicate that >30% of all isolates surviving counterselection represented the intended recombinants and that recombinants can be dramatically enriched using counterselection alone.
To confirm temperature-sensitive loss of the plasmid system, we tested colonies containing both vectors for their retention of vector-conferred antibiotic resistance phenotypes after growth on nonselective media overnight under temperatures that were permissive (32°C) or nonpermissive (43°C) for plasmid replication. All 10 colonies grown at the permissive temperature retained resistance to both chloramphenicol and erythromycin, whereas all 10 of the colonies grown at the nonpermissive temperature were unable to grow in the presence of either antibiotic, indicating effective loss of both plasmids after brief passaging at elevated temperatures.
Recombineering of a mutS gene deletion allows high-efficiency recombineering of single nucleotide substitutions.
To expand the utility of our genome editing system in S. aureus, and to demonstrate recombineering of a large deletion lacking a selectable phenotype, we sought to generate an MMR-deficient strain which would be capable of recombineering single base changes with high efficiency (46). We therefore engineered into ATCC 29213 a 2,484-bp (828-amino-acid), in-frame mutS deletion (residues 16 to 843) using a mutagenic oligonucleotide matching 45 bp of flanking sequence on either side of the change, with concurrent counterselection performed against a PAM within the deleted region.
Viable colonies carrying the counterselection vector were obtained at a rate of 13 ± 9.1 recombinants (average ± standard deviation) per 108 cells, roughly one-quarter the frequency obtained when recombineering base substitution mutations. Successful editing events were evaluated by fragment length analysis PCR (Fig. 4A). Of the 10 colonies screened, three (~30%) carried a size shift consistent with the deletion. Half (50%) were wild type and thus likely resulted from spontaneous mutation of the Cas9 target site or the counterselection plasmid itself. The remaining two colonies failed to amplify with primers exterior to the mutS gene, suggesting a deletion of the locus beyond the intended boundaries.
FIG 4 .
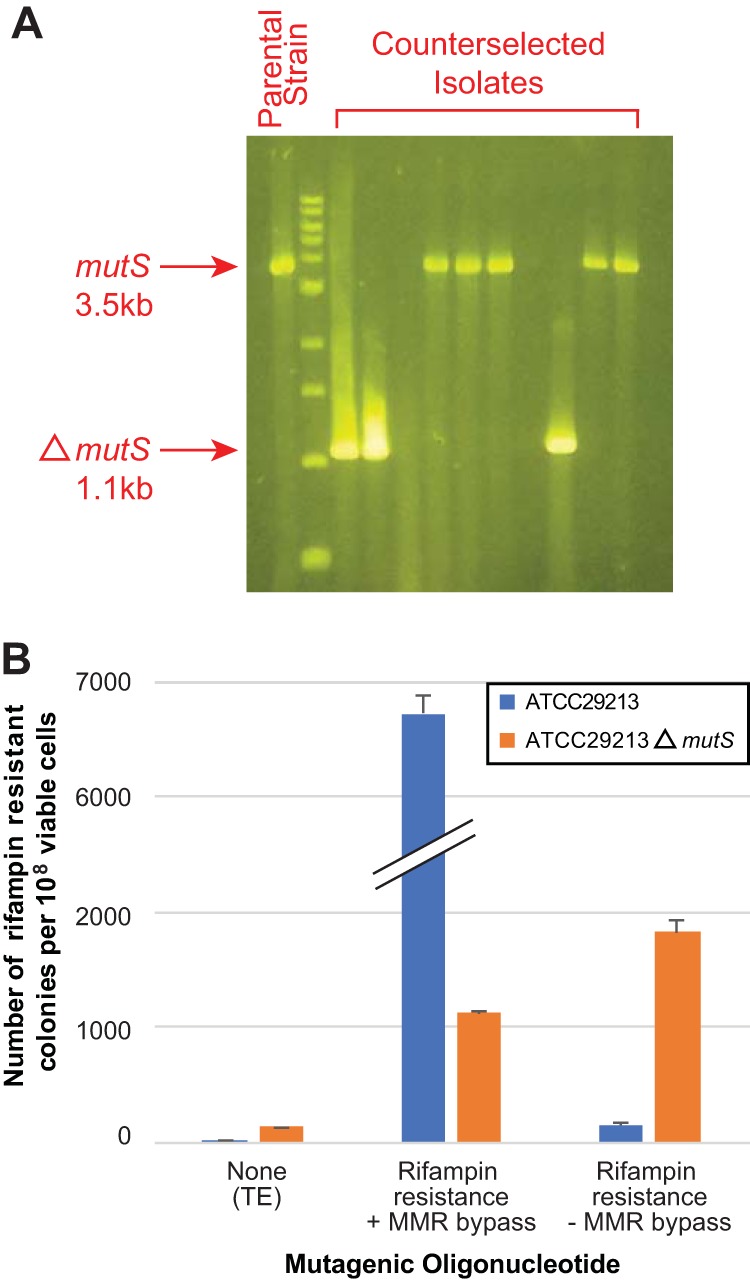
Recombineering of a 2.4-kb mutS deletion strain and effects of mutS deletion on subsequent recombineering. (A) PCR amplification of the mutS locus from representative colonies surviving counterselection. The wild-type allele corresponds to an amplicon 3.5 kb in size, whereas the engineered deletion results in a product at 1.1 kb. A 1-kb ladder is shown. (B) Recombineering efficiencies of the mutS deletion strain and mutS-intact parental strain compared to paired, mock-transformation controls lacking mutagenic oligonucleotide. Recombineering oligonucleotides encode rifampin resistance and either incorporate or lack silent mutations promoting bypass of MMR pathway repair. Error bars indicate standard errors of the means.
As a final step, we evaluated the ability of the mutS-deficient strain to recombineer mutagenic oligonucleotides either containing or lacking silent mutations designed to evade MMR repair when carrying the recombineering vector (Fig. 4B). Surprisingly, the mutS deletion mutant was able to recombineer an oligonucleotide encoding rpoB H481Y and MMR-bypassing synonymous mutations with only one-sixth the efficiency of the parental strain (P = 8.7 × 10−6 [two-tailed t test]). However, while the parental strain could incorporate oligonucleotides lacking MMR bypass mutations at rates of ~150 per 108 cells, only modestly (P = 0.014 [two-tailed t test]) higher than the background mutation frequency, the mutS deletion strain produced recombinants with significantly higher rates (P = 1.4 × 10−5 [two-tailed t test]) approaching 2,000 per 108 viable cells. These rates were also markedly greater (P = 4.6 × 10−6 [two-tailed t test]) than those seen for the mutS deletion strain when recombineering was performed using oligonucleotides containing MMR-bypassing synonymous mutations, likely reflecting the decreased number of mismatches between the mutagenic oligonucleotide and the genome. As expected from the role of mutS in repairing errors arising during DNA replication, we observed more than a 7-fold increase in the rate of spontaneous rifampin-resistant isolates for the mutS deletion strain compared to the wild type (P = 1.5 × 10−4 [two-tailed t test]) when mock transformation was performed, consistent with a mutator phenotype (47). We conclude that the mutS-deficient background allows robust recombineering of single base mismatches, at the expense of an increased background mutation rate.
Efficiency of gene deletion by recombineering.
To explore the editing efficiencies of additional genes across the S. aureus genome, we recombineered in-frame deletions for seven additional factors (aroE, baeS, cyoD, nupC, purR, ypfH, and yhcF) in ATCC 29213, performing counterselection and screening as before. There was substantial variability in the performance of the individual editing events (see Fig. S1 in the supplemental material), in terms of both the fraction of counterselected colonies carrying the desired edit (range, 0.1 to 0.875) and the rate of successful editing events per viable cell (3.2 × 10−9 to 2.3 × 10−7). Consistent with recombineering studies in E. coli (48), we observed a negative correlation between the size of the engineered deletion and the efficiency with which it could be generated. However, even deletions of similar sizes could display marked differences in editing efficiency, suggesting that other factors, such as local genomic architecture and recombineering oligonucleotide properties, have agency (48).
Efficiency of constructing in-frame deletion mutants. Success rates for constructing the desired in-frame deletions are indicated as a function of deletion size. Results are expressed as the fraction of counterselected colonies carrying the desired edit (left y axis, blue data series) and the rate of successful editing events per viable cell (right y axis, orange data series). Gene names are indicated above relevant data points. Download FIG S1, PDF file, 0.99 MB (345.3KB, pdf) .
Copyright © 2018 Penewit et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Here we describe a simple, precise, efficient, scalable, rapid, and inexpensive method for engineering directed genomic changes in S. aureus, related to systems which have been developed for other organisms (27–32). We have demonstrated its utility in generating both point mutations (Fig. 1) and large gene deletions (Fig. 4). Our method (Fig. 2) entails successively transforming S. aureus with a recombineering vector to make a conditionally recombinogenic strain and then performing recombineering in the presence of a locus-specific CRISPR/Cas9-mediated counterselection vector, which results in the elimination of unedited cells. By virtue of being encoded on temperature-sensitive elements, this system allows genome editing to be performed without the permanent carriage of selectable markers or genome editing machinery, and it can consequently be used to produce truly isogenic S. aureus strains. In contrast to existing genome modification systems in S. aureus (17, 18, 20–22), a major advantage of our approach is that it both generates and selects for desired genetic changes using inexpensive, commercially synthesized synthetic single-stranded DNA oligonucleotides, which can be manufactured in bulk. As such, this technology is amenable to scaling and therefore provides opportunities for high-throughput genome engineering in S. aureus. Moreover, recombineering has also proven highly efficient, typically yielding hundreds to thousands of recombinants per transformation (Fig. 1C) or 10 to 20 recombinants during cotransformation of counterselection constructs (Fig. 3). Recombineering can be generally performed in S. aureus lineages which can be made electrocompetent, and have we have shown functionality in both laboratory and clinical strains (Fig. 1C). We have found that recombineering efficiencies can vary markedly from strain to strain, based on contributions of host factors which have not yet been identified. Counterselection results in populations which are highly enriched for successful recombinants (Fig. 3B), which accounted for ~30% of the surviving population when point mutations or a large gene deletion were engineered.
The workflow of our approach is swift, even when it is performed in a S. aureus strain for the first time. Counterselection vectors are readily constructed in advance from synthetic oligonucleotides and are cloned directly into transgenic E. coli strains that modify plasmids to escape S. aureus restriction systems (15) (day 1). Successful clones are rapidly identified by fragment length analysis PCR (day 2), and plasmid is harvested (day 3). Preparing electrocompetent cells for a given S. aureus strain and transformation with the recombineering vector are performed only once (day 1), after which transgenic electrocompetent cells are prepared (day 2). This single batch of electrocompetent cells can enable transformation of many different mutagenic oligonucleotides and counterselection vectors (day 2), with screening of recombinants possible the following day (day 3).
Because expression of CRISPR/Cas9 targeted to the genome is lethal in wild-type strains, effective regulation of counterselection constructs is critical (34). We initially attempted to achieve regulation of counterselection using an inducible system (34) but were unable to achieve complete repression of Cas9 activity even when using a mutagenized promoter that minimized leakiness (49). Regardless, the high efficiencies achievable by our system allowed simultaneous introduction of the recombineering oligonucleotide and the counterselection construct to achieve concurrent genome editing and counterselection. This configuration also eliminates the need to prepare electrocompetent lines after introduction of each counterselection vector, facilitating higher throughput.
Our work has revealed several interesting features of S. aureus biology as relevant to recombineering. First, we were unable to identify a recombinase originating from S. aureus that functioned with detectable efficiency when expressed in that background. The activity of phage- or host-encoded recombinase genes is considered to be highly dependent on the overall biology of an organism, including the presence of other contributory host factors and exonuclease systems (25). As such, it is generally thought that recombinases function with the highest efficiency in their background of origin (50). Our failure to identify a S. aureus recombinase which conforms to these expectations suggests either that recombinases native to S. aureus lack activity for ssDNA or that S. aureus recombinases which are optimal for recombineering are markedly dissimilar in sequence and structure to those which have been previously defined (51). We also found that certain S. aureus strains are naturally recombinogenic with mutagenic oligonucleotides introduced through electroporation. Although spontaneous, low-frequency ssDNA recombineering has been previously observed in E. coli (52), the substantial differences in spontaneous recombineering efficiency among S. aureus strains (Fig. 1C) suggest contributions of differing host genetic factors or physiologies in mediating this process.
Recombineering with CRISPR/Cas9 counterselection provides a novel tool for testing the phenotypic consequences of large- and small-scale genetic variation in the medically important organism S. aureus. Although robust in its current incarnation, future improvements to this technology may further improve or expand functionality. Definition and incorporation of factors which potentiate recombineering function in particular strain backgrounds could lead to further improved efficiencies of recombineering. Use of chemically modified oligonucleotides could enable bypass of the MMR system (53) without the need for accessory mutations or a mutS-deficient background (Fig. 4), the latter of which results in a mutator phenotype (47). Relatedly, expression of dominant-negative MMR proteins from the genome editing vectors could enable enjoyment of these benefits without necessitating disruption of chromosomal DNA repair genes (54). Development of a counterselection vector containing high-specificity mutants of Cas9 (55) could improve the specificity of counterselection for the related motifs which appear multiply in the S. aureus genome, whereas use of CRISPR effector protein Cpf1, which recognizes a 5′-TTTV-3′ PAM (56, 57), could expand counterselection to A/T-rich regions. Lastly, integration of functional elements from the recombineering and counterselection plasmids into a single element could further streamline the workflow, although the reduced transformation efficiency of that significantly larger plasmid could prove limiting (58).
In summary, we have developed a rapid and precise technology for genome editing in S. aureus which incorporates the use of recombineering, reported here for the first time in that organism. Leveraging recent advancements which enable exogenous DNA to be efficiently introduced into S. aureus strains (15, 16), this approach is broadly applicable across lineages, including poorly defined clinical isolates. In contrast to existing approaches, our technique is unique in utilizing commercially synthesized synthetic DNA oligonucleotides as substrates for introducing precise genomic modifications and performing counterselection, making it possible to scalably and inexpensively engineer desired changes. This method will facilitate studies seeking to address a variety of issues about the function of particular genes and specific mutations in S. aureus.
MATERIALS AND METHODS
Strains, plasmids, and whole-genome analysis.
Strains used in this study are summarized in Table S1 in the supplemental material. S. aureus strain ATCC 29213 (37) was obtained from the American Type Culture Collection and S. aureus N315 (40) from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources). S. aureus strain N315ex w/o ϕ, from which the ϕ phage has been deleted (43), was a generous gift of Kazuya Morikawa (University of Tsukuba). Clinical S. aureus strains, each originating from a different patient, were obtained from the University of Washington Clinical Microbiology Laboratory as deidentified specimens. Escherichia coli SA08B (16) was obtained from Lucigen. E. coli DC10B (15) and S. aureus strain Newman (39) were generous gifts from Daniel Wolter (University of Washington).
Vectors pCN50 and pCN33 (36) were obtained from BEI Resources, and pCAS9 (27) and pCL52.2 (59) were purchased from Addgene.
All strains were maintained using LB media unless noted otherwise. E. coli carrying ampicillin resistance-encoding shuttle vectors was cultured at 37°C in the presence of 100 μg/ml antibiotic, S. aureus carrying chloramphenicol resistance vectors at 32°C in the presence of 10 μg/ml antibiotic, and S. aureus carrying erythromycin resistance vectors at 32°C in the presence of 10 μg/ml antibiotic. S. aureus strains carrying multiple vectors were maintained using both antibiotics. Rifampin resistance was selected using 25 μg/ml antibiotic.
Clinical strains were subjected to whole-genome sequencing and assembly as described elsewhere (60) and were assigned to sequence types using MLST v1.8 (61) and subsequently to clonal complexes using eBURST v3 (62).
Vector construction.
Vectors generated in this study are summarized in Table S1. All oligonucleotides and synthetic gene sequences (Gblocks) were synthesized by IDT (Table S2). All restriction enzymes were obtained from New England Biolabs.
Summary of oligonucleotides used. Download TABLE S2, XLSX file, 0.03 MB (32.1KB, xlsx) .
Copyright © 2018 Penewit et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The BsaI restriction site of pCN50 was first eliminated using site-directed mutagenesis with primers pCN_AMP_mut_F and pCN_AMP_mut_R to produce vector pCN50-BSAI.
Recombinases were synthesized as gBlocks under the control of the constitutive P23 promoter (63), and were codon optimized for S. aureus. gBlocks were digested with XmaI and NarI and cloned into similarly digested pCN50-BSAI to yield vectors pCN50-EF2132, pCN50-gp20, pCN50-recTS2, pCN50-recTS3, and pCN50-bet. Orientation of the recombinase as concordant with the pCN50 transcriptional terminator was confirmed by restriction digestion.
To generate vector pCN-EF2132tet, a S. aureus codon-optimized tetR cassette (64) under the control of the P23 promoter was synthesized as a gBlock and then amplified with primers tetR-Gibson_pcn50F and tetR-Gibson_pcn50R and Gibson assembled (65) into XhoI-digested pCN50-EF2132. Due to our inability to fully repress Cas9 expression under the influence of the tetR element, however, this feature was not relevant to the final implementation of our methods.
To construct vector pCAS9counter, the chloramphenicol resistance cassette of pCN50-BSAI was replaced with an erythromycin resistance cassette by amplifying ermC from vector pCN33 using primers 675_ermC_F and 675_ermC_R and cloning into ApaI and XhoI pCN50-BSAI restriction sites to generate plasmid pCN50-BSAI-emrc. The existing S. aureus replicon of pCN50-BSAI-ermc was replaced with the temperature-sensitive E194ts replicon of pCL52.2 by amplifying pCL52.2 with pE194ts_replicon_gibson_F and pE194ts_replicon_gibson_R, amplifying pCN50-BSAI-ermc with pcn50-ermc_repliconoverlap_F and pcn50-ermc_ repliconoverlap_R, and Gibson assembling the products to yield vector pCN50-ermc-E194ts. A minimal SsrA tag (34, 66) was introduced at the terminus of Cas9 in pCAS9 using site-directed mutagenesis with primers Cas9_ssRA_SDM_F and Cas9_ssRA_SDM_R, and then Cas9 was amplified from vector pCAS9 and placed under the control of the PRAB17 (49) promoter using primers cas9toprab17 and CAS9_r_V3, followed by cloning into pCN50-ermc-E194ts digested with XmaI and SbfI to yield pCN50-ermc-Cas9. Synthetic sgRNA (45) under the control of the PRAB17 promoter with a downstream transcriptional terminator was synthesized as a gBlock and cloned into the NarI site of pCN50-ermc-Cas9 to produce vector pCAS9counter.
pCAS9counter guide RNAs were designed using the program CRISPRscan (67). Guide RNAs were synthesized as ssDNA oligonucleotides (tailed 5′-AGCTC-3′ upstream and 5′-G-3′ downstream) and their reverse complements (tailed 5′-AAAAC-3′ upstream and 5′-G-3′ downstream), with tails allowing their ligation into the vector’s BsaI cut site. Oligonucleotides were phosphorylated and annealed in a reaction mixture containing using T4 PNK (New England Biolabs) in T4 ligase buffer heated to 37°C for 40 min and then to 95°C for 5 min and then gradually cooled to 20°C over 42 min. Annealed oligonucleotides were ligated into BsaI-digested pCAS9counter. The presence of targeting oligonucleotides was confirmed by fragment size shift (loss of ~100 bp) after amplifying with primers sgRNA_check_F and sgRNA_check_R.
S. aureus transformation, recombineering, and counterselection.
Plasmids were made restriction compatible with S. aureus prior to electroporation by passaging in E. coli SA08B or E. coli DC10B. Electrocompetent cells were prepared essentially as described elsewhere (15), except that strains were cultured in either LB or B2 medium as necessary to support their growth.
For transformation of strains with recombineering vectors, electrocompetent cells were combined with 1 μg plasmid precipitated with pellet paint (Novagen) as described previously (15). Electroporation was performed using 1-mm-gap cuvettes and a Bio-Rad MicroPulser set to 2.3 kV and a 2.5-ms time constant. Following electroporation, strains were incubated in 950 μl recovery medium (15) at 32°C for 2 h before plating onto appropriate media was performed.
For recombineering experiments, mutagenic oligonucleotide was introduced into electrocompetent cells previously transformed with the recombineering vector. Mutagenic oligonucleotide (200 pmol in a 2-µl total volume) was combined with competent cells, and the cells were electroporated and grown as described above.
When recombineering was performed concurrently with counterselection, 200 pmol mutagenic oligonucleotide was used to resuspend 1 μg precipitated plasmid (15) in a 2-μl total volume, which was electroporated as described above into electrocompetent cells previously transformed with the recombineering vector.
Data availability.
Genome assemblies from the six clinical isolates examined in this study have been deposited in NCBI GenBank under accession numbers PNPA00000000 through PNPF00000000.
ACKNOWLEDGMENTS
We thank E. A. Salipante and J. A. Salipante for helpful discussions.
This work was supported by a grant from the Cystic Fibrosis Foundation (to S.J.S.).
Footnotes
Citation Penewit K, Holmes EA, McLean K, Ren M, Waalkes A, Salipante SJ. 2018. Efficient and scalable precision genome editing in Staphylococcus aureus through conditional recombineering and CRISPR/Cas9-mediated counterselection. mBio 9:e00067-18. https://doi.org/10.1128/mBio.00067-18.
REFERENCES
- 1.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK; Active Bacterial Core surveillance (ABCs) MRSA Investigators . 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 2.Wertheim HFL, Vos MC, Ott A, van Belkum A, Voss A, Kluytmans JAJW, van Keulen PHJ, Vandenbroucke-Grauls CMJE, Meester MHM, Verbrugh HA. 2004. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet 364:703–705. doi: 10.1016/S0140-6736(04)16897-9. [DOI] [PubMed] [Google Scholar]
- 3.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, J. Rybak M, Talan DA, Chambers HF. 2011. Clinical practice guidelines by the infectious diseases society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis 52:285–292. doi: 10.1093/cid/cir034. [DOI] [PubMed] [Google Scholar]
- 4.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 5.Conlon BP. 2014. Staphylococcus aureus chronic and relapsing infections: evidence of a role for persister cells: an investigation of persister cells, their formation and their role in S. aureus disease. Bioessays 36:991–996. doi: 10.1002/bies.201400080. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed MI, Mukherjee S. 2016. Treatment for chronic methicillin-sensitive Staphylococcus aureus pulmonary infection in people with cystic fibrosis. Cochrane Database Syst Rev 3:CD011581. doi: 10.1002/14651858.CD011581.pub2. [DOI] [PubMed] [Google Scholar]
- 7.McAdam PR, Holmes A, Templeton KE, Fitzgerald JR. 2011. Adaptive evolution of Staphylococcus aureus during chronic endobronchial infection of a cystic fibrosis patient. PLoS One 6:e24301. doi: 10.1371/journal.pone.0024301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah PL, Mawdsley S, Nash K, Cullinan P, Cole PJ, Wilson R. 1999. Determinants of chronic infection with Staphylococcus aureus in patients with bronchiectasis. Eur Respir J 14:1340–1344. doi: 10.1183/09031936.99.14613409. [DOI] [PubMed] [Google Scholar]
- 9.Cullen L, McClean S. 2015. Bacterial adaptation during chronic respiratory infections. Pathogens 4:66–89. doi: 10.3390/pathogens4010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zecconi A, Scali F. 2013. Staphylococcus aureus virulence factors in evasion from innate immune defenses in human and animal diseases. Immunol Lett 150:12–22. doi: 10.1016/j.imlet.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 11.Powers ME, Bubeck Wardenburg J. 2014. Igniting the fire: Staphylococcus aureus virulence factors in the pathogenesis of sepsis. PLoS Pathog 10:e1003871. doi: 10.1371/journal.ppat.1003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oogai Y, Matsuo M, Hashimoto M, Kato F, Sugai M, Komatsuzawa H. 2011. Expression of virulence factors by Staphylococcus aureus grown in serum. Appl Environ Microbiol 77:8097–8105. doi: 10.1128/AEM.05316-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon CP, Williams P, Chan WC. 2013. Attenuating Staphylococcus aureus virulence gene regulation: a medicinal chemistry perspective. J Med Chem 56:1389–1404. doi: 10.1021/jm3014635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saïd-Salim B, Dunman PM, McAleese FM, Macapagal D, Murphy E, McNamara PJ, Arvidson S, Foster TJ, Projan SJ, Kreiswirth BN. 2003. Global regulation of Staphylococcus aureus genes by Rot. J Bacteriol 185:610–619. doi: 10.1128/JB.185.2.610-619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monk IR, Shah IM, Xu M, Tan MW, Foster TJ. 2012. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3:e00277-11. doi: 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monk IR, Tree JJ, Howden BP, Stinear TP, Foster TJ. 2015. Complete bypass of restriction systems for major Staphylococcus aureus lineages. mBio 6:e00308-15. doi: 10.1128/mBio.00308-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindberg M, Sjöström JE, Johansson T. 1972. Transformation of chromosomal and plasmid characters in Staphylococcus aureus. J Bacteriol 109:844–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu D, Zhao L, Xue T, Sun B. 2012. Staphylococcus aureus autoinducer-2 quorum sensing decreases biofilm formation in an icaR-dependent manner. BMC Microbiol 12:288. doi: 10.1186/1471-2180-12-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prax M, Lee CY, Bertram R. 2013. An update on the molecular genetics toolbox for staphylococci. Microbiology 159:421–435. doi: 10.1099/mic.0.061705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brückner R. 1997. Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS Microbiol Lett 151:1–8. [DOI] [PubMed] [Google Scholar]
- 21.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Chen W, Zhang Y, Yeo WS, Bae T, Ji Q. 2017. Rapid and efficient genome editing in Staphylococcus aureus by using an engineered CRISPR/Cas9 system. J Am Chem Soc 139:3790–3795. doi: 10.1021/jacs.6b13317. [DOI] [PubMed] [Google Scholar]
- 23.Liu Q, Jiang Y, Shao L, Yang P, Sun B, Yang S, Chen D. 2017. CRISPR/Cas9-based efficient genome editing in Staphylococcus aureus. Acta Biochim Biophys Sin 49:764–770. doi: 10.1093/abbs/gmx074. [DOI] [PubMed] [Google Scholar]
- 24.Schneewind O, Missiakas D. 2014. Genetic manipulation of Staphylococcus aureus. Curr Protoc Microbiol 32:Unit 9C.3. doi: 10.1002/9780471729259.mc09c03s32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Datta S, Costantino N, Zhou X, Court DL. 2008. Identification and analysis of recombineering functions from Gram-negative and Gram-positive bacteria and their phages. Proc Natl Acad Sci U S A 105:1626–1631. doi: 10.1073/pnas.0709089105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selle K, Barrangou R. 2015. Harnessing CRISPR-Cas systems for bacterial genome editing. Trends Microbiol 23:225–232. doi: 10.1016/j.tim.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang H, Zheng G, Jiang W, Hu H, Lu Y. 2015. One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta Biochim Biophys Sin 47:231–243. doi: 10.1093/abbs/gmv007. [DOI] [PubMed] [Google Scholar]
- 29.Altenbuchner J. 2016. Editing of the Bacillus subtilis genome by the CRISPR-Cas9 system. Appl Environ Microbiol 82:5421–5427. doi: 10.1128/AEM.01453-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mougiakos I, Bosma EF, de Vos WM, van Kranenburg R, van der Oost J. 2016. Next generation prokaryotic engineering: the CRISPR-Cas toolkit. Trends Biotechnol 34:575–587. doi: 10.1016/j.tibtech.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 31.Barrangou R, van Pijkeren JP. 2016. Exploiting CRISPR-Cas immune systems for genome editing in bacteria. Curr Opin Biotechnol 37:61–68. doi: 10.1016/j.copbio.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 32.Xu T, Li Y, Shi Z, Hemme CL, Li Y, Zhu Y, Van Nostrand JD, He Z, Zhou J. 2015. Efficient genome editing in Clostridium cellulolyticum via CRISPR-Cas9 nickase. Appl Environ Microbiol 81:4423–4431. doi: 10.1128/AEM.00873-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellis HM, Yu D, DiTizio T, Court DL. 2001. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc Natl Acad Sci U S A 98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reisch CR, Prather KLJ. 2015. The no-SCAR (Scarless Cas9 Assisted Recombineering) system for genome editing in Escherichia coli. Sci Rep 5:15096. doi: 10.1038/srep15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe Y, Cui L, Katayama Y, Kozue K, Hiramatsu K. 2011. Impact of rpoB mutations on reduced vancomycin susceptibility in Staphylococcus aureus. J Clin Microbiol 49:2680–2684. doi: 10.1128/JCM.02144-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Charpentier E, Anton AI, Barry P, Alfonso B, Fang Y, Novick RP. 2004. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl Environ Microbiol 70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cowan ST, Shaw C, Williams RE. 1954. Type strain for Staphylococcus aureus Rosenbach. J Gen Microbiol 10:174–176. doi: 10.1099/00221287-10-1-174. [DOI] [PubMed] [Google Scholar]
- 38.Sawitzke JA, Costantino N, Li XT, Thomason LC, Bubunenko M, Court C, Court DL. 2011. Probing cellular processes with oligo-mediated recombination and using the knowledge gained to optimize recombineering. J Mol Biol 407:45–59. doi: 10.1016/j.jmb.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duthie ES, Lorenz LL. 1952. Staphylococcal coagulase; mode of action and antigenicity. J Gen Microbiol 6:95–107. doi: 10.1099/00221287-6-1-2-95. [DOI] [PubMed] [Google Scholar]
- 40.Kuwahara-Arai K, Kondo N, Hori S, Tateda-Suzuki E, Hiramatsu K. 1996. Suppression of methicillin resistance in a mecA-containing pre-methicillin-resistant Staphylococcus aureus strain is caused by the mecI-mediated repression of PBP 2′ production. Antimicrob Agents Chemother 40:2680–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. 2008. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J Bacteriol 190:300–310. doi: 10.1128/JB.01000-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soni I, Chakrapani H, Chopra S. 2015. Draft genome sequence of methicillin-sensitive Staphylococcus aureus ATCC 29213. Genome Announc 3:e01095-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morikawa K, Takemura AJ, Inose Y, Tsai M, Nguyen Thi le T, Ohta T, Msadek T. 2012. Expression of a cryptic secondary sigma factor gene unveils natural competence for DNA transformation in Staphylococcus aureus. PLoS Pathog 8:e1003003. doi: 10.1371/journal.ppat.1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mosberg JA, Gregg CJ, Lajoie MJ, Wang HH, Church GM. 2012. Improving lambda red genome engineering in Escherichia coli via rational removal of endogenous nucleases. PLoS One 7:e44638. doi: 10.1371/journal.pone.0044638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Costantino N, Court DL. 2003. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc Natl Acad Sci U S A 100:15748–15753. doi: 10.1073/pnas.2434959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Denamur E, Matic I. 2006. Evolution of mutation rates in bacteria. Mol Microbiol 60:820–827. doi: 10.1111/j.1365-2958.2006.05150.x. [DOI] [PubMed] [Google Scholar]
- 48.Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. 2009. Programming cells by multiplex genome engineering and accelerated evolution. Nature 460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Helle L, Kull M, Mayer S, Marincola G, Zelder M-E, Goerke C, Wolz C, Bertram R. 2011. Vectors for improved Tet repressor-dependent gradual gene induction or silencing in Staphylococcus aureus. Microbiology 157:3314–3323. doi: 10.1099/mic.0.052548-0. [DOI] [PubMed] [Google Scholar]
- 50.Sun Z, Deng A, Hu T, Wu J, Sun Q, Bai H, Zhang G, Wen T. 2015. A high-efficiency recombineering system with PCR-based ssDNA in Bacillus subtilis mediated by the native phage recombinase GP35. Appl Microbiol Biotechnol 99:5151–5162. doi: 10.1007/s00253-015-6485-5. [DOI] [PubMed] [Google Scholar]
- 51.Van Pijkeren JP, Neoh KM, Sirias D, Findley AS, Britton RA. 2012. Exploring optimization parameters to increase ssDNA recombineering in Lactococcus lactis and Lactobacillus reuteri. Bioengineered 3:209–217. doi: 10.4161/bioe.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomason LC, Costantino N, Court DL. 2016. Examining a DNA replication requirement for bacteriophage λ Red- and Rac prophage RecET-promoted recombination in Escherichia coli. mBio 7. doi: 10.1128/mBio.01443-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang HH, Xu G, Vonner AJ, Church G. 2011. Modified bases enable high-efficiency oligonucleotide-mediated allelic replacement via mismatch repair evasion. Nucleic Acids Res 39:7336–7347. doi: 10.1093/nar/gkr183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nyerges Á, Csörgő B, Nagy I, Bálint B, Bihari P, Lázár V, Apjok G, Umenhoffer K, Bogos B, Pósfai G, Pál C. 2016. A highly precise and portable genome engineering method allows comparison of mutational effects across bacterial species. Proc Natl Acad Sci U S A 113:2502–2507. doi: 10.1073/pnas.1520040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, Doudna JA. 2017. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550:407–410. doi: 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F. 2015. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim HK, Song M, Lee J, Menon AV, Jung S, Kang YM, Choi JW, Woo E, Koh HC, Nam JW, Kim H. 2017. In vivo high-throughput profiling of CRISPR-Cpf1 activity. Nat Methods 14:153–159. doi: 10.1038/nmeth.4104. [DOI] [PubMed] [Google Scholar]
- 58.Lee JC. 1995. Electrotransformation of staphylococci. Methods Mol Biol 47:209–216. doi: 10.1385/0-89603-310-4:209. [DOI] [PubMed] [Google Scholar]
- 59.Sau S, Sun J, Lee CY. 1997. Molecular characterization and transcriptional analysis of type 8 capsule genes in Staphylococcus aureus. J Bacteriol 179:1614–1621. doi: 10.1128/jb.179.5.1614-1621.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salipante SJ Sr, SenGupta DJ, Cummings LA, Land TA, Hoogestraat DR, Cookson BT. 2015. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J Clin Microbiol 53:1072–1079. doi: 10.1128/JCM.03385-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, Jelsbak L, Sicheritz-Pontén T, Ussery DW, Aarestrup FM, Lund O. 2012. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol 50:1355–1361. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. 2004. EBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol 186:1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van der Vossen JM, van der Lelie D, Venema G. 1987. Isolation and characterization of Streptococcus cremoris Wg2-specific promoters. Appl Environ Microbiol 53:2452–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corrigan RM, Foster TJ. 2009. An improved tetracycline-inducible expression vector for Staphylococcus aureus. Plasmid 61:126–129. doi: 10.1016/j.plasmid.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 65.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 66.Donegan NP, Marvin JS, Cheung AL. 2014. Role of adaptor TrfA and ClpPC in controlling levels of SsrA-tagged proteins and antitoxins in Staphylococcus aureus. J Bacteriol 196:4140–4151. doi: 10.1128/JB.02222-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moreno-Mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, Giraldez AJ. 2015. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods 12:982–988. doi: 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of strains and plasmids used. Download TABLE S1, XLSX file, 0.02 MB (20.6KB, xlsx) .
Copyright © 2018 Penewit et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Efficiency of constructing in-frame deletion mutants. Success rates for constructing the desired in-frame deletions are indicated as a function of deletion size. Results are expressed as the fraction of counterselected colonies carrying the desired edit (left y axis, blue data series) and the rate of successful editing events per viable cell (right y axis, orange data series). Gene names are indicated above relevant data points. Download FIG S1, PDF file, 0.99 MB (345.3KB, pdf) .
Copyright © 2018 Penewit et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Summary of oligonucleotides used. Download TABLE S2, XLSX file, 0.03 MB (32.1KB, xlsx) .
Copyright © 2018 Penewit et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
Genome assemblies from the six clinical isolates examined in this study have been deposited in NCBI GenBank under accession numbers PNPA00000000 through PNPF00000000.