

Abstract
HnRNP D, better known as AUF1, is an extensively studied protein that controls a variety of cellular pathways. Consequently, its expression has to be tightly regulated to prevent the onset of pathologies. In contrast, the cellular functions and regulation of its ubiquitously expressed paralog hnRNP DL are barely explored. Here, we present an intricate crosstalk between these two proteins. Both hnRNP D and DL are able to control their own expression by alternative splicing of cassette exons in their 3′UTRs. Exon inclusion produces mRNAs degraded by nonsense-mediated decay. Moreover, hnRNP D and DL control the expression of one another by the same mechanism. Thus, we identified two novel ways of how hnRNP D expression is controlled. The tight interconnection of expression control directly links hnRNP DL to hnRNP D-related diseases and emphasizes the importance of a systematic analysis of its cellular functions.
Keywords: splicing, autoregulation, hnRNP, NMD, cassette exon
INTRODUCTION
The family of hnRNPs (heterogeneous nuclear ribonucleoproteins) is a diverse group of RNA-binding proteins (RBPs) involved in all aspects of an mRNA's life, e.g., controlling processing steps, translation efficiency, and stability. All hnRNPs contain at least one RNA-binding domain, mostly of the RRM (RNA recognition motif) type. In addition, they often contain auxiliary domains of low complexity, like proline-rich, acidic or glycine-rich regions (for reviews, see Han et al. 2010; Busch and Hertel 2012; Geuens et al. 2016). Together with the family of SR (serine/arginine-rich) proteins, hnRNPs are the most prominent regulators of alternative splicing decisions. Genome-wide analyses uncovered a plethora of target genes for individual hnRNPs and a high degree of interconnection, thus showing cooperative as well as competitive effects (Huelga et al. 2012).
Although ubiquitously expressed and clearly connected to human diseases, some hnRNPs yet await further characterization. One such poorly studied family member is hnRNP DL (D-like). HnRNP DL is a paralog of hnRNP D, better known as AUF1 (AU-rich element binding factor 1). HnRNP DL was found to be up-regulated in patient samples with prostate cancer or chronic myeloid leukemia and promotes proliferation of cell lines derived from the respective cancer types (Wu et al. 2008; Zhou et al. 2014). Heterozygous mutations in the C-terminal glycine-rich domain of hnRNP DL are causative for limb girdle muscular dystrophy (LGMD) type 1G (Vieira et al. 2014), a rare muscle-wasting disease. How hnRNP DL promotes these diseases is unclear. So far only one endogenous target gene, the NFκB repressing factor (NKRF), has been verified (Reboll et al. 2007; Omnus et al. 2011).
HnRNP D, in contrast, is a well-studied factor, mainly known for its function in mRNA decay. Among other functions, hnRNP D recognizes AU-rich elements (AREs) in 3′UTRs and mediates degradation of its target mRNAs (for reviews, see White et al. 2013, 2017; Moore et al. 2014). It is important for the destabilization of several mRNAs encoding cell cycle regulators, proto-oncoproteins and proinflammatory factors. As shown in knockout mice, hnRNP D is crucial for the fast turnover of cytokines, thus promoting the resolution of inflammatory responses and ultimately, preventing severe endotoxic shock (Lu et al. 2006). It is unclear if hnRNP DL participates in ARE-mediated decay as well. The high sequence similarity between the RNA binding domains of hnRNP D and DL suggests that the two RBPs recognize similar target sequences (Doi et al. 1998). However, the interplay between hnRNP DL and hnRNP D and possible coregulation of cellular processes has not been studied so far.
Due to the versatile functions of hnRNPs (and RBPs in general), tight regulation of their expression is crucial to prevent the onset and progression of pathological processes. Homeostasis in protein levels is often achieved by negative feedback loops. Thus, the RBP controls its expression by direct binding to its own transcript. Reduced expression is achieved by diverse mechanisms, most often by shifting pre-mRNA splicing toward the production of noncoding isoforms (Sun et al. 2010; Änko et al. 2012; Rösel-Hillgärtner et al. 2013; Bergeron et al. 2015). One prevalent mechanism for such a regulation is AS-NMD. It couples alternative splicing (AS) to nonsense-mediated decay (NMD) by deliberate production of mRNA isoforms harboring NMD features, i.e., a termination codon >50–55 nt upstream of an exon-junction complex (EJC).
AS-NMD is not only used for autoregulation, but also for cross-regulation between RBPs. Two examples are the hnRNP paralogous pairs of hnRNP L/LL and PTB/nPTB. Here, the abundantly expressed paralogs hnRNP L and PTB not only autoregulate their own expression, but also restrict the expression of their “minor” paralog by AS-NMD (Spellman et al. 2007; Rossbach et al. 2009). So far, it is not known if hnRNP D and DL are likewise able to autoregulate their own expression by AS-NMD. Although NMD-sensitive isoforms for hnRNP D have been reported, it is not known if hnRNP D is capable to induce their production (Banihashemi et al. 2006). Furthermore, as both hnRNP D and DL are ubiquitously expressed, it is unclear what kind of cross-regulation, if any, is to be expected. Precise knowledge of the regulation of their expression, however, is essential to understand the contribution of these hnRNPs to both physiological and pathological processes. In the present study, we explored the auto- and cross-regulation of hnRNP D and DL to shed light on their interconnection, revealing a role of hnRNP DL in the function of hnRNP D in human physiology and health.
RESULTS AND DISCUSSION
An ultraconserved element in the hnRNP DL gene contains an NMD-sensitive exon
The hnRNP DL gene contains an ultraconserved element (uc.144) that is 205 bp in length (Lomonaco et al. 2014). The element covers the sequence of a cassette exon (exon 8) and is the highest conserved region of the gene (Fig. 1A;Ni et al. 2007). Inclusion of exon 8 into the 3′UTR of the hnRNP DL mRNA introduces two exon junctions, with the second one located >55 nt downstream from the natural termination codon. Therefore, the exon inclusion isoform is predicted to be a target for NMD. In contrast, the single exon junction introduced by skipping of exon 8 is only 22 nt downstream from the termination codon and therefore not expected to induce NMD. In consequence, only the isoform in which exon 8 is skipped is predicted to produce a protein despite the fact that both isoforms encode the same open reading frame.
FIGURE 1.

The ultraconserved element in the 3′UTR of hnRNP DL contains an NMD-sensitive exon. (A) Exon cluster of the hnRNP DL gene. Constitutive exons are shown in black, the alternative exon 8 in blue. Lines indicate introns, big boxes show coding exons, while smaller boxes show untranslated exons. Arrows indicate the location of oligonucleotides used for RT-PCR in B. The sequence conservation of 100 vertebrates is shown below. An ultraconserved element (uc. 144) is located in the 3′UTR and covers exon 8. (B) RT-PCR after puromycin treatment (puro) in HeLa cells. Cells were treated for 4 h with 200 µg/mL puromycin. The short isoform 7/9 corresponds to the exon 8 exclusion isoform, the long isoform 7/8/9 to exon 8 inclusion. RPLP0 (P0) is shown as loading control. n = 2. (C) Transient knockdown of the NMD factor UPF1. (Left) RT-qPCR quantification of UPF1 mRNA after siRNA-mediated knockdown. (Right) RT-qPCR quantification of isoforms hnRNP DL 7/9 and hnRNP DL 7/8/9 after UPF1 knockdown. Expression values are normalized to the housekeeping gene RPLP0. n = 4, (**) P-value <0.01.
We tested whether the mRNA isoform containing exon 8 is subject to NMD by treating HeLa cells with puromycin, inhibiting translation and as a consequence NMD. Exon 8 inclusion was visualized by RT-PCR using oligonucleotides that amplify both isoforms (Fig. 1B). Exon 8 inclusion into the mRNA of hnRNP DL increased after treatment with 200 µg/mL puromycin. Additionally, UPF1 an essential part of the NMD machinery was transiently reduced by RNAi. Quantification of the two mRNA isoforms of hnRNP DL by RT-qPCR using isoform-specific oligonucleotides, showed an increase of the exon 8 containing isoform, while levels of the exon skipping isoform were unaffected (Fig. 1C). Therefore, we conclude that the mRNA including exon 8 indeed represents a target for NMD.
HnRNP DL protein activates inclusion of the poison exon in its own mRNA
To test whether hnRNP DL itself impacts on exon 8 inclusion, we fused its entire 3′UTR to a luciferase reporter gene (Fig. 2A). This allows the simultaneous observation of splicing changes and protein output. We cotransfected the luciferase-3′UTR-fusion construct together with an hnRNP DL or, as a control, a GFP expression plasmid into HeLa cells and analyzed the splicing changes and luciferase activity, respectively (Fig. 2B–D). Upon hnRNP DL overexpression, we observed an increase of exon 8 inclusion, accompanied by a decrease of the exon skipping isoform by RT-PCR (Fig. 2C). Luciferase activity is fivefold reduced upon hnRNP DL overexpression (Fig. 2D).
FIGURE 2.

HnRNP DL promotes inclusion of the poison exon in a minigene system. (A) Scheme of the luciferase-hnRNPDL-3′UTR minigene. The entire 3′UTR of hnRNP DL was fused to the luc2 gene in a dual luciferase vector (Luc_DL_UTR). The cassette exon 8 is indicated in blue. The big box indicates the protein coding region, small boxes indicate untranslated regions, lines indicate intronic sequences. Arrows indicate the location of oligonucleotides used for RT-PCR in C. (B) Western blot against hnRNP DL (DL). HnRNP DL or GFP as a control were transiently transfected into HeLa cells. Anti-β-actin was used as a loading control. n = 2. (C) RT-PCR of Luc_DL_UTR after overexpression of hnRNP DL (DL) or GFP as control (GFP). The short isoform 7/9 corresponds to the exon 8 exclusion isoform, the long isoform 7/8/9 to exon 8 inclusion. n = 2. (D) Luciferase activity of Luc_DL_UTR after overexpression of hnRNP DL (DL) or GFP (GFP) as a control. Firefly luciferase activity was normalized to Renilla luciferase as an internal control. Values are normalized to an empty vector control, without hnRNP DL 3′UTR sequences. n = 4, (**) P-value <0.01.
To test whether exon inclusion influences luciferase expression independent of AS-NMD, we inserted exon 8, either alone or plus 20 nt of the flanking exons 7 and 9, into the 3′UTR of a luciferase reporter gene. Both insertions did not change basal luciferase expression and showed no response to hnRNP DL overexpression (Supplemental Fig. S1). Thus exon 8 does not affect autoregulation independent of pre-mRNA splicing. This confirms that hnRNP DL is able to reduce gene expression by shifting pre-mRNA splicing of its own 3′UTR toward an NMD-sensitive isoform.
We further tested if alternative splicing of the poison exon is sufficient to trigger AS-NMD in an unrelated transcript. For this purpose, hnRNP DL exon 8 together with introns 7 and 8 and parts of the flanking constitutive exons 7 and 9 were transferred to the 3′UTR of DPP4 (Supplemental Fig. S2). The DPP4 3′UTR is coded by one single exon. It thus represents the regular gene organization of human genes. Fusion of the DPP4 3′UTR alone to a luciferase reporter gene does not allow for hnRNP DL-dependent regulation. However, insertion of hnRNP DL exon 8 along with its flanking sequences is enough to reduce luciferase activity 2.4-fold after hnRNP DL overexpression. The observed reduction is accompanied by an increase in exon 8 inclusion. Thus, alternative splicing of exon 8 renders the expression of unrelated transcripts dependent on hnRNP DL.
To validate the autoregulation of hnRNP DL in a chromosomal context, we analyzed the endogenous hnRNP DL protein level after overexpression of a GFP-hnRNP DL fusion protein. Fusion to GFP increases protein size in comparison with the endogenous protein and therefore allows the simultaneous detection by western blot. To overcome the background of untransfected cells, we stably integrated the GFP-hnRNP DL fusion into the genome of HeLa cells (Supplemental Fig. S3). We observed a twofold reduction of the endogenous hnRNP DL protein (Fig. 3A). Quantification of the two splicing isoforms by RT-qPCR revealed that the exon skipping isoform is decreased, whereas exon 8 inclusion increases (Fig. 3B).
FIGURE 3.
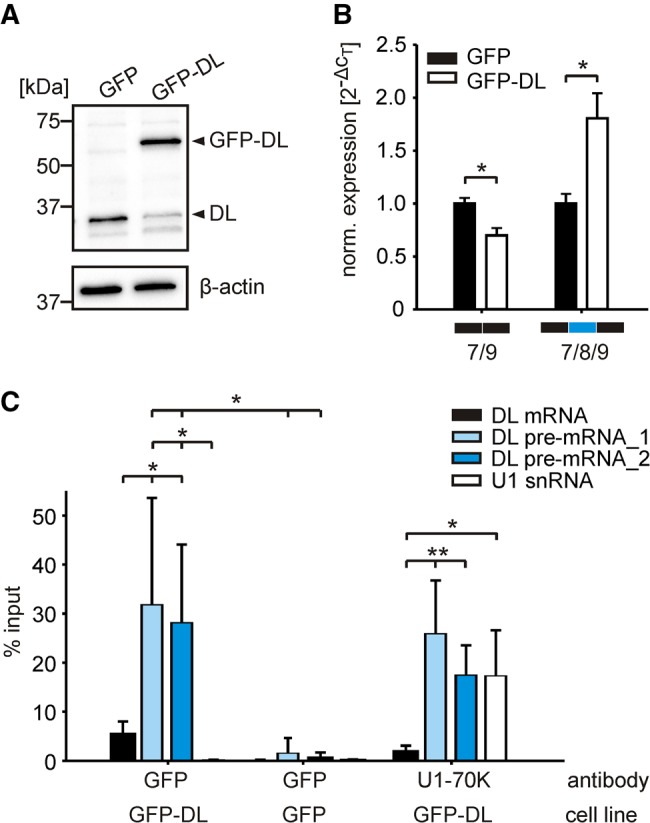
HnRNP DL regulates its own expression in a negative feedback loop. (A) Western blot of endogenous hnRNP DL protein levels after integration of a GFP-hnRNP DL fusion protein. Anti-hnRNP DL was used to simultaneously detect endogenous hnRNP DL (DL) and the integrated GFP-hnRNP DL fusion protein (GFP-DL). Anti-β-actin was used as a loading control. n = 3. (B) RT-qPCR quantification of isoforms hnRNP DL 7/9 and hnRNP DL 7/8/9 in GFP-hnRNP DL expressing cells (GFP-DL). A GFP expression cell line (GFP) was used as control. Expression values are normalized to the housekeeping gene RPLP0. n = 3. (C) RNA immunoprecipitation using antibodies against the GFP-hnRNP DL fusion protein and GFP or U1-70K as controls. Detected by RT-qPCR were fully spliced, mature hnRNP DL mRNA (DL mRNA), hnRNP DL pre-mRNA (DL pre-mRNA_1: oligonucleotides located in exon 8 and intron 8, DL pre-mRNA_2: both oligonucleotides located in intron 8), and U1 snRNA. n = 5. (**) P-value <0.01; (*) P-value <0.05.
RNA immunoprecipitation, with either the GFP-hnRNP DL fusion protein or GFP alone, confirmed a direct interaction of hnRNP DL with its own pre-mRNA (Fig. 3C). Notably, the hnRNP DL pre-mRNA was preferentially enriched over the fully spliced, mature hnRNP DL mRNA or the unrelated U1 snRNA. All tested RNAs were virtually absent in the control immunoprecipitation with GFP alone. Western blot analysis confirmed that similar amounts of GFP-hnRNP DL fusion protein and GFP were precipitated, excluding that the RNAs are differentially enriched, because of unequal protein precipitation efficiency (Supplemental Fig. S4). We also performed RNA immunoprecipitation with U1-70K, a U1 snRNP component, as positive control. Along with U1 snRNA, an integral component of the U1 snRNP, the hnRNP DL pre-mRNA was highly enriched. Consistently, the fully spliced hnRNP DL mRNA was not enriched. In sum, the data confirm that hnRNP DL regulates its own expression by AS-NMD.
Cross-regulation between the paralogs hnRNP D and DL
HnRNP D contains a similarly highly conserved region in its 3′UTR (Supplemental Fig. S5A). A cassette exon (exon 9) is located in this conserved region and was shown to target the mRNA for degradation by NMD (Banihashemi et al. 2006). We tested if hnRNP D is also able to modulate its own expression by AS-NMD via inclusion of exon 9. Further, we tested if any cross-regulation occurs between hnRNP D and DL in either direction. The hnRNP D gene produces four different protein isoforms by alternative splicing of exons 2 and 7 (Supplemental Fig. S4B). Previous reports show unique functions for the different isoforms in target gene regulation. Therefore, we tested splicing regulation for all four isoforms individually.
Similar to our approach for hnRNP DL, we fused the complete 3′UTR of hnRNP D to a luciferase reporter gene. Luciferase-3′UTR-fusions of hnRNP D and DL were then cotransfected with expression plasmids for GFP, hnRNP DL or one of the four different hnRNP D protein isoforms (D45, D42, D40, D37) (Fig. 4A; Supplemental Fig. S5B). Luciferase measurements and RT-PCR analyses showed that hnRNP DL drives not only splicing of its own transcript, but also the splicing of the hnRNP D pre-mRNA toward inclusion of the respective poison exon. This is also true for the isoforms D45 and D42, which increased exon inclusion and decrease luciferase expression (Fig. 4B,C). Therefore, we conclude that both hnRNP D and DL regulate their own and each other's expression by AS-NMD.
FIGURE 4.

Cross-regulation between hnRNP D and DL. (A) Western blot of hnRNP DL and hnRNP D overexpression. GFP, hnRNP DL, or hnRNP D isoforms (D45, D42, D40, D37) were transiently transfected into HeLa cells. Anti-hnRNP DL (above) and anti-hnRNP D (middle) were used to verify the respective overexpression. Anti-HSP60 (below) was used as loading control. (B) Luciferase activity of Luc_DL_UTR and Luc_D_UTR after overexpression of GFP as a control (GFP), hnRNP DL (DL), or hnRNP D isoforms (D45, D42, D40, D37). Luciferase activity was normalized to Renilla luciferase as internal control. Values are normalized to an empty vector control, without hnRNP D or DL 3′UTR sequences. n = 3. (C) RT-PCR of Luc_DL_UTR (left) and Luc_D_UTR (right) after overexpression with GFP as a control (GFP), hnRNP DL (DL), or hnRNP D isoforms (D45, D42, D40, D37). The short isoforms 7/9 and 8/10 correspond to exon exclusion isoforms, the long isoforms 7/8/9 and 8/9/10 to exon inclusion isoforms of hnRNP DL and D, respectively. n = 2. (D,E) RT-qPCR quantification of isoforms (D) hnRNP D 8/10 and hnRNP D 8/9/10 or (E) hnRNP DL 7/9 and hnRNP DL 7/8/9 after siRNA-mediated knockdown of hnRNP DL or D, respectively. Expression values are normalized to the housekeeping gene RPLP0. n = 5. (F) Western blot of hnRNP D and hnRNP DL after siRNA-mediated knockdown. Anti-hnRNP D (above) and anti-hnRNP DL (below) were used to verify the respective knockdown. Anti-HSP60 was used as loading control. n = 3. (**) P-value <0.01. (*) P-value <0.05.
It is interesting to note that only two of the four hnRNP D isoforms, D45 and D42, affect pre-mRNA splicing. Both isoforms contain exon 7 and have been shown to be mostly restricted to the nucleus, whereas the smaller isoforms D40 and D37 that do not contain exon 7 efficiently shuttle to the cytoplasm (Zhang et al. 1993; Arao et al. 2000; Inoue et al. 2003; Wilson et al. 2003). This disparity in subcellular distribution could explain the observed differences in splicing regulation. It also suggests that changes in isoform expression and/or subcellular distribution affect auto- and cross-regulation. Differences in hnRNP D isoform expression are obvious when comparing various tissues (Lu and Schneider 2004) and can also be sex-specific (Sheflin and Spaulding 2000). Their subcellular distribution is changed in response to stress (Laroia et al. 1999) and dysregulated in diverse cancers (for review, see Zucconi and Wilson 2011). Such changes will thus feedback on the overall levels of hnRNP D and DL.
In addition, overexpression of the smallest hnRNP D isoform D37 significantly reduces luciferase activity for both hnRNP D and DL constructs, although without a clear impact on pre-mRNA splicing, which hints at an additional layer of regulation (Fig. 4B,C). One possible mechanism for this regulation might be mRNA destabilization. Wilson et al. (1999) showed that D37 can bind AREs within intron 9 in vitro, suggesting ARE-mediated decay of intron 9–containing isoforms. However, we neither detected such intron 9–containing isoforms, nor a decrease in total mRNA amount in our RT-PCR experiments. Thus, we did not detect any evidence for an autoregulatory mechanism involving mRNA destabilization. However, hnRNP D is also capable of controlling protein levels by regulating translation efficiency (Liao et al. 2007; Yoon et al. 2014). Translation regulation does not necessitate changes in mRNA levels and is therefore a candidate for the effect observed with D37 overexpression.
To verify cross-regulation between the cellular proteins, we performed RNAi experiments of hnRNP D and DL and monitored changes in alternative splicing and protein levels of the respective paralog (Fig. 4D–F). Knockdown of both, hnRNP D and DL, led to reduced exon inclusion in their paralog. Consequently, the respective protein-coding mRNA isoforms, without an NMD-sensitive exon in their 3′UTR, are increased, resulting in increased protein levels. Thus the RNAi experiments confirmed that hnRNP D and DL regulate expression of one another by AS-NMD.
The change in protein output is considerably higher for hnRNP DL after hnRNP D knockdown compared to vice versa. This might be a result of the lower inclusion level of hnRNP D exon 9 in HeLa cells, when compared to hnRNP DL exon 8. Thus, a further reduction of exon inclusion after hnRNP DL knockdown only moderately changes hnRNP D protein levels. This suggests that differences in basal exon inclusion levels in different cell types or tissues will affect the extent of cross-regulation between these two proteins. Moreover, the observed pronounced increase in hnRNP DL protein levels after hnRNP D knockdown emphasizes the importance to investigate its participation in hnRNP D-regulated processes.
In sum, we demonstrated that both hnRNP D and DL negatively regulate both their own and the expression of one another by AS-NMD. Moreover, the different competence of the four hnRNP D protein isoforms to mediate AS-NMD adds an additional layer of complexity to these feedback regulations. Apart from that, our findings directly link hnRNP DL to ARE-mediated decay. More importantly, we demonstrate that this little-known and mostly overlooked hnRNP presents a novel angle to study dysregulation of hnRNP D in diseases. Future work should explore if and to what extent target genes are shared between these two hnRNPs to shed further light on the interconnections of hnRNP D and DL in controlling (patho-)physiological processes. In essence, our study establishes hnRNP DL as a disease-relevant gene that should be the focus of systematic and in-depth research.
MATERIALS AND METHODS
Cell culture and transfection
HeLa cells (Leibniz-Institute DSMZ, DSMZ no.: ACC 57) were cultured in T75 flasks in Dulbecco's modified Eagle's medium (Sigma-Aldrich) supplemented with 10% FBS Superior (Biochrom), 1 mM sodium pyruvate (Thermo Fisher Scientific) and Pen Strep (Thermo Fisher Scientific).
For plasmid transfection, cells were seeded to a confluency of 80%–90%. 24 h after seeding, cells were transfected using Lipofectamine 3000 (Thermo Fisher Scientific). For minigene analysis with subsequent RNA or protein isolation, cells were transfected in a 12-well format with 200 ng of minigene construct (pDL) and 600 ng of overexpression plasmid (pCMV-GFP or pCMV-hnRNPD or DL). Samples were prepared 24 h post transfection. For dual luciferase assays, HeLa cells were transfected in triplicates in a 24-well format, each well with 100 ng pDL constructs and 300 ng overexpression plasmid (pCMV-GFP or pCMV-hnRNPD or DL). Luciferase activity was measured 24 h after transfection using the Dual-Glo luciferase assay system (Promega, Mannheim). For each well, firefly values were normalized to the Renilla values of the same well.
For siRNA transfection, cells were seeded to a confluency of 60%–80%. 24 h after seeding, cells were transfected using Lipofectamine RNAiMAX (Thermo Fisher Scientific). For subsequent RNA or protein isolation, cells were transfected in a 12-well format with 4 pmol of a nonsilencing control siRNA (5′-UUCUCCGAACGUGUCACGU[dT][dT]-3′), an siRNA targeting hnRNP DL (5′-GGGUAUAACUAUGGGAACU[dT][dT]-3′) or a pool of three different siRNAs targeting hnRNP D (5′-GGGUCCCUCUGAAGUUUAA[dT][dT]-3′, 5′-AGACUGCACUCUGAAGUUA[dT][dT]-3′, 5′-GAAGGUGAUUGAUCCUAAA[dT][dT]-3′). Samples were prepared 48 h post transfection.
For puromycin treatment HeLa cells were seeded to a confluency of 80%–90% in a 12-well plate. Medium was changed after 24 h, 2 h later puromycin (Sigma-Aldrich) was added to a final concentration of 200 µg/mL for 4 h.
The HeLa-derived Flp-In host cell line HF1-3 was cultured as described for HeLa cells, but with 100 µg/mL Zeocin (Invivogen). The cell line HF1-3 was generated as described for the cell line HF1-1 (Berens et al. 2006). Both were obtained from the same selection and are independent sister cell lines. The integration site was described to be independent from positional effects.
The Flp-In system (Thermo Fisher Scientific) was used to generate cells that stably express GFP or a GFP-hnRNP DL fusion protein. The pFRT constructs were cotransfected with pOG44 Flp-recombinase expression vector (Thermo Fisher Scientific) in a molar ratio of 1:9 into the HF1-3 cells using Lipofectamine 2000 (Thermo Fisher Scientific). The following selection steps were performed as described in the manufacturer's protocol. After 2 wk of cultivating the cells with 150 µg/mL hygromycin B (Invivogen), RNA and protein were prepared and analyzed. To avoid biases based on clonal variability, we did not analyze single clones, but the complete pool of cells after genomic integration.
Plasmid construction
Overexpression plasmids: pCMV-GFP, pCMV-hnRNPDL, pCMV-hnRNPD
The eGFP CDS was amplified from the pEGFP-N1 vector (Clontech). The hnRNP DL CDS was amplified from cDNA (Ensembl Transcript ID: ENST00000602300.5). AgeI and XbaI restriction sites were used for ligation into the pCMV-MS vector (Kemmerer and Weigand 2014).
The CDS of the four hnRNP D proteins was amplified from cDNA using two pairs of oligonucleotides. The upstream (exon 1 to exon 3/4) and downstream (exon 3/4 to exon 8) parts of the CDS were amplified and two different fragments were obtained for each oligonucleotide pair. The amplified fragments were joined by overlap extension PCR to generate the four different CDS. The transcripts were hnRNP D45 (Ensembl Transcript ID: ENST00000313899.11), hnRNP D42 without exon 2 (Ensembl Transcript ID: ENST00000352301.8), hnRNP D40 without exon 7 (Ensembl Transcript ID: ENST00000353341.8) and hnRNP D37 without exon 2 and exon 7 (RefSeq ID: NP_001003810.1). XhoI and AgeI restriction sites were used for ligation into the pCMV-MS vector (Kemmerer and Weigand 2014).
Integration plasmids: pFRT-GFP, pFRT-GFP-hnRNPDL
The pFRT-GFP plasmid was used for the stable integration of GFP into HF1-3 cells (Beilstein et al. 2015). A GFP-hnRNP DL fusion protein was generated for integration. The CDS sequence of GFP was amplified from the pCMV-GFP plasmid, the CDS of hnRNP DL was amplified from the pCMV-hnRNPDL plasmid. Both fragments were joined by overlap extension PCR, separated by an SG4 linker sequence. GFP is N-terminally fused to hnRNP DL. The fragment was ligated into the pFRT backbone using BsiWI and SacI as restriction sites.
Luciferase minigenes: pDL-hnRNPDL, pDL-hnRNPD, pDL-DPP4, pDL-DPP4_DL, pDL-e8, pDL-e8 + 20 nt
The complete 3′ UTR sequences of the hnRNP DL gene (exons 7–9) (gene ID: ENSG00000152795), the hnRNP D gene (exons 8–10) (gene ID: ENSG00000138668) and the DPP4 gene (gene ID: ENSG00000197635) plus 100 nt downstream from the respective genomic context were cloned behind the CDS of the firefly luciferase gene (luc2) in the dual luciferase vector pDL (Kemmerer and Weigand 2014). The fragments were amplified from gDNA and ligated into the pDL plasmid using NotI and SalI (pDL-hnRNPD and pDL-hnRNPDL) or NotI and ClaI (pDL-DPP4) as restriction sites. For cloning of pDL-DPP4_DL hnRNP DL intron 7, exon 8 and intron 8 plus 3 nt of exon 7 and 45 nt of exon 9 were amplified from pDL-hnRNPDL and inserted at position +20 into the DPP4 3′UTR by overlap extension PCR.
The hnRNP DL exon 8 sequence was assembled from two oligonucleotides. The sequence of hnRNP DL exon 8 plus 20 nt of the flanking exons 7 and 9 was amplified from pDL-hnRNPDL. Both inserts were ligated into the pDL plasmid using NotI and HindIII restriction sites. The complete vector sequences are available upon request.
RNA isolation
Total RNA from HeLa cells was isolated using TRIzol reagent (Invitrogen), followed by TURBO DNase (Thermo Fisher Scientific) treatment. Total RNA from HF1-3 cells was isolated using the miRNeasy Mini kit (Qiagen), including the optional on-column DNA digestion with the RNase-Free DNase Set (Qiagen). After isolation, 1 µg RNA was quality checked on a 1% agarose gel.
RT- and RT-qPCR
RT-PCR analysis and RT-qPCR quantification was performed as in (Weigand et al. 2012). Oligonucleotide sequences are listed in Supplemental Table S1. All PCR products were verified by sequencing. Additionally, the specificity of isoform-specific oligonucleotides was verified by qPCR using plasmids coding for either the exon inclusion or exclusion isoforms of hnRNP D or DL, respectively.
Western blot
For western blot analyses, cells were lysed in lysis buffer (137 mM NaCl, 10% glycerol, 20 mM Tris–HCl pH 8.0, 2 mM EDTA pH 8.0, 1% Igepal, 5 µL protease inhibitor cocktail [Sigma-Aldrich]) for 20 min on ice. After centrifugation (15 min at 17,000g, 4°C) the protein content of the samples was determined in three technical replicates according to the Bradford method. 10–20 µg protein were loaded onto precast gels and blotted onto PVDF membranes (both Bio-Rad). Primary antibodies targeting hnRNP DL (sc-133699, Santa Cruz), hnRNP D (07-260 Merck Millipore), GFP (118144600, Roche), β-actin (A-5441, Sigma-Aldrich), or HSP60 (ab6530, Abcam) were used. Horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (Jackson ImmunoResearch) were used as secondary antibodies. Blots were developed with the ECL system (Bio-Rad) or Amersham ECL select (GE Healthcare) for weaker signals. Images were detected using the ChemiDoc Imaging System (Bio-Rad).
RNA immunoprecipitation (RIP)
RIP was performed according to the manual using the Magna RIP kit (Merck Millipore), followed by DNase I digestion (Sigma-Aldrich). 4 × 106 cells (HF1-3-GFP or HF1-3-GFP-DL) were used per RIP. 10% of each precipitate was used for western blot analyses. Mouse IgG and anti-snRNP70 antibody were included in the kit. A primary antibody targeting GFP (see above) was used for RIP and western blot.
Statistical analysis
All bar graphs are reported as mean values ± standard deviation. Statistical analysis was done using Student's t-test.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Professor Dr. Beatrix Süß for helpful discussions and Britta Kluge for excellent technical assistance. This work was supported by the Dr. Ing. Wilhelm und Maria Kirmser-Stiftung and the Deutsche Forschungsgemeinschaft (SFB902/B14).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.063420.117.
REFERENCES
- Änkö ML, Müller-McNicoll M, Brandl H, Curk T, Gorup C, Henry I, Ule J, Neugebauer KM. 2012. The RNA-binding landscapes of two SR proteins reveal unique functions and binding to diverse RNA classes. Genome Biol 13: R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arao Y, Kuriyama R, Kayama F, Kato S. 2000. A nuclear matrix-associated factor, SAF-B, interacts with specific isoforms of AUF1/hnRNP D. Arch Biochem Biophys 380: 228–236. [DOI] [PubMed] [Google Scholar]
- Banihashemi L, Wilson GM, Das N, Brewer G. 2006. Upf1/Upf2 regulation of 3′ untranslated region splice variants of AUF1 links nonsense-mediated and A+U-rich element-mediated mRNA decay. Mol Cell Biol 26: 8743–8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilstein K, Wittmann A, Grez M, Suess B. 2015. Conditional control of mammalian gene expression by tetracycline-dependent hammerhead ribozymes. ACS Synth Biol 4: 526–534. [DOI] [PubMed] [Google Scholar]
- Berens C, Lochner S, Löber S, Usai I, Schmidt A, Drueppel L, Hillen W, Gmeiner P. 2006. Subtype selective tetracycline agonists and their application for a two-stage regulatory system. Chembiochem 7: 1320–1324. [DOI] [PubMed] [Google Scholar]
- Bergeron D, Pal G, Beaulieu YB, Chabot B, Bachand F. 2015. Regulated intron retention and nuclear pre-mRNA decay contribute to PABPN1 autoregulation. Mol Cell Biol 35: 2503–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, Hertel KJ. 2012. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA 3: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi A, Shiosaka T, Takaoka Y, Yanagisawa K, Fujita S. 1998. Molecular cloning of the cDNA encoding A+U-rich element RNA binding factor. Biochim Biophys Acta 1396: 51–56. [DOI] [PubMed] [Google Scholar]
- Geuens T, Bouhy D, Timmerman V. 2016. The hnRNP family: insights into their role in health and disease. Hum Genet 135: 851–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SP, Tang YH, Smith R. 2010. Functional diversity of the hnRNPs: past, present and perspectives. Biochem J 430: 379–392. [DOI] [PubMed] [Google Scholar]
- Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, Yan BY, Donohue JP, Shiue L, Hoon S, Brenner S, et al. 2012. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep 1: 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Arao Y, Omori A, Ichinose S, Nishio K, Yamamoto N, Kinoshita Y, Mita S. 2003. Identification of S1 proteins B2, C1 and D1 as AUF1 isoforms and their major role as heterogeneous nuclear ribonucleoprotein proteins. Biochem J 372: 775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemmerer K, Weigand JE. 2014. Hypoxia reduces MAX expression in endothelial cells by unproductive splicing. FEBS Lett 588: 4784–4790. [DOI] [PubMed] [Google Scholar]
- Laroia G, Cuesta R, Brewer G, Schneider RJ. 1999. Control of mRNA decay by heat shock-ubiquitin-proteasome pathway. Science 284: 499–502. [DOI] [PubMed] [Google Scholar]
- Liao B, Hu Y, Brewer G. 2007. Competitive binding of AUF1 and TIAR to MYC mRNA controls its translation. Nat Struct Mol Biol 14: 511–518. [DOI] [PubMed] [Google Scholar]
- Lomonaco V, Martoglia R, Mandreoli F, Anderlucci L, Emmett W, Bicciato S, Taccioli C. 2014. UCbase 2.0: ultraconserved sequences database (2014 update). Database (Oxford) 2014: bau062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JY, Sadri N, Schneider RJ. 2006. Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev 20: 3174–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JY, Schneider RJ. 2004. Tissue distribution of AU-rich mRNA-binding proteins involved in regulation of mRNA decay. J Biol Chem 279: 12974–12979. [DOI] [PubMed] [Google Scholar]
- Moore AE, Chenette DM, Larkin LC, Schneider RJ. 2014. Physiological networks and disease functions of RNA-binding protein AUF1. Wiley Interdiscip Rev RNA 5: 549–564. [DOI] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O'Brien G, Shiue L, Clark TA, Blume JE, Ares M Jr. 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21: 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omnus DJ, Mehrtens S, Ritter B, Resch K, Yamada M, Frank R, Nourbakhsh M, Reboll MR. 2011. JKTBP1 is involved in stabilization and IRES-dependent translation of NRF mRNAs by binding to 5′ and 3′ untranslated regions. J Mol Biol 407: 492–504. [DOI] [PubMed] [Google Scholar]
- Reboll MR, Oumard A, Gazdag AC, Renger I, Ritter B, Schwarzer M, Hauser H, Wood M, Yamada M, Resch K, et al. 2007. NRF IRES activity is mediated by RNA binding protein JKTBP1 and a 14-nt RNA element. RNA 13: 1328–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rösel-Hillgärtner TD, Hung LH, Khrameeva E, Le Querrec P, Gelfand MS, Bindereif A. 2013. A novel intra-U1 snRNP cross-regulation mechanism: alternative splicing switch links U1C and U1-70K expression. PLoS Genet 9: e1003856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossbach O, Hung LH, Schreiner S, Grishina I, Heiner M, Hui J, Bindereif A. 2009. Auto- and cross-regulation of the hnRNP L proteins by alternative splicing. Mol Cell Biol 29: 1442–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheflin LG, Spaulding SW. 2000. Testosterone and dihydrotestosterone regulate AUF1 isoforms in a tissue-specific fashion in the mouse. Am J Physiol Endocrinol Metab 278: E50–E57. [DOI] [PubMed] [Google Scholar]
- Spellman R, Llorian M, Smith CW. 2007. Crossregulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol Cell 27: 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Zhang Z, Sinha R, Karni R, Krainer AR. 2010. SF2/ASF autoregulation involves multiple layers of post-transcriptional and translational control. Nat Struct Mol Biol 17: 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira NM, Naslavsky MS, Licinio L, Kok F, Schlesinger D, Vainzof M, Sanchez N, Kitajima JP, Gal L, Cavaçana N, et al. 2014. A defect in the RNA-processing protein HNRPDL causes limb-girdle muscular dystrophy 1G (LGMD1G). Hum Mol Genet 23: 4103–4110. [DOI] [PubMed] [Google Scholar]
- Weigand JE, Boeckel JN, Gellert P, Dimmeler S. 2012. Hypoxia-induced alternative splicing in endothelial cells. PLoS One 7: e42697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EJ, Brewer G, Wilson GM. 2013. Post-transcriptional control of gene expression by AUF1: mechanisms, physiological targets, and regulation. Biochim Biophys Acta 1829: 680–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EJ, Matsangos AE, Wilson GM. 2017. AUF1 regulation of coding and noncoding RNA. Wiley Interdiscip Rev RNA 8: e1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GM, Lu J, Sutphen K, Sun Y, Huynh Y, Brewer G. 2003. Regulation of A+U-rich element-directed mRNA turnover involving reversible phosphorylation of AUF1. J Biol Chem 278: 33029–33038. [DOI] [PubMed] [Google Scholar]
- Wilson GM, Sun Y, Sellers J, Lu H, Penkar N, Dillard G, Brewer G. 1999. Regulation of AUF1 expression via conserved alternatively spliced elements in the 3′ untranslated region. Mol Cell Biol 19: 4056–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YY, Li H, Lv XY, Wei Q, Li X, Liu XY, Zhou Q, Wei YQ. 2008. Overexpression of JKTBP1 induces androgen-independent LNCaP cell proliferation through activation of epidermal growth factor-receptor (EGF-R). Cell Biochem Funct 26: 467–477. [DOI] [PubMed] [Google Scholar]
- Yoon JH, De S, Srikantan S, Abdelmohsen K, Grammatikakis I, Kim J, Kim KM, Noh JH, White EJ, Martindale JL, et al. 2014. PAR-CLIP analysis uncovers AUF1 impact on target RNA fate and genome integrity. Nat Commun 5: 5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wagner BJ, Ehrenman K, Schaefer AW, DeMaria CT, Crater D, DeHaven K, Long L, Brewer G. 1993. Purification, characterization, and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol Cell Biol 13: 7652–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Ge Y, Sun L, Ma W, Wu J, Zhang X, Hu X, Eaves CJ, Wu D, Zhao Y. 2014. Growth arrest specific 2 is up-regulated in chronic myeloid leukemia cells and required for their growth. PLoS One 9: e86195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucconi BE, Wilson GM. 2011. Modulation of neoplastic gene regulatory pathways by the RNA-binding factor AUF1. Front Biosci (Landmark Ed) 16: 2307–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.