

ABSTRACT
There is a growing awareness that molecular diagnostics for detect-to-treat applications will soon need a highly multiplexed mutation detection and identification capability. In this study, we converted an open-amplicon microarray hybridization test for multidrug-resistant (MDR) Mycobacterium tuberculosis into an entirely closed-amplicon consumable (an amplification microarray) and evaluated its performance with matched sputum and sediment extracts. Reproducible genotyping (the limit of detection) was achieved with ∼25 M. tuberculosis genomes (100 fg of M. tuberculosis DNA) per reaction; the estimated shelf life of the test was at least 18 months when it was stored at 4°C. The test detected M. tuberculosis in 99.1% of sputum extracts and 100% of sediment extracts and showed 100% concordance with the results of real-time PCR. The levels of concordance between M. tuberculosis and resistance-associated gene detection were 99.1% and 98.4% for sputum and sediment extracts, respectively. Genotyping results were 100% concordant between sputum and sediment extracts. Relative to the results of culture-based drug susceptibility testing, the test was 97.1% specific and 75.0% sensitive for the detection of rifampin resistance in both sputum and sediment extracts. The specificity for the detection of isoniazid (INH) resistance was 98.4% and 96.8% for sputum and sediment extracts, respectively, and the sensitivity for the detection of INH resistance was 63.6%. The amplification microarray reported the correct genotype for all discordant phenotype/genotype results. On the basis of these data, primary sputum may be considered a preferred specimen for the test. The amplification microarray design, shelf life, and analytical performance metrics are well aligned with consensus product profiles for next-generation drug-resistant M. tuberculosis diagnostics and represent a significant ease-of-use advantage over other hybridization-based tests for diagnosing MDR tuberculosis.
KEYWORDS: amplification microarray, closed amplicon, drug resistance, in vitro diagnostic, microfluidic, Mycobacterium tuberculosis
INTRODUCTION
The global rollout of the Xpert MTB/RIF system unquestionably improved multidrug-resistant (MDR) tuberculosis (TB) case detection and the time to diagnosis but also brought to light several health care, infrastructure, technology, genotyping, and cost considerations for high-priority diagnostics (1–4). At the same time, Mycobacterium tuberculosis whole-genome sequencing efforts involving hundreds or thousands of drug-resistant M. tuberculosis isolates continue to identify new genes and mutations that cause or are correlated with mono-, multi-, or extensive drug resistance or compensate for known mutations (5–12). Host genetic factors are also known to play a role in M. tuberculosis infection and disease (13). Given the genetic complexity and heterogeneous evolution of M. tuberculosis drug resistance (14), whole-genome sequencing rather than biomarker discovery is now being considered for M. tuberculosis diagnostic purposes (15–19). While it is not yet clear if or when whole-genome sequencing will satisfy high-priority M. tuberculosis diagnostic technical product profiles (3), it is evident that the number of genes and mutations necessary to diagnose drug resistance is increasing and that the identification (as opposed to the detection) of single-nucleotide polymorphisms (SNP) will become increasingly important for prescribing a patient-specific treatment regimen that accounts for patient genetics/drug metabolism and minimizes the emergence of new drug-resistant M. tuberculosis phenotypes.
Microarrays were originally invented as a DNA sequencing technology (20–22) and can address the multiple-gene, multiple-mutation challenge of diagnosing drug-resistant TB (23–30). Microarrays have yet to impact clinical practice in the same way as real-time PCR, in part because of poor reproducibility and repeatability, complex work flows (inclusive of sample preparation), user subjectivity, and a host of related technical and nontechnical issues, even in developed countries and centralized testing laboratories (31–34). Considering the clinical user and operating requirements for TB diagnostics (1–3), it becomes clear that microarray (and sequencing) platforms need to be developed from an entirely distinct perspective to harmonize competing user needs and product requirements for MDR-TB and extremely drug resistant (XDR) TB diagnostics.
One way to overcome the inherent complexity of microarray-based diagnostics for routine use (especially in lower-resource settings) is to combine amplification and microarray hybridization within a single microfluidic chamber, confine amplification products within the consumable (i.e., a closed-amplicon device), simplify the total number of biochemical steps necessary to detect mutations, and optimize the assay for use with low-cost, field-portable microarray imagers (35, 36). The objectives of this study were to advance these concepts and develop a closed-amplicon, microarray-based consumable for the detection of MDR-TB, characterize the device's analytical and shelf-life behavior, and evaluate the test on matched primary sputum and N-acetyl-l-cysteine (NALC)–NaOH-decontaminated sediment extracts from confirmed or suspected M. tuberculosis-positive patients.
MATERIALS AND METHODS
Genomic DNA, isolates, and positive controls.
Purified M. tuberculosis H37Ra DNA was acquired from the American Type Culture Collection (ATCC; Manassas, VA) and quantified on a NanoDrop 3000 fluorometer before use. Materials from the Special Programme for Research and Training in Tropical Diseases (TDR) Tuberculosis Strain Bank (37) (now integrated under BCCM/TIM) were provided as heat-killed crude lysates. Cell lysates were further processed through a BD GeneOhm bead lysis kit (catalogue number 441243; San Diego, CA), and genomic DNA was purified with a Qiagen DNA minikit (catalogue number 51304; Germantown, MD) per the manufacturers' respective instructions, except that samples were incubated at 56°C for 30 min instead of Qiagen's recommended 10 min. Purified nucleic acids were quantified by real-time PCR (see below) relative to the amounts on an external standard curve prepared with M. tuberculosis H37Ra DNA. M13mp18 single-stranded DNA was purchased from New England BioLabs (Ipswich, MA) and diluted to 750 pg ml−1 in 10 mM Tris, 1 mM Na2 EDTA (pH 7.5). Purified nucleic acids were stored at −20°C until use.
Primary sputum and decontaminated sediment samples.
This study utilized retrospective, banked sputum and sediment samples that were collected for a different research objective. Samples were originally derived from patients in public-sector clinics in Lima, Peru, if they had symptoms consistent with M. tuberculosis infection and a prior primary sputum sample testing positive for acid-fast bacilli (AFB) by Ziehl-Neelsen staining. No identifying information or additional clinical data were collected from the study participants. All participants completed written informed consent, and the parent study was approved by the Institutional Review Board of the Harvard Medical School and the Ethics Committee of the Peru National Institute of Health. Thereafter, 2 ml of each sputum sample was decontaminated with 2% NaOH and 0.25% NALC for 15 min, neutralized by adding enough saline phosphate buffer to reach a 50-ml total volume, and centrifuged at 3,000 × g for 30 min. The sediment was resuspended in 1.5 ml of phosphate-buffered saline, and 0.2 ml of each sample was used to inoculate two Löwenstein-Jensen (LJ) slants. Culture tubes were incubated at 37°C and monitored for growth for up to 8 weeks. Ziehl-Neelsen staining and an M. tuberculosis identification test were performed on the culture-positive sputum specimens to confirm the presence of M. tuberculosis in the primary specimen. All remaining sputum specimens and their paired sediment specimens were stored at −80°C until use in the experiments reported here.
Before use in the amplification microarray experiments reported here, we performed follow-up Ziehl-Neelsen acid-fast staining, some of which generated a smear-negative result for AFB (see Table S1 in the supplemental material), even though the original sample was smear positive for AFB. Clinical samples assigned a scanty AFB smear status upon retesting were considered smear positive for this purpose of this study.
Standard drug susceptibility tests (DSTs) were retrospectively performed on positive LJ cultures that were still viable at the time that this study was initiated.
Automated DNA extraction from sputum and sediment.
Total genomic DNA was extracted from the primary sputum and NALC-NaOH-decontaminated sediment using an Akonni TruTip automated workstation, 1.2 ml SPT TruTips, and preloaded reagent plates. Briefly, 500 μl primary sputum was mixed with 80 μl Akonni liquefaction buffer, and the mixture was incubated at 56°C for 20 min with intermittent mixing. Thereafter, a batch of seven sputum or sediment samples and one water blank (500 μl) were loaded onto the workstation and processed in parallel with an automated protocol consisting of 10 min of magnetically induced vortexing, a 10-min incubation at 56°C, total nucleic acid binding to the matrix, washing and drying, and DNA elution in 100 μl 10 mM Tris-HCl (pH 8.0). Purified DNA was stored at −20°C until use.
IS6110-specific quantitative PCR.
M. tuberculosis-specific DNA in nucleic acid extracts was amplified by real-time PCR using a Roche LightCycler 480 instrument and the IS6110 insertion element as a proxy for M. tuberculosis in the primary specimen (38). Briefly, 5 μl of each nucleic acid extract was combined with 20 μl master mix in a 96-well plate to achieve a final reaction composition of 1× LightCycler FastStart DNA Master HybProbe buffer and enzyme (Roche), 2.5 mM MgCl2, 0.45 μM forward primer (5′-GGG-TAG-CAG-ACC-TCA-CCT-ATG), 1.35 μM reverse primer (5′-AGC-GTA-GGC-GTC-GGT-GA), and 25 nM minor groove binding internal probe (5′ 6FAM-TCG-CCT-ACG-TGG-CCT-TT-MGB, where 6FAM is 6-carboxyfluorescein). The microtiter plates were loaded onto the thermal cycler, denatured for 10 min at 95°C, and cycled for 45 cycles of 95°C for 15 s and 60°C for 60 s.
Phenotypic drug susceptibility testing.
M. tuberculosis isolates recovered from positive LJ slants were tested for drug susceptibility using a Bactec MGIT 960 system (Becton, Dickinson, Sparks, MD) according to the manufacturer's instructions. The final antibiotic concentrations in MGIT tubes were 0.1 and 1.0 μg ml−1 for isoniazid (INH) and rifampin (RIF), respectively.
Microfluidic amplification microarray primers, probes, and synthetic DNA standards.
Microarray primers and probes were designed against M. tuberculosis mutations known to confer an RIF and INH resistance phenotype (Table 1). Five PCR primer pairs were designed to work together in a multiplex, asymmetric master mix. One of the primers in each pair was synthesized with a Cy3 label and incorporated into the multiplex reaction mixture at 5 to 10 times the concentration of the unlabeled primer. The PCR primer and microarray probe sequences are identical to those of the PCR primers and microarray probes used in a previous study (36). PCR primers were synthesized by the use of standard phosphoramidite chemistry at Akonni Biosystems, purified by high-performance liquid chromatography (HPLC), and quantified by UV absorption before use. The resulting Cy3-labeled amplicons ranged from 92 to 139 nucleotides in length.
TABLE 1.
Integrated microfluidic amplification microarray genetic coveragea
| Drug | Gene | Amplicon size (nt) | Targeted mutation, target, or description |
|---|---|---|---|
| RIF | rpoB | 139 | 507DEL, Q510H, L511P, L511R, S512T, S512R, Q513L, Q513K, Q513P, M515I, D516E, D516Y, D516G, D516V, S522L, L524S, H526D, H526R, H526L, H526Q (CAA), H526Q (CAG), H526C, H526N, H526P, H526Y, S531W, S531L, S531Q, S531C, L533P |
| INH | katG | 127 | S315T (ACC), S315T (ACA), S315N |
| INH | inhA promoter | 106 | −8A, −8C, −15T, −17T |
| NA | IS6110 | 99 | M. tuberculosis complex |
| NA | M13 | 92 | Internal positive control |
nt, number of nucleotides; DEL, deletion; NA, not applicable.
Microarray probes were synthesized by Akonni with a custom 3′ linker and purified to reach >90% purity by HPLC. Probe purity was measured and confirmed by electrospray ionization mass spectrometry. Microarrays contained at least one universal hybridization probe for each resistance-associated gene and primer pair to verify that M. tuberculosis gene targets were amplified from each sample. At least one matched pair of microarray probes (wild type [WT] and single-nucleotide mutant [MU]) was included for each mutation of interest. Control probes included a Cy3 beacon for manufacturing quality control and positional reference, a probe for an M13 internal positive amplification and inhibition control (internal positive control [IPC]), and control probes for rpoB, katG, and inhA amplification and detection.
Amplification microarray manufacture and quality control.
Gel element arrays were manufactured on custom-coated glass substrates using a 4% copolymer, essentially as described in reference 39. Cy3 fiducial markers were resuspended in the gel precursor at 1 μM, and all other probes were resuspended at a 50 μM concentration before printing. The photopolymerized, washed, and dried microarrays were stored at 4°C for up to 1 week until assembly. All microarrays were visually inspected for gel element presence, a uniform 3-dimensional morphology, and Cy3 beacon fluorescence/uniformity (acceptance criteria, a coefficient of variation of <4% within and between arrays in a production run) before use.
The microarray consumable design is substantially similar to that described elsewhere (35, 40). Essential features of the microfluidic design are that target amplification and microarray hybridization occur simultaneously within a single microfluidic chamber (75 μl), and all amplified products and wash solutions are retained within an integrated waste chamber during and after the wash step (i.e., a closed-amplicon consumable). Microfluidic spacers and cover films were precut, aligned to the microarray substrate with custom jigs, and loosely joined with a pressure roller before permanent lamination in a controlled press. Preassembled waste chambers, inlet ports, and inlet port cover seals were then aligned and loosely affixed before permanent lamination. All finished assemblies were visually inspected for gel element array damage, gross structural defects, adhesive or plastic debris, fluorescent particles that might interfere with automated microarray image analysis, and uniform Cy3 beacon fluorescence (as described above). Integrated consumables that passed final inspection were stored in a vacuum-sealed slide box at 4°C until use (for <2 months, except in the shelf-life study described below).
Amplification microarray shelf life.
Replicate amplification microarray consumables were manufactured over the course of 2 days, individually wrapped, placed inside vacuum-sealed boxes, and stored at 4°C. At 1, 3, 6, and 18 months, replicate amplification microarrays were removed from storage and processed with 10 pg M. tuberculosis H37Ra genomic DNA per reaction mixture, as described below.
Amplification, hybridization, washing, and detection.
Purified nucleic acid extract (approximately 21 μl) was combined with the PCR master mix to achieve an 80-μl total reaction volume consisting of 1× Qiagen HotStar Taq Plus buffer and enzyme, 7.6% formamide, 5% dimethyl sulfoxide, 1 mg ml−1 nonacetylated bovine serum albumin, 4 units of additional HotStar Taq (Qiagen), 750 fg internal positive control, and each primer at a final concentration of 0.04 to 1.2 μM. Seventy-five microliters of each reaction mixture was loaded into an amplification microarray, and the inlet port was sealed with a pierceable foil cover. Thereafter, the amplification microarrays were placed on a Quanta (Hain Life Science, UK) QB-96 flat block thermal cycler and subjected to a touchdown thermal cycling program consisting of an initial denaturation for 5 min at 89°C; 30 cycles of 89°C for 45 s, 60 to 55°C (touchdown) for 1 min, and 65°C for 30 s; 20 cycles of 89°C for 45 s, 55°C for 1 min, and 65°C for 30 s; a final extension at 65°C for 3 min; and a postamplification hybridization at 55°C for 3 h. An external positive control (10 pg purified genomic DNA of known genotype) and negative control (water blank) were run with each batch of amplification microarrays.
After amplification and hybridization, the amplification microarrays were washed by piercing the foil seal with a 1-ml pipette tip and flushing 1 ml 1× SSPE (1× SSPE is 0.18 M NaCl, 10 mM NaH2PO4, and 1 mM EDTA [pH 7.7])–0.01% Triton X-100 through the reaction chamber in a single bolus. The microfluidic design is such that the wash buffer imbibes into the waste chamber, effectively rendering the microarray chamber dry and ready for immediate imaging.
Automated image and data analysis.
Washed and intact amplification microarray consumables were imaged for 0.2 s on a prototype Akonni Dx2000 imager consisting of a high-intensity green light-emitting diode (LED), custom optics, a noncooled charge-coupled-device camera, and Akonni automated gridding, segmentation, and data analysis software. An integrated signal intensity and a local background signal were acquired for each gel element on the array. The standard deviation of each local background was calculated, and then an average was taken for all local backgrounds. Noise was then calculated as 3 · (average standard deviation for all local backgrounds) · 2 · Rint, where Rint is the radius of the fixed circle cell used to acquire the signal. A test was declared valid if the internal positive-control probe generated signal-to-noise (SNR) values of ≥3 or the IS6110 target was detected at an SNR value of ≥10. Otherwise, the test was deemed invalid and the test results were not reported. If IS6110 was detected at an SNR value of ≥10, then the outputs from the internal control probes were reported as not applicable. Deferring the interpretation of internal control probes in the event of an “M. tuberculosis detected” result is based on the fact that the internal positive control is included at a concentration very near its limit of detection (LoD). In those cases where there is abundant M. tuberculosis DNA in the asymmetric PCR, there may be preferential amplification of the M. tuberculosis genes such that IPC amplification is limited and the IPC SNR value is <3 (i.e., it is not detected).
Positive detection of the IS6110 target at an SNR value of ≥10 triggered the automated analysis of universal rpoB, katG, and inhA probes. For a universal probe(s) with an SNR value of <3, the susceptibility or resistance report for the associated drug was deemed indeterminate and there was no further analysis of wild-type or mutant probe signals. Otherwise, universal probe SNR values of ≥3 triggered the analysis of wild-type and mutant probe discrimination ratios (D). For the rpoB gene, both universal probes needed an SNR value of ≥3 to advance the analysis. If at least one of the two paired (wild-type or mutant) probes had an integrated signal intensity greater than the noise floor (defined above), then D = (SNRWT − SNRMU)/(SNRWT + SNRMU), where SNRWT is the SNR value for the wild type and SNRMU is the SNR value for the mutant. Otherwise, D was not calculated and the output for the specific mutation was deemed indeterminate. Any value of D of <0 was reported as a mutation at the targeted nucleotide position with a “resistance detected” output and an itemized list of the associated mutation(s) from Table 1. If all gene-specific probes generated D values of ≥0, then the gene was reported to be the wild type and led to a “resistance not detected” output for the associated drug. Thus, the algorithm and software report on mono- or multidrug resistance. Near the limits of detection and depending on multiplex amplification efficiency, the software may also report susceptibility or resistance for only one antibiotic (RIF or INH), while the drug resistance profile for the other antibiotic is reported as “indeterminate.”
Discrepant samples.
For those samples in which there was a difference between the results of phenotypic DST and amplification microarray genotyping, the corresponding rpoB, katG, or inhA gene (n = 26 total reactions) was amplified from the corresponding extract and subjected to bidirectional DNA sequencing (Eurofins MWG Operon, Louisville, KY).
RESULTS
Clinical sample characteristics.
There were 146 sputum samples available for this study. Those samples that generated a contaminated culture were excluded from further analysis, and the results for the samples are not reported here. The remaining 130 samples were categorized by smear and culture status, and the results are shown in Table 2. One sputum sample was consumed in its entirety during NALC-NaOH decontamination, resulting in 129 paired sputum and sediment samples plus one unpaired sediment sample (289 total amplification microarray tests). RIF and INH phenotypic drug susceptibility data were available for only 74 of the 114 culture-positive clinical specimens. Complete smear, culture, DST, and amplification microarray data are itemized in Table S1 in the supplemental material.
TABLE 2.
Amplification microarray M. tuberculosis and resistance-associated gene detection rate
| Sampleb | Target | Detection rate (%) |
|||
|---|---|---|---|---|---|
| Smear-positive specimens |
Smear-negative specimens |
||||
| Culture-positive specimens (n = 107 or 108a) | Culture-negative specimens (n = 8) | Culture-positive specimens (n = 5) | Culture-negative specimens (n = 9) | ||
| Sputum extracts | M. tuberculosis | 100 | 100 | 80 | 89 |
| rpoB | 100 | 100 | 100 | 100 | |
| katG | 100 | 88 | 100 | 100 | |
| inhA | 100 | 100 | 100 | 100 | |
| Sediment extracts | M. tuberculosis | 100 | 100 | 100 | 67 |
| rpoB | 99 | 100 | 100 | 83 | |
| katG | 100 | 100 | 100 | 80 | |
| inhA | 100 | 100 | 80 | 100 | |
There were 107 and 108 smear- and culture-positive sputum and sediment extracts, respectively.
Resistance-associated genes were scored only if M. tuberculosis (the IS6110 element) was detected by the amplification microarray.
Analytical specificity and limits of detection.
Microarray probe specificity against clinical M. tuberculosis isolates was largely established in prior work (36), and DNAs from 25 well-characterized (i.e., sequenced) M. tuberculosis isolates were again used here as external positive controls throughout the study (n = 28 batch runs). All positive and negative (reagent blank) controls behaved as expected, with no false-positive or false-negative results and correct SNP genotyping and identification.
Analytical limits of detection for the test were estimated by analyzing triplicate dilutions of wild-type M. tuberculosis H37Ra genomic DNA. Average signal-to-noise ratios (SNR values) for the three replicates are plotted in Fig. 1 for the internal positive controls (IPCs) and universal, resistance-associated gene probes. All tests were valid, and the IS6110 marker was detected at an SNR value of >10 for all dilutions and replicates down to and including 100 fg of genomic DNA. At 50 fg DNA input, one IS6110 signal did not exceed the SNR value threshold for positive detection, which, according to the preestablished decision logic, terminated the analysis of other probe signals. Positive amplification and positive detection were achieved for the universal, resistance-associated gene probes for all replicates down to and including the 100-fg DNA input, but the rpoB probes were undetectable in one replicate at 50 fg DNA. On the basis of the SNR values, the katG gene appeared to be preferentially amplified over all other targets and had an average SNR value of 120 even with the 50-fg DNA input, perhaps to the detriment of inhA amplification. Correct genotyping and the correct drug susceptibility determination were achieved for all replicates where the test was valid and the universal, resistance-associated gene probes were detected at an SNR value of ≥3, including those replicates at 50 fg that met all predetermined decision logic criteria. We therefore estimated the analytical limit of detection to be ∼100 fg M. tuberculosis genomic DNA per reaction, or ∼25 cell equivalents, assuming 4.4 × 106 bp (41) and ∼4 fg genomic DNA per cell.
FIG 1.
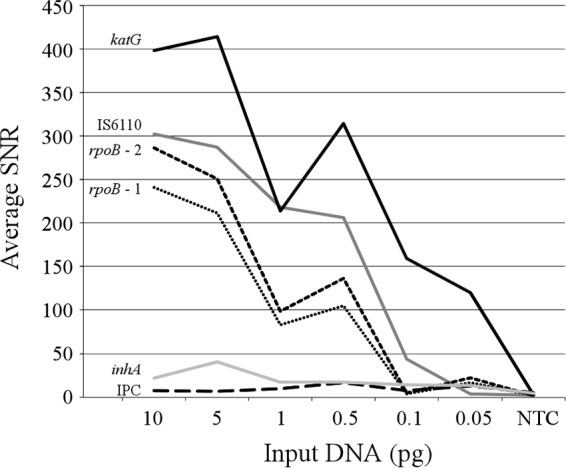
Analytical LoD. IPC, internal positive control; NTC, no-template control.
Amplification microarray shelf life.
The average, integrated signal intensities, SNR values, and discrimination ratios (D) for internal positive controls, universal resistance-associated gene probes, and selected mutations are shown in Fig. 2. After an initial 30 to 40% decrease in average intensity, all probe responses were stable to 18 months of storage. There was no appreciable increase in background noise, genotyping results were correct for all tests, and the behavior of all other probes on the array was substantially similar to that shown in Fig. 2 (not shown).
FIG 2.
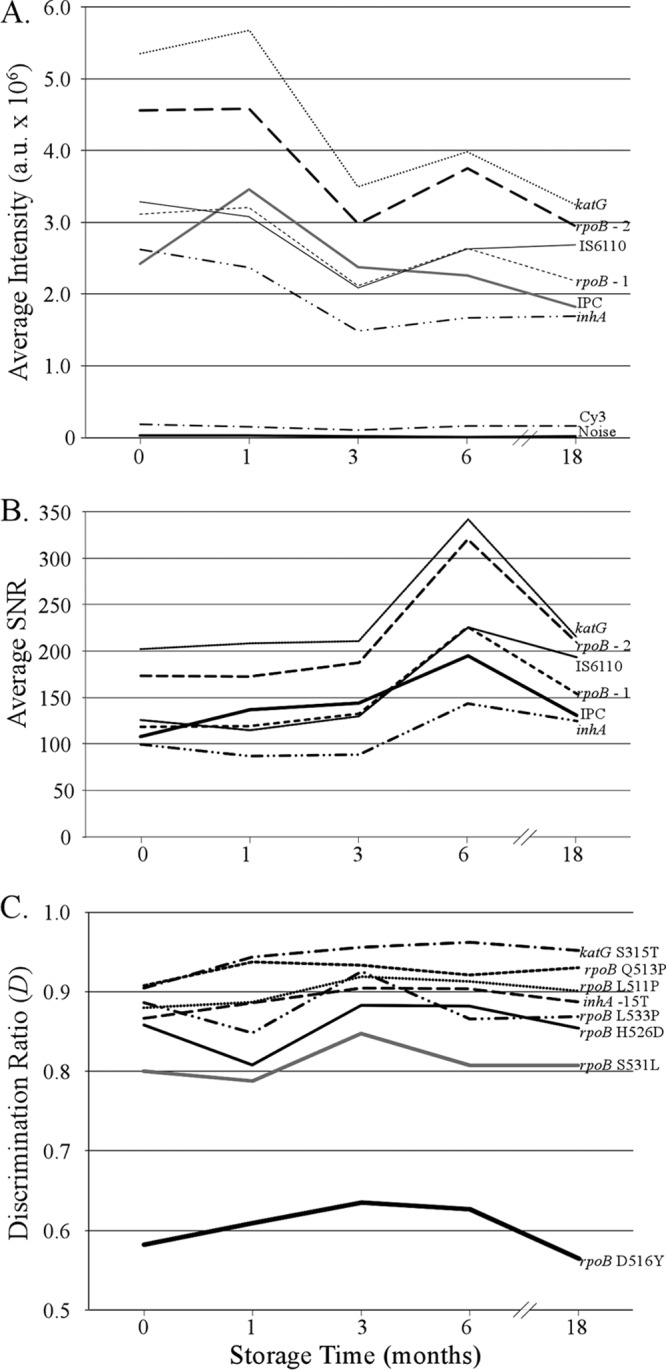
Average fluorescent intensity (A), signal-to-noise ratios (B), and discrimination ratios (C) after amplification microarray storage at 4°C. a.u., absorbance units; IPC, internal positive control; Cy3, fluorescently labeled fiducial marker.
Nucleic acid recovery and M. tuberculosis detection.
Real-time PCR detected the IS6110 element in all samples (average threshold cycle [CT] value, ≤37) except for one sputum extract and two sediment extracts that were smear negative and culture negative (S−/C−). Two primary sputum samples and 8 sediment extracts resulted in an IS6110 CT value of >35, and all but one of these samples occurred among smear-negative samples. These results reflect the presence of a relatively low concentration of M. tuberculosis in the original sample. Excluding S−/C− samples, the automated sample preparation recovered significantly more M. tuberculosis DNA from the primary sputum than from the matched sediments (average CT = 22.6 versus 25.2; P ≪ 0.0001).
M. tuberculosis and gene detection efficacy.
M. tuberculosis detection efficacy relative to smear and culture status is summarized in Table 2. Excluding the nine S−/C− samples for which the true state of M. tuberculosis infection could not be determined, the amplification microarray detected M. tuberculosis in 99.1% (119/120) of sputum extracts and 100% (127/127) of sediment extracts. There was one failure to detect M. tuberculosis in a culture-positive sputum sample (Table S1, sample 91), but the sample was also M. tuberculosis negative by real-time PCR. The results of real-time PCR and the amplification microarray were 99.1% concordant for M. tuberculosis detection (229/231 amplification reactions), with the single discrepancy occurring in an S−/C− sample (Table S1, sample 116). The concordance between amplification microarray M. tuberculosis and resistance-associated gene detection was 99.1% (119/120) and 98.4% (125/127) for sputum and sediment extracts, respectively. There were no cases where a gene probe was detected in the absence of an IS6110 signal, and a failure to detect the resistance-associated genes was always associated with low M. tuberculosis DNA quantities in the extract (Table S1).
Amplification microarray genotyping relative to DST.
Of the 129 paired sputum and sediment extracts, the amplification microarray test identified the isolates in 6 to be INH monoresistant, the isolates in 4 to be RIF monoresistant, and the isolates in 14 to be MDR. Of the 74 culture-based DST results, 8 indicated that the isolates were INH monoresistant, 1 indicated that the isolate was RIF monoresistant, and 3 indicated that the isolates were MDR. Relative to the results of the culture-based DST, the amplification microarray test was 97.1% specific and 75.0% sensitive for the detection of RIF resistance in the isolates in both sputum and sediment extracts. The specificity for the detection of INH resistance was 98.4% and 96.8% for sputum and sediment extracts, respectively, and the sensitivity for the detection of INH resistance was 63.6%. Amplification microarray genotyping results were 100% concordant between sputum and sediment extracts for the subset of 74 samples for which a phenotypic DST result was available. Any DST and amplification microarray genotyping outcomes were resolved by bidirectional DNA sequencing rather than repeat DST because of concerns about the integrity of the isolates (which were not banked or frozen) and the age of the banked sputum and sediment samples. In all cases, the microarray genotype matched the corresponding DNA sequence.
Relative to the results of DST, there were three false-resistant (FR) genotype calls for RIF and INH resistance (Table S1, samples 34, 106, and 109). One of those results (the sample 109 sediment extract) could be explained by poor DNA recovery relative to that for its paired sputum specimen, with the concentration of available DNA being at or near the LoD of the test (CT = 36.0). Otherwise, the FR results may be explained by a failure of the MGIT DST system to detect antibiotic resistance (42). There was one RIF-false-susceptible result from a smear-negative, culture-positive sample. Again, the microarray signals were strong, the sediment and sputum extracts generated the same genotype, and the amplification microarray genotypes matched the corresponding DNA sequence. The RIF-false-susceptible genotype could therefore represent RIF resistance arising from a mutation elsewhere in the genome (9, 10, 14, 43). The four INH-false-susceptible results, all of which were correct from the perspective of the DNA sequence, are most likely a consequence of limited gene and SNP coverage on the amplification microarray relative to the number of genes and SNPs now linked to INH resistance (5–10).
Genotyping in sputum versus sediments.
Excluding the S−/C− extracts, the genotyping results from the sputum and sediment extracts were 99.1% concordant (119/120 samples). The single discrepancy (Table S1, sample 109) was a culture-positive sample with a smear result of +1, where the results of genotyping for RIF and INH resistance for the sediment (CT = 36.0) were indeterminate, whereas the matched sputum extract (CT = 24.7) reported RIF and INH susceptibility. This result may be explained by the loss of M. tuberculosis cells during NALC-NaOH decontamination and sedimentation and, subsequently, poor DNA recovery from the sediment.
DISCUSSION
Amplification microarrays.
The genetic complexity of M. tuberculosis drug resistance and the host response indicate that next-generation M. tuberculosis diagnostics will require relatively high levels of multiplexing and an ability to specifically identify resistance-conferring or compensatory mutations that are present in the isolates in a specimen. For this reason, microarrays remain a potentially useful platform for M. tuberculosis diagnostics and personalized medicine, provided that they can be configured to meet known, high-priority product requirements. In this context, the premise of an amplification microarray is to simplify microarray-based biochemistry and the work flow for clinical practice, ideally (and eventually) near the point of use. In this study, we converted a previously described open-amplicon MDR-TB microarray test (36) into an entirely closed-amplicon consumable, a work-flow transition that is conceptually similar to the conversion of PCR into real-time PCR. The simplifying microfluidic principle(s) includes geometries and materials to pin the contact line of the liquid meniscus to confine the amplification reagent mixture to the gel element array amplification chamber during thermal cycling. A closed-amplicon consumable is maintained by using a hydrophilic absorbent to help imbibe all liquids into the waste chamber during the wash step without the need for active pumps or valves. Relative to other hybridization-based tests for the detection of drug-resistant M. tuberculosis (24–27, 30, 44–47), the amplification microarray described here combines up to seven manual steps and processes into a single step in a single microfluidic reaction chamber. It is also important to note that the resulting amplification microarray differs from seemingly related PCR array technologies in that the amplification microarray is based on a homogeneous multiplexed reaction, whereas PCR arrays split the sample into multiple reaction wells, droplets, or channels. Sample splitting can become limiting when the diagnostic objective requires a very stringent limit of detection for many targets and SNPs, as is the case for drug-resistant M. tuberculosis.
In addition to the data in Fig. 2, anecdotal evidence from numerous shipments and collaborative efforts (not shown) further indicates that there is no degradation of the amplification microarray signals or performance when the amplification microarray is shipped at ambient temperature by air freight, including when the amplification microarray is shipped overseas and subjected to multiweek, uncontrolled storage while shipments pass through customs. Taken together, the amplification microarray microfluidic design and shelf-life data show that the amplification microarray represents a significant integration step toward aligning microarray technology with high-priority M. tuberculosis target product profiles (3).
Amplification microarray performance.
The amplification microarray analytical specificity against 25 external positive controls was consistent with that found in prior work, and the lower limit of detection and reproducible genotyping (25 cell equivalents of genomic DNA per reaction) represent a fourfold improvement over those of the predecessor (open-amplicon) test described elsewhere (36). The analytical sensitivity is therefore on par with that of real-time PCR or isothermal amplification technologies, a conclusion further supported by the 99.1% concordance rate between real-time PCR and amplification microarray detection of the IS6110 element. Given that 20% of the nucleic acid extract was included in each amplification reaction, the implication is that TruTip (or other sample preparation technology) needs to generate only approximately 0.5 pg M. tuberculosis DNA per extraction (and elution into 100 μl) to generate a reproducible MDR M. tuberculosis genotyping result.
That the automated workstation recovered significantly more M. tuberculosis DNA from primary sputum than from decontaminated sediments is significant in itself, as the consensus opinion is that primary sputum is the preferred (or required) sample type for new M. tuberculosis molecular diagnostics (3). While the total number of smear-negative samples in this study was purposely low, the data also suggest that the combined sample preparation and amplification microarray method has the analytical sensitivity needed to detect M. tuberculosis and the drug resistance genotype in smear-negative sputum specimens. Confirmation of this hypothesis will require a new study with an expanded set of smear-negative samples. In the interim, the analytical performance data suggest that the method can meet the diagnostic sensitivity requirements for patient triage (or referral) and simultaneously provide the detailed SNP identification and reporting that are (or will be) required to initiate a patient-specific treatment regimen.
The M. tuberculosis detection and genotyping concordance data indicate that the amplification microarray consumable and test are reproducible and are equally efficacious when primary sputum or decontaminated sediments are used as the test input. When one considers that the automated workstation consistently extracted more M. tuberculosis DNA from primary sputum than from the paired sediment and that smear-negative sputum extracts were more likely than sediment extracts to give a strong output of RIF or INH resistance (Table S1), one could argue that primary sputum is the preferred sample type for the TruTip/amplification microarray test.
The clinical samples for this study were intentionally selected to estimate amplification microarray genotyping specificity. With only 74 DST results and four phenotypically RIF-resistant samples, however, there are not enough data to draw conclusions about amplification microarray genotyping sensitivity. We do know that the INH-reporting probes used here provide limited coverage of INH resistance-conferring mutations (48, 49); the power of an amplification microarray is the ability to increase the number of INH resistance-conferring genes and SNPs into the test (e.g., see reference 12).
Summary.
This study integrated a complex MDR-TB microarray work flow into an entirely closed-amplicon, integrated microfluidic device (the amplification microarray) and demonstrated its efficacy and substantial equivalence to other methods for M. tuberculosis detection and genotyping for RIF and INH resistance with primary sputum and decontaminated sediments. The microfluidic simplification, shelf life, and analytical performance of the amplification microarray are well aligned with the consensus product profiles for new M. tuberculosis diagnostics and represent a significant ease-of-use advantage over other (manual) hybridization-based tests for MDR-TB. We are now able to expand the test coverage for additional resistance-conferring mutations, integrate primers and probes that are predictive of an XDR phenotype (6, 11), and combine the TruTip and amplification microarray technologies into a sample-to-answer system.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH) under Center of Excellence in Translational Research (CETR) grant U19 AI109755.
We are grateful for the outstanding work of Gustavo Pariona, Carmen Contreras, Julia Coit, Milagros Mendoza, Segundo Leon, Jerome Galea, Rosina Reinoso, Erika Zavala, Nancy Mendoza, and Gaby Tunque to collect and process primary specimens in Peru. We further acknowledge Max Salfinger and staff at National Jewish Health for assistance in reculturing selected culture-negative sediments.
Crude extracts from the TDR Tuberculosis Strain Bank were provided by the United Nations Children's Fund/United Nations Development Programme/World Bank/World Health Organization Special Programme for Research and Training in Tropical Diseases (TDR), Geneva, Switzerland.
All Akonni-affiliated authors are employees of Akonni Biosystems Inc. Y.L., C.K., N.T., R.H., A.K., C.G.C., and D.P.C. are also Akonni shareholders.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JCM.01652-17.
REFERENCES
- 1.Albert H, Nathavitharana RR, Isaacs C, Pai M, Denkinger CM, Boehme CC. 2016. Development, roll-out and impact of Xpert MTB/RIF for tuberculosis: what lessons have we learnt and how can we do better? Eur Respir J 48:516–525. doi: 10.1183/13993003.00543-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kik SV, Denkinger CM, Casenghi M, Vadnais C, Pai M. 2014. Tuberculosis diagnostics: which target product profiles should be prioritised? Eur Respir J 44:537–540. doi: 10.1183/09031936.00027714. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. 2014. High-priority target product profiles for new tuberculosis diagnostics: report of a consensus meeting. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 4.World Health Organization. 2014. Xpert MTB/RIF implementation manual. Technical and operational ‘how-to’: practical considerations. World Health Organization, Geneva, Switzerland. [PubMed] [Google Scholar]
- 5.Valafar F, Ramirez-Busby SM, Torres J, Paul LV, Rodwell TC, Victor TC, Rodrigues C, Gler MT, Crudu V, Catanzaro T. 2015. Prognostic significance of novel katG mutations in Mycobacterium tuberculosis. Int J Mycobacteriol 4(Suppl 1):S51–S52. doi: 10.1016/j.ijmyco.2014.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farhat MR, Sultana R, Iartchouk O, Bozeman S, Galagan J, Sisk P, Stolte C, Nebenzahl-Guimaraes H, Jacobson K, Sloutsky A, Kaur D, Posey J, Kreiswirth BN, Kurepina N, Rigouts L, Streicher EM, Victor TC, Warren RM, van Soolingen D, Murray M. 2016. Genetic determinants of drug resistance in Mycobacterium tuberculosis and their diagnostic value. Am J Respir Crit Care Med 194:621–630. doi: 10.1164/rccm.201510-2091OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker TM, Kohl TA, Omar SV, Hedge J, Elias CDO, Bradley P, Iqbal Z, Feverriegel S, Niehaus KE, Wilson DJ, Clifton DA, Kapatai G, Ip CLC, Bowden R, Drobniewski FA, Allix-Beguec C, Gaudin C, Parkhill J, Diel R, Supply P, Crook DW, Smith EG, Walker AS, Ismail N, Niemann S, Peto TE, Modernizing Medical Microbiology (MMI) Informatics Group. 2015. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect Dis 15:1193–1202. doi: 10.1016/S1473-3099(15)00062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen KA, Abeel T, McGuire AM, Desjardins CA, Munsamy V, Shea TP, Walker BJ, Bantubani N, Almeida DV, Alvarado L, Chapman SB, Mvelase N, Duffy EY, Fitzgerald MG, Govender P, Gujja S, Hamilton S, Howarth C, Larimer JD, Maharaj K, Pearson MD, Priest ME, Zeng Q, Padayatchi N, Grosset J, Young SK, Wortman J, Milisana KP, O'Donnell MR, Birren BW, Bishai WR, Pym AS, Earl AM. 2015. Evolution of extensively drug-resistant tuberculosis over four decades: whole genome sequencing and dating analysis of Mycobacterium tuberculosis isolates from KwaZulu-Natal. PLoS Med 12:e100880. doi: 10.1371/journal.pmed.1001880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, Galagan J, Niemann S, Gagneux S. 2011. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet 44:106–110. doi: 10.1038/ng.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Vos M, Müller B, Borrell S, Black P, van Helden P, Warren R, Gagneux S, Victor T. 2013. Putative compensatory mutations in the rpoC gene of rifampin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob Agents Chemother 57:827–832. doi: 10.1128/AAC.01541-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodwell TC, Valafar F, Douglas J, Qian L, Garfein RS, Chawla A, Torres J, Zadorozhny V, Kim MS, Hoshide M, Catanzaro D, Jackson L, Lin G, Desmond E, Rodrigues C, Eisenach KD, Victor TC, Ismail N, Crudu V, Gler MR, Catanzaro A. 2014. Predicting extensively drug-resistant Mycobacterium tuberculosis phenotypes with genetic mutations. J Clin Microbiol 52:781–789. doi: 10.1128/JCM.02701-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torres JN, Paul LV, Rodwell TC, Victor TC, Amallraja AM, Elghraoui A, Goodmanson AP, Ramirez-Busby SM, Chawla A, Zadorozhny V, Streicher EM, Sirgel FA, Catanzaro D, Rodrigues C, Gler MT, Crudu V, Catanzaro A, Valafar F. 2015. Novel katG mutations causing isoniazid resistance in clinical M. tuberculosis isolates. Emerg Microbes Infect 4:e42. doi: 10.1038/emi.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith CM, Proulx MK, Olive AJ, Laddy D, Mishra BB, Moss C, Guiterrez NM, Bellerose MM, Barreira-Silva P, Phuah JY, Baker RE, Behar SM, Kornfeld H, Evans TG, Beamer B, Sassetti CM. 2016. Tuberculosis susceptibility and vaccine protection are independently controlled by host genotype. mBio 7:e01516-16. doi: 10.1128/mBio.01516-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Müller B, Borrell S, Rose G, Gagneux S. 2013. The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis. Trends Genet 29:160–169. doi: 10.1016/j.tig.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown AC, Bryant JM, Einer-Jensen K, Holdstock J, Houniet DT, Chan JZM, Depledge DP, Nikolayevskyy V, Broda A, Stone MJ, Christiansen MT, Williams R, McAndrew MB, Tutill H, Brown J, Melzer M, Rosmarin C, McHugh TD, Shorten RJ, Drobniewski F, Speight G, Breuer J. 2015. Rapid whole-genome sequencing of Mycobacterium tuberculosis isolates directly from clinical samples. J Clin Microbiol 53:2230–2237. doi: 10.1128/JCM.00486-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sekiguchi J- I, Miyoshi-Akiyama T, Augustynowicz-Kopec E, Zwolska Z, Kirikae F, Toyota E, Kobayashi I, Morita K, Kudo K, Kato S, Kuratsuji T, Mori T, Kirikae T. 2007. Detection of multidrug resistance in Mycobacterium tuberculosis. J Clin Microbiol 45:179–192. doi: 10.1128/JCM.00750-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gan M, Liu Q, Yang C, Gao Q, Luo T. 2016. Deep whole-genome sequencing to detect mixed infection of Mycobacterium tuberculosis. PLoS One 11:e0159029. doi: 10.1371/journal.pone.0159029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee RS, Behr MA. 2016. The implications of whole-genome sequencing in the control of tuberculosis. Ther Adv Infect Dis 32:47–62. doi: 10.1177/2049936115624630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Witney AA, Cosgrove CA, Arnold A, Hinds J, Stoker NG, Butcher PD. 2016. Clinical use of whole genome sequencing for Mycobacterium tuberculosis. BMC Med 14:46. doi: 10.1186/s12916-016-0598-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drmanac R, Labat I, Brukner I, Crkvenjakov R. 1989. Sequencing of megabase plus DNA by hybridization: theory of the method. Genomics 4:114–128. doi: 10.1016/0888-7543(89)90290-5. [DOI] [PubMed] [Google Scholar]
- 21.Fodor SP, Read JL, Pirrung MC, Stryer L, Lu AT, Solas D. 1991. Light-directed, spatially addressable parallel chemical synthesis. Science 251:767–773. doi: 10.1126/science.1990438. [DOI] [PubMed] [Google Scholar]
- 22.Khrapko KR, Lysov YP, Khorlyn AA, Shick VV, Florentiev VL, Mirzabekov AD. 1989. An oligonucleotide hybridization approach to DNA sequencing. FEBS Lett 256:118–122. doi: 10.1016/0014-5793(89)81730-2. [DOI] [PubMed] [Google Scholar]
- 23.Troesch A, Nguyen H, Miyada CG, Desvarenne S, Gingeras TR, Kaplan PM, Cros P, Mabilat C. 1999. Mycobacterium species identification and rifampin resistance testing with high-density DNA probe arrays. J Clin Microbiol 37:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao C, Zhu T, Li Y, Zhang L, Zhang B, Huang J, Fu W. 2010. Detection of rpoB, katG and inhA gene mutations in Mycobacterium tuberculosis clinical isolates from Chongqing as determined by microarray. Clin Microbiol Infect 16:1639–1643. doi: 10.1111/j.1469-0691.2010.03267.x. [DOI] [PubMed] [Google Scholar]
- 25.Volokhov DV, Chizhikov VE, Denkin S, Zhang Y. 2009. Molecular detection of drug-resistant Mycobacterium tuberculosis with a scanning-frame oligonucleotide microarray. Methods Mol Biol 465:395–417. doi: 10.1007/978-1-59745-207-6_26. [DOI] [PubMed] [Google Scholar]
- 26.Bergval I, Sengstake S, Brankova N, Levterova V, Abadia E, Tadumaze N, Bablishvili N, Akhalaia M, Tuin K, Schuitema AR, Panaiotov S, Bachiyska E, Kantardjiev T, de Zwaan R, Schürch A, van Soolingen D, van't Hoog A, Cobelens F, Aspindzelashvili R, Sola C, Klaster P, Anthony R. 2012. Combined species identification, genotyping, and drug resistance detection of Mycobacterium tuberculosis cultures by MLPA on a bead-based array. PLoS One 7:e43240. doi: 10.1371/journal.pone.0043240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caoili JC, Mayorova A, Sikes D, Hickman L, Plikaytis BB, Shinnick TM. 2006. Evaluation of the TB-Biochip oligonucleotide microarray system for rapid detection of rifampin resistance in Mycobacterium tuberculosis. J Clin Microbiol 44:2378–2381. doi: 10.1128/JCM.00439-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu PL, Lin YC, Yang YC, Jou R, Huang SC, Jenh YS, Huang HH, Chang TC. 2013. Evaluation of a membrane array for detection of Mycobacterium tuberculosis complex and nontuberculous mycobacteria in positive liquid cultures. Diagn Microbiol Infect Dis 75:337–341. doi: 10.1016/j.diagmicrobio.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 29.Denkin S, Volokhov D, Chizhikov V, Zhang Y. 2005. Microarray-based pncA genotyping of pyrazinamide-resistant strains of Mycobacterium tuberculosis. J Med Microbiol 54:1127–1131. doi: 10.1099/jmm.0.46129-0. [DOI] [PubMed] [Google Scholar]
- 30.Zimenkov DV, Antonova OV, Kuz'min AV, Isaeva YD, Krylova LY, Popov SA, Zasedatelev AS, Michailovich VM, Gryadunov DA. 2013. Detection of second-line drug resistance in Mycobacterium tuberculosis using oligonucleotide microarrays. BMC Infect Dis 13:e240. doi: 10.1186/1471-2334-13-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yauk CL, Berndt ML. 2007. Review of the literature examining the correlation among DNA microarray technologies. Environ Mol Mutagen 48:380–394. doi: 10.1002/em.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Draghici S, Khatri P, Eklund AC, Szallasi Z. 2006. Reliability and reproducibility issues in DNA microarray measurements. Trends Genet 22:101–109. doi: 10.1016/j.tig.2005.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi L, Tong W, Goodsaid FM, Frueh FW, Fang H, Han T, Fuscoe JC, Casciano DA. 2004. QA/QC: challenges and pitfalls facing the microarray community and regulatory agencies. Exp Rev Mol Diagn 4:761–777. doi: 10.1586/14737159.4.6.761. [DOI] [PubMed] [Google Scholar]
- 34.Jordan BR. 2010. Is there a niche for DNA microarrays in molecular diagnostics? Expert Rev Mol Diagn 10:875–882. doi: 10.1586/erm.10.74. [DOI] [PubMed] [Google Scholar]
- 35.Chandler DP, Bryant L, Griesemer SB, Gu R, Knickerbocker C, Kukhtin A, Parker J, Zimmerman C, St George K, Cooney CG. 2012. Integrated amplification microarrays for infectious disease diagnostics. Microarrays 1:107–124. doi: 10.3390/microarrays1030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linger YL, Kukhtin A, Golova J, Perove A, Lambarqui A, Bryant L, Rudy GB, Dionne K, Fisher SL, Parrish N, Chandler DP. 2014. Simplified microarray system for simultaneously detecting rifampin, isoniazid, ethambutol, and streptomycin resistance markers in Mycobacterium tuberculosis. J Clin Microbiol 52:2100–2107. doi: 10.1128/JCM.00238-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vincent V, Rigouts L, Nduwamahoro E, Holmes B, Cunningham J, Guillerm M, Nathanson CM, Moussy F, De Jong B, Portaels F, Ramsay A. 2012. The TDR tuberculosis strain bank: a resource for basic science, tool development and diagnostic services. Int J Tuberc Lung Dis 16:24–31. doi: 10.5588/ijtld.11.0223. [DOI] [PubMed] [Google Scholar]
- 38.Savelkoul PH, Catsburg A, Mulder S, Oostendorp L, Schirm J, Wilke H, van der Zanden AG, Noordhoek GT. 2006. Detection of Mycobacterium tuberculosis complex with real time PCR: comparison of different primer-probe sets based on the IS6110 element. J Microbiol Methods 66:177–180. doi: 10.1016/j.mimet.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 39.Golova JB, Chernov BK, Perov AN, Reynolds J, Linger YL, Kukhtin A, Chandler DP. 2012. Non-volatile copolymer compositions for fabricating gel element microarrays. Anal Biochem 421:526–533. doi: 10.1016/j.ab.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cooney CG, Sipes D, Thakore N, Holmberg R, Belgrader P. 2012. A plastic, disposable microfluidic flow cell for coupled on-chip PCR and microarray detection of infectious agents. Biomed Microdevices 14:45–53. doi: 10.1007/s10544-011-9584-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream M-A, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BB. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 42.Rigouts L, Gumusboga M, de Rijk WB, Nduwamahoro E, Uwizeye C, de Jong B, Van Deun A. 2013. Rifampin resistance missed in automated liquid culture system for Mycobacterium tuberculosis isolates with specific rpoB mutations. J Clin Microbiol 51:2641–2645. doi: 10.1128/JCM.02741-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun G, Luo T, Yang C, Dong X, Li J, Zhu Y, Zheng H, Tian W, Wang S, Barry CE III, Mei J, Gao Q. 2012. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J Infect Dis 206:1724–1733. doi: 10.1093/infdis/jis601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitarai S, Kato S, Ogata H, Aono A, Chikamatsu K, Mizuno K, Toyota E, Sejimo A, Suzuki K, Yoshida S, Saito T, Moriya A, Fujita A, Sato S, Matsumoto T, Ano H, Suetake T, Kondo Y, Kirikae T, Mori T. 2012. Comprehensive multicenter evaluation of a new line probe assay kit for identification of Mycobacterium species and detection of drug-resistant Mycobacterium tuberculosis. J Clin Microbiol 50:884–890. doi: 10.1128/JCM.05638-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun AH, Fan XL, Li LW, Wang LF, Ans WY, Yan J. 2009. Rapid detection of rpoB gene mutations in RIF-resistant M. tuberculosis isolates by oligonucleotide microarray. Biomed Environ Sci 22:253–258. doi: 10.1016/S0895-3988(09)60053-2. [DOI] [PubMed] [Google Scholar]
- 46.Zimenkov DV, Kulagina EV, Antonova OV, Krasnova MA, Chernyaeva EN, Zhuravlev VY, Kuz'min AV, Popov SA, Zasedatelev AS, Gryadunov DA. 2015. Evaluation of a low-density hydrogel microarray technique for mycobacterial species identification. J Clin Microbiol 53:1103–1114. doi: 10.1128/JCM.02579-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zimenkov DV, Kulagina EV, Antonova OV, Zhuravlev VY, Gryadunov DA. 2016. Simultaneous drug resistance detection and genotyping of Mycobacterium tuberculosis using a low-density hydrogel microarray. J Antimicrob Chemother 71:1520–1531. doi: 10.1093/jac/dkw015. [DOI] [PubMed] [Google Scholar]
- 48.Kim SY, Park YK, Song E, Jang H, Kim C, Yoo J, Kang SJ. 2006. Evaluation of the CombiChip mycobacteria drug-resistance detection DNA chip for identifying mutations associated with resistance to isoniazid and rifampin in Mycobacterium tuberculosis. Diagn Microbiol Infect Dis 54:203–210. doi: 10.1016/j.diagmicrobio.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 49.Madania A, Habous M, Zarzour H, Ghoury I, Hebbo B. 2012. Characterization of mutations causing rifampicin and isoniazid resistance of Mycobacterium tuberculosis in Syria. Pol J Microbiol 61:23–32. doi: 10.1099/jmm.0.027052-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.