

Abstract
Objective
Cigarette smoke exposure (CSE) is a risk factor for cerebral aneurysm (CA) formation, but the molecular mechanisms are unclear. Although CSE is known to contribute to excess reactive oxygen species (ROS) generation, the role of oxidative stress on vascular smooth muscle cell (VSMC) phenotypic modulation and pathogenesis of CAs is unknown. The goal of this study was to investigate if CSE activates a NAPDH oxidase (NOX) dependent pathway leading to VSMC phenotypic modulation and CA formation and rupture.
Approach and Results
In cultured cerebral VSMCs, CSE increased expression of NOX1 and ROS which preceded upregulation of pro-inflammatory/matrix remodeling genes (MCP-1, MMPs, TNF-α, IL-1β, NF-κB, KLF4) and down-regulation of contractile genes (SM-α-actin, SM-22α, SM-MHC) and myocardin. Inhibition of ROS production and knock-down of NOX1 with siRNA or antisense decreased CSE-induced upregulation of NOX1 and inflammatory genes and downregulation of VSMC contractile genes and myocardin. p47phox−/−, NOX knockout mice or pre-treatment with NOX inhibitor, apocynin, significantly decreased CA formation and rupture compared to controls. NOX1 protein and mRNA expression were similar in p47phox−/− mice or those pre-treated with apocynin, but was elevated in unruptured and ruptured CAs. CSE increased CA formation and rupture, which was diminished with apocynin pre-treatment. Similarly, NOX1 protein and mRNA, and ROS were elevated by CSE, and in unruptured and ruptured CAs.
Conclusions
CSE initiates oxidative stress-induced phenotypic modulation of VSMCs and CA formation and rupture. These molecular changes implicate oxidative stress in the pathogenesis of CAs and may provide a potential target for future therapeutic strategies.
Keywords: Smooth muscle, phenotypic modulation, cigarette, smoke, cerebrovascular, atherosclerosis, stroke, aneurysm, subarachnoid hemorrhage, rupture, reactive, oxygen, species, oxidative, stress
Subject Codes: Cerebral Aneurysms, Intracranial Hemorrhage, Ischemic Stroke, Vascular Biology, Hypertension, Inflammation, Cerebrovascular Disease/Stroke
INTRODUCTION
Cigarette smoking exposure (CSE) is a major risk factor for cerebral vascular injury, including atherosclerosis,1–5 a key process underlying cerebral aneurysm (CA) formation.6–10 Additionally, CSE is the most significant modifiable risk factor associated with CA formation, progression, and rupture.11–24 Although CSE has been demonstrated to cause significant alterations in the cerebral immune system and inflammatory response,25–35 the means by which CSE contributes to the pathogenesis of CAs has not been elucidated.
In vitro and in vivo studies have shown that CSE results in endothelial dysfunction in nearly every vascular territory.27–32 This may be secondary to activation of immune cells, upregulation of pro-inflammatory mediators, and production of reactive oxygen species (ROS).27 The key mechanisms which lead to CSE induced vascular dysfunction remain unclear.
Unlike terminally differentiated cardiac or skeletal muscle cells, vascular smooth muscle cells (VSMC) retain remarkable plasticity.36,37 In response to environmental stimuli, VSMCs can undergo changes from cells principally concerned with contraction to cells that are primarily involved in inflammation and matrix remodeling.36 This phenotypic modulation is defined by decreased expression of key VSMC contractile proteins (smooth muscle myosin heavy chain [SM-MHC], SM-α-actin, and SM-22α) with concomitant increased expression of inflammatory mediators.36,38–43 VSMC phenotypic modulation is known to be important in the response to vascular injury41 and the development and progression of atherosclerosis42, both critical processes behind CA pathogenesis.43 Although a role has been postulated for the significant pathological effects of CSE, studies have not assessed its role in VSMC phenotypic modulation or the mechanisms directly contributing to CA formation, progression, or rupture.
Therefore, the aims of the present study are: (1) To evaluate the effect of CSE in inducing phenotypic modulation of cultured cerebral VSMCs through a NAPDH oxidase (NOX) dependent pathway; (2) To assess in vivo whether phenotypic modulation of cerebral VSMCs during CA formation is accompanied by increased free radical generation and oxidative stress; (3) To determine if CSE further increases oxidative stress along with downregulation of VSMC marker genes and upregulation of inflammatory genes during CA formation and rupture. 4) To assess whether CSE-induced oxidative stress leads to increased CA formation and rupture and whether genetic and pharmacological inhibition of NOX reverses CSE initiation of oxidative stress, induction of phenotypic modulation, and CA formation and rupture.
MATERIALS AND METHODS
Materials and Methods are available in the online-only Data Supplement.
Statistical Analysis
Please see supplemental methods for power analysis. All experiments were performed with a minimum of triplicate samples and were conducted in 3 to 6 independent experiments unless otherwise indicated. Data are presented as mean and range for continuous variables, and as frequency for categorical variables. Analysis was carried out using Chi-square, Fisher’s exact, Wilcoxon rank sum, and Kruskal-Wallis test with Wilcoxon rank sum post hoc test as appropriate. Secondary assessment of risk of rupture was carried out by Kaplan-Meier survival analysis with Cox regression analysis to assess hazard ratios (HR). Error bars represent standard error of the mean (SEM). Statistical significance was considered defined as P<0.05.
RESULTS
CSE-Induced Expression of NOX and Oxidative Stress in VSMC
As cigarette smoke has been shown to increase inflammation and ROS levels in VSMCs, 27 we sought to determine the role of CSE on NOX expression and on the phenotypic modulation of cultured cerebral VSMCs. VSMC were treated with either 10 or 40 μg/ml CSE for up to 48 h. CSE exposure increased expression of NOX1 mRNA in a dose and time dependent fashion with maximal expression occurring 4 h after exposure followed by a gradual decrease in expression over time (Figure 1A). In contrast, NOX2, NOX3, and NOX4 expression were not significantly changed by CSE (Figure 1 B–D).
Figure 1. CSE-Induced expression of NOX1 and ROS in VSMCs.

Cultured cerebral VSMCs were treated with the indicated concentrations of CSE. NOX1, NOX2, NOX3 and NOX4 mRNA expression was quantified using RT-PCR and normalized to 18S rRNA, and expressed as fold increase over vehicle A–D). Values represent mean ± SEM of n = 3–6. *P<0.05 vs control (vehicle) using Kruskal-Wallis analysis with Wilcoxon rank sum post hoc test. E) Differential time-course for CSE-induced ROS production in cerebral VSMCs assessed by lucigenin-enhanced chemiluminescence. Data represent the mean ± SEM of n=4 to 6 experiments; *P<0.05 vs time 0 using Kruskal-Wallis analysis with Wilcoxon rank sum post hoc test. F) Following stimulation with vehicle or CSE (10 or 40 μg/ml) cerebral VSMCs were incubated with dihydroethidium and then imaged by fluorescence microscopy.
To assess if CSE was inducing oxidative stress, we exposed VSMC to 10 or 40 μg/ml CSE and ROS levels were measured for up to 48 h following CSE using lucigenin chemiluminescence as described in the methods. As shown in Figure 1E, ROS levels were increased significantly 30 min following CSE exposure and they continued to increase reaching maximal levels of approximately 5 fold of controls by 4 h which was followed by a gradual decease in ROS levels reaching approximately 2 fold of controls at 48 h (Figure 1E). Similarly, in situ superoxide imaging of VSMC with dihydroethidium demonstrated an increase in free radical expression following CSE exposure (Figure 1F).
CSE-Induces a Pro-inflammatory & Matrix Remodeling Phenotypic Modulation of VSMCs
To determine if CSE altered the gene expression profile of VSMC, we exposed VSMC to CSE for 24 h and measured gene expression by RT-PCR. CSE significantly increased expression of the pro-inflammatory and matrix remodeling genes MCP-1, MMP-3, MMP-9, TNF-α, IL-1β, iNOS, NF-κB, and KLF-4 (Figure 2). Gene expression was generally maximal at 24 h, at a time point after maximal NOX1 expression. Additionally, pretreatment with the antioxidant enzyme, SOD, abrogated CSE-induced upregulation of pro-inflammatory and matrix remodeling genes (Figure 2A).
Figure 2. CSE Induces pro-inflammatory & matrix remodeling phenotypic modulation of VSMCs and represses myocardin and SMC marker gene expression in cultured cerebral VSMC.

Cultured cerebral VSMCs were treated for 24 h with the indicated concentration of CSE with or without SOD pre-treatment. RT-PCR was performed, normalized to 18S rRNA, and expressed as fold increase over vehicle. Pro-inflammatory and matrix remodeling gene expression is shown in A) with myocardin and SMC marker gene expression shown in B). *P<0.05 vs control (vehicle) using Kruskal-Wallis analysis with Wilcoxon rank sum post hoc test. C) SM-MHC and SM- α-actin promoter-luciferase constructs were transiently transfected into cerebral VSMCs for 24 hours followed by overexpression of NOX1 with adenovirus for 24 hours. Luciferase activity was measured and normalized to total protein content and then expressed as fold increase over control (empty adenovirus). *P<0.05 vs control using Wilcoxon rank sum. The effects of NOX1 overexpression on SMC marker genes (Myocardin, SM-22α, SM-α-actin and SM-MHC levels is shown in D) with pro-inflammatory and matrix remodeling genes expression levels shown in E). All RT-PCR data was normalized to 18S rRNA and expressed as fold increase over control (empty adenovirus). *P<0.05 vs control using Wilcoxon rank sum. F) Cultured VSMC were treated with CSE (40 μg/ml) and infected with NOX1 adenovirus (AD) or anti-sense NOX1 adenovirus (AS) for 24 h. RT-PCR was performed to measure the mRNA expression levels of myocardin and the SMC marker gene, SM-MHC and the pro-inflammatory and matrix remodeling genes KLF4 and MCP1 which were normalized to 18S rRNA and expressed as fold increase over control (empty adenovirus). *P<0.05 vs control using Kruskal-Wallis analysis with Wilcoxon rank sum post hoc test. G) Cultured VSMC were transfected with siRNA specific to NOX1 or siNeg (GFP) and treated with CSE (40 μg/ml) for 24 h. mRNA expression of indicated genes were analyzed with RT-PCR and normalized to 18S rRNA. *P<0.05 vs siNeg+ Vehicle using Kruskal-Wallis analysis with Wilcoxon rank sum post hoc test. For all experiments values represent mean ± SEM of n = 3 to 6.
CSE Exposure Repressed Myocardin & Phenotypic Marker Gene Expression in VSMC
Although a number of factors control VSMC differentiation, myocardin has been shown to be a key transcriptional regulator of SMC differentiation in peripheral vascular beds.36 To assess the effects of CSE on phenotypic modulation, we exposed VSMC to CSE and analyzed expression of VSMC marker genes by RT-PCR. CSE exposure significantly reduced expression of myocardin mRNA with a maximal suppression at 24 h (Figure 2B). Similarly, CSE significantly downregulated SM-α-actin, SM-MHC and SM-22α mRNA expression (Figure 2B). CSE-dependent suppression of myocardin and SMC marker gene expression was prevented by pretreatment with SOD (Figure 2B).
NOX1 Decreases Promoter Activity and Expression of VSMC Marker Genes
Our data indicates that the phenotypic modulation of VSMC by CSE is dependent upon oxidative stress. Since we show in Figure 1 that NOX1, an inducer of oxidative stress, is upregulated early following CSE, we were interested in determining if NOX1 regulates VSMC gene expression. To assess the role of NOX1 on the expression of VSMC marker genes, VSMCs were co-transfected with VSMC promoter-reporter constructs and with a NOX1 expression vector. Overexpression of NOX1 significantly decreased SM-α-actin and SM-MHC promoter activity (Figure 2C). The reduced promotor activity in NOX1 overexpressing VSMC was paralleled with a significant reduction in endogenous gene expression of SM- α-actin, SM-22α, SM-MHC, and myocardin levels (Figure 2D). Similarly overexpression of NOX1 significantly increased expression of pro-inflammatory and matrix remodeling genes (KLF-4, MCP-1 MMP2, MMP3, MMP9, IL-1β, NF-κB, iNOS; Figure 2E). To determine if NOX1 is involved in CSE mediated VSMC gene expression, we knocked-down NOX1 protein expression by adenovirus delivered anti-sense NOX1 and analysed CSE changes in gene expression. As demonstrated in Figure 2F, when cells were infected with control anti-sense and CSE, there was a significant decrease in myocardin and SM-MHC and an increase in MCP-1 and KLF-4 expression. In contrast, knock-down of NOX1 reversed CSE-induced decrease of myocardin and SMC-MHC and increase of MCP-1 and KLF-4 (Figure 2F).
To confirm a role of NOX1 in CSE gene regulation, cultured VSMC were transfected with siRNA specific to NOX1 or GFP for 24 hours and treated with CSE (40 μg/ml) for 24 hours. CSE treatment significantly increased the expression of NOX1, KLF-4, IL-1β and VCAM when treated with control (siGFP), but not when cells were treated with CSE and NOX1spedific siRNA (Figure 2G).
Cerebral Aneurysm Formation
CA formation was carried out as previously described.44 Based upon dose assessment, experimental planning, and power analysis, mice were injected with vehicle (N=10) or a single stereotactic elastase injection (3.5 mU; N=10, 17.5 mU; N=10, and 35 mU; N=10) and aneurysm formation, defined as 1.5 times the diameter of the parent vessel, determined 28 days later. Cerebral aneurysms were not observed in sham treated animals but were formed in a dose dependent fashion with 10%, 55%, and 80% of animals developing aneurysm formation, respectively (Figure 3A).
Figure 3. Cerebral aneurysm formation in mice varies with elastase dose, pretreatment with apocynin, and genetic inactivation of NOX1.
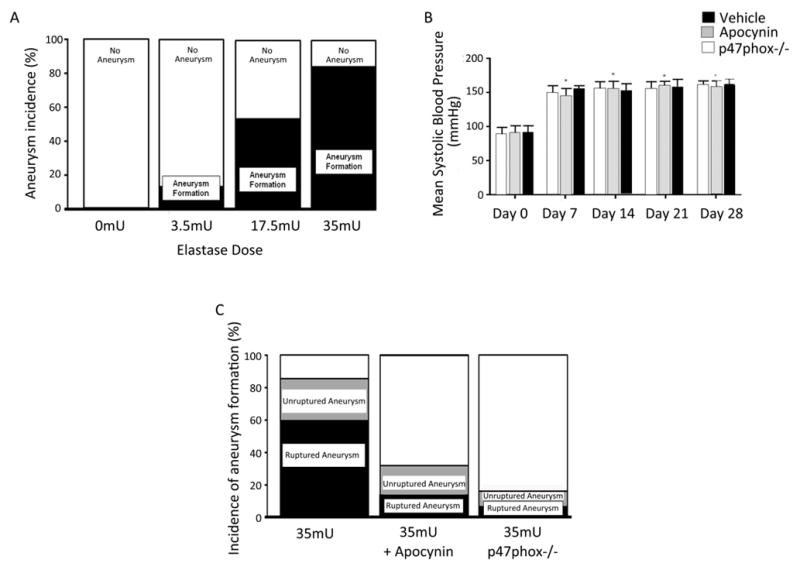
Cerebral aneurysms were induced in mice and aneurysm formation determined 28 days later. Cerebral aneurysms formation occurred in 0 mice receiving vehicle (n=10), 10% receiving 3.5 mU of elastase (n=10), 55% receiving 17.5 mU of elastase (n=10), 80% receiving 35 mU (n=10) A). B) Blood pressure was elevated one week after aneurysm induction, but was not significantly different between vehicle, apocynin treated or p47phox−/− mice at any single time period. C) CA formation occurred in 21 of 25 animal (84%) controls, 8 of 25 animals (32.0%) treated with apocynin, and 2 of 12 (16.7%) p47phox −/− mice (P<0.05, apocynin using Chi-square analysis and p47phox−/− using Fisher’s exact test versus control). Aneurysm rupture occurred in 15 of 25 (60.0%) controls, 3 of 25 (12%) animals treated with apocynin, and 1 of 12 (8.3%) of p47phox −/− mice (P<0.05, apocynin and p47phox−/− using Fisher’s exact test versus control)..
Role of NOX in the Formation of Cerebral Aneurysms
Based on our results and other published data,45 we sought to assess the role of NOX in cerebral aneurysm formation in vivo. To test the role of NOX in CA formation, animals began treatment with the NOX inhibitor apocynin three days prior to elastase injection and were compared to CA formation in vehicle treated mice and p47phox−/− mice, which lack NOX activity. Mice pre-treated with apocynin, p47phox−/− mice, and those treated only with vehicle undergoing CA induction had significant increases in systolic blood pressure 7 days after elastase injection (35 mU) that was sustained until 28 days, but was not significantly different between the 3 cohorts at any time point (Figure 3B).
Analysis of CA formation reveled aneurysm formation in 21 of 25 animals (84%) receiving 35 mU of elastase and vehicle as compared to 8 of 25 animals (32%, p<0.001) treated with apocynin (Figure 3C). The incidence of ruptured aneurysm was also significantly increased in vehicle treated mice, 15 of 25 (60%), compared to those receiving apocynin (3 of 25, 12%, p=0.001; Figure 3C). CA formation was associated with the development of neurological deficits (score 1–5) and aneurysmal SAH between day 7 and 21. Survival analysis demonstrated that animals treated with apocynin were 7.2 times (95% CI 2.1–25.3, p=0.002) less likely to have CA rupture compared to those receiving vehicle. Next we induced CA formation in p47phox−/− mice, which lack NOX activity. As compared to wild-type mice, p47phox−/− mice were significantly less likely to form CAs (2 of 12; 16.7%; p=0.001; Figure 3C), and had a lower incidence of CA rupture (1 of 12; 8.3%; p=0.004; Figure 3C). Similarly, survival analysis demonstrated that p47phox−/− mice were also less likely to experience CA rupture (HR=10.3; 95% CI 1.4–78.2, p=0.024). As the incidence of CA formation and rupture was low in p47phox−/− mice, these experiments were aborted after the first cohort of 12 mice. These results suggest that NOX plays an important role in CA formation.
NOX Expression in Unruptured and Ruptured Intracranial Aneurysms
Histological assessment of the CAs demonstrated structural changes that were similar to those found in human CAs (Figure 4, an enlarged image is shown in supplemental Figure I).46,47 Hematoxylin and Eosin and trichrome staining of cerebral vessels from sham operated mice demonstrated two to three layers of smooth muscle cells and a single layer of continuous endothelial cells (Figure 4; A1–2). Trichrome staining demonstrated one layer of elastic lamina (A2).47 In CA, there were layers of discontinuous endothelial cells and scattered VSMCs (Figure 4; B1–2, unruptured and C1–2; ruptured). Trichrome staining revealed disorganized elastic lamina (B2 and C2).
Figure 4. Histological and mRNA expression of cerebral aneurysms demonstrates NOX-Induced VSMC Phenotypic Modulation in Unruptured and Furthermore Ruptured Cerebral Aneurysms.

Hematoxylin and Eosin (panel 1), and trichrome staining (panel 2) of cerebral vessels from sham operated mice (A1–2), CA unruptured (B1–2), and CA ruptured (C1–2). NOX1 immunostaining (green) is show in in panel 3 and 4 (A-sham, B-unruptured CA; C-ruptured CA), samples were co-stained with either SMC-22α for SMCs (red) in panel 3 or CD68 for macrophages (red) in panel 4. In situ superoxide imaging, as carried out with dihydroethidium is shown in panel 5 (A-sham, B-unruptured CA; C-ruptured CA). NOX1, NOX2, NOX3, and NOX4 mRNA expression was quantified using RT-PCR and normalized to 18S rRNA, and expressed as fold increase over vehicle (D–G). *P<0.05 versus sham surgery+vehicle controls using Kruskal-Wallis analysis with Wilcoxon rank sum post hoc test. RT-PCR analysis of VSMC marker gene expression in (H) and pro-inflammatory and matrix remodeling genes in (I) in sham, p47phox−/− mice, apocynin treated mice and in unruptured and ruptured CA. *P<0.05 versus sham surgery+vehicle controls using Kruskal-Wallis analysis with Wilcoxon rank sum post hoc test. For all experiments values represent mean ± SEM of n = 3 to 6.
As compared to sham mice, there was no difference in immunofluorescence reactivity for NOX1 when comparing p47phox−/− mice or those treated with apocynin (not shown). NOX1 immunoreactivity was significantly higher in unruptured CAs, and highest in ruptured CAs (Figure 4; A–C3 and A–C4). Next we sought to determine the cellular source of NOX1. To localize NOX1 expression, samples were co-stained with either SMC-22α for SMCs or CD68 for macrophages and NOX1 (Figure 4). NOX1 expression was observed to co-localize with both SMCs and macrophages in both unruptured (Figure 4; B3–4) and ruptured CAs (Figure 4; C3–4). SM-22α but not with CD68 was observed in normal-sized cerebral blood vessels adjacent to CAs (C3). There were more macrophages observed in ruptured CAs (C4) as compared to unruptured CAs (B4), and an absence of macrophages in sham operated mice (A4).
To assess whether an increase in NOX1 expression leads to increased oxidative stress during CA formation, we measured superoxide production with dihydroethidium as described in methods. As shown in Figure 4 (panel A5) there was a low level of free radical formation observed in sham operated mice. In contrast free radical expression was increased in unruptured CAs (panel B5) and further increased in ruptured CAs (panel C5) as compared to sham operated mice (A5). There was limited expression of free radicals in p47phox−/− mice or those treated with apocynin (not shown), suggesting a possible connection between NOX1 expression and free radical production in CA formation.
To confirm that NOX1 is expressed during CA formation, we analyzed NOX1 expression by RT-PCR. As compared with sham operated mice, RT- PCR analysis demonstrated no significant difference in NOX1 mRNA expression in cerebral blood vessels from p47phox −/− mice or those treated with apocynin (Figure 4D). NOX1 mRNA extracted from unruptured, and even more so from ruptured CAs, was significantly elevated as compared with sham operated mice. NOX2, NOX3, and NOX4 expression were not significantly changed in the various cohorts (Figure 4E–G). As a secondary control, β-actin expression was assessed and found to be not significantly different among sham operated mice, p47phox −/− mice, or those treated with apocynin, unruptured aneurysm and ruptured CAs (data not shown).
NOX Regulates SMC Phenotypic Modulation in Unruptured and Ruptured Cerebral Aneurysms
Our in vitro VSMC studies reveal a NOX1 dependent regulation of VSMC phenotypic gene expression. Therefore we were interested in determining if NOX also regulated VSMC gene expression, in vivo. To test this possibility we first assessed whether VSMC phenotypic modulations occur in our CA model system, by examining alterations in expression of VSMC marker genes and pro-inflammatory, and matrix remodeling genes. As shown in Figure 4H, SM-α-actin, SM-MHC, SM-22α mRNA levels where decreased in ruptured CAs as compared to unruptured CAs. As compared to both unruptured and ruptured CA, sham operated mice, p47phox −/− mice, and those treated with apocynin had significantly higher levels of the VSMC marker genes (Figure 4H). Additionally, myocardin was decreased in unruptured CAs, and further decreased in ruptured CAs (Figure 4H).
Similar to our in vitro gene expression studies we found that the pro-inflammatory and matrix remodeling genes, MCP-1, MMP-3, MMP-9, TNF-α, IL-1β, iNOS, TNF-α, NF-κB, VCAM and KLF-4 were increased in unruptured CAs, and further increased in ruptured CAs (Figure 4I).
The Role of CSE in Cerebral Aneurysm Formation and Rupture
To assess the effects of CSE in CA development and rupture, mice underwent daily CSE for 7 days prior to nephrectomy. Mice then underwent CA induction surgery (3.5 mU elastase) as described in the Methods. CSE control mice and mice exposed to CSE, and CSE + apocynin pre-treatment had significant increases in systolic blood pressure 7 days after elastase injection that was sustained until day 28, but was not significantly different between the 3 cohorts at any time point (Figure 5A).
Figure 5. CSE increases the incidence of CA formation and rupture, and NOX1 mRNA expression in vivo and induces phenotypic modulation.

To assess the role of CSE in CA development and rupture, mice underwent daily CSE starting 7 days prior to nephrectomy followed by CA induction surgery (3.5 mU elastase) as described in methods. The effect of CA induction on blood pressure is shown in A) *P<0.05 versus Day 0 using Kruskal-Wallis analysis with Wilcoxon rank sum post hoc test. The incidence of CA formation and rupture in animals treated with vehicle, CSE but pre-treated with apocynin, and CSE is shown in B). The effects of CSE, apocynin, CA formation and rupture on NOX1, NOX2, NOX3, and NOX4 mRNA expression are shown in C–F) and free radical expression is shown in G). *P<0.05 versus sham surgery controls using Kruskal-Wallis analysis with Wilcoxon rank sum post hoc test. mRNA expression of pro-inflammatory, matrix remodeling and VSMC phenotypic genes are shown in H) and I) respectively following indicated treatments. *P<0.05 versus sham surgery controls using Kruskal-Wallis analysis with Wilcoxon rank sum post hoc test. For all experiments values represent mean ± SEM of n = 3 to 6.
Analysis of CA formation revealed that the incidence of CA formation was significantly higher in mice receiving CSE (13 of 30; 43.3%) when compared to mice receiving CSE with pre-treatment of apocynin (6 of 30; 20%), or control mice not receiving CSE (3 of 30; 10%; p=0.011, Figure 5B). Similarly, the rate of CA rupture was also significantly increased in mice receiving CSE alone (9 of 30; 30%) versus those receiving CSE, but pre-treated with apocynin (2 of 30; 6.7%) and control mice that did not receive CSE (0 of 30; 0%; p=0.001; Figure 5B). CA formation was not observed in animals receiving CSE and undergoing sham surgery (data not shown).
Since CSE enhances both CA formation and rupture we were then interested in determining if CSE altered NOX1 expression in both unruptured and ruptured CAs. Therefore, we assessed NOX1 mRNA levels following CA induction. As demonstrated in Figure 5C, RT-PCR analysis revealed that CSE increased expression of NOX1 in unruptured CAs, which was further increased in ruptured CA. In contrast there was no change in NOX2, NOX3, or NOX4 expression in any of the cohorts (Figure 5D–F). The elevated expression of NOX1 in CA of CSE treated mice was also increased over animals that underwent sham surgery but received CSE and furthermore over a control cohort that underwent sham surgery but did not receive CSE or a cohort undergoing surgery with CSE but pretreated with apocynin. The increase in NOX1 expression in CA following CSE was paralleled with a similar increase in superoxide production, as assessed by dihydroethidium imaging. Exposing mice to CSE alone did not substantially increase dihydroethidium fluorescence when compared to controls, whereas pretreatment with apocynin, which inhibits NOX activity, reduced the dihydroethidium fluorescence. (Figure 5G). Superoxide production was found to be increased in unruptured and furthermore in ruptured aneurysms (Figure 5G. an enlarged image is shown in supplemental Figure II).
CSE Initiates NOX-Dependent VSMC Phenotypic Modulation in Cerebral Aneurysm Formation and Rupture
To assess whether VSMC phenotypic modulation occurs in the formation and rupture of CAs following CSE, we assessed alterations in mRNA expression of VSMC marker genes and pro-inflammatory and matrix remodeling genes. SM-α-actin, SM-MHC, SM-22α mRNA, and myocardin expression where found to be decreased in ruptured CAs as compared to unruptured CAs, and as compared to those pre-treated with apocynin (Figure 5H). Additionally, animals receiving CSE that underwent sham surgery, had decreased levels of SM-α-actin, SM-MHC, SM-22α mRNA as compared to those pre-treated with apocynin or a sham surgery group that was not exposed to cigarette smoke (Figure 5H).
Analysis of inflammatory and matrix remodeling genes revealed increased expression of MCP-1, MMP-3, MMP-9, TNF-α, IL-1β, iNOS, TNF-α, NF-κB, VCAM and KLF4 in unruptured CAs, which were further increased in ruptured CAs receiving CSE (Figure 5I). This was also increased over animals that underwent sham surgery, but received CSE and furthermore over cohorts either pretreated with apocynin or those only receiving sham surgery.
Direct Application of CSE Alters NOX1 Expression and VSMC Phenotype
To access if CSE directly affects NOX1 expression and modulates phenotypic gene expression, in vivo, or whether other systemic factors are required, we directly applied CSE to the adventitial surface of rat carotid arteries using a pluronic gel as detailed in Methods. Consistent with our previous findings NOX1 mRNA expression was significantly increased 6 h following CSE application when compared to application of pluronic gel alone (Figure 6A). The increase in NOX1 was followed with a significant decrease in mRNA expression of the VSMC marker genes (SM-22α, SM-αActin, and SM-MHC) and myocardin and with a parallel increase in pro-inflammatory and matrix remodeling genes, KLF4, MCP1, MMP3, MMP9, TNF-α, and IL-1β (Figure 6B–C) 12 h following CSE application.
Figure 6. Direct effects of CSE on NOX1 mRNA expression and phenotypic modulation.
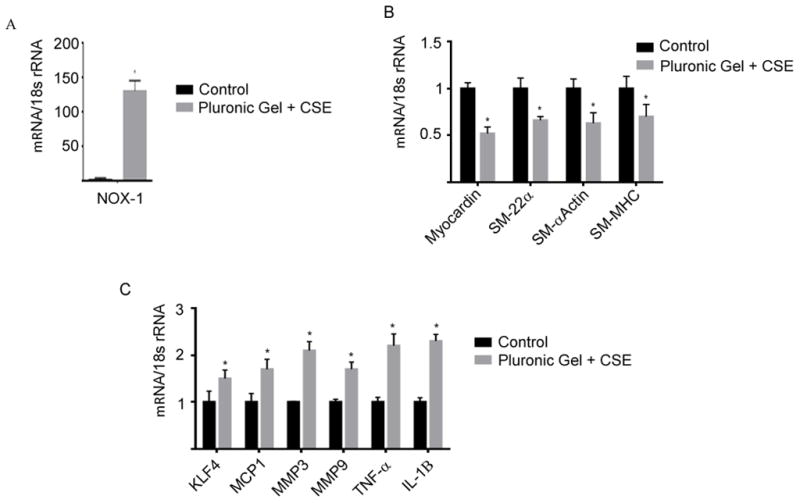
CSE was directly applied to the adventitial surface of rat carotid arteries using a pluronic gel containing 0.8 mg/ml CSE. Changes in NOX1 mRNA expression 6–8 h following direct application of CSE is shown in A). mRNA expression of VSMC phenotypic genes, and pro-inflammatory and matrix remodeling genes are shown in B) and C) respectively following 12 h of direct CSE application. All values represent mean ± SEM of n =6. *P<0.05 versus control using Wilcoxon rank sum.
DISCUSSION
Smoking is a key modifiable risk factor for CA formation and rupture. However the precise mechanisms of CSE induced vascular alteration and injury in the cerebral circulation remain unclear.11–24 CSE can cause significant alterations in the cerebral immune system and inflammatory pathways, resulting in direct vascular injury and production of ROS.25–35 Additionally, prior studies have found that CSE results in endothelial dysfunction, leading to attraction of pro-inflammatory mediators and immune cells, resulting in further SMC injury.27–32 As these are key mechanisms underlying CA pathogenesis, it has been hypothesized that CSE 8, oxidative stress 48, and SMC phenotypic modulation 49 contribute to a pro-inflammatory environment 7, which promotes CA formation and rupture. In this study, we have found that NOX and oxidative stress contributes to CA formation and rupture. Additionally, CSE leads to upregulation of NOX1 and free radical oxidative stress both in vitro in cultured cerebral VSMCs and in vivo in a mouse model of CA formation and rupture. Excess free radical production leads to VSMC phenotypic modulation, with alteration of VSMC function from a role primarily concerned with contraction to that of inflammation and matrix remodeling. Both pharmacological and genetic inhibition of NOX-induced oxidative stress prevents VSMC phenotypic modulation and decreases both CA formation and rupture. CSE increases oxidative stress and CA formation and rupture, and pharmacological inhibition of NOX is protective against CSE-induced oxidative stress, VSMC phenotypic modulation, and CA formation and rupture.
Increased ROS have been previously demonstrated in human CAs, which may arise from a number of potential sources.50 In this study, we found upregulation of NOX1 and oxidative stress in an in vivo model of CA formation and rupture that involves induced hypertension. We, and others, have also found that NOX expression and ROS formation is increased in an in vivo model of CA formation that involves induced hypertension and hemodynamic stress prior to CA formation.50,51 We also demonstrate that CSE further incites NOX1 expression and induces oxidative stress, and further contributes to CA formation and rupture. Hypertension, hemodynamic stress, and CSE are significant risk factors for CA formation and rupture, and likely all contribute to ROS formation 7,8,48. CSE is a major potential cause of CA induction, which exert its effects through activation of NOX and production of ROS 52,53. The observed effects of CSE may arise directly from the gas/tar phase, activated inflammatory cells, or endogenous sources of ROS, such as xanthine oxidase, uncoupled nitric oxide, and the mitochondrial electron transport chain.32,53 Once activated, VSMCs and activated macrophages can further produce ROS within CAs.
In this study, we found that CSE results in NOX-induced VSMC phenotypic modulation both in vivo and in vitro. CSE-induced NOX1 upregulation and ROS formation was preceded by downregulation of VSMC marker genes. Additionally, inhibition of free radical formation reversed NOX1-induced VSMC phenotypic modulation. Similarly, NOX-induced VSMC phenotypic modulation occurred in CA formation and rupture, and was similarly abrogated by both genetic and pharmacological inhibition of NOX. In the peripheral circulation, myocardin is a key transcriptional promotor of SMC differentiation through transcriptional upregulation of SMC marker genes 36,38–40. Myocardin was found to be decreased both in cultured cerebral VSMCs and CAs. Results from the present experiments support that peripheral phenotypic modulation in VSMC pathology occurs through similar mechanisms within CAs.
Increased NOX1 mRNA expression was observed in both isolated VSMC and in CA tissue following CSE and was paralleled with a robust increase in NOX1 protein expression in unruptured and ruptured CA tissue. This increased in NOX1 protein levels following CA induction was primarily observed in VSMC, and to a degree, in macrophages. This is in contrast to NOX2, NOX3, and NOX4 which were not significantly increased by CSE. Our in vitro results demonstrate a link between NOX1 expression and VSM gene regulation. However because apocynin and p47phox null mice inhibit other NOX family members besides NOX1 we cannot fully rule out the possibility that altered NOX2, NOX3, or NOX4 protein activity, and not gene expression, also contributed to the observed effects of CSE on aneurysm pathogenesis.
Immune cells and inflammatory markers have been found to be systemically increased in cigarette smokers.33–35 Studies in cell culture and animal models have demonstrated increased immune modulating cells and cytokines following CSE.27–32 In the present study CSE resulted in a pro-inflammatory, matrix remodeling phenotype in a NOX-dependent fashion. Both in vivo and in vitro CSE resulted in downregulation of myocardin, SM-α-actin, SM-MHC and SM-22α, and upregulation of MCP-1, MMPs, TNF-α, IL-1β, and NF-κB. These alterations have been found to be critical steps in the initiation of atherosclerosis, vascular disease, and CA pathogenesis. We previously found that TNF-α expression is increased in CA formation and rupture, and that genetic and pharmacological inhibition of TNF-α decreases formation and rupture of CAs. ROS have been shown to activate TNF-α which, as an immune modulator, can activate further pro-inflammatory cascades in macrophages as well as phenotypic modulation in VSMCs. MCP-1 has been found to be increased in CA walls, and genetic inhibition of MCP-1 decreases MMP production and the incidence of CA formation. This may work through a variety of mechanisms, including chemoattraction of macrophages into CAs leading to further production of ROS, TNF-α and MMPs.54 MMPs degrade the extracellular matrix55 and have been found to be increased in human CAs.56 Inhibition of MMPs has also been shown to decrease the incidence of CA formation and progression in animals.47,57 NF-κB has also been shown to help produce ROS, and prior studies have demonstrated that it is necessary for CA formation. Additionally, IL-1β promotes SMC phenotypic modulation through increased expression of pro-inflammatory genes and has been found to be a key mediator of atherosclerosis and vascular injury.
It currently remains unclear whether CA rupture is the end result of CA formation, or if molecular mediators are activated after CA formation and are required for CA progression and eventual rupture. In the current study, CSE-initiated oxidative stress induced VSMC phenotypic modulation leading to both CA formation and rupture. Oxidative stress can result from increased production and/or decreased removal of free radicals, and can result in DNA damage, cellular toxicity, and apoptosis 58. Other key inflammatory mediators, such as TNF-α, help regulate apoptosis and phagocytosis of VSMCs and may lead to CA progression and rupture.44,59–61 Following exposure to CSE, VSMCs change from a contractile phenotype to one characterized by pro-inflammatory characteristics and matrix remodeling, which may eventually lead to apoptosis, with loss of both phenotypes and eventual CA rupture.
Further assessment of the molecular mechanisms underlying both CA formation and progression is indicated. We have previously found that epigenetic control mechanisms, including alterations in histone modifications characteristic of transcriptional suppression,62–64 provided evidence that KLF4 regulates cerebral VSMC phenotypic modulation through inhibition of myocardin-mediated activation of SMC genes.41,65 KLF4 is a pluripotency factor involved in the reprogramming of somatic cells.66 Prior studies have also shown that KLF4 is required for SMC phenotypic modulation following vascular injury41 and experimental atherosclerosis.42 The SMC MHC gene39 and myocardin promoter38 also possess KLF4 binding sites, and are inhibited during SMC phenotypic switching to an inflammatory phenotype. KLF4 has also been found to be integral to the activation of macrophages, an inflammatory cell necessary for CA formation.54,67 In the present study, KLF4 was increased and myocardin was decreased following CSE, but after NOX1 upregulation and formation of ROS.
Although oxidative stress and VSMC phenotypic modulation are likely key elements for CA pathogenesis, it is probably a multifactorial process that leads to CA pathogenesis. In this study we focused upon the effects of CSE on VSM phenotypic modulation, a characteristic of aneurysm pathophysiology, and on CA pathogenesis. However, CSE would have a global effect on the cellular vascular environment, affecting not only VSM but also endothelial cell, and infiltrating macrophages which would all culminate in CA formation, progression and rupture. Interestingly CSE increases intracellular communication between VSM, and endothelial cells, and macrophages leading to altered modulation of signaling molecules associated with abdominal aortic aneurysm formation68. The ability of CSE to induce endothelial dysfunction in cerebral arteries is well documented 69. Therefore it is likely that the CSE induced aneurysm formation and rupture observed in this study is the result of the accumulative effects of VSM, endothelial cells and macrophages dysfunction by CSE.
In conclusion, this is the first study to assess how CSE induces oxidative stress and cerebral VSMC phenotypic modulation in CA pathogenesis. It provides novel in vitro and in vivo evidence that CSE profoundly suppresses expression of mature cerebral VSMC marker genes and promotes matrix remodeling/inflammatory gene expression in CA formation and rupture. These processes are, at least in part, regulated by CSE induction of NOX1-associated oxidative stress. This provides important mechanisms whereby CSE may contribute to CA pathogenesis and provide pathways for protective therapies.
Supplementary Material
Table 1.
Real-time PCR Primer Sequences
| Forward Primer (Rat) | Reverse Primer (Rat) | |
|---|---|---|
| 1 | SM-α-Actin | |
| AGTCGCCATCAGGAACCTCGAG | ATCTTTTCGATGTCGTCCCAGTTG | |
| 2 | SM-MHC | |
| CAGTTGGACACTATGTCAGGGAAA | ATGGAGACAAATGCTAATCAGCC | |
| 3 | SM-22α | |
| GCATAAGAGGGAGTTCACAGACA | GCCTTCCCTTTCTAACTGATGATC | |
| 4 | Myocardin | |
| CGGATTCGAAGCTGTTGTCTT | AAACCAGGCCCCCTTCC | |
| 5 | KLF4 | |
| CTTTCCTGCCAGACCAGATG | GGTTTCTCGCCTGTGTGAGT | |
| 6 | 18S | |
| CGGCTACCACATCCAAGGAA | AGCTGGAATTACCGCGGC | |
| 7 | MMP3 | |
| GCCAATGCTGAAGCTTTGATGTAC | GGGAGGTCCATAGAGGGATTGAAT | |
| 8 | IL-1β | |
| TTGTGCAAGTGTCTGAAGCA | TGTCAGCCTCAAAGAACAGG | |
| 9 | MCP1 | |
| CTCAGCCAGATGCAGTTAATGC | TCTCCAGCCGACTCATTG G | |
| 10 | TNF-α | |
| AAAGCATGATCCGAGATGT | AGCAGGAATGAGAAGAGG C | |
| 11 | NF-κB (IκB-α) | |
| GCTGAAGAAGGAGCGCTACT | TCGTACTCCTCGTCTTTCATGGA | |
| 12 | MMP9 | |
| AAGCCTTGGTGTGGCACGAC | TGGAAATACGCAGGGTTTGC | |
| NOX1 | ||
| TTCCCTGGAACAAGAGATGG | GACGTCAGTGGCTCTGTCAA | |
| NOX2 | ||
| AGCTATGAGGTGGTGATGTTAGTGG | CACAATATTTGTACCAGACAGACTTGAG | |
| NOX3 | ||
| GCTGGCTGCACTTTCCAAAC | AAGGTGCGGACTGGATTGAG | |
| NOX4 | ||
| CTGGAAGAACCCAAGTTCCA | CGGATGCATCGGTAAAGTCT |
Highlights.
Cigarette smoke exposure increases expression of NOX1 and ROS in vascular smooth muscle cells.
Cigarette smoke exposure induces NOX1 and ROS dependent vascular smooth muscle phenotypic modulation.
NOX1 and ROS are increased in aneurysms and even further in ruptured aneurysms.
Inhibition of NOX reduces aneurysm formation and rupture.
Cigarette smoke exposure increases cerebral aneurysm formation and rupture.
Acknowledgments
SOURCES OF FUNDING
This work was supported by National Institutes of Health (NIH) grants K08NS067072 to AD, R03NS079227-01A1 and K08NS082363-01A1 to DH, R01 HL57353, R01 HL098538, and R01 HL087867 to GO, and R01HL110737, R01HL084275, R01HL094849, R01HL107110 and UM1HL113460 grant to JH. This work was supported by the Neurosurgery Research Education Foundation and by a Cerebrovascular Section Young Clinician Investigator Award to RMS.
Nonstandard Abbreviations and Acronyms
- BAPN
Beta-aminoproprionitrile
- CA
Cerebral aneurysm
- CSE
Cigarette smoking exposure
- DOCA
deoxycorticosterone acetate
- GFP
Green fluorescent protein
- FBS
Fetal bovine serum
- HBSS
Hank’s Balanced Salt Solution
- KLF4
Kruppel-like factor four
- MOI
Multiplicity of infection
- NOX
NADP
- PBS
Phosphate buffered saline
- ROS
Reactive oxygen species
- RT-PCR
Real-time polymerase chain reaction
- SEM
Standard error of the mean
- SM22α
Smooth muscle 22 alpha
- SM-MHC
Smooth muscle myosin heavy chain
- SM-α-actin
Smooth muscle alpha actin
- SMC
Smooth Muscle Cells
- VSMC
vascular smooth muscle cells
Footnotes
DISCLOSURES
The authors have no disclosures to report. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.He J, Vupputuri S, Allen K, Prerost MR, Hughes J, Whelton PK. Passive smoking and the risk of coronary heart disease--a meta-analysis of epidemiologic studies. N Engl J Med. 1999;340(12):920–926. doi: 10.1056/NEJM199903253401204. [DOI] [PubMed] [Google Scholar]
- 2.You RX, Thrift AG, McNeil JJ, Davis SM, Donnan GA. Ischemic stroke risk passive exposure to spouses’ cigarette smoking. Melbourne Stroke Risk Factor Study (MERFS) Group. Am J Public Health. 1999;89(4):572–575. doi: 10.2105/ajph.89.4.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sacco RL, Adams R, Albers G, et al. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co-sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline. Stroke. 2006;37(2):577–617. doi: 10.1161/01.STR.0000199147.30016.74. [DOI] [PubMed] [Google Scholar]
- 4.Diez-Roux AV, Nieto FJ, Comstock GW, Howard G, Szklo M. The relationship of active and passive smoking to carotid atherosclerosis 12–14 years later. Prev Med. 1995;24(1):48–55. doi: 10.1006/pmed.1995.1007. [DOI] [PubMed] [Google Scholar]
- 5.Inoue T, Oku K, Kimoto K, et al. Relationship of cigarette smoking to the severity of coronary and thoracic aortic atherosclerosis. Cardiology. 1995;86(5):374–379. doi: 10.1159/000176904. [DOI] [PubMed] [Google Scholar]
- 6.Andersen KK, Olsen TS, Dehlendorff C, Kammersgaard LP. Hemorrhagic and ischemic strokes compared: stroke severity, mortality, and risk factors. Stroke. 2009;40(6):2068–2072. doi: 10.1161/STROKEAHA.108.540112. [DOI] [PubMed] [Google Scholar]
- 7.Chalouhi N, Ali MS, Jabbour PM, et al. Biology of intracranial aneurysms: role of inflammation. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2012;32(9):1659–1676. doi: 10.1038/jcbfm.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chalouhi N, Ali MS, Starke RM, et al. Cigarette smoke and inflammation: role in cerebral aneurysm formation and rupture. Mediators of inflammation. 2013;2012:271582. doi: 10.1155/2012/271582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldstein LB, Bushnell CD, Adams RJ, et al. Guidelines for the primary prevention of stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2010;42(2):517–584. doi: 10.1161/STR.0b013e3181fcb238. [DOI] [PubMed] [Google Scholar]
- 10.Jha P, Ramasundarahettige C, Landsman V, et al. 21st-century hazards of smoking and benefits of cessation in the United States. N Engl J Med. 2013;368(4):341–350. doi: 10.1056/NEJMsa1211128. [DOI] [PubMed] [Google Scholar]
- 11.Brown RD, Jr, Huston J, Hornung R, et al. Screening for brain aneurysm in the Familial Intracranial Aneurysm study: frequency and predictors of lesion detection. J Neurosurg. 2008;108(6):1132–1138. doi: 10.3171/JNS/2008/108/6/1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Connolly ES, Jr, Poisik A, Winfree CJ, et al. Cigarette smoking and the development and rupture of cerebral aneurysms in a mixed race population: implications for population screening and smoking cessation. J Stroke Cerebrovasc Dis. 1999;8(4):248–253. doi: 10.1016/s1052-3057(99)80074-3. [DOI] [PubMed] [Google Scholar]
- 13.Nahed BV, Bydon M, Ozturk AK, Bilguvar K, Bayrakli F, Gunel M. Genetics of intracranial aneurysms. Neurosurgery. 2007;60(2):213–225. doi: 10.1227/01.NEU.0000249270.18698.BB. discussion 225–216. [DOI] [PubMed] [Google Scholar]
- 14.Wermer MJ, van der Schaaf IC, Velthuis BK, Algra A, Buskens E, Rinkel GJ. Follow-up screening after subarachnoid haemorrhage: frequency and determinants of new aneurysms and enlargement of existing aneurysms. Brain. 2005;128(Pt 10):2421–2429. doi: 10.1093/brain/awh587. [DOI] [PubMed] [Google Scholar]
- 15.Clarke M. Systematic review of reviews of risk factors for intracranial aneurysms. Neuroradiology. 2008;50(8):653–664. doi: 10.1007/s00234-008-0411-9. [DOI] [PubMed] [Google Scholar]
- 16.Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke. 2005;36(12):2773–2780. doi: 10.1161/01.STR.0000190838.02954.e8. [DOI] [PubMed] [Google Scholar]
- 17.Isaksen J, Egge A, Waterloo K, Romner B, Ingebrigtsen T. Risk factors for aneurysmal subarachnoid haemorrhage: the Tromso study. J Neurol Neurosurg Psychiatry. 2002;73(2):185–187. doi: 10.1136/jnnp.73.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Juvela S, Porras M, Poussa K. Natural history of unruptured intracranial aneurysms: probability and risk factors for aneurysm rupture. Neurosurg Focus. 2000;8(5) Preview 1. [PubMed] [Google Scholar]
- 19.Juvela S, Porras M, Poussa K. Natural history of unruptured intracranial aneurysms: probability of and risk factors for aneurysm rupture. J Neurosurg. 2008;108(5):1052–1060. doi: 10.3171/JNS/2008/108/5/1052. [DOI] [PubMed] [Google Scholar]
- 20.Kissela BM, Sauerbeck L, Woo D, et al. Subarachnoid hemorrhage: a preventable disease with a heritable component. Stroke. 2002;33(5):1321–1326. doi: 10.1161/01.str.0000014773.57733.3e. [DOI] [PubMed] [Google Scholar]
- 21.Koskinen LO, Blomstedt PC. Smoking and non-smoking tobacco as risk factors in subarachnoid haemorrhage. Acta Neurol Scand. 2006;114(1):33–37. doi: 10.1111/j.1600-0404.2006.00591.x. [DOI] [PubMed] [Google Scholar]
- 22.Okamoto K, Horisawa R, Ohno Y. The relationships of gender, cigarette smoking, and hypertension with the risk of aneurysmal subarachnoid hemorrhage: a case-control study in Nagoya, Japan. Ann Epidemiol. 2005;15(10):744–748. doi: 10.1016/j.annepidem.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 23.Rinkel GJ. Natural history, epidemiology and screening of unruptured intracranial aneurysms. Rev Neurol (Paris) 2008;164(10):781–786. doi: 10.1016/j.neurol.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 24.Weir BK, Kongable GL, Kassell NF, Schultz JR, Truskowski LL, Sigrest A. Cigarette smoking as a cause of aneurysmal subarachnoid hemorrhage and risk for vasospasm: a report of the Cooperative Aneurysm Study. J Neurosurg. 1998;89(3):405–411. doi: 10.3171/jns.1998.89.3.0405. [DOI] [PubMed] [Google Scholar]
- 25.Arnson Y, Shoenfeld Y, Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun. 2010;34(3):J258–265. doi: 10.1016/j.jaut.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 26.Mazzone P, Tierney W, Hossain M, Puvenna V, Janigro D, Cucullo L. Pathophysiological impact of cigarette smoke exposure on the cerebrovascular system with a focus on the blood-brain barrier: expanding the awareness of smoking toxicity in an underappreciated area. Int J Environ Res Public Health. 2010;7(12):4111–4126. doi: 10.3390/ijerph7124111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orosz Z, Csiszar A, Labinskyy N, et al. Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: role of NAD(P)H oxidase activation. Am J Physiol Heart Circ Physiol. 2007;292(1):H130–139. doi: 10.1152/ajpheart.00599.2006. [DOI] [PubMed] [Google Scholar]
- 28.Perlstein TS, Lee RT. Smoking, metalloproteinases, and vascular disease. Arterioscler Thromb Vasc Biol. 2006;26(2):250–256. doi: 10.1161/01.ATV.0000199268.27395.4f. [DOI] [PubMed] [Google Scholar]
- 29.Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res. 2011;91(2):142–149. doi: 10.1177/0022034511421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol. 2002;2(5):372–377. doi: 10.1038/nri803. [DOI] [PubMed] [Google Scholar]
- 31.Hossain M, Mazzone P, Tierney W, Cucullo L. In vitro assessment of tobacco smoke toxicity at the BBB: do antioxidant supplements have a protective role? BMC Neurosci. 12:92. doi: 10.1186/1471-2202-12-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43(10):1731–1737. doi: 10.1016/j.jacc.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 33.Schwartz J, Weiss ST. Cigarette smoking and peripheral blood leukocyte differentials. Ann Epidemiol. 1994;4(3):236–242. doi: 10.1016/1047-2797(94)90102-3. [DOI] [PubMed] [Google Scholar]
- 34.Tracy RP, Psaty BM, Macy E, et al. Lifetime smoking exposure affects the association of C-reactive protein with cardiovascular disease risk factors and subclinical disease in healthy elderly subjects. Arterioscler Thromb Vasc Biol. 1997;17(10):2167–2176. doi: 10.1161/01.atv.17.10.2167. [DOI] [PubMed] [Google Scholar]
- 35.Mendall MA, Patel P, Asante M, et al. Relation of serum cytokine concentrations to cardiovascular risk factors and coronary heart disease. Heart. 1997;78(3):273–277. doi: 10.1136/hrt.78.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75(3):487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 37.Yoshida T, Owens GK. Molecular determinants of vascular smooth muscle cell diversity. Circ Res. 2005;96(3):280–291. doi: 10.1161/01.RES.0000155951.62152.2e. [DOI] [PubMed] [Google Scholar]
- 38.Creemers EE, Sutherland LB, McAnally J, Richardson JA, Olson EN. Myocardin is a direct transcriptional target of Mef2, Tead and Foxo proteins during cardiovascular development. Development. 2006;133(21):4245–4256. doi: 10.1242/dev.02610. [DOI] [PubMed] [Google Scholar]
- 39.Madsen CS, Hershey JC, Hautmann MB, White SL, Owens GK. Expression of the smooth muscle myosin heavy chain gene is regulated by a negative-acting GC-rich element located between two positive-acting serum response factor-binding elements. J Biol Chem. 1997;272(10):6332–6340. doi: 10.1074/jbc.272.10.6332. [DOI] [PubMed] [Google Scholar]
- 40.Pidkovka NA, Cherepanova OA, Yoshida T, et al. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res. 2007;101(8):792–801. doi: 10.1161/CIRCRESAHA.107.152736. [DOI] [PubMed] [Google Scholar]
- 41.Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest. 2000;106(9):1139–1147. doi: 10.1172/JCI10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wamhoff BR, Hoofnagle MH, Burns A, Sinha S, McDonald OG, Owens GK. A G/C element mediates repression of the SM22alpha promoter within phenotypically modulated smooth muscle cells in experimental atherosclerosis. Circ Res. 2004;95(10):981–988. doi: 10.1161/01.RES.0000147961.09840.fb. [DOI] [PubMed] [Google Scholar]
- 43.Chalouhi N, Ali MS, Jabbour PM, et al. Biology of intracranial aneurysms: role of inflammation. J Cereb Blood Flow Metab. doi: 10.1038/jcbfm.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Starke RM, Chalouhi N, Jabbour PM, et al. Critical role of TNF-alpha in cerebral aneurysm formation and progression to rupture. J Neuroinflammation. 2014;11(1):77. doi: 10.1186/1742-2094-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Starke RM, Ali MS, Jabbour PM, et al. Cigarette smoke modulates vascular smooth muscle phenotype: implications for carotid and cerebrovascular disease. PloS one. 2013;8(8):e71954. doi: 10.1371/journal.pone.0071954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schievink WI. Intracranial aneurysms. N Engl J Med. 1997;336(1):28–40. doi: 10.1056/NEJM199701023360106. [DOI] [PubMed] [Google Scholar]
- 47.Nuki Y, Tsou TL, Kurihara C, Kanematsu M, Kanematsu Y, Hashimoto T. Elastase-induced intracranial aneurysms in hypertensive mice. Hypertension. 2009;54(6):1337–1344. doi: 10.1161/HYPERTENSIONAHA.109.138297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Starke RM, Chalouhi N, Ali MS, et al. The role of oxidative stress in cerebral aneurysm formation and rupture. Curr Neurovasc Res. 2013;10(3):247–255. doi: 10.2174/15672026113109990003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Starke RM, Chalouhi N, Ding D, et al. Vascular Smooth Muscle Cells in Cerebral Aneurysm Pathogenesis. Transl Stroke Res. 2013 doi: 10.1007/s12975-013-0290-1. [DOI] [PubMed] [Google Scholar]
- 50.Aoki T, Nishimura M, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Reactive oxygen species modulate growth of cerebral aneurysms: a study using the free radical scavenger edaravone and p47phox(−/−) mice. Lab Invest. 2009;89(7):730–741. doi: 10.1038/labinvest.2009.36. [DOI] [PubMed] [Google Scholar]
- 51.Ali MS, Starke RM, Jabbour PM, et al. TNF-alpha induces phenotypic modulation in cerebral vascular smooth muscle cells: implications for cerebral aneurysm pathology. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33(10):1564–1573. doi: 10.1038/jcbfm.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asano H, Horinouchi T, Mai Y, et al. Nicotine- and tar-free cigarette smoke induces cell damage through reactive oxygen species newly generated by PKC-dependent activation of NADPH oxidase. J Pharmacol Sci. 2012;118(2):275–287. doi: 10.1254/jphs.11166fp. [DOI] [PubMed] [Google Scholar]
- 53.Talukder MA, Johnson WM, Varadharaj S, et al. Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice. American journal of physiology. Heart and circulatory physiology. 2011;300(1):H388–396. doi: 10.1152/ajpheart.00868.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanematsu Y, Kanematsu M, Kurihara C, et al. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke. 2010;42(1):173–178. doi: 10.1161/STROKEAHA.110.590976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39(3):279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- 56.Kim SC, Singh M, Huang J, et al. Matrix metalloproteinase-9 in cerebral aneurysms. Neurosurgery. 1997;41(3):642–666. doi: 10.1097/00006123-199709000-00027. discussion 646–647. [DOI] [PubMed] [Google Scholar]
- 57.Makino H, Tada Y, Wada K, et al. Pharmacological stabilization of intracranial aneurysms in mice: a feasibility study. Stroke. 2012;43(9):2450–2456. doi: 10.1161/STROKEAHA.112.659821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part I: basic mechanisms and in vivo monitoring of ROS. Circulation. 2003;108(16):1912–1916. doi: 10.1161/01.CIR.0000093660.86242.BB. [DOI] [PubMed] [Google Scholar]
- 59.Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74:13–40. doi: 10.1146/annurev-physiol-012110-142315. [DOI] [PubMed] [Google Scholar]
- 60.Legein B, Temmerman L, Biessen EA, Lutgens E. Inflammation and immune system interactions in atherosclerosis. Cell Mol Life Sci. 2013 doi: 10.1007/s00018-013-1289-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stoll G, Bendszus M. Inflammation and atherosclerosis: novel insights into plaque formation and destabilization. Stroke. 2006;37(7):1923–1932. doi: 10.1161/01.STR.0000226901.34927.10. [DOI] [PubMed] [Google Scholar]
- 62.McDonald OG, Owens GK. Programming smooth muscle plasticity with chromatin dynamics. Circ Res. 2007;100(10):1428–1441. doi: 10.1161/01.RES.0000266448.30370.a0. [DOI] [PubMed] [Google Scholar]
- 63.McDonald OG, Wamhoff BR, Hoofnagle MH, Owens GK. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest. 2006;116(1):36–48. doi: 10.1172/JCI26505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trojer P, Reinberg D. Histone lysine demethylases and their impact on epigenetics. Cell. 2006;125(2):213–217. doi: 10.1016/j.cell.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 65.Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J Biol Chem. 2005;280(10):9719–9727. doi: 10.1074/jbc.M412862200. [DOI] [PubMed] [Google Scholar]
- 66.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 67.Aoki T, Kataoka H, Morimoto M, Nozaki K, Hashimoto N. Macrophage-derived matrix metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke. 2007;38(1):162–169. doi: 10.1161/01.STR.0000252129.18605.c8. [DOI] [PubMed] [Google Scholar]
- 68.Ghosh A, Pechota LV, Upchurch GR, Jr, Eliason JL. Cross-talk between macrophages, smooth muscle cells, and endothelial cells in response to cigarette smoke: the effects on MMP2 and 9. Molecular and cellular biochemistry. 2015;410(1–2):75–84. doi: 10.1007/s11010-015-2539-3. [DOI] [PubMed] [Google Scholar]
- 69.Chalouhi N, Ali MS, Starke RM, et al. Cigarette smoke and inflammation: role in cerebral aneurysm formation and rupture. Mediators of inflammation. 2012;2012:271582. doi: 10.1155/2012/271582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.