

Abstract
Over the course of the past decade, genome‐wide association studies (GWAS) have revolutionised our understanding of complex disease genetics. One of the diseases that has benefitted most from this technology has been Crohn's disease (CD), with the identification of autophagy, the IL‐17/IL‐23 axis and innate lymphoid cells as key players in CD pathogenesis. Our increasing understanding of the genetic architecture of CD has also highlighted how a failure to suppress aberrant immune responses may contribute to disease development – a realisation that is now being incorporated into the design of new treatments. However, despite these successes, a significant proportion of disease heritability remains unexplained. Similarly, most of the causal variants at associated loci have not yet been identified, and even fewer have been functionally characterised. Because of the inarguable rise in the incidence of CD in regions of the world that previously had low disease rates, GWAS studies will soon have to shift from a largely Caucasian focus to include populations from other ethnic backgrounds. Future studies should also move beyond conventional studies of disease susceptibility into phenotypically driven ‘within‐cases’ analyses in order to explore the role of genetics in other important aspects of disease biology. These studies are likely to include assessments of prognosis and/or response to treatments and may be critical if personalised medicine is ever to become a reality.
Keywords: Crohn's disease, GWAS, Pharmacogenetics, prognosis, susceptibility
Introduction
Crohn's disease (CD) and ulcerative colitis (UC), collectively termed Inflammatory Bowel Disease (IBD), are some of the most extensively and successfully studied diseases in complex disease genetics. However, genetic studies in CD were not always successful. Initial efforts to identify the genetic determinants of CD were performed using linkage mapping and were largely disappointing, often producing weak or inconsistent signals.1 One exception to this was the discovery of NOD2 as a major CD susceptibility gene2, 3, 4 – a finding that represents one of the few successes of linkage mapping across all diseases. The development of genome‐wide association studies (GWAS), which facilitate a hypothesis‐free comparison of allele frequencies at thousands of single nucleotide polymorphisms (SNPs) between cases and controls (Figure 1), transformed the study of CD genetics and led to the discovery of many CD susceptibility SNPs. Nonetheless, the overall number of hits identified in early, single cohort GWAS studies was typically modest (between 1 and 10) due to relatively small sample sizes and the resulting limitations in study power.5, 6, 7, 8 Fortunately, the combination of a high disease heritability and a strong collaborative spirit between research groups from around the world meant that subsequent meta‐analyses could include much larger numbers of samples and were accordingly far more successful in identifying the genetic determinants of IBD.9, 10, 11, 12 For example, in 2008, the first international meta‐analysis was performed by combining GWAS data from the UK, US and Franco‐Belgian IBD Genetics consortia13 – studies that had individually identified a total of 10 susceptibility loci.5, 6, 8 The gain in power that was afforded by combining these data sets led to the identification of 32 associated loci13 and highlighted the value of collaborating to create larger data sets for analysis. This study was unsurprisingly followed 2 years later by an even larger meta‐analysis – incorporating ~22 000 cases and ~29 000 controls from 13 different countries – in which 71 susceptibility loci were identified.9 Subsequent larger studies, which have taken advantage of more affordable genotyping approaches (such as the Illumina Immunochip), have increased this number even further, with 241 IBD susceptibility loci being confirmed in the most recent analysis.14 Such advances clearly demonstrate the power of GWAS to provide novel insights into disease pathogenesis, particularly when applied to an ever‐increasing sample size.15 Interestingly, the majority of these loci increase the risk of both CD and UC,14 and many of them alter susceptibility to other immune‐mediated diseases as well – including diseases that are known to be related to IBD, such as primary sclerosing cholangitis, psoriasis and ankylosing spondylitis, and others that were previously thought to be unrelated, such as type 1 diabetes and multiple sclerosis.16 Despite this undoubted success, however, it is important to note that most GWAS hits are only proxies for the true causal variant(s) at each locus (with which they are inherited through linkage disequilibrium). Indeed, it was the realisation that entire haplotypes could be interrogated by genotyping just one of their constituent SNPs that helped make GWAS possible in the first place, as it meant that genome‐wide genetic variation could be captured using a tractable number of variants (that could be incorporated into a single genotyping chip) (Figure 1). As such, to identify the functional consequences of these associated haplotypes – and the mechanism by which they alter disease risk – it will be important to identify the causal variant(s) that is ultimately responsible for the association. In some cases, this may be possible using statistical fine mapping – such as has recently been performed by the International Inflammatory Bowel Disease Genetic Consortium (IIBDGC)17 – but in others, some form of functional characterisation of the downstream effects of each SNP in a locus will be necessary.
Figure 1.
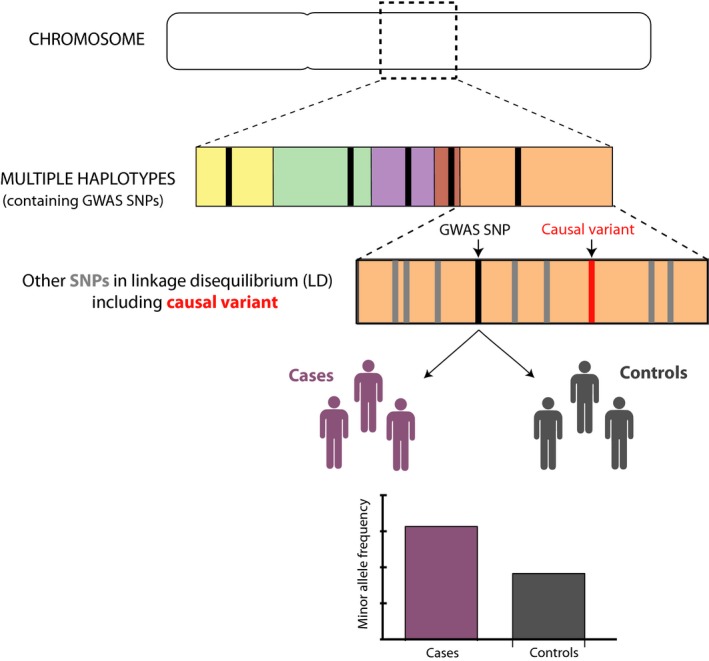
Basic principles of GWAS. GWAS has been made possible because of the haplotype structure of the human genome. Every chromosome consists of multiple haplotypes – regions that are inherited together during meiosis. Within each haplotype, there are typically many SNPs, which are co‐inherited within the larger genetic region, and thus, their alleles are inherited nonrandomly (i.e. they are in linkage disequilibrium). This means that it is possible to infer the genotypes at multiple SNPs within the haplotype (shown in grey) if the genotype at one or more SNPs is known. GWAS SNPs (shown in black) are selected so as to tag each haplotype, but where association is observed, they are unlikely to be the causal variant at the locus (shown in red). By genotyping SNPs from each haplotype in the genome in disease cases and healthy controls, it is possible to identify SNPs where the allele frequency is significantly different between the cases and controls, and which are associated with the disease.
Genetics alone will not provide all of the answers, but if combined with careful study of the downstream biology, it seems likely that new pathways involved in disease biology will be discovered – improving our understanding of disease pathogenesis and potentially providing new opportunities for therapeutic intervention.
Unravelling disease pathogenesis – the genetic contribution
Although the pathophysiology of CD is not fully understood, the current hypothesis assumes a complex interplay between environmental, genetic and intestinal microbial factors, all of which interact with the host's immune system to result in a pathological auto‐inflammatory response directed towards the intestine.18 During the past decade, GWASs have identified multiple SNPs that – by virtue of their proximity to genes known to be involved in specific pathways – have led to the implication of these pathways in disease development. Some of these proposed pathophysiological mechanisms will now be discussed in more detail – especially those that have made important contributions to our current understanding of CD pathogenesis (Table 1).
Table 1.
Summary of IBD GWAS hits involved in autophagy, the IL‐17/IL‐23 axis/type 3 innate lymphoid cells and the failure to suppress of aberrant immune responses
| Chromosome | SNP | P value | OR | AF | Candidate gene in/near locus | |
|---|---|---|---|---|---|---|
| Autophagy | 2 | rs6752107 | 1.42E−73 | 1.25 | 0.55 | ATG16L1 |
| 5 | rs11741861 | 2.94E−37 | 1.25 | 0.05 | IRGM | |
| 12 | rs11564258 | 6.38E−29 | 1.33 | 0.025 | LRRK2 | |
| 16 | rs2066844 | 2.27E−217 | 2.00 | 0.06 | NOD2 | |
| IL‐23/IL‐17 Axis Type 3 innate lymphoid cells | 1 | rs11581607 | 8.76E−175 | 0.46 | 0.05 | IL23R, IL12RB2 |
| 1 | rs4845604 | 1.21E−17 | 0.88 | 0.13 | RORC | |
| 5 | rs56167332 | 7.17E−50 | 1.17 | 0.35 | IL12B | |
| 6 | rs1819333 | 6.76E−21 | 1.08 | 0.48 | CCR6 | |
| 9 | rs75900472 | 4.70E−48 | 1.16 | 0.13 | JAK2 | |
| 12 | rs11614178 | 2.22E−32 | 1.19 | 0.4 | IL22, IFNG | |
| 17 | rs12942547 | 5.51E−22 | 1.1 | 0.55 | STAT3, STAT5A, STAT5B | |
| 19 | rs11879191 | 5.27E−20 | 0.89 | 0.2 | TYK2 | |
| 21 | rs7282490 | 2.35E−26 | 1.1 | 0.39 | ICOSLG | |
| Failure to suppress aberrant immune responses | 1 | rs3024505 | 2.99E−50 | 1.22 | 0.19 | IL10 |
| 4 | rs7657746 | 3.00E−13 | 1.1 | 0.20 | IL2 | |
| 7 | rs1077773 | 5.96E−9 | 0.93 | 0.48 | AHR | |
| 10 | rs12722515 | 3.76E−10 | 1.1 | 0.85 | IL2RA | |
| 15 | rs17293632 | 2.71E−20 | 1.11 | 0.14 | SMAD3 | |
| 16 | rs529866 | 1.73E−16 | 1.12 | 0.81 | SOCS1 | |
| 17 | rs12942547 | 5.51E−22 | 1.1 | 0.55 | STAT3, STAT5A, STAT5B | |
| 18 | rs7240004 | 1.01E−10 | 0.94 | 0.34 | SMAD7 | |
| 21 | rs2284553 | 2.14E−16 | 1.12 | 0.60 | IL10RB |
Autophagy
Prior to genetic studies that identified ATG16L1 and IRGM as susceptibility loci for CD,7, 19 a role for autophagy in disease pathogenesis had not been envisaged. Autophagy is a well‐described and evolutionarily conserved process whereby cytoplasmic components are engulfed by double‐membraned vesicles (autophagosomes) that then fuse with lysozymes in order for their contents (protein aggregates and damaged organelles) to be degraded and recycled.20 Given the importance of intestinal microbes in CD pathogenesis, it is notable that autophagy also plays a role in clearance of intracellular micro‐organisms (a process known as ‘xenophagy’).21, 22 Moreover, autophagy has also been linked to endoplasmic reticulum (ER) stress23 – another process that has been implicated in intestinal inflammation and IBD.24 ER stress has been shown to induce autophagy, and autophagy reciprocally plays a role in resolving ER stress.25 Interestingly, Crohn's disease risk SNPs seem to be able to disrupt this balance, which can lead to intestinal inflammation.26
When autophagy genes were first associated with CD, the mechanism by which autophagy altered disease risk was unknown. Several groups therefore sought to investigate this by genetically targeting ATG16L1 27, 28, 29 – the first autophagy gene to be identified.7 Most of these studies used loss‐of‐function approaches (i.e. ATG16L1 knockout or knock‐down) and while they provided interesting insights into the role of autophagy in intestinal inflammation, it was unclear as to how the results might relate to the risk variant identified by GWAS. This was because unlike most GWAS hits, which are typically noncoding and thus presumed to affect gene expression, the risk SNP in ATG16L1 is a missense mutation that leads to a threonine to alanine substitution at position 300 (rs2241880, Thr300Ala or T300A).7 Missense mutations are generally assumed to affect protein structure or function due to the effect that an amino acid substitution could have on protein folding – but would not be expected to directly affect the amount of the protein, as are typically modelled with loss‐of‐function approaches. However, in this case, it was subsequently shown that the missense mutation does primarily affect the amount of ATG16L1 protein within cells, rather than the function of the protein per se, as the alanine substitution makes ATG16L1 protein more susceptible to caspase‐3‐mediated degradation.30 This in turn reduces ATG16L1 levels, and hence autophagy, during periods of cellular stress (e.g. inflammation) and results in defective clearance of intracellular pathogens and increased inflammatory cytokine production.30 A similar effect on intracellular pathogen clearance was also reported at the IRGM locus. Here, the original GWAS association (rs1000113 C>T) was shown to be in perfect linkage disequilibrium with a synonymous exonic SNP (rs10065172 C>T) that is located within a binding site (‘seed sequence’) for a microRNA, miR‐196. This microRNA was found to be highly expressed within the intestinal epithelia of patients with CD, and its expression was shown to anti‐correlate with IRGM mRNA levels – consistent with a physiological role in repressing IRGM.31 Interestingly, the disease‐associated variant (T) at rs10065172 disrupted the miR‐196 seed sequence, so that miR‐196 could not bind and resulted in carriers of the CD‐associated variant being unable to repress IRGM mRNA levels, which led to impaired autophagy and a failure to clear intracellular bacteria.31 These examples, where the functional consequences of a risk allele have been elucidated, are relatively uncommon, but highlight how the study of genetic variants can uncover specific pathway defects that contribute to CD pathogenesis.
In addition to helping resolve the role of autophagy in CD pathogenesis, the identification of genetic risk variants in known autophagy genes also led to the realisation that other susceptibility genes, which had not previously been linked to autophagy, might actually be involved in this process. For example, NOD2 – the first CD susceptibility gene to be discovered2, 3, 4 – was principally thought to be an intracellular pattern recognition receptor involved in defence against bacteria,32 but it was subsequently shown that NOD2 also interacts with ATG16L1 during autophagosome formation.33, 34, 35
The identification of autophagy genes, and specifically ATG16L1, also helped uncover the important role that Paneth cells are thought to play in Crohn's disease. Paneth cells are located at the base of intestinal crypts and are known to be involved in mucosal defence, producing large amounts of anti‐microbial peptides, including defensins.36 Early studies into the role of autophagy in intestinal inflammation revealed that mice with reduced Atg16l1 expression had morphologically abnormal Paneth cells with reductions in the number and size of secretory granules.37 Similar defects were also noted in CD patients who were homozygous for the ATG16L1 T300A variant.38 Interestingly, Paneth cell dysfunction has also been reported in mice following deletion of a transcription factor (Xbp1), which is involved in the unfolded protein response (UPR) and thereby helps to resolve ER stress.24 Indeed, it was subsequently shown that CD‐like ileitis spontaneously develops in mice if the UPR and autophagy were simultaneously compromised in Paneth cells, suggesting that the balance between ER stress and autophagy is likely to set an important threshold for initiating intestinal inflammation.39
The realisation that autophagy plays an important role in CD has also led to questions about whether pharmacological manipulation of autophagy could be used therapeutically. Studies involving mouse models have shown that rapamycin (sirolimus), which upregulates autophagy by inhibiting mTOR, can ameliorate experimental colitis.40, 41 A beneficial effect of sirolimus has also been reported in a case study of an adult with refractory CD42 and a retrospective case series of children with refractory IBD,43 although no randomised placebo‐controlled trials have yet been performed. This, however, remains an active area of research, not least because sirolimus is also known to also have anti‐fibrotic effects that could be beneficial in CD – a trial investigating this is currently underway (NCT02675153).
The IL‐23/IL‐17 axis, bridging the innate and adaptive immune system
Early studies into the pathogenesis of IBD identified differences in the ratio of T helper (TH)1/TH2 cells in the intestinal mucosa of patients with CD or UC, and corresponding differences in their cytokine profiles.44, 45, 46 CD appeared to be a TH1‐associated disease with increased levels of interferon‐γ and IL‐12, while UC appeared to more TH2‐like – being characterised by increased IL‐5 and IL‐13 production (although not IL‐4). Several animal models supported this hypothesis, with an anti‐IL‐4 antibody showing therapeutic benefit in oxazolone‐induced colitis,47 which histologically resembles UC, while the transmural, CD‐like colitis induced by 2,4,6 trinitrobenzene sulphonic acid could be abrogated by an anti‐IL‐12 antibody48 – a molecule that was subsequently found to be effective in human CD trials.49 However, this hypothesis ultimately proved to be overly simplistic, and it was soon realised that the anti‐IL‐12 antibody would have also blocked IL‐23 due to its target antigen being a common subunit (p40) that is shared between these cytokines.50 Indeed, it was later shown that the effect upon IL‐23, a cytokine important in the development and maintenance of mucosal effector TH17 cells, was responsible for the therapeutic benefit in animal models.51 Activated TH17 cells could also be identified in the blood and intestine of CD patients.52 Around this time, genetics provided further support for a possible role of TH17 cells in CD and UC, with numerous susceptibility variants being identified within or near to genes implicated in TH17 biology (e.g. RORC, IL23R, IL12B, TYK2, JAK2, STAT3, CCR6 and ICOSLG, Table 1).9, 10 However, while these genetic associations are consistent with a role for TH17 cells in CD pathogenesis, it is important to note that the functional consequences of the risk alleles at most of these loci have not yet been elucidated. As such, we still need to confirm that these genes are directly affected by the disease‐associated SNPs at these loci, and also that the functional consequences of the risk alleles specifically affect IL‐23/IL‐17 signalling, as several of these genes are also involved in other pathways (e.g. STAT3 is critical for IL‐10 signalling). Accordingly, the precise role of TH17 cells in CD pathogenesis still needs to be fully characterised, and in this context, it is notable that antibodies which target a key TH17 cytokine, IL‐17, are ineffective in CD53 and yet are highly effective in other TH17‐driven diseases.54
Another reason why the exact role of TH17 cells remains obscure, despite several lines of evidence appearing to implicate them in CD pathogenesis, is because of the discovery of innate lymphoid cells (ILCs). These cells belong to the lymphoid lineage but do not express antigen‐specific receptors and are therefore not part of the adaptive immune system.55 Nonetheless, subsets of ILCs exist that mirror T‐cell subsets in terms of their cytokine secretion profiles.55 Type 3 ILC cells (ILC3s), for example, correspond to TH17 cells in that they can express RORγt, produce IL‐17 and IL‐22, and are responsive to IL‐23.55 Moreover, these cells have been shown to be responsible for bacteria‐driven colitis in mice that lack T cells56 and to selectively accumulate in the intestine of patients with active CD.57 Nevertheless, their specific contribution to CD pathogenesis – as distinct from that of TH17 cells – is difficult to ascertain because not only are they greatly outnumbered by T cells in the intestine, but many of the methods used to deplete them will also affect T cells (and vice versa) making it difficult to disentangle their relative contributions. To complicate things further, it appears that other ILC subsets may also play a role in CD pathogenesis. For example, type 1 ILCs (ILC1s), which resemble TH1 cells in that they express the transcription factor T‐bet, respond to IL‐12 and can produce interferon‐γ, are substantially increased in the intestine of patients with CD.58 For this reason, although the exact role of ILCs in IBD pathogenesis has not been fully established, this remains an active area of research – especially as they might provide tissue‐specific therapeutic targets for some patients.
The failure to suppress aberrant immune responses
It has been estimated that the human body contains ~40 trillion bacteria, of which the vast majority are located within the intestine.59 This means that there are actually more bacteria in our bodies than human cells and illustrates why the intestine presents such a challenge to the immune system. In order to be able to mount an appropriate immune response to intestinal pathogens, for example, there have to also be mechanisms to prevent aberrant responses to nonpathogenic gut commensals in order to preserve health. Disrupting such control mechanisms is known to lead to colitis in animal models – for example, if the Il10 gene is deleted60 or if regulatory T cells are absent.61 Results from GWAS studies have demonstrated that disruption of these anti‐inflammatory mechanisms is also likely to be involved in the development of CD. For example, several genes involved in IL‐10 signalling (the anti‐inflammatory cytokine which when deleted leads to colitis in animal models) lie near to GWAS hits, including IL10, IL10RB, STAT3 and TYK2.11, 62 Although the functional consequences of these SNPs have not been determined, the notion that IL‐10 signalling may be defective in IBD is supported by other genetic studies. For example, private missense mutations in genes encoding components of the IL‐10 receptor (IL10RA or IL10RB) have also been shown to cause a severe, early‐onset form of IBD in consanguineous families due to abrogation of IL‐10 signalling.63 Other anti‐inflammatory cytokines – and genes involved in their signalling pathways – have also been identified within CD‐associated genetic loci, including IL22 (another member of the IL‐10 cytokine family) and SMAD3 and SMAD7, which are important components of TGFβ signalling. Indeed, SMAD7, which inhibits TGFβ signalling and is expressed at high levels in the intestines of patients with IBD,64 has been shown to be an effective therapeutic target via an antisense oligonucleotide (Mongersen) that binds to and degrades SMAD7 mRNA.65
Although several cell types are known to play a role in suppressing exuberant immune responses, including M‐2 macrophages, certain ILC populations and regulatory B cells, by far the most studied immunosuppressive cell type are regulatory T cells (Tregs). Several GWAS hits also highlight the importance of Tregs in IBD pathogenesis. For example, it is known that IL‐2 signalling is critical for maintaining Treg numbers66 and for mediating their suppressor function via activation of STAT5,67 and GWASs have identified IBD‐associated loci that contain IL2, IL2RA (part of the IL‐2 receptor) and STAT5.11 Moreover, IL‐10 signalling – discussed earlier – is also important in conferring Tregs with the ability to suppress TH17‐mediated inflammation68 and in mediating that suppression.69 Additionally, there is a growing realisation that plasticity exists within T‐cell subsets and particularly between Tregs and TH17 cells, with evidence that TH17 cells can transdifferentiate into Tregs as inflammation resolves – a process that involves signalling through the aryl‐hydrocarbon receptor (AHR), which also harbours a genetic association with IBD.12 Further evidence of the role of Tregs in intestinal inflammation can also be found in patients who carry rare missense mutations in FOXP3, which encodes the master transcription factor for naturally occurring Tregs. The resulting syndrome, termed ‘immunodysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome’ or ‘IPEX’,70 is characterised by an autoimmune enteropathy as well as several other severe autoimmune phenomena.
Based on these and other insights, Tregs are now also being studied as potential therapies in IBD. Different strategies have been proposed including inducing Tregs ex vivo from naïve T cells using an unrelated antigen71 or isolating thymically derived Tregs and expanding them before re‐infusing them back into patients.72 These expanded Tregs have been shown to be able to suppress the activation and proliferation of intestinal T cells from CD patients in vitro and to have an epigenetically stable FOXP3 locus, theoretically limiting the possibility for TH17 conversion.72 However, full clinical trials using these approaches have yet to be performed, and it should be noted that in other disease areas, infused regulatory T cells have not shown the same efficacy as had been observed in vitro or in animal models.73
Overlap with other immune‐mediated diseases – shared features of autoimmunity?
In addition to highlighting pathways that are likely to be important in the development of CD, GWAS has also provided insights into how the genetic architecture of CD relates to that of other autoimmune diseases.16 For example, while most IBD susceptibility loci are shared between CD and UC, it is notable that SNPs that are implicated in autophagy seem to be specific to CD, while SNPs that are located in, or near to, genes involved in epithelial barrier function tend to be more UC specific.74 Interesting overlaps have also been noted in other nonintestinal diseases. This is perhaps best exemplified by the IL‐23/IL‐17 axis, to which multiple autoimmune diseases have been linked, including psoriasis (SNPs in or near to IL23R, IL12B, TYK2 and STAT3), ankylosing spondylitis (SNPs in or near to IL23R, IL12B, TYK2 and JAK2) and multiple sclerosis (SNPs in or near to IL12B, IL12A, TYK2 and STAT3). Overlap with some of these diseases, such as psoriasis or ankylosing spondylitis, was unsurprising since these diseases share certain clinical and pathogenic features with CD, although overlap with other diseases, such as multiple sclerosis, was less expected. However, it is important to note that interpreting this overlap is not straightforward. Indeed, just because the same gene is implicated in several diseases does not mean that common genetic haplotypes (and biological mechanisms) are responsible for these associations. For example, although both CD and psoriasis are both associated with multiple SNPs in TYK2, these signals are largely distinct.11, 75 Indeed, the only shared susceptibility SNP at this locus (rs12720356, a missense variant) shows discordant effects, with the risk allele for CD being protective for psoriasis. Discordant effects at the same SNP are surprisingly common in autoimmune disease genetics and present an additional challenge to understanding how genetic variation can influence disease susceptibility. For example, a SNP in STAT3, rs744166, is associated with both CD and multiple sclerosis, but the risk allele for CD is protective for multiple sclerosis and vice versa.13, 76 As such, while it is undoubtedly interesting that overlapping associations are detectable across distinct diseases – either at the SNP or gene level – much more work needs to be done to determine the true extent of any biological overlap. This work will be particularly important given emerging evidence that drugs targeting these ‘shared’ pathways may have very different effects between diseases, such as the anti‐IL17 antibody secukinumab, which is highly effective in psoriasis but entirely ineffective in CD.53, 54
More work to do
Despite the successes described above, much more work needs to be done to fulfil the original goal of GWAS in Crohn's disease – to learn how genetic variation contributes to disease biology. For example, because our ability to identify disease‐associated variants has far outstripped our ability to characterise their functional effects, we still do not know the biological consequences for most of the associated variants. This bottleneck has led to the temptation to assume that we know which gene is mediating the effect at a particular locus – often because one particular gene might be involved in a familiar aspect of disease biology – but the reality is that there is often little or no evidence to support this. Indeed, in many cases, we are not even certain of which SNP – or combination of SNPs – is responsible for driving the genetic association since the SNPs included on GWAS chips are simply tags for haplotypes that may contain hundreds of associated variants (Figure 1). Fine‐mapping studies have sought to resolve the genetic associations within GWAS regions, with some success,11, 17 although strong linkage disequilibrium has prevented the identification of a single causal variant for most loci.17 Similarly, there have been several efforts to understand how genetic variation at a locus might affect the expression of nearby genes – especially since most GWAS hits (and their associated haplotypes) do not appear to affect the coding sequence of genes. This has commonly been done by combining genotypic data with transcriptomic data in order to identify correlations between SNP genotypes and gene expression. However, even when genetic variants are identified that correlate with gene expression (termed expression quantitative trait loci or eQTLs), strong linkage disequilibrium usually means that it is not possible to identify which of the SNPs within the haplotype is responsible for the observed effect.77, 78 As such, while these studies can be useful for identifying the genes that are affected by genotype at a particular SNP, they usually cannot identify the causal SNP within the haplotype. Moreover, these studies have revealed that many eQTL effects are only detectable in a specific cell type and/or in the presence of a specific stimulus,77, 78 meaning that a causal variant could easily be overlooked if the wrong tissue or condition is being examined.79
For these reasons, a major goal of future studies should be to functionally dissect the consequences of disease‐associated genetic variation at each locus in order to avoid incorrectly attributing an association with a gene that is not involved and/or overlooking an important gene that is involved in an unknown aspect of disease biology because it might appear irrelevant. This is no small task, as it will need to include identification of both the cell types in which the effect is relevant and also the context (e.g. during a particular stage in development or in the presence of a particular stimulus). Examples of such studies have been performed in other fields,80, 81, 82 but will need to be embraced by IBD researchers in order to understand the underlying mechanisms and functional consequences of disease‐associated noncoding genetic variation (Figure 2).
Figure 2.
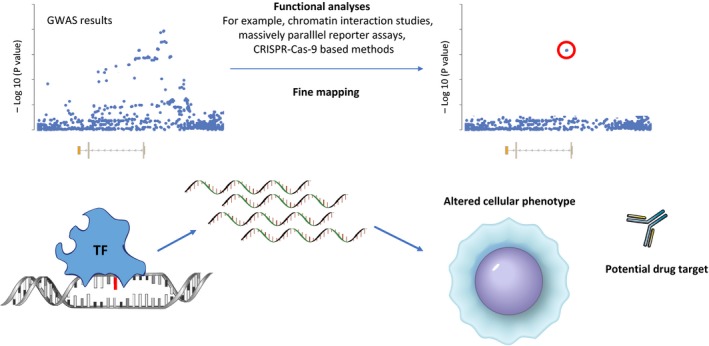
Moving from SNPs to biology. Due to linkage disequilibrium, it is not possible to know which is the causal SNP at an associated locus without additional studies such as fine‐mapping or functional analyses to systematically dissect the effects of each variant. Once this is known the mechanism by which the effect occurs and the downstream cellular consequences can be determined – for example, a SNP might introduce a binding site for a tissue‐specific transcription factor (TF) and the resulting effect on gene expression could confer an altered cellular phenotype that could provide new insights into disease biology or provide an opportunity for therapy.
Another ongoing challenge in CD genetics is to understand why the genetic variants identified to date account for only a small proportion of the variance of CD (the portion of heritability attributable to genetic variation). It has been estimated that even with such a high number of disease‐associated SNPs, we have only accounted for 26% of CD variance.83 The remainder, which has been termed ‘missing heritability’, still has to be accounted for, and there are several hypotheses that might explain this. First, because GWAS studies perform a univariate analysis, they cannot account for gene–gene interactions (epistasis) that might substantially alter the effect size of a given mutation. For example, in psoriasis, an epistatic interaction has been identified between genetic variants in ERAP1 and MHC‐C, which has a much larger effect size that either of the variants in isolation.84 Second, gene–environment interactions would similarly be expected to alter the effect size attributed to a particular variant if the environmental condition specifically interacted with the pathway affected by a causal SNP. For example, tobacco smoke – a known environmental risk factor for CD – has been shown to modify the association between 64 SNPs and CD, including NOD2 and the HLA region.85 Third, despite the name, most GWAS chips do not actually provide genome‐wide coverage. As such, it is likely that some of the missing heritability simply lies in regions that have not been analysed to date, either because they are difficult to impute or because they are poorly covered on GWAS chips.15 For instance, GWAS studies in CD have not yet analysed any SNPs on the sex chromosomes, even though these are likely to contain some disease‐associated variants.86 Indeed, one of the few studies to include the chromosome‐X was a meta‐analysis of 10 paediatric diseases, including CD and UC, which identified a SNP in CD40LG, an X chromosome gene, that was associated with CD, UC and coeliac disease.87 Similarly, some regions, such as the Fc receptor gene cluster or the KIR genes, contain highly repetitive sequences are difficult to genotype using standard approaches. Fourth, it has been proposed that rare variants – which are not included on GWAS chips but which could have much larger effect sizes – might account for some of the missing heritability. However, several studies have now been performed to attempt to identify rare disease‐associated variants, but without much success88, 89 – mirroring the situation in other diseases.90 Fifth, because most GWAS SNPs are proxies for the true causal variant, estimates of variance based on these are likely to underestimate the true attributable risk at each locus. With the introduction of fine‐mapping studies, though, this problem should soon be resolved.
All of these possibilities seek to address the issue of missing heritability from the perspective of identifying missing disease risk. However, it is equally possible that some of the missing heritability might be a consequence of our heritability calculations being incorrect. For instance, estimates of SNP heritability – the proportion of disease variance accounted for by risk SNPs – assume that gene–gene interactions do not occur, even though it is widely accepted that they probably do. If heritability calculations are performed using a model that allows epistasis, then it has been shown that up to 80% of the missing heritability in CD can be accounted for.91 Similarly, if the effects of minor allele frequency (MAF), LD and genotype certainty are factored into calculations of SNP heritability, this also tends to lead to higher estimates.92
GWAS in different populations
During most of the twentieth century, the incidence of CD rose steadily in the Western world but was relatively low in developing countries.93 However, recently the incidence of CD in newly industrialised regions has been rising rapidly – particularly in South East Asia, South America and the Middle East – such that previously noted ethnic differences appear to be narrowing.93 This increase in the incidence of CD is also detectable when comparing first‐ and second‐generation immigrants from low risk areas who emigrate to countries with higher incidence of IBD94 and highlights the importance of environmental factors in CD development. Notably, the vast majority of GWAS studies in IBD have been performed in Caucasian populations (indeed GWAS chips were mostly designed based on Caucasian haplotype structure). Of the few studies that have been performed in individuals of non‐Caucasian descent, the results suggest that there may be some important population‐based differences in the genetic risk of CD. For example, a meta‐analysis of Asian genetic studies revealed that variants in ATG16L1 and NOD2, which are associated with CD in Caucasian populations, were not associated in Han Chinese, Japanese, South Korean, Indian and Malaysian CD populations.95 Similarly, a GWAS in Ashkenazi Jewish CD patients identified five novel genetic regions not previously found in non‐Jewish Caucasian CD populations.96 A recent trans‐ethnic GWAS incorporating 86 640 individuals of European descent and 9846 individuals of East‐Asian, Indian or Iranian descent also highlighted a shared genetic risk across European and non‐European cohorts for most IBD risk loci, although genetic heterogeneity was observed – both in terms of allele frequency and/or effect size – between at several loci, including NOD2, TNFSF15, ATG16L1, IL23R and IRGM.12 In contrast, a GWAS in 2345 African Americans with IBD confirmed known risk loci but did not reveal any new variants associated with CD susceptibility.97 Clearly, larger studies will be necessary to better explore the commonalities and differences in the genetic variance of CD between populations, and these should ideally be performed using GWAS chips that are predicated upon the haplotype structure of the population being studied or whole genome sequencing, so as not to overlook the contribution of variants that are specific to non‐Caucasian populations.
Pharmacogenetics
Aside from disease susceptibility, GWASs have also been used to try to identify genetic variants that are associated with specific side effects to medications commonly used to treat CD. Interest in this area was initially driven by studies in other fields that identified low‐frequency variants with large effect sizes that were responsible for well‐recognised side effects98 and by previous work that had demonstrated the clinical value of genotyping for mutations in TPMT that alter the risk of thiopurine‐induced myelosuppression.99 GWAS studies have since identified genetic variants in NUDT15 that alter the risk of thiopurine‐induced myelosuppression,100 and in the MHC that alter the risk of thiopurine‐induced pancreatitis (HLA‐DQA1*02:01‐HLA‐DRB1*07:01)101 and 5‐ASA‐induced nephrotoxicity (HLA‐DRB1*03:01).102 Interestingly, the HLA‐DRB1*03 allele has also been associated with the development of antibodies against infliximab – an anti‐TNF‐α monoclonal antibody that is one of the most effective treatments for CD. Patients carrying the risk haplotype were almost seven times more likely to develop antibodies, which could potentially neutralise the drug and lead to a loss of efficacy.103
In addition to studying the genetic contribution to drug side effects, efforts have also been made to identify a genetic contribution to treatment response. So far, however, most of these studies have used a candidate gene approach, investigating individual genes or groups of genes – often those linked to CD susceptibility – rather than adopting a genome‐wide approach.104 Similar to disease susceptibility, it seems unlikely that a single variant will determine the efficacy of CD therapies, and therefore, a genome‐wide approach will be important. Moreover, there is no reason to suspect that variants influencing response to treatment would be the same as those that alter disease susceptibility, and thus, it was unsurprising when a polygenic risk score based on 140 CD risk loci did not associate with response to anti‐TNF therapy.105 To date, a genome‐wide study of response to anti‐TNF therapy has not been performed in IBD, although a study based on the Illumina immunochip (a custom designed genotyping chip containing variants that have been associated with autoimmune diseases) did provide some encouraging results,106 though further validation and larger independent discovery cohorts will be important. In rheumatoid arthritis (RA), such a study has already been performed and identified eight loci that were associated with response to infliximab.107 A similar study has also been reported in abstract form for ustekinumab, an anti‐IL12/23 antibody, in which two loci were shown to be associated with response to therapy in CD patients.108
In summary, although attempts to understand the genetic contribution to side effects or treatment response are far less advanced than equivalent studies in disease susceptibility, early studies have provided plenty of reason for optimism. Indeed, if genome‐wide approaches were to be adopted in suitably powered cohorts, it seems probable that genetic variants will be discovered that could both shed light on the biology responsible for responses to treatment and also potentially provide useful biomarkers for screening.
Subphenotypes: moving beyond disease susceptibility
Despite the success of GWAS studies into the genetic contribution to disease susceptibility, there are several other aspects of CD biology that have been proposed to have a genetic component but which – until recently – had not been studied by GWAS. For example, the distribution of Crohn's disease within the intestine and the clinical course of disease following diagnosis (prognosis) have both been shown to follow similar patterns within families109 but have been largely overlooked in favour of successively larger meta‐analyses into disease susceptibility. Part of the reason for this has been the assumption that susceptibility variants would also influence other aspects of disease.110 For example, it was assumed that the total burden of susceptibility variants would determine disease course, so that individuals who carry more risk SNPs would experience a worse prognosis. This hypothesis has been shown to be correct for qualitative traits such as height,111 but in CD – and other diseases – it would only be correct if susceptibility and prognosis were on a continuous spectrum and thus influenced by the same variants. If, however, this assumption is incorrect, then it would be very unlikely for SNPs, which influence disease prognosis to be identified through comparison of unstratified cases with controls.112 Instead, it would be necessary to directly compare subsets of patients based on phenotypic differences – a so‐called within‐cases analysis (Figure 3).
Figure 3.
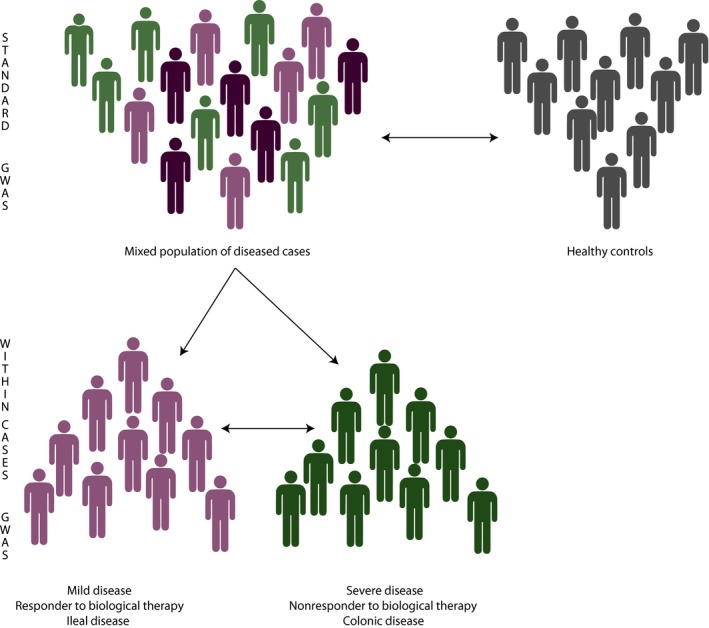
The difference between a conventional GWAS analysis and ‘within‐cases’ analysis. Schematic depicting the different analytical strategies employed by a conventional (susceptibility) GWAS in which the genetic profiles of cases and ethnically matched controls are compared, and the design of ‘within‐cases’ GWAS in which the genetic profiles of patients with distinct (and ideally contrasting) subphenotypes of a disease are compared.
To date, there have only been a few examples of using within‐cases analyses in CD genetics. The largest study was performed by the International IBD Genetics Consortium and investigated the contribution of a large number of autoimmune disease‐associated SNPs to CD distribution, age of onset and behaviour (i.e. inflammatory, stricturing or fistulating). Although this identified little or no genetic association with disease behaviour, it did identify three loci that were associated with disease location (NOD2, MHC and MST1) and showed that a genetic risk score representing the sum of all known risk alleles for IBD showed a strong association with disease location.113
Despite the importance of CD prognosis in determining patient well‐being, most of the studies into the genetic contribution to prognosis have focused on small numbers of susceptibility SNPs in relatively small numbers of patients and have unsurprisingly provided inconsistent results.114, 115, 116 One of the few replicable associations identified was between variants in NOD2 and need for surgery,117 although this was subsequently shown to be entirely driven by the association between NOD2 and ileal disease (which is most commonly treated with surgery). Indeed, if this analysis is stratified by disease location, then no association between disease course and NOD2 variants is detectable.113 For this reason, we elected to investigate the genetics of CD prognosis using a within‐cases analysis in which the genetic profiles of patients with contrasting courses of CD would be compared. This was first done as a candidate gene study and identified a noncoding SNP in FOXO3 that was not a disease susceptibility variant, but which was associated with a milder course of CD118 – an association that has since been replicated.119, 120 Functional studies into the biological consequences of this SNP, which does not affect disease susceptibility, identified a FOXO3‐driven pathway that abrogated inflammatory responses in monocytes via TGFβ1 and led to reduced TNF‐α and IL‐6 production in carriers of the mild CD‐associated allele.118 Interestingly, this SNP was also shown to associate with good prognosis in RA (another TNF‐α‐driven disease121) and with poor prognosis in malaria (in which TNF‐α is anti‐parasitic).118 Since then, this variant has also been independently associated with clinical outcome in tuberculosis,122 another disease in which TNF‐α is known to be important. Based on this result, we then extended the analysis to a genome‐wide level and identified a further three loci that were significantly associated with prognosis in CD (XACT, a region upstream of IGFBP1 and the MHC region).120 Strikingly, none of these variants were associated with disease susceptibility, and conversely, none of the CD susceptibility variants were associated with prognosis in our analysis, either individually or collectively. This demonstrated that the genetic contribution to prognosis comes from loci that are distinct from those that drive disease development, which in turn has important implications for our understanding of disease pathogenesis and could provide new opportunities for drug development and/or personalised medicine. In the future, larger within‐cases analyses will be important to further resolve the genetic contribution to clinically important disease subphenotypes, but the success of these will critically depend on there being detailed, high‐quality and consistent phenotypic data, else they are likely to provide (falsely) negative results.
Conclusions
During the past decade, our understanding of CD pathogenesis has benefitted greatly from the advent of GWAS – success that has been possible thanks to large‐scale international collaboration. This has resulted in several novel insights into disease biology, which are now being investigated as therapeutic targets. However, it is important not to think that the job is done. There is much work left to do in order to fulfil the original goals of GWAS and learn more about disease biology, from determining the functional consequences at each associated locus to exploring genetic risk in other populations to identifying the genetic contribution to other important aspects of disease biology, such as prognosis. Much has been accomplished since the first CD GWAS 10 years ago, but much more should be accomplishable in the next 10 years if we make the most of the discoveries to date.
Acknowledgments
BV is a doctoral fellow of the Research Foundation Flanders, Belgium (11Y9418N). KGCS is an NIHR Senior Investigator. JCL is supported by a Wellcome Trust Intermediate Clinical Fellowship (105920/Z/14/Z).
Conflict of interest
We declare no conflict of interests.
References
- 1. Umicevic Mirkov M, Verstockt B, Cleynen I. Genetics of inflammatory bowel disease: beyond NOD2. Lancet Gastroenterol Hepatol 2017; 2: 224–234. [DOI] [PubMed] [Google Scholar]
- 2. Hugot JP, Laurent‐Puig P, Gower‐Rousseau C et al Mapping of a susceptibility locus for Crohn's disease on chromosome 16. Nature 1996; 379: 821–823. [DOI] [PubMed] [Google Scholar]
- 3. Hugot JP, Chamaillard M, Zouali H et al Association of NOD2 leucine‐rich repeat variants with susceptibility to Crohn's disease. Nature 2001; 411: 599–603. [DOI] [PubMed] [Google Scholar]
- 4. Ogura Y, Bonen DK, Inohara N et al A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 2001; 411: 603–606. [DOI] [PubMed] [Google Scholar]
- 5. Wellcome Trust Case Control Consortium . Genome‐wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007; 447: 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Libioulle C, Louis E, Hansoul S et al Novel Crohn disease locus identified by genome‐wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet 2007; 3: e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hampe J, Franke A, Rosenstiel P et al A genome‐wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 2007; 39: 207–211. [DOI] [PubMed] [Google Scholar]
- 8. Duerr RH, Taylor KD, Brant SR et al A genome‐wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006; 314: 1461–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Franke A, McGovern DP, Barrett JC et al Genome‐wide meta‐analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet 2010; 42: 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anderson CA, Boucher G, Lees CW et al Meta‐analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet 2011; 43: 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jostins L, Ripke S, Weersma RK et al Host‐microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491: 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu JZ, van Sommeren S, Huang H et al Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015; 47: 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barrett JC, Hansoul S, Nicolae DL et al Genome‐wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet 2008; 40: 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Lange KM, Moutsianas L, Lee JC et al Genome‐wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 2017; 49: 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee JC, Parkes M. Genome‐wide association studies and Crohn's disease. Brief Funct Genomics 2011; 10: 71–76. [DOI] [PubMed] [Google Scholar]
- 16. Parkes M, Cortes A, van Heel DA et al Genetic insights into common pathways and complex relationships among immune‐mediated diseases. Nat Rev Genet 2013; 14: 661–673. [DOI] [PubMed] [Google Scholar]
- 17. Huang H, Fang M, Jostins L et al Fine‐mapping inflammatory bowel disease loci to single‐variant resolution. Nature 2017; 547: 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol 2016; 13: 13–27. [DOI] [PubMed] [Google Scholar]
- 19. Parkes M, Barrett JC, Prescott NJ et al Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet 2007; 39: 830–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pavel M, Rubinsztein DC. Mammalian autophagy and the plasma membrane. FEBS J 2017; 284: 672–679. [DOI] [PubMed] [Google Scholar]
- 21. Levine B. Eating oneself and uninvited guests: autophagy‐related pathways in cellular defense. Cell 2005; 120: 159–162. [DOI] [PubMed] [Google Scholar]
- 22. Gutierrez MG, Master SS, Singh SB et al Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119: 753–766. [DOI] [PubMed] [Google Scholar]
- 23. Yorimitsu T, Nair U, Yang Z et al Endoplasmic reticulum stress triggers autophagy. J Biol Chem 2006; 281: 30299–30304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaser A, Lee AH, Franke A et al XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008; 134: 743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kruse KB, Brodsky JL, McCracken AA. Autophagy: an ER protein quality control process. Autophagy 2006; 2: 135–137. [DOI] [PubMed] [Google Scholar]
- 26. Tschurtschenthaler M, Adolph TE, Ashcroft JW et al Defective ATG16L1‐mediated removal of IRE1alpha drives Crohn's disease‐like ileitis. J Exp Med 2017; 214: 401–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cadwell K, Patel KK, Maloney NS et al Virus‐plus‐susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell 2010; 141: 1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saitoh T, Fujita N, Jang MH et al Loss of the autophagy protein Atg16L1 enhances endotoxin‐induced IL‐1beta production. Nature 2008; 456: 264–268. [DOI] [PubMed] [Google Scholar]
- 29. Sorbara MT, Ellison LK, Ramjeet M et al The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy‐independent manner. Immunity 2013; 39: 858–873. [DOI] [PubMed] [Google Scholar]
- 30. Murthy A, Li Y, Peng I et al A Crohn's disease variant in Atg16 l1 enhances its degradation by caspase 3. Nature 2014; 506: 456–462. [DOI] [PubMed] [Google Scholar]
- 31. Brest P, Lapaquette P, Souidi M et al A synonymous variant in IRGM alters a binding site for miR‐196 and causes deregulation of IRGM‐dependent xenophagy in Crohn's disease. Nat Genet 2011; 43: 242–245. [DOI] [PubMed] [Google Scholar]
- 32. Kobayashi KS, Chamaillard M, Ogura Y et al Nod2‐dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005; 307: 731–734. [DOI] [PubMed] [Google Scholar]
- 33. Cooney R, Baker J, Brain O et al NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 2010; 16: 90–97. [DOI] [PubMed] [Google Scholar]
- 34. Homer CR, Richmond AL, Rebert NA et al ATG16L1 and NOD2 interact in an autophagy‐dependent antibacterial pathway implicated in Crohn's disease pathogenesis. Gastroenterology 2010; 139: 1630–1641, 1641 e1631‐1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Travassos LH, Carneiro LA, Ramjeet M et al Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 2010; 11: 55–62. [DOI] [PubMed] [Google Scholar]
- 36. Ayabe T, Satchell DP, Wilson CL et al Secretion of microbicidal alpha‐defensins by intestinal Paneth cells in response to bacteria. Nat Immunol 2000; 1: 113–118. [DOI] [PubMed] [Google Scholar]
- 37. Cadwell K, Liu JY, Brown SL et al A key role for autophagy and the autophagy gene Atg16 l1 in mouse and human intestinal Paneth cells. Nature 2008; 456: 259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deuring JJ, Fuhler GM, Konstantinov SR et al Genomic ATG16L1 risk allele‐restricted Paneth cell ER stress in quiescent Crohn's disease. Gut 2014; 63: 1081–1091. [DOI] [PubMed] [Google Scholar]
- 39. Adolph TE, Tomczak MF, Niederreiter L et al Paneth cells as a site of origin for intestinal inflammation. Nature 2013; 503: 272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Farkas S, Hornung M, Sattler C et al Rapamycin decreases leukocyte migration in vivo and effectively reduces experimentally induced chronic colitis. Int J Colorectal Dis 2006; 21: 747–753. [DOI] [PubMed] [Google Scholar]
- 41. Macias‐Ceja DC, Cosin‐Roger J, Ortiz‐Masia D et al Stimulation of autophagy prevents intestinal mucosal inflammation and ameliorates murine colitis. Br J Pharmacol 2017; 174: 2501–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Massey DC, Bredin F, Parkes M. Use of sirolimus (rapamycin) to treat refractory Crohn's disease. Gut 2008; 57: 1294–1296. [DOI] [PubMed] [Google Scholar]
- 43. Mutalib M, Borrelli O, Blackstock S et al The use of sirolimus (rapamycin) in the management of refractory inflammatory bowel disease in children. J Crohns Colitis 2014; 8: 1730–1734. [DOI] [PubMed] [Google Scholar]
- 44. Breese E, Braegger CP, Corrigan CJ et al Interleukin‐2‐ and interferon‐gamma‐secreting T cells in normal and diseased human intestinal mucosa. Immunology 1993; 78: 127–131. [PMC free article] [PubMed] [Google Scholar]
- 45. Niessner M, Volk BA. Altered Th1/Th2 cytokine profiles in the intestinal mucosa of patients with inflammatory bowel disease as assessed by quantitative reversed transcribed polymerase chain reaction (RT‐PCR). Clin Exp Immunol 1995; 101: 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fuss IJ, Neurath M, Boirivant M et al Disparate CD4 + lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN‐gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL‐5. J Immunol 1996; 157: 1261–1270. [PubMed] [Google Scholar]
- 47. Boirivant M, Fuss IJ, Chu A et al Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J Exp Med 1998; 188: 1929–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Neurath MF, Fuss I, Kelsall BL et al Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med 1995; 182: 1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mannon PJ, Fuss IJ, Mayer L et al Anti‐interleukin‐12 antibody for active Crohn's disease. N Engl J Med 2004; 351: 2069–2079. [DOI] [PubMed] [Google Scholar]
- 50. Yen D, Cheung J, Scheerens H et al IL‐23 is essential for T cell‐mediated colitis and promotes inflammation via IL‐17 and IL‐6. J Clin Invest 2006; 116: 1310–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hue S, Ahern P, Buonocore S et al Interleukin‐23 drives innate and T cell‐mediated intestinal inflammation. J Exp Med 2006; 203: 2473–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kleinschek MA, Boniface K, Sadekova S et al Circulating and gut‐resident human Th17 cells express CD161 and promote intestinal inflammation. J Exp Med 2009; 206: 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hueber W, Sands BE, Lewitzky S et al Secukinumab, a human anti‐IL‐17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double‐blind placebo‐controlled trial. Gut 2012; 61: 1693–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Langley RG, Elewski BE, Lebwohl M et al Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med 2014; 371: 326–338. [DOI] [PubMed] [Google Scholar]
- 55. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol 2016; 17: 765–774. [DOI] [PubMed] [Google Scholar]
- 56. Buonocore S, Ahern PP, Uhlig HH et al Innate lymphoid cells drive interleukin‐23‐dependent innate intestinal pathology. Nature 2010; 464: 1371–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Geremia A, Arancibia‐Carcamo CV, Fleming MP et al IL‐23‐responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med 2011; 208: 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bernink JH, Peters CP, Munneke M et al Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 2013; 14: 221–229. [DOI] [PubMed] [Google Scholar]
- 59. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 2016; 14: e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rennick D, Davidson N, Berg D. Interleukin‐10 gene knock‐out mice: a model of chronic inflammation. Clin Immunol Immunopathol 1995; 76: S174–S178. [DOI] [PubMed] [Google Scholar]
- 61. Powrie F, Leach MW, Mauze S et al Phenotypically distinct subsets of CD4 + T cells induce or protect from chronic intestinal inflammation in C. B‐17 scid mice. Int Immunol 1993; 5: 1461–1471. [DOI] [PubMed] [Google Scholar]
- 62. Moore KW, de Waal MR, Coffman RL et al Interleukin‐10 and the interleukin‐10 receptor. Annu Rev Immunol 2001; 19: 683–765. [DOI] [PubMed] [Google Scholar]
- 63. Glocker EO, Kotlarz D, Boztug K et al Inflammatory bowel disease and mutations affecting the interleukin‐10 receptor. N Engl J Med 2009; 361: 2033–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Monteleone G, Kumberova A, Croft NM et al Blocking Smad7 restores TGF‐beta1 signaling in chronic inflammatory bowel disease. J Clin Invest 2001; 108: 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Monteleone G, Neurath MF, Ardizzone S et al Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn's disease. N Engl J Med 2015; 372: 1104–1113. [DOI] [PubMed] [Google Scholar]
- 66. Fontenot JD, Rasmussen JP, Gavin MA et al A function for interleukin 2 in Foxp3‐expressing regulatory T cells. Nat Immunol 2005; 6: 1142–1151. [DOI] [PubMed] [Google Scholar]
- 67. Chinen T, Kannan AK, Levine AG et al An essential role for the IL‐2 receptor in Treg cell function. Nat Immunol 2016; 17: 1322–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chaudhry A, Samstein RM, Treuting P et al Interleukin‐10 signaling in regulatory T cells is required for suppression of Th17 cell‐mediated inflammation. Immunity 2011; 34: 566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huber S, Gagliani N, Esplugues E et al Th17 cells express interleukin‐10 receptor and are controlled by Foxp3(−) and Foxp3 + regulatory CD4 + T cells in an interleukin‐10‐dependent manner. Immunity 2011; 34: 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bennett CL, Christie J, Ramsdell F et al The immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001; 27: 20–21. [DOI] [PubMed] [Google Scholar]
- 71. Desreumaux P, Foussat A, Allez M et al Safety and efficacy of antigen‐specific regulatory T‐cell therapy for patients with refractory Crohn's disease. Gastroenterology 2012; 143: 1207–1217 e1201‐1202. [DOI] [PubMed] [Google Scholar]
- 72. Canavan JB, Scotta C, Vossenkamper A et al Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for Crohn's disease. Gut 2016; 65: 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bluestone JA, Buckner JH, Fitch M et al Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 2015; 7: 315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Barrett JC, Lee JC, Lees CW et al Genome‐wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet 2009; 41: 1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stuart PE, Nair RP, Tsoi LC et al Genome‐wide association analysis of psoriatic arthritis and cutaneous psoriasis reveals differences in their genetic architecture. Am J Hum Genet 2015; 97: 816–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jakkula E, Leppa V, Sulonen AM et al Genome‐wide association study in a high‐risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am J Hum Genet 2010; 86: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ye CJ, Feng T, Kwon HK et al Intersection of population variation and autoimmunity genetics in human T cell activation. Science 2014; 345: 1254665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fairfax BP, Humburg P, Makino S et al Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 2014; 343: 1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Peters JE, Lyons PA, Lee JC et al Insight into Genotype‐Phenotype Associations through eQTL Mapping in Multiple Cell Types in Health and Immune‐Mediated Disease. PLoS Genet 2016; 12: e1005908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ulirsch JC, Nandakumar SK, Wang L et al Systematic functional dissection of common genetic variation affecting red blood cell traits. Cell 2016; 165: 1530–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Oldridge DA, Wood AC, Weichert‐Leahey N et al Genetic predisposition to neuroblastoma mediated by a LMO1 super‐enhancer polymorphism. Nature 2015; 528: 418–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Canver MC, Smith EC, Sher F et al BCL11A enhancer dissection by Cas9‐mediated in situ saturating mutagenesis. Nature 2015; 527: 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chen GB, Lee SH, Brion MJ et al Estimation and partitioning of (co)heritability of inflammatory bowel disease from GWAS and immunochip data. Hum Mol Genet 2014; 23: 4710–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Genetic Analysis of Psoriasis Consortium , the Wellcome Trust Case Control Consortium , Strange A et al A genome‐wide association study identifies new psoriasis susceptibility loci and an interaction between HLA‐C and ERAP1. Nat Genet 2010; 42: 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yadav P, Ellinghaus D, Remy G et al Genetic factors interact with tobacco smoke to modify risk for inflammatory bowel disease in humans and mice. Gastroenterology 2017; 153: 550–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Vermeire S, Satsangi J, Peeters M et al Evidence for inflammatory bowel disease of a susceptibility locus on the X chromosome. Gastroenterology 2001; 120: 834–840. [DOI] [PubMed] [Google Scholar]
- 87. Li YR, Li J, Zhao SD et al Meta‐analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat Med 2015; 21: 1018–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hunt KA, Mistry V, Bockett NA et al Negligible impact of rare autoimmune‐locus coding‐region variants on missing heritability. Nature 2013; 498: 232–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Luo Y, de Lange KM, Jostins L et al Exploring the genetic architecture of inflammatory bowel disease by whole‐genome sequencing identifies association at ADCY7. Nat Genet 2017; 49: 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Fuchsberger C, Flannick J, Teslovich TM et al The genetic architecture of type 2 diabetes. Nature 2016; 536: 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zuk O, Hechter E, Sunyaev SR et al The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci U S A 2012; 109: 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Speed D, Cai N, Johnson MR, Nejentsev S, Balding DJ, UCLEB Consortium . Reevaluation of SNP heritability in complex human traits. Nat Genet 2017; 49: 986–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Loftus EV Jr. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology 2004; 126: 1504–1517. [DOI] [PubMed] [Google Scholar]
- 94. Li X, Sundquist J, Hemminki K et al Risk of inflammatory bowel disease in first‐ and second‐generation immigrants in Sweden: a nationwide follow‐up study. Inflamm Bowel Dis 2011; 17: 1784–1791. [DOI] [PubMed] [Google Scholar]
- 95. Ng SC, Tsoi KK, Kamm MA et al Genetics of inflammatory bowel disease in Asia: systematic review and meta‐analysis. Inflamm Bowel Dis 2012; 18: 1164–1176. [DOI] [PubMed] [Google Scholar]
- 96. Kenny EE, Péer I, Karban A et al A genome‐wide scan of Ashkenazi Jewish Crohn's disease suggests novel susceptibility loci. PLoS Genet 2012; 8: e1002559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Brant SR, Okou DT, Simpson CL et al Genome‐wide association study identifies African‐specific susceptibility loci in African Americans With Inflammatory Bowel Disease. Gastroenterology 2017; 152: 206–217 e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Daly AK, Donaldson PT, Bhatnagar P et al HLA‐B*5701 genotype is a major determinant of drug‐induced liver injury due to flucloxacillin. Nat Genet 2009; 41: 816–819. [DOI] [PubMed] [Google Scholar]
- 99. Winter J, Walker A, Shapiro D et al Cost‐effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20: 593–599. [DOI] [PubMed] [Google Scholar]
- 100. Yang SK, Hong M, Baek J et al A common missense variant in NUDT15 confers susceptibility to thiopurine‐induced leukopenia. Nat Genet 2014; 46: 1017–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Heap GA, Weedon MN, Bewshea CM et al HLA‐DQA1‐HLA‐DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat Genet 2014; 46: 1131–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Heap GA, So K, Weedon M et al Clinical features and HLA association of 5‐aminosalicylate (5‐ASA)‐induced nephrotoxicity in inflammatory bowel disease. J Crohns Colitis 2016; 10: 149–158. [DOI] [PubMed] [Google Scholar]
- 103. Billiet T, Vande Casteele N, Van Stappen T et al Immunogenicity to infliximab is associated with HLA‐DRB1. Gut 2015; 64: 1344–1345. [DOI] [PubMed] [Google Scholar]
- 104. Linares‐Pineda TM, Canadas‐Garre M, Sanchez‐Pozo A et al Pharmacogenetic biomarkers of response in Crohn's disease. Pharmacogenomics J 2017; [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 105. Billiet T, Papamichael K, de Bruyn M et al A matrix‐based model predicts primary response to infliximab in Crohn's Disease. J Crohns Colitis 2015; 9: 1120–1126. [DOI] [PubMed] [Google Scholar]
- 106. Barber GE, Yajnik V, Khalili H et al Genetic markers predict primary non‐response and durable response to anti‐TNF biologic therapies in Crohn's Disease. Am J Gastroenterol 2016; 111: 1816–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Umicevic Mirkov M, Cui J, Vermeulen SH et al Genome‐wide association analysis of anti‐TNF drug response in patients with rheumatoid arthritis. Ann Rheum Dis 2013; 72: 1375–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hart A, Li K, Gasink C et al DOP046 Genome‐wide association study of baseline disease characteristics and response to Ustekinumab in moderate to severe Crohn's disease. JCC 2017; 11: S54. [Google Scholar]
- 109. Satsangi J, Grootscholten C, Holt H et al Clinical patterns of familial inflammatory bowel disease. Gut 1996; 38: 738–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Plomin R, Haworth CM, Davis OS. Common disorders are quantitative traits. Nat Rev Genet 2009; 10: 872–878. [DOI] [PubMed] [Google Scholar]
- 111. Marouli E, Graff M, Medina‐Gomez C et al Rare and low‐frequency coding variants alter human adult height. Nature 2017; 542: 186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lee JC, Smith KG. Prognosis in autoimmune and infectious disease: new insights from genetics. Clin Transl Immunology 2014; 3: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Cleynen I, Boucher G, Jostins L et al Inherited determinants of Crohn's disease and ulcerative colitis phenotypes: a genetic association study. Lancet 2016; 387: 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ananthakrishnan AN, Huang H, Nguyen DD et al Differential effect of genetic burden on disease phenotypes in Crohn's disease and ulcerative colitis: analysis of a North American cohort. Am J Gastroenterol 2014; 109: 395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Cleynen I, Gonzalez JR, Figueroa C et al Genetic factors conferring an increased susceptibility to develop Crohn's disease also influence disease phenotype: results from the IBDchip European Project. Gut 2013; 62: 1556–1565. [DOI] [PubMed] [Google Scholar]
- 116. Jung C, Colombel JF, Lemann M et al Genotype/phenotype analyses for 53 Crohn's disease associated genetic polymorphisms. PLoS ONE 2012; 7: e52223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Helio T, Halme L, Lappalainen M et al CARD15/NOD2 gene variants are associated with familially occurring and complicated forms of Crohn's disease. Gut 2003; 52: 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lee JC, Espeli M, Anderson CA et al Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3‐regulated pathway. Cell 2013; 155: 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Alonso A, Domenech E, Julia A et al Identification of risk loci for Crohn's disease phenotypes using a genome‐wide association study. Gastroenterology 2015; 148: 794–805. [DOI] [PubMed] [Google Scholar]
- 120. Lee JC, Biasci D, Roberts R et al Genome‐wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn's disease. Nat Genet 2017; 49: 262–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Viatte S, Lee JC, Fu B et al Association between genetic variation in FOXO3 and reductions in inflammation and disease activity in inflammatory polyarthritis. Arthritis Rheumatol 2016; 68: 2629–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lu Y, Zhu Y, Wang X et al FOXO3 rs12212067: T>G association with active tuberculosis in Han Chinese population. Inflammation 2016; 39: 10–15. [DOI] [PubMed] [Google Scholar]