

Allostery is the process by which biological macromolecules (mostly proteins) transmit the regulatory effects induced by the binding of a ligand at one site to a different, often distant, functional site. This process governs the function of almost all metabolism and gene regulation (1, 2). Despite the importance of this process, a general understanding of the structural and energetic basis for allostery has been elusive. On page 685 of this issue, Bai et al. (3) shed light on a key obstacle to understanding allosteric control by showing how proteins existing as ensembles of multiple conformations affect the allosteric signal transduction process.
For the past 40 years, allostery has usually been interpreted through either of two classic models: the concerted model (4) or the sequential model (5). The former model treats the conformational coupling between different parts of the molecule as absolute, whereas the latter assumes a fixed coupling between ligand binding and conformational changes (6). The practical consequence of these assumptions is that without knowing how probable the allosterically activated states are in the absence of the allosteric ligand, it is difficult to know what structural and/or energetic features of a protein must be present in order for signal transduction to occur.
Attempts to resolve this problem have relied most heavily on high-resolution structural analysis (such as x-ray crystallography) of the various liganded species (7, 8). Although such analysis can show what bonds are formed and lost between the liganded and unliganded species, the underlying assumption is that the functional states of the molecules—the so-called tensed (T) or relaxed (R) states—are well represented by single static structures. This may not be the case, as Bai et al. show in their analysis of allostery in the bacterial flagellar motor (3). Their work shows that the presence of conformational heterogeneity among the subunits in the native state ensemble of the protein is key both to the allosteric mechanism and to the cooperativity of the motor. Thus, a functional state cannot be obligatorily associated with a single static structural state of the protein.
Recent studies investigating the importance of conformational dynamics (9–12), local unfolding (13–15), and intrinsic disorder (16) to allosteric signaling support the conclusion that regional changes in the conformational heterogeneity of proteins often accompany allosteric transitions. Thus, allosteric proteins may often rely on a mechanism in which multiple conformations must retain significant probabilities in order to function and transmit signal. In such cases, structural studies that rely on a single static structure to describe the activated or inactivated states of the protein may only provide limited insight into allosteric mechanisms. This potential setback, however, is accompanied by a more important opportunity: Knowledge of the conformational states in the native state ensemble can provide vital clues to understanding the ground rules governing allosteric control, giving insight into how nature designs allosteric proteins, and how these same principles can be applied to the rational design of signaling systems.
The important role played by the ensemble in determining allostery is demonstrated in the figure, which shows the allostery between an effector (I) and a functional (II) subunit. Depending on the energetic cost of interconverting from the active to the inactive state of each subunit, each state in the ensemble has a different stability (3, 16–18), which determines how much time the protein spends in that state. Addition of the effector ligand redistributes the ensemble and—provided that the ensemble equilibria are suitably poised—activates the functional domain.
Figure. The ensemble model of allostery reveals the energetic basis of coupling.
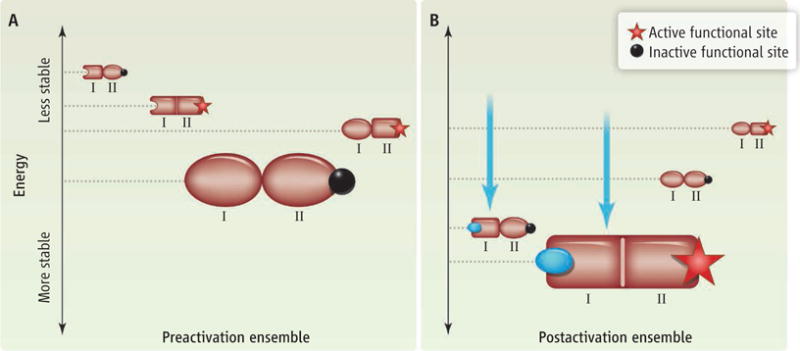
The ensemble for a protein with an effector (I) and a functional (II) subunit is shown (A) before and (B) after activation with an effector ligand (blue circle). Prior to activation, the most probable state has an inactive functional subunit (II with black circle). Stabilization of the active conformation of the effector subunit (blue arrows) populates the active conformation of the functional subunit (II with red star).
Although conceptually simple, this ensemble view has important ramifications. Specifically, the ability of a protein to facilitate site-to-site allosteric coupling can be understood in terms of the stabilities of the states in the native ensemble (3, 16–18), without requiring detailed insights about the mechanical or structural pathways that connect the coupled sites (16). Of course, this does not mean that understanding allostery at the atomic level should not be an ultimate goal. To the contrary, the ensemble view suggests that the multitude of sequence and structural permutations that exist for a particular allosteric protein can be reconciled within a common framework.
The implications of an ensemble view of allostery are twofold. First, it provides mechanistic insight into, for example, how amino acid substitutions distal from the binding sites may affect allosteric coupling, or how different combinations of amino acid changes can combine to maintain coupling. Second, and perhaps more important, the potential robustness of coupling revealed in the ensemble model suggests that the evolution and design of allosteric systems may be much more straightforward than a detailed analysis of the structural interactions might suggest; this speculation has recent experimental support (19, 20).
Within the context of the ensemble model, evolving (or designing) allosteric coupling between subunits can be reduced to achieving two separate, albeit related goals. First, mutations must be introduced that position the equilibria between the active and inactive states of the different subunits in a regime that is suitable to facilitate coupling (3, 16). Second, mutations must be introduced that maintain a substantial interaction energy between one or the other (that is, active or inactive) states of each subunit (3, 16). In this respect, the ensemble model (see the figure) provides a framework for articulating the problem in energetic terms, providing the ground rules that govern coupling between two (or even many) sites. These ground rules may serve as the foundation for future design strategies targeting the development of signaling-competent systems.
Acknowledgments
Supported by grants from the NIH (GM63747), NSF (MCB-0446050), and the Robert A. Welch Foundation (H-1461). I thank T. Oas (Duke University) for helpful discussions and suggestions.
References and Notes
- 1.Monod J, Changeux JP, Jacob F. J Mol Biol. 1963;6:306. doi: 10.1016/s0022-2836(63)80091-1. [DOI] [PubMed] [Google Scholar]
- 2.Changeux JP, Edelstein SJ. Science. 2005;308:1424. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 3.Bai F, et al. Science. 2010;327:685. doi: 10.1126/science.1182105. [DOI] [PubMed] [Google Scholar]
- 4.Monod J, Wyman J, Changeux P. J Mol Biol. 1965;12:88. [Google Scholar]
- 5.Koshland DE, Nemethy G, Filmer D. Biochemistry. 1966;5:365. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 6.Wyman J. Curr Top Cell Regul. 1972;6:207. [Google Scholar]
- 7.Kantrowitz ER, Lipscomb WN. Science. 1988;241:669. doi: 10.1126/science.3041592. [DOI] [PubMed] [Google Scholar]
- 8.Perutz M. Cooperativity and Allosteric Regulation in Proteins. Cambridge Univ Press; Cambridge, UK: 1990. [Google Scholar]
- 9.Gunasekaran K, Ma B, Nussinov R. Prot Struct Funct Bioinf. 2004;57:433. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 10.Popovych N, Sun S, Ebright RE, Kalodimos CG. Nat Struct Mol Biol. 2006;13:831. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarkson MW, Gilmore SA, Edgell MH, Lee AL. Biochemistry. 2006;45:7693. doi: 10.1021/bi060652l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smock RG, Gierasch LM. Science. 2009;324:198. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan H, Lee JC, Hilser VJ. Proc Natl Acad Sci USA. 2000;97:12020. doi: 10.1073/pnas.220240297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyashita O, Onuchic JN, Wolynes PG. Proc Natl Acad Sci USA. 2003;100:12570. doi: 10.1073/pnas.2135471100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laine O, Streaker ED, Nabavi M, Fenselau CC, Beckett D. J Mol Biol. 2008;381:89. doi: 10.1016/j.jmb.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hilser VJ, Thompson EB. Proc Natl Acad Sci USA. 2007;104:8311. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duke TAJ, Le Novère N, Bray D. J Mol Biol. 2001;308:541. doi: 10.1006/jmbi.2001.4610. [DOI] [PubMed] [Google Scholar]
- 18.Duke TAJ, Bray D. Proc Natl Acad Sci USA. 1999;96:10104. doi: 10.1073/pnas.96.18.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ostermeier M. Protein Eng Des Sel. 2005;18:359. doi: 10.1093/protein/gzi048. [DOI] [PubMed] [Google Scholar]
- 20.Strickland D, Moffat K, Sosnick TR. Proc Natl Acad Sci USA. 2008;105:10709. doi: 10.1073/pnas.0709610105. [DOI] [PMC free article] [PubMed] [Google Scholar]