

Abstract
Hypoglycemia is commonly associated with insulin therapy, limiting both its safety and efficacy. The concept of modifying insulin to render its glucose-responsive release from an injection depot (of an insulin complexed exogenously with a recombinant lectin) was proposed approximately 4 decades ago but has been challenging to achieve. Data presented here demonstrate that mannosylated insulin analogs can undergo an additional route of clearance as result of their interaction with endogenous mannose receptor (MR), and this can occur in a glucose-dependent fashion, with increased binding to MR at low glucose. Yet, these analogs retain capacity for binding to the insulin receptor (IR). When the blood glucose level is elevated, as in individuals with diabetes mellitus, MR binding diminishes due to glucose competition, leading to reduced MR-mediated clearance and increased partitioning for IR binding and consequent glucose lowering. These studies demonstrate that a glucose-dependent locus of insulin clearance and, hence, insulin action can be achieved by targeting MR and IR concurrently.
Keywords: Endocrinology, Metabolism
Keywords: Diabetes, Glucose metabolism, Insulin
Mannosylated insulin controls blood glucose with reduced hypoglycemic risk by engaging mannose receptor in a glucose-responsive manner.
Introduction
Insulin is a required treatment for type 1 diabetes mellitus (T1DM) and often is needed to control hyperglycemia in type 2 diabetes mellitus (T2DM). Progressive development of rapid-acting and long-acting insulin analogs has continued to refine pharmacokinetic (PK) and pharmacodynamic (PD) profiles to more closely match the profiles of endogenous insulin secretion during prandial and postabsorptive conditions, respectively (1). However, these insulin analogs retain the narrow therapeutic index of native insulin, manifesting a steep dose response for reducing plasma glucose and a narrow margin between an efficacious dose and one that can induce a dangerous episode of hypoglycemia (2). Confronted daily with the dilemma that only a small increase in insulin dose or that a change of meal content or physical activity holds the risk to provoke hypoglycemia, patients must be cautious and vigilant in selecting insulin dose, making it challenging to optimize glycemic control and reduce risk for late-stage comorbidities (notably cardiovascular disease, retinopathy, nephropathy, and neuropathy) (3). To unleash the full therapeutic potential of insulin, there is an aspiration to create an analog molecule with an improved therapeutic index, an insulin that enables patients to achieve tight glycemic control yet with a reduced risk for hypoglycemia. Ideally, an innovative insulin analog would modulate its action in coordination with changes in blood glucose, being fully functional under hyperglycemic conditions, but have scalable attenuation of insulin action as blood glucose approaches euglycemia. The concept of a glucose-responsive insulin (GRI) was first proposed in 1979 (4). In this seminal work, it was shown that a glycosylated insulin, i.e., insulin that has been conjugated to maltose and polymerized with concanavalin A (ConA), could serve as a GRI reservoir, increasing the rate of insulin release in response to a rise of glucose (5). This provocative discovery and concept has since been expanded by exploring the combination of insulin with diverse matrices, such as glucose-responsive gel, microparticle or nanoparticle, bioinorganic membrane, and micelle, some of which were taken into preclinical studies for in vivo proof of concept (6, 7). Alternative approaches that can serve as the glucose-sensing trigger to modulate insulin release have also been attempted, such as glucose oxidase (8) and phenylboronate-based glucose sensing (9) and a GRI patch (10). Admirable progress to identify a GRI has been made (7, 11), yet many obstacles remain in the creation of insulin analogs that can continuously sense and respond to blood glucose fluctuation in a minimally invasive manner. As one example of these challenges, the Brownlee and Cerami approach, starts with a polymer complex exogenously formed of glycosylated insulin with ConA (which preferably binds to α-D-mannosyl and α-D-glucosyl residues) (4, 5). The release and clearance kinetics of glycoinsulin by this and similar designs have not been optimized to recapitulate an endogenous profile of insulin secretion. In addition, ConA is not a viable option for clinical use due to its mitogenic and immunogenic nature (12). Safe and cost-effective choices for alternative recombinant lectins are not currently available. An alternative approach, however, could be to utilize endogenous lectin(s) that can bind to and have capacity to clear an appropriately glycosylated insulin in a glucose-responsive manner. Such an approach would not be designed to serve as a depot of insulin and modulate its release; rather the intention would be to engineer by glycosylation onto insulin an additional pathway of clearance, such that the rate of clearance by a lectin-based pathway is glucose responsive. This alternative strategy for creating a GRI, by exploiting lectin-based clearance that might be glucose responsive in its kinetics, was initially observed by Zion and colleagues (13). The studies to be presented in this report are efforts by our laboratory (14) to further develop and refine the concept and to identify the specific endogenous lectin involved.
Lectins are sugar-binding proteins in circulation or on cell membranes that recognize and bind specific carbohydrates (15). Key functions of most animal lectins are to bind endogenous glycoproteins “earmarked” for degradation, to recognize cell surface carbohydrates on pathogens and facilitate their removal, and to play a role in both the innate and adaptive immune systems (16). Common classes of lectins found in animals include S-, P-, and C-type lectins, which can be further separated into subclasses based on structure and binding specificity. The family of C-type lectins encompasses soluble lectins, such as mannose-binding lectin (MBL), surfactant proteins A and D (SP-A or SP-D), and dendritic cell–specific intercellular adhesion molecule-3–grabbing nonintegrin (DC-SIGN, also known as CD209), which have overlapping but different sugar structural preference and distinct physiological functions (16–19). Mannose receptor C type 1 (MRC1, also known as CD206) is a prototypical member of the MR family of transmembrane lectins, a family that also includes PLA2 receptor, DEC-205, and ENDO180 (also known as MRC2) (20–22). The main function of MR is to recognize endogenous “waste” proteins tagged for destruction or pathogens via their surface glycan, through preferential binding to terminal mannose (e.g., paucimannose, including Man3) and, to a lesser extent, L-fucose and N-acetylglucosamine (18, 20, 23–25). MR was first discovered in macrophages. In the liver, MR is highly expressed in Kupffer cells as well as liver sinusoidal endothelial cells (LSECs) (21, 26, 27). MR constitutively recycles between cell surface and endosome, delivering the ligand for lysosome degradation (27, 28). Though regarded as a component of the innate immunity system, the role of MR in antigen presentation, stimulating a cytokine, or inflammatory response is less clearly defined. Mice with MR deletion have been shown to retain an intact host defense, even when challenged with common pathogens, but do manifest increased circulating levels of endogenous glycoproteins that would otherwise be cleared by the MR-mediated pathway (23, 24, 29).
To use an endogenous lectin as part of the GRI concept, the lectin system must have sufficient capacity to rapidly remove the glycosylated insulin from the circulation and to have the lectin-mediated removal rate be modulated by glucose across the physiological range of blood glucose excursions experienced in T1DM and T2DM. MR is recognized as a high capacity lectin with a rapid cycling rate (28). Interestingly, it had been observed that some glycoproteins can manifest differences in clearance in animals in relation to blood glucose levels (26, 30), and the site of this action was attributed to hepatic endothelial cells. Accordingly, we sought to engineer a mannosylated insulin that recognized both MR and the insulin receptor (IR), with affinities that would enable the desired GRI and lead to glucose lowering in diabetics with low risk of hypoglycemia. The studies reported herein outline testing of this concept and the effort to identify which endogenous lectin system is engaged.
Results
Mannosylated insulin analogs lower glucose in a glucose-responsive manner.
To generate a mannosylated insulin, trimannose was conjugated to recombinant human insulin (RHI) via a diantennary or triantennary linker at Lys-B29 and/or A1 positions (Figure 1A). One resulting compound (GRI1) was found to lower glucose in mice after a single s.c. administration (Figure 1B), albeit at higher doses compared with RHI. Interestingly, efficacy of GRI1 increased with an initial doubling of dose, from 42 to 84 nmol/kg; but with an additional dose increase (compare doses of 84 and 168 nmol/kg, Figure 1B), the glycemic response reached an apparent plateau, at a level above hypoglycemia (~60 mg/dl in mice). In contrast, doubling of an efficacious RHI dose (6–12 nmol/kg) caused hypoglycemia without evidence of leveling the glycemic response (Figure 1, B and C). These findings were reproducible, as a similar pattern of response was observed using the same or similar mannosylated insulin analogs and in other animal species. For example, in diabetic rats induced by streptozotocin (STZ), inspection of individual responses indicated substantially less propensity to induce hypoglycemia after a single dose of GRI1, chosen to yield an equivalent efficacy to RHI, compared with RHI (average glucose in Figure 2A; individual animal glucose values in Figure 2, B and C). This putative glucose-responsive modulation of insulin action evident during a single injection of GRI1 was also discerned when GRI1 and RHI were administered to diabetic mice via s.c. pumps for 7 continuous days at respective rates resulting in equal glucose-lowering efficacy (average glucose in Figure 2D). As revealed in the plots of individual animal glucose values (Figure 2, E and F), regardless of glycemic equipoise attained at onset of pump delivery of respective insulins, during sustained pump administration at a constant rate, RHI administration was associated with more hypoglycemia, as indicated by glucose values below 60 mg/dl.
Figure 1. Mannosylated insulin analog shows differential glucose-lowing profile compared with RHI.

(A) Schematics of GRI1, GRI2, and GRI3 molecules with their sugar and linker structures. (B) GRI1-induced glucose lowering in C57BL/6 mice after a single s.c. injection. (C) Glucose AUC was calculated, and fold changes over vehicle are shown. n = 4–6 per group. *P < 0.05, ***P < 0.001 vs. vehicle group by 2-way ANOVA with Dunnett’s test. Results are shown as mean ± SEM of values and represent 3 independent experiments.
Figure 2. Mannosylated insulin analog GRI1 lowers glucose effectively without hypoglycemic incidence.

(A) STZ-induced diabetic rats received a single bolus of RHI or GRI1 with equivalent glucose-lowering potencies. n = 6 per group. (B and C) Scatter plots of individual animal glucose values corresponding to glucose values shown in A. (D) STZ-induced diabetic rats were implanted with a s.c. pump to deliver continuous infusion of optimized doses of GRI1 and RHI for 7 days to reach comparable glucose-lowering efficacy to each other. n = 8–9 per group. (E and F) Scatter plots of individual animal glucose values corresponding to glucose values shown in D. Results are shown as mean ± SEM of values and represent 3 independent experiments.
These preliminary in vivo data prompted a more controlled approach, using glucose clamps, to examine glucose-responsive modulation of GRI1 action. GRI1 was administered (via s.c. bolus) to nondiabetic or STZ-induced diabetic rats under conditions of a glucose clamp at 100 or 400 mg/dl, respectively, to assess glucose modulation of its time-action profile and dose response (see Figure 3, A and B, for representative glucose and glucose infusion rate data). A dose of RHI (12 nmol/kg) was chosen to be approximately equal in its PD effect, represented by AUC of glucose infusion rate, to that of 168 nmol/kg GRI1 in the euglycemic clamp (Figure 3C). In these euglycemic clamp studies, a 2-fold increase in GRI1 from the 168 nmol/kg dose and successive 2-fold reductions revealed dose-dependent changes in its PD effect (Figure 3C). Repeating these doses of GRI1 under hyperglycemic clamp conditions revealed marked augmentation of the PD effect of GRI1 relative to comparable doses in euglycemic clamp studies, whereas the PD effect of RHI was modestly reduced in the STZ-induced diabetic rats at hyperglycemia compared with its effect in healthy rats at euglycemia (Figure 3D vs. Figure 3C). This striking contrast of the glycemic modulation of PD action of GRI1 relative to RHI was associated with differences in GRI1’s circulating concentrations under hyperglycemic versus euglycemic clamp. For example, the AUC of GRI1 concentration for the 336 nmol/kg dose was 4,660 ± 1,019 nM·min at euglycemia, and it increased more than 2-fold to 11,659 ± 1,263 nM·min during hyperglycemic clamps, a significant group difference in PK (P < 0.01). Under the same conditions, the corresponding AUC for RHI (12 nmol/kg) was 307 ± 241 nM·min and 50 ± 17 nM·min, respectively, in euglycemic and hyperglycemic groups (P = 0.14). Acknowledging a potential limitation of comparing PK and PD in healthy versus STZ-induced diabetic rats, the pronounced changes in GRI1 PK and PD at hyperglycemia versus euglycemia support the hypothesis that a mannosylated insulin analog like GRI1 undergoes an alteration in its PK, yielding a shift in PD effect in a glucose-dependent manner.
Figure 3. GRI1 is more efficacious under hyperglycemia in diabetics compared with nondiabetic rats.

(A) RHI and GRI1 were administered as a single bolus s.c. to normal SD rats or STZ-induced diabetic SD rats, followed by glucose clamped at 100 mg/dl (representative groups only; n = 4–7) or 400 mg/dl (n = 2–12), respectively. (B) RHI and GRI1 potencies were assessed by glucose infusion rate (GIR) (representative groups corresponding to A). (C and D) GIR AUC of normal SD rats or STZ-induced diabetic SD rats groups. *P < 0.05, **P < 0.01 vs. RHI group by 2-way ANOVA with Dunnett’s test. Results are shown as mean ± SEM of values, where appropriate, and represent 2–3 independent experiments. Numbers in graphs represent fold changes over the corresponding RHI-treated group.
Based on the observations that mannosylated insulin can manifest glucose-responsive modulation of PK and PD, we postulated that a mannose-preferring lectin was responsible. This hypothesis was examined by assessing the effect on GRI in vivo action during a coadministration of methyl-α-D-mannopyranoside (α-methyl mannose [α-MM]). α-MM has high affinity to mannose-preferring C-type lectins and is often used to inhibit mannosylprotein-lectin binding (31). When GRI1 was coadministered with α-MM in mice, an apparently “weak” dose (only nominal PD effect) became significantly more potent in glucose lowering (Figure 4A). Correspondingly, GRI1 plasma levels were dramatically elevated in the presence of α-MM (Figure 4B). These data suggest the likelihood that an endogenous mannose-binding lectin or lectins is responsible for rapid GRI1 clearance under euglycemia, and in the presence of α-MM (which imposes a potent inhibitory effect on lectin-mediated clearance), GRI1 manifests higher plasma levels and a greater effect to lower glucose.
Figure 4. Cellular MR levels correlate with GRI clearance.

(A) Glucose lowering by GRI1 in C57BL/6 mice was assessed in the absence or presence of α-MM coinjection, and corresponding GRI1 plasma levels are shown in B. n = 8 per group. **P < 0.01, ***P < 0.001 vs. GRI1 alone group by 1-way ANOVA with Tukey’s test. (C) High-content imaging analysis of cellular uptake of Alexa488-labeled GRI1 and GRI2 in the presence or absence of 10 mg/ml mannan in cultured rat macrophage NR8383 cells. n = 2–5 per group. **P < 0.01 corresponding GRI vs. GRI + mannan by Student’s t test. (D) Cellular uptake of GRI2 and lysosome staining by LysoTracker are measured in cultured rat macrophage NR8383 cells. Original magnification, ×40. (E) siRNA-mediated knockdown of MR and corresponding GRI uptake in NR8383 cells. n = 3 per group. *P < 0.05 vs. scrambled siRNA group by Student’s t test. A 57% reduction of GRI2 uptake corresponded to a 65% reduction of MR protein. Western blot detection of MR protein is shown in E. (F) Overexpression of human MR in HEK293 cells and corresponding GRI1 and GRI2 uptake are shown in the presence or absence of 10 mg/ml mannan. n = 2–3 per group. **P < 0.01 vs. corresponding GRI uptake in parental HEK cells by Student’s t test. (G) Representative FACS analysis of GRI2 and OVA uptake in primary human liver cell mixture is shown. The x axis represents medium fluorescence intensity of Alexa488-labeled GRI2/OVA in each cell. The y axis represents medium fluorescence intensity of APC-labeled MR/IgG antibodies only detecting membrane-expressing MR in each cell. Results are shown as mean ± SEM (A and B) or mean ± SD (C, E, and F). Results represent 2 (F) or 3 independent experiments (A, B, C, E, and G).
Cellular MR levels correlate with GRI internalization.
Many of the mannose-binding lectins in animals belong to C-type lectins that include circulating (e.g., MBL and DC-SIGN) and transmembrane (e.g., MR, ENDO180, and DEC-205) proteins (16). To investigate whether membrane-bound lectins can mediate internalization of GRIs, Alexa Fluor 488–labeled (Alexa488-labeled) GRI1 or GRI2 was added to cultured NR8383 cells, a rat macrophage cell line. Using high content imaging analysis, it is demonstrated that NR8383 cells had robust uptake of GRI1 and GRI2, which can be inhibited by mannan (Figure 4C). Among many tested GRI molecules, GRI2 had the highest uptake signals, so it is often used as a representative GRI to investigate mechanisms of action in cell-based assays. As shown in Figure 4D, GRI2 was internalized to an intracellular site that costained with LysoTracker Red (Thermo Fisher Scientific), indicating that GRI2 was targeted to lysosomes, which would be consistent with MR biology (20, 28). Similar uptake can be demonstrated in primary mouse macrophage cells, and uptake was completely inhibited in the presence of mannan (Supplemental Figure 2A; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.97476DS1).
Among the membrane-bound C-type lectins, MR is well-known for its role in glycoprotein clearance, as it targets glycoproteins for lysosome degradation (16, 20). The high clearance capacity and rapid lysosome targeting of GRI2 resembled MR-mediated glycoprotein clearance (28). To investigate whether MR is the major receptor responsible for GRI clearance, GRI2 internalization studies were conducted in NR8383 cells transfected with MR siRNA, which resulted in an approximately 65% reduction of MR mRNA levels in NR8383 cells relative to the effect of nontargeting siRNA transfection in these cells (Supplemental Figure 2B). The knockdown of MR was confirmed by Western analysis, and a tight correlation between MR knockdown and reduction of GRI2 uptake was shown (Figure 4E). In contrast, in the same study, 80% knockdown of a control gene, CycB, had no effect on GRI2 uptake and MR expression (Figure 4E and Supplemental Figure 2B). Mannan, a C-type lectin-binding inhibitor, completely blocked GRI2 internalization in these studies. Similarly, MR knockdown in human blood PBMC-differentiated macrophages resulted in a corresponding reduction of GRI2 uptake (Supplemental Figure 2C). In another set of experiments, labeled GRI2 showed no uptake in parental HEK293 cells, which do not express MR. However, these cells obtained robust capacity for GRI2 internalization upon transfection of human MR (Figure 4F). These cellular assays demonstrate that MR is capable of and sufficient for mediating GRI uptake in vitro.
Besides macrophages, there are other cell types, notably LSECs, that express high levels of MR as well as other lectins and contribute to the clearance of mannosylated proteins (32). Since liver is a major organ for the clearance of glycosylated proteins, freshly isolated human liver cells were prepared (a mixture that included macrophages, LSECs, and hepatocytes) for GRI uptake assays. MR expression level was measured in the FACS studies. As shown in Figure 4G, approximately 60% of the cells (fourth panel, top right, Figure 4G) demonstrated that OVA (a glycosylated egg protein) uptake expressed MR, while approximately 40% of non-MR-expressing cells (fourth panel, lower right, Figure 4G) also took up OVA. In contrast, only MR-positive cells internalized GRI2 (third panel, Figure 4G). Similar results were obtained in studies of a liver cell mixture prepared from various animal species, including mouse, dog, minipig, and monkey (data not shown). These results demonstrate that MR is the major receptor responsible for uptake of GRI in human and animals.
GRI binds to MR specifically and in a glucose-dependent manner.
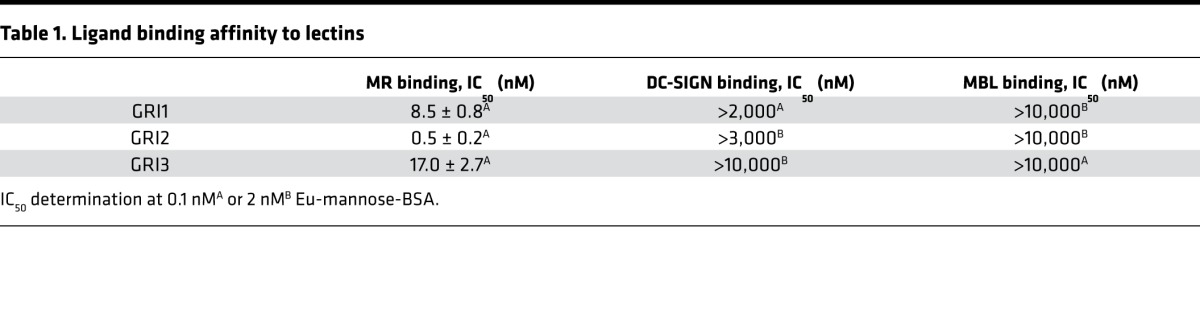
To determine the binding affinity of GRI to MR and examine the specificity of this binding to MR, a set of lectin-binding assays were developed based on the Delfia platform (18). The primary assay investigated recombinant MR protein dose-responsive binding to GRI. MR-binding affinity was determined by assessing GRI inhibition of the interaction between europium-labeled (Eu-labeled) mannose-BSA and immobilized MR (see Figure 5A for assay design). This assay examined binding to human MR by GRI analogs (Table 1). Similar assays were also built for additional lectins, such as DC-SIGN and MBL, which demonstrated much lower binding affinity for GRI analogs relative to MR (Table 1). In contrast, mannan was revealed to manifest nondifferentiating inhibition of Eu-labeled mannose-BSA binding to these lectins, with an average IC50 for MR and DC-SIGN of 70 and 9 ng/ml, respectively.
Figure 5. GRIs selectively bind to MR in a glucose-dependent manner.

(A) Plate-based Delfia lectin binding assays were developed to quantify the ability of GRI to inhibit the binding between lectins and Eu2+-Mannose-BSA. (B) Modified assay replacing Eu2+-mannose-BSA with individual Eu2+-GRIs was applied to determine glucose inhibition of MR binding to Eu2+-GRI.
Table 1. Ligand binding affinity to lectins.
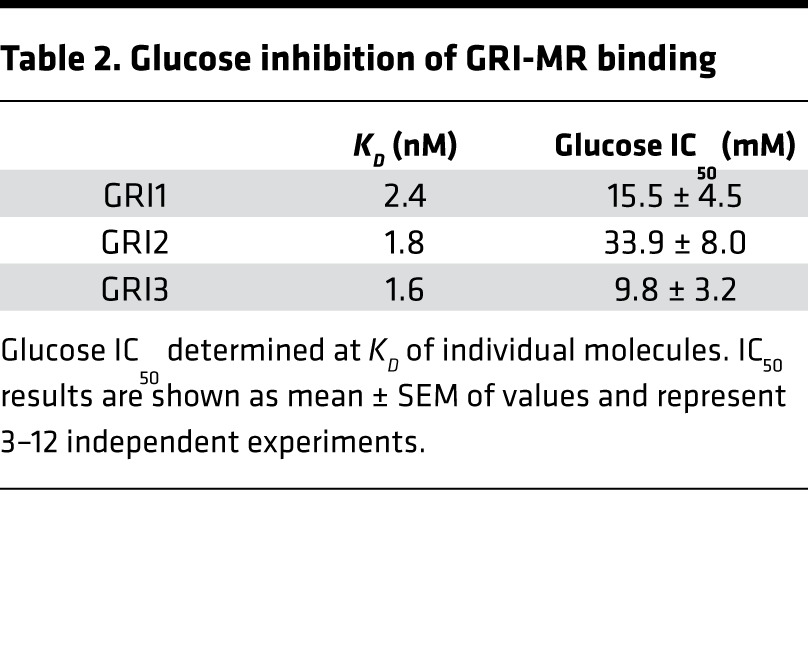
To assess glucose-responsive modulation of GRI binding to MR, a modified version of the Delfia assay was applied, in which Eu-labeled GRI replaced Eu-labeled mannose-BSA. This modification enabled direct measurement of glucose competition of Eu-GRI binding to MR (Figure 5B). The assay demonstrated that selected GRIs have glucose IC50 in the desirable range of physiological relevance (Table 2). GRI’s in vitro binding affinity to IR was also determined (IC50 of 0.2 nM for RHI; CHO-IR cell-based competition binding), which yielded IC50 values of 0.8, 17.6, and 6.6 nM, respectively, for GRI1, GRI2, and GRI3. The GRI-IR binding was not affected by glucose levels tested in MR assays (data not shown). Collectively, the results support the notion that the observed in vivo glucose responsiveness action of GRI is likely mediated by MR.
Table 2. Glucose inhibition of GRI-MR binding.
MR plays a predominant role in GRI clearance in vivo.
To further investigate the effect of MR selectivity on GRI clearance in vivo, studies were conducted in mice with homozygous MR deletion (MR KO mice; ref. 23). GRI1 was administered during an insulin tolerance test performed in WT and MR KO mice. In WT mice, GRI1 reduced glucose slightly more than vehicle and less than an equimolar dose of RHI. However, in MR KO mice, GRI1 lowered glucose more substantially and equally well as RHI (Figure 6, A and B). Corresponding PK analyses demonstrated a very large increase of plasma GRI1 levels in MR KO mice compared with WT mice (Figure 6, C and D). Mitogenic activities of GRIs were evaluated in a Saos2.B10 cell proliferation assay using [3H]-thymidine uptake. Relative to IGF-1 and RHI (IC50 of 0.27 and 32.8 nM, respectively), GRIs show weak activities, with IC50 values of 165, 432, or 546 nM, respectively, for GRI1, GRI2, or GRI3. This is in line with the observation that no cell or tissue toxicity was evident in animals receiving chronic GRI dosing (data not shown).
Figure 6. MR plays a predominant role in GRI clearance in mice.

MR depletion in mice abolishes GRI clearance and completely restores its glucose-lowering capacity. (A–D) Equal molar RHI and GRI1 were tested in WT and MR KO mice via acute s.c. administration. Glucose-lowering (A and B) and plasma drug levels (C and D) in WT (A and C) and KO mice (B and D) are shown. n = 8. *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle control by 2-way ANOVA with Dunnett’s test. Results are shown as mean ± SEM of values and represent 3 independent experiments. (E) Detection of MR-dependent GRI endocytosis in liver by immunofluorescence colocalization of GRI with MR. Liver samples collected 15 minutes after GRI1 dosing were paraffin embedded and sectioned, followed by staining with insulin (green with Alexa Fluor 647 goat anti-HRP) and MR (Cy3 donkey anti-rabbit) antibodies sequentially, as described in Methods. Original magnification, ×40. (F) FACS analysis of GRI1 uptake in freshly isolated WT or MR KO mouse liver cell mixture. The x axis represents medium fluorescence intensity of Alexa488-labeled GRI1 in each cell. The y axis represents the forward scatter sorting of individual cell size. Results represent 2 independent experiments. (G) Schematics showing differential MR-mediated GRI clearance as glucose level changes. Consequently, IR-positive cells are exposed to higher GRI at hyperglycemia than under euglycemic or hypoglycemic conditions.
Since a major site of MR expression is within LSECs and Kupffer cells, immunofluorescence assays were developed to detect endocytosed GRI in MR-positive hepatic tissues as a further validation test of the dependency of in vivo GRI uptake and clearance on MR. In liver sections from WT mice dosed with GRI1, internalized GRI1 was shown to colocalize with MR-positive cells (Kupffer and LSECs), as shown in Figure 6E. However, in MR KO mice that received the same dose of GRI1, there was no detection of MR or of internalized GRI. Furthermore, in an isolated liver cell mixture prepared from WT or MR KO mice, uptake of GRI1 was not observed in the latter and could be substantially blocked in the WT cell mixture by addition of mannan, indicating again that MR is the receptor responsible for GRI uptake and clearance (Figure 6F). In this assay, GRI2 and GRI3 showed similar uptake in cells from WT and MR KO mice to GRI1 (GRI1, GRI2, and GRI3 uptake: 2,494, 2,473, and 2,586 relative fluorescence units (RFU) in WT mice and 447, 118, and 232 RFU in MR KO mice, respectively). Taken together, these results provide strong evidence that MR is capable and sufficient to mediate the clearance of GRI analogs used in the current studies.
Discussion
The current studies were undertaken with a practical purpose to explore whether it is feasible to engineer glucose-responsive properties into insulin by modifying its structure to engage an endogenous lectin. This is an approach initially suggested by Zion and Lancaster (13). Our efforts to put to practice this provocative concept demonstrate that insulin conjugated with mannose motifs can engage endogenous lectin and be taken up and delivered for lysosomal degradation and that this process can be modulated by ambient glucose. Although conjugation of saccharides to native insulin does influence its binding affinity to IR, these chimeric insulin analogs exert classic insulin actions, notably lowering of blood glucose in diabetic rodents. Yet, these analogs, at similar and higher doses than those that reduce hyperglycemia, manifest an attenuated effect when blood glucose in a diabetic rodent has been lowered to euglycemia. This portends an increased therapeutic index of these GRI analogs relative to native insulin. Using in vitro and in vivo assays, including MR KO mice, it was found that, among endogenous lectins, these GRIs preferentially target MR and confirmed that binding to MR can be competitively inhibited by hyperglycemia. In vivo studies indicate that flux of GRI1 into the MR system is of sufficient amplitude to clearly influence GRI1 PK and that glucose responsiveness of changes in PK translate into meaningfully effect on GRI1 governance of glucose homeostasis.
GRI1 is a semisynthetic insulin analog prepared by saccharide conjugation to RHI via a linker. The sugar moieties of GRI1 confer selective and tight binding to MR at low glucose levels, mediating cellular uptake followed by lysosomal clearance, thus diminishing availability of this insulin chimera to engage with the IR. Under hyperglycemia, blood glucose competes with GRI1 for binding to MR, diminishing MR-based uptake and thereby partitioning more GRI1 to engage with IR and elicit glucose lowering. Other analogs, such as GRI2 and GRI3, illustrated a similar glucose-dependent autoregulatory property for regulating PK and PD. These concepts are illustrated in Figure 6G. It can be rightly asserted that the PD action of native insulin is influenced by ambient glucose; at a given level of circulating insulin, its PD effect is more pronounced at hyperglycemia (33). These mass-action influences are evident in modulating rates of insulin-stimulated glucose transport during hyperglycemia but are insufficient to mitigate risk of insulin-induced hypoglycemia as blood glucose values return to euglycemia. Native insulin and all insulin preparations currently available to treat patients with diabetes manifest a steep slope for dose-responsive glucose lowering (34). This steep dose response translates into a substantial challenge in precisely selecting a dose that tightly controls blood glucose within a near normal range, because relatively modest increases of an insulin dose can precipitate hypoglycemia. Conversely, relatively minor decreases of dose can fail to control hyperglycemia (35). This challenge has been recognized by practitioners and patients since the discovery of insulin nine decades ago and remains an urgent and daily challenge (2, 3, 36).
Contemporary efforts focused on a closed-loop artificial pancreas for delivery of insulin in a manner that reduces hypoglycemia while simultaneously achieving tight control of blood glucose (37). The approach explored in the current studies represents a clear difference in strategy from this and the alternative attempts following Brownlee and Cerami’s proposal of the autoregulated “glucose control” of insulin delivery from an injected depot (4, 5). We endeavored to introduce directly onto insulin an autoregulatory modulus of control once the insulin analog is circulating in blood and interstitial tissues by targeting an endogenous lectin. Many animal lectins serve critical physiological roles, such as immune surveillance, clearance of glycoproteins (endogenous or surface protein on invading agents), and cell-cell communication. It is undesirable to engage lectins with a GRI that will evoke these responses. MR is a possible exception from the safety perspective, as recent data suggest its main function is to remove endogenous senescent proteins for destruction (23, 24, 29). Recombinant β-glucocerebrosidase (e.g., Cerezyme), which is decorated with trimannose similar to those used for GRI, targets macrophage MR as an enzyme replacement therapy and has been used safely for many years to treat patients with type 1 Gaucher’s disease (38). As a scavenger lectin, MR has a very large clearance capacity, making it feasible to efficiently remove GRI (28). Our in vitro data indicate minimal off-target binding (vs. MR) of GRIs to human MBL and DC-SIGN (Table 1) and no detectable binding to lectins, including ENDO 180, DEC-205, or PLA2R (data not shown). In addition, knockout mouse studies have ruled out MBL and SP-D as endogenous lectin targets for GRI (data not shown).
In summary, studies presented here demonstrate that GRIs consisting of RHI conjugated to specific mannose moieties are capable of targeting both IR and MR and undergoing glucose-responsive clearance within the physiologic range of glucose concentrations (Figure 6G). Representative GRIs lower glucose with a RHI-like time action profile that is compatible for prandial glycemic control. The glucose dependency of the GRI (which can be adjusted by choice of sugar affinity, valency, insulin scaffold, and conjugation site) can be achieved by the carbohydrate conjugations that compete with glucose for binding to MR. Under euglycemic/hypoglycemic conditions, GRI is primarily cleared from circulation by an MR-dependent process, with kinetics that are more rapid than the removal of a nonglycosylated insulin. At hyperglycemia, via slowdown of MR-mediated GRI clearance and consequent GRI accumulation, glucose levels fall rapidly through increased activation of IR. The characteristics of GRI uncovered here can also be expanded to the design of additional stimuli-responsive therapeutics.
Methods
Reagents and chemicals.
All chemicals and reagents were procured from commercial sources, except for GRI1, GRI2, and GRI3, which were synthesized in-house. GRI1, GRI2, and GRI3 were prepared by selectively acylating either the ε-amino group of B29 Lys or both the α-amino group of A1 Gly and the ε-amino group of B29 Lys of RHI using corresponding mannose-based sugar cluster–containing N-hydroxysuccimide–activated ester in good yields (scheme 1, Supplemental Figure 1). Selective acylation is achieved by exploiting the reactivity and basicity difference among the steric hindrance of the ε-amino group of B29 Lys and the α-amino groups of A1 Gly and B1 Phe (39).
Animal studies.
Male SD rats were obtained from Taconic and male C57BL/6 mice were obtained from Taconic Farm. All animals were housed in a room maintained at constant temperature (25°C) and humidity, with ad libitum access to autoclaved water and diet. To generate STZ-induced diabetic mice/rats, animals were treated with a single i.p. injection of STZ (MilliporeSigma), followed by grouping based on body weight and ambient glucose levels prior to studies. The MR KO mouse was generated previously and was purchased from The Jackson Laboratory.
Evaluations of antihyperglycemic efficacy in mice and rats.
To test glucose-lowering effects of the insulin analogs, animals were fasted for 2 hours prior to s.c. injection of GRI or RHI at doses indicated in the Results. Glucose was measured in blood from tail bleeds using an OneTouch glucometer (Lifescan) at specified time points after insulin injection. Plasma were collected at 0, 30, 60, 120, and 180 minutes after dosing. Insulin/GRI levels were measured by ELISA according to the manufacturer’s protocol (Mercodia). Concentration of insulin analogs were calculated based on standard curves derived from the specific compounds. In some experiments, α-MM (21 mg/ml) was administered at the same time with insulin/GRI. In a separate cohort of mice, plasma samples were collected 0, 30, and 60 minutes after insulin dosing.
In the minipump insulin infusion study, male C57BL/6 mice received a single i.p. injection of 120 mg/kg STZ and were grouped based on glucose (average value ~450 mg/dl) 7–10 days after injection. A surgically planted 7-day Alzet minipump model 2001 contained vehicle, RHI, or GRI1 in final volume of 225 μl. Blood glucose was measured at 9 AM and 4 PM daily.
To conduct euglycemia and hyperglycemia clamps, vein and artery cannulated SD rats were allowed to recovery for 5–7 days. After overnight fasting, basal blood samples were obtained at –60 and 0 minutes. The rats then received bolus s.c. injection of vehicle, RHI, or GRI1 at time 0. Blood glucose was frequently measured to guide glucose infusion rate adjustment in order to maintain glycemic level of 100 mg/dl (euglycemia) or 400 mg/dl (hyperglycemia).
Cellular GRI uptake measurements and the effect of MR knockdown or overexpression.
Rat macrophage cells, NR8383 cells (ATCC, CRL2192), were cultured in F12K medium containing 15% FBS. Cells were plated in 24-well collagen-coated plates to reach 80% confluency. Cells were washed with PBS and transfected with rat MR siRNA, rat CYCB siRNA, or control nontargeting siRNA (Dharmacon) using Mirus TransItTKO reagent (MIR2150) according to the manufacturer’s protocol. After 48 hours, GRI uptake was measured and cells were collected for mRNA or protein analysis. All GRIs were labeled with Alexa488 dye using a protein-labeling kit (Invitrogen, A30006). For uptake measurements, siRNA-transfected cells were washed with PBS and incubated with 250 nM Alexa488-labeled GRIs in assay buffer (HEPES-buffered saline pH 7.4 + 1% BSA + 0.1% HI FBS + 2 mM CaCl2 + 0.5 mM MgSO4) at 37°C for 1 hour. Cells were washed with ice-cold HSSB buffer containing 1% BSA and 0.1% FBS and fixed with 4% paraformaldehyde for 20 minutes. Cells were then washed with PBS and stored at 4°C (in the dark) for further analysis. Fluorescence of internalized GRIs was measured by high-content image analysis (ArrayScan VTI from Thermo Fisher Scientific). Western blot analysis used MR antibody (Abcam, ab64693) for MR protein detection in NR8383 cells.
In human primary macrophage studies, freshly isolated PBMCs from blood were plated on collagen-coated T75 flasks and cultured in RPMI1640 (Invitrogen, 21870) medium containing 10% FBS for 3 hours. Cells were washed with PBS and incubated with macrophage differentiation medium (RPMI1640 with 10% FBS containing 50 ng/ml MCSF, R&D Systems) for 3 days. Cells were further incubated with fresh differentiation medium for another 2 days. The differentiated macrophages were removed from T75 flasks, and siRNA transfection was performed by electroporation using Ingenio solution (Mirus, 50112) as described by the manufacturer’s protocol. Cells were plated into 96-well or 24-well plates and incubated in differentiation medium for 2 days for further analysis. GRI uptake assay was conducted as described above.
Flow cytometry analysis was used to quantify cell membrane MR protein using APC-conjugated MR antibody (BD Biosciences, 550889) as well as the GRI uptake. Freshly isolated human hepatocytes were purchased from Celsis In Vitro Technologies. Freshly isolated mouse liver cell mix, including hepatocytes, Kupffer cells, LSECs, and others, were prepared by the collagenase perfusion method, as described in the Lonza Amaxa mouse hepatocytes nucleofactor kit (Lonza, VAPL-1004). Hepatocytes and nonhepatocyte liver cells were further separated by centrifugation at 50 g for 3 minutes. OVA is a highly glycosylated protein used as a nonspecific control for lectin-mediated cellular uptake. Liver cell mix was first gated by nuclear staining followed by light scatter gating to separate single nuclear cells into 4 categories. By measuring Alexa488 fluorescence intensity, only one population showed strong uptake of GRI or OVA compared with no dye–treated control cells. Medium intensity of the Alexa488 in each cell was calculated to represent the amount of the GRI uptake.
To overexpress MR in HEK293 cells, the human MRC1 construct was obtained from OriGene. HEK293 cells were plated onto 96-well collagen-coated plates in DMEM growth medium with 10% FBS. After reaching 90% confluence, cells were transfected with human MRC1 using Lipofectamine 2000. Twenty-four hours after transfection, GRI uptake assay was conducted as described above in the presence or absence of 10 mg/ml mannan (MilliporeSigma, M7504) at 37°C for 1 hour.
GRI-lectin-binding assay and determination of glucose responsiveness.
In the Delfia binding assay (Figure 5A), a Pierce protein G–coated black plate (Thermo scientific, 15157) was washed with wash buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 5 mM CaCl2, and 1 mM MgCl2 + 0.1% Tween 20). 200 ng MR antibody (R&D Systems, AF2534) in 100 μl PBS buffer + 1% BSA (stabilizer [DTPA] purified BSA 7.5%, PerkinElmer, CR84-100) was then added to each well and shaken at room temperature for 1 hour. The plate was washed with wash buffer followed by incubation with 100 ng/well MR protein (R&D Systems, 2534-MR) in 100 μl PBS plus 1% BSA and shaken for 1 hour at room temperature. After wash, 100 μl Eu-Man-BSA (prepared in house) and its competitors (10 mg/ml mannan, MilliporeSigma, 10 μM GRIs, or mannose BSA) in binding buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 5 mM CaCl2, 1 mM MgCl2 0.2% BSA + protease inhibitor cocktail) were added to each well for 1 hour at room temperature. Plates were incubated with 100 μl/well Delfia inducer solution (PerkinElmer, 4013-0010) for 15 minutes at room temperature and read on Envision monochrometer (Ex 340 m/Em 615 m). For the GRI glucose-responsiveness assay, a similar protocol was used as in the Delfia-binding assay described above, with the exception that Eu-GRI replaces Eu-Man-BSA and the binding competitor is glucose (Figure 5B).
MR and GRI colocalization studies.
Liver samples collected 15 minutes after GRI1 s.c. dosing were paraffin embedded and sectioned. Antigen retrieval with buffer (100×-Tris-EDTA Buffer, pH 9.0; Thermo Scientific, TA-125-PM4X) was followed by incubation with insulin antibody (Invitrogen, 180067; 30 μg/ml) for 1 hour at room temperature. This is followed by incubation with secondary goat anti-GP antibody (Jackson, 106-006-003) at 5 μg/ml for 30 minutes. SuperPicture HRP-polymer (Invitrogen, 879363) was incubated with sectioned liver samples for 15 minutes and with Alexa Fluor 647 goat anti-HRP (Jackson, 123-605-021; 1:200) for 30 minutes at room temperature. The slides were then incubated in Protein Block Serum-Free solution with 20 μg/ml donkey IgG for 30 minutes, followed by 0.5 μg/ml MR antibody (Abcam, ab64693) for 1 hour at room temperature. Secondary antibody Cy3 donkey anti-rabbit (Jackson, 711-166-152) was incubated at room temperature 1:200 for 30 minutes. The slides were then stained with DAPI and mounted. Slides were washed 3 times (5 minutes each time) in between each step.
Statistics.
Data analyses were performed in GraphPad Prism. Calculations of P values were based on ANOVA and the unpaired 2-tailed Student’s t test, as applicable. Statistical significance was defined as P < 0.05.
Study approval.
All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the research laboratories of Merck & Co. Inc. The Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011) was followed in the conduct of the animal studies. Veterinary care was given to any animals requiring medical attention.
Author contributions
RY, MW, SL, RPN, XL, TK, MDE, DEK, and JM designed the research; RY, MW, XL, LY, GD, YQ, QDY, PAF, YC, XS, PH, and DDF performed the research; RY, MW, TK, YQ, PAF, and YC analyzed the data; and RY, MW, SL, MDE, DEK, and JM wrote the manuscript.
Supplementary Material
Acknowledgments
The authors are thankful for helpful discussions with Bei Zhang, Nancy Thornberry, Todd Zion, and Tom Lancaster during study design. The authors are also thankful for the technical and scientific support from Joseph A. Clemas, Xiaoping Zhang, Yonghua Zhu, Yuli Chen, Zuliang Yao, Xiaoli Ping, and Anka G. Ehrhardt. The research is solely sponsored by Merck.
Version 1. 01/11/2018
Electronic publication
Footnotes
JM’s present address is: Merck Research Laboratories, South San Francisco, California, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(1):e97476. https://doi.org/10.1172/jci.insight.97476.
Contributor Information
Ruojing Yang, Email: ruojing_yang@merck.com.
Margaret Wu, Email: margaret.s.wu@gmail.com.
Songnian Lin, Email: songnian_lin@merck.com.
Xinghai Li, Email: lixinghai999@gmail.com.
Lin Yan, Email: lin_yan@merck.com.
Ge Dai, Email: daige3268@gmail.com.
Ying Qian, Email: ying_qian@merck.com.
Qing Dallas-yang, Email: qing_yang@merck.com.
Yan Cui, Email: yan_cui2@merck.com.
Xiaolan Shen, Email: xiaolan_shen@merck.com.
Pei Huo, Email: pei_huo@merck.com.
Danqing Dennis Feng, Email: danqing_feng@merck.com.
James Mu, Email: yingjun_mu@merck.com.
References
- 1.Cahn A, Miccoli R, Dardano A, Del Prato S. New forms of insulin and insulin therapies for the treatment of type 2 diabetes. Lancet Diabetes Endocrinol. 2015;3(8):638–652. doi: 10.1016/S2213-8587(15)00097-2. [DOI] [PubMed] [Google Scholar]
- 2.Oyer DS. The science of hypoglycemia in patients with diabetes. Curr Diabetes Rev. 2013;9(3):195–208. doi: 10.2174/15733998113099990059. [DOI] [PubMed] [Google Scholar]
- 3.Kumar A, Kesavadev J, Sethi B, Jain SM, Guruprasad CS, Shah SN. Intensifying insulin therapy in type 2 diabetes: Choices & challenges. J Assoc Physicians India. 2015;63(5 Suppl):8–14. [PubMed] [Google Scholar]
- 4.Brownlee M, Cerami A. A glucose-controlled insulin-delivery system: semisynthetic insulin bound to lectin. Science. 1979;206(4423):1190–1191. doi: 10.1126/science.505005. [DOI] [PubMed] [Google Scholar]
- 5.Brownlee M, Cerami A. Glycosylated insulin complexed to concanavalin A. Diabetes. 1983;32(6):499–504. doi: 10.2337/diab.32.6.499. [DOI] [PubMed] [Google Scholar]
- 6.Wu Q, Wang L, Yu H, Wang J, Chen Z. Organization of glucose-responsive systems and their properties. Chem Rev. 2011;111(12):7855–7875. doi: 10.1021/cr200027j. [DOI] [PubMed] [Google Scholar]
- 7.Zaykov AN, Mayer JP, DiMarchi RD. Pursuit of a perfect insulin. Nat Rev Drug Discov. 2016;15(6):425–439. doi: 10.1038/nrd.2015.36. [DOI] [PubMed] [Google Scholar]
- 8.Traitel T, Cohen Y, Kost J. Characterization of glucose-sensitive insulin release systems in simulated in vivo conditions. Biomaterials. 2000;21(16):1679–1687. doi: 10.1016/S0142-9612(00)00050-8. [DOI] [PubMed] [Google Scholar]
- 9.Chou DH, et al. Glucose-responsive insulin activity by covalent modification with aliphatic phenylboronic acid conjugates. Proc Natl Acad Sci U S A. 2015;112(8):2401–2406. doi: 10.1073/pnas.1424684112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu J, et al. Microneedle-array patches loaded with hypoxia-sensitive vesicles provide fast glucose-responsive insulin delivery. Proc Natl Acad Sci U S A. 2015;112(27):8260–8265. doi: 10.1073/pnas.1505405112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webber MJ, Anderson DG. Smart approaches to glucose-responsive drug delivery. J Drug Target. 2015;23(7-8):651–655. doi: 10.3109/1061186X.2015.1055749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mackler BF, Wolstencroft RA, Dumonde DC. Concanavalin A as an inducer of human lymphocyte mitogenic factor. Nat New Biol. 1972;239(92):139–142. doi: 10.1038/newbio239139a0. [DOI] [PubMed] [Google Scholar]
- 13. Zion TC, Lancaster TC, inventors; Smartcells, Inc., assignee. Conjugate based systems for controlled drug delivery. US patent US9050370 B2. June 9, 2015. [Google Scholar]
- 14. Nussbaum A. Merck Agrees to Purchase Insulin Maker SmartCells. Bloomberg. http://www.bloomberg.com/news/articles/2010-12-02/merck-to-acquire-smartcells.Published December 2, 2010 Accessed December 18, 2017.
- 15.Loris R. Principles of structures of animal and plant lectins. Biochim Biophys Acta. 2002;1572(2-3):198–208. doi: 10.1016/s0304-4165(02)00309-4. [DOI] [PubMed] [Google Scholar]
- 16.Kilpatrick DC. Animal lectins: a historical introduction and overview. Biochim Biophys Acta. 2002;1572(2-3):187–197. doi: 10.1016/s0304-4165(02)00308-2. [DOI] [PubMed] [Google Scholar]
- 17.East L, Isacke CM. The mannose receptor family. Biochim Biophys Acta. 2002;1572(2-3):364–386. doi: 10.1016/s0304-4165(02)00319-7. [DOI] [PubMed] [Google Scholar]
- 18.Lee RT, Hsu TL, Huang SK, Hsieh SL, Wong CH, Lee YC. Survey of immune-related, mannose/fucose-binding C-type lectin receptors reveals widely divergent sugar-binding specificities. Glycobiology. 2011;21(4):512–520. doi: 10.1093/glycob/cwq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol. 2005;23:901–944. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Pomares L. The mannose receptor. J Leukoc Biol. 2012;92(6):1177–1186. doi: 10.1189/jlb.0512231. [DOI] [PubMed] [Google Scholar]
- 21.Stahl P, Schlesinger PH, Sigardson E, Rodman JS, Lee YC. Receptor-mediated pinocytosis of mannose glycoconjugates by macrophages: characterization and evidence for receptor recycling. Cell. 1980;19(1):207–215. doi: 10.1016/0092-8674(80)90402-X. [DOI] [PubMed] [Google Scholar]
- 22.Taylor PR, Gordon S, Martinez-Pomares L. The mannose receptor: linking homeostasis and immunity through sugar recognition. Trends Immunol. 2005;26(2):104–110. doi: 10.1016/j.it.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Lee SJ, et al. Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science. 2002;295(5561):1898–1901. doi: 10.1126/science.1069540. [DOI] [PubMed] [Google Scholar]
- 24.Lee SJ, Zheng NY, Clavijo M, Nussenzweig MC. Normal host defense during systemic candidiasis in mannose receptor-deficient mice. Infect Immun. 2003;71(1):437–445. doi: 10.1128/IAI.71.1.437-445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor ME, Bezouska K, Drickamer K. Contribution to ligand binding by multiple carbohydrate-recognition domains in the macrophage mannose receptor. J Biol Chem. 1992;267(3):1719–1726. [PubMed] [Google Scholar]
- 26.Summerfield JA, Vergalla J, Jones EA. Modulation of a glycoprotein recognition system on rat hepatic endothelial cells by glucose and diabetes mellitus. J Clin Invest. 1982;69(6):1337–1347. doi: 10.1172/JCI110573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wileman TE, Lennartz MR, Stahl PD. Identification of the macrophage mannose receptor as a 175-kDa membrane protein. Proc Natl Acad Sci U S A. 1986;83(8):2501–2505. doi: 10.1073/pnas.83.8.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magnusson S, Berg T. Extremely rapid endocytosis mediated by the mannose receptor of sinusoidal endothelial rat liver cells. Biochem J. 1989;257(3):651–656. doi: 10.1042/bj2570651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Napper CE, Taylor ME. The mannose receptor fails to enhance processing and presentation of a glycoprotein antigen in transfected fibroblasts. Glycobiology. 2004;14(10):7C–12C. doi: 10.1093/glycob/cwh109. [DOI] [PubMed] [Google Scholar]
- 30.Pizzo SV, Lehrman MA, Imber MJ, Guthrow CE. The clearance of glycoproteins in diabetic mice. Biochem Biophys Res Commun. 1981;101(2):704–708. doi: 10.1016/0006-291X(81)91315-2. [DOI] [PubMed] [Google Scholar]
- 31.Firon N, Ashkenazi S, Mirelman D, Ofek I, Sharon N. Aromatic alpha-glycosides of mannose are powerful inhibitors of the adherence of type 1 fimbriated Escherichia coli to yeast and intestinal epithelial cells. Infect Immun. 1987;55(2):472–476. doi: 10.1128/iai.55.2.472-476.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elvevold K, Simon-Santamaria J, Hasvold H, McCourt P, Smedsrod B, Sorensen KK. Liver sinusoidal endothelial cells depend on mannose receptor-mediated recruitment of lysosomal enzymes for normal degradation capacity. Hepatology. 2008;48(6):2007–2015. doi: 10.1002/hep.22527. [DOI] [PubMed] [Google Scholar]
- 33.Yki-Jarvinen H. Action of insulin on glucose metabolism in vivo. Baillieres Clin Endocrinol Metab. 1993;7(4):903–927. doi: 10.1016/S0950-351X(05)80239-3. [DOI] [PubMed] [Google Scholar]
- 34.Rizza RA, Mandarino LJ, Gerich JE. Dose-response characteristics for effects of insulin on production and utilization of glucose in man. Am J Physiol. 1981;240(6):E630–639. doi: 10.1152/ajpendo.1981.240.6.E630. [DOI] [PubMed] [Google Scholar]
- 35.McCall AL. Insulin therapy and hypoglycemia. Endocrinol Metab Clin North Am. 2012;41(1):57–87. doi: 10.1016/j.ecl.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cryer PE. Severe hypoglycemia predicts mortality in diabetes. Diabetes Care. 2012;35(9):1814–1816. doi: 10.2337/dc12-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovatchev B, Tamborlane WV, Cefalu WT, Cobelli C. The artificial pancreas in 2016: A digital treatment ecosystem for diabetes. Diabetes Care. 2016;39(7):1123–1126. doi: 10.2337/dc16-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charrow J. Enzyme replacement therapy for Gaucher disease. Expert Opin Biol Ther. 2009;9(1):121–131. doi: 10.1517/14712590802573395. [DOI] [PubMed] [Google Scholar]
- 39. Friesen HL. The relative reactivity of insulin amino groups as an indicator of structural accessibility and its use for synthetic approaches for structure-function studies. In: Brandenburg D, Wollmer A, eds. Insulin: Chemistry, structure and function of insulin and related hormones. New York, NY: Walter de Gruyter; 1980:125–133. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.