

Abstract
Monocytosis and neutrophilia are frequent events in atherosclerosis. These phenomena arise from the increased proliferation of hematopoietic stem and multipotential progenitor cells (HSPCs) and HSPC mobilization from the bone marrow to other immune organs and circulation. High cholesterol and inflammatory signals promote HSPC proliferation and preferential differentiation to the myeloid precursors (i.e., myelopoiesis) that than give rise to pro‐inflammatory immune cells. These cells accumulate in the plaques thereby enhancing vascular inflammation and contributing to further lesion progression. Studies in animal models of atherosclerosis showed that manipulation with HSPC proliferation and differentiation through the activation of LXR‐dependent mechanisms and restoration of cholesterol efflux may have a significant therapeutic potential.
Keywords: atherosclerosis, monocyte, neutrophil, chemokines, inflammation, atherosclerotic plaque
| • Introduction |
| • High‐cholesterol levels increase chemokine signalling |
| • Role of different monocyte subsets in atherosclerosis |
| • NR4A1‐deficient animal model |
| • Factors that associate impaired fat metabolism with the inflammatory responses in atherosclerosis |
| ‐ Liver X receptors |
| ‐ High cholesterol and hematopoietic progenitor cells |
| • The repertoire of hematopoietic progenitor cells is biased towards the preferential myeloid production in atherogenesis |
| • Mechanisms of monocyte release from the bone marrow |
| • Local macrophage proliferation is a major source of lesional macrophages |
| • Contribution of monocytes to plaque progression |
| • Factors that regulate macrophage retention or emigration from the plaque |
| • Neutrophils and their role in atherosclerosis |
| ‐ Function and characteristics of neutrophils |
| ‐ Components of neutrophil granules |
| ‐ Neutrophilia and hypercholesterolaemia |
| ‐ Mediators of neutrophil recruitment to lesions |
| ‐Neutrophil granule components in atherosclerosis |
| ‐ NETs in atherosclerosis |
| • Conclusion |
| • Acknowledgement |
| • Conflict of interest |
Introduction
Atherosclerotic disease is a chronic process associated with the arterial inflammation, accumulation of lipids in the vessel wall, plaque formation, thrombosis and late mortal complications such as myocardial infarction and ischaemic stroke 1. High‐blood cholesterol content and increased level of low‐density lipoprotein (LDL) were shown to primarily contribute to atherogenesis both in humans and in animal experimental models 2. During atherogenesis, cholesterol, oxidized LDL (oxLDL) and other lipids accumulate in the vascular wall contributing to the formation of the lipid core and then necrotic core of the plaque. Treatment with statins, which is focused on lowering serum lipid levels results in significant reduction of circulating lipid levels and, in general, in a one‐third decrease in the incidence of acute cardiovascular events 3. Furthermore, experimental animals, such as rodents, have innately low‐LDL content in the blood (about 50 mg/dl or less) 4 and are invulnerable to atherogenesis 5. Together, these observations indicate hypercholesterolaemia as an independent risk factor for atherosclerotic disease.
In addition to lipid deposits, the lipid core of the lesion also contains multiple types of infiltrated leucocytes. There are neutrophils, T cells, mast cells and dendritic cells 6, 7, 8. However, the majority of intraplaque immune cells are monocytes and macrophages that are derived from them. The monocytes exhibit heterogeneity: the most frequent subset (up to 90%) in humans is CD16−CD14+, while in mice there are Ly6Chigh cells termed as classical monocytes that are differentiated to pro‐inflammatory macrophages during inflammation. The minor monocyte population (called patrol, resident or non‐classical monocytes) is represented by the CD16+CD14dim subset in people and by the Ly6Clow cells in mice 9. Of these two subpopulations, the classical monocytes tend to accumulate in the plaques and give rise to pro‐inflammatory (M1) macrophages 10. These findings therefore underline the key role of both inflammation and dyslipidaemia in the atherogenic process 11, 12.
Monocytosis is an elevation of monocyte counts circulating in the blood. Commonly, monocytosis occurs in acute and chronic inflammatory conditions including atherosclerosis. Early epidemiological studies showed that increased numbers of leucocytes in the circulation can indicate higher risk of cardiovascular disease 13. Among leucocytes, further studies underlined the association of elevated levels of monocytes and neutrophils with atherosclerotic progression 14, 15, 16. In experimental murine models of atherogenesis, numbers of blood neutrophils and Ly6Chigh monocytes were shown to correlate with atherosclerotic progression 10, 17, 18, 19. In humans, a direct correlation between the high‐cholesterol levels and increased counts of peripheral mononuclear phagocytes was also found therefore reflecting the stimulating contribution of lipids on the levels of monocytes and neutrophils in the circulation 20. Treatment with lipid‐lowering agents caused decrease in levels of both neutrophils and monocytes 10, 21 thereby suggesting for the positive link between hypercholesterolaemia and pro‐atherogenic monocytosis/neutrophilia.
In this review, we discuss the mechanisms by which hypercholesterolaemia is associated with the atherogenic process.
High‐cholesterol levels increase chemokine signalling
Increase in the numbers of blood neutrophils may arise from the mechanisms that stimulate departure of neutrophils from the bone marrow and weaken their tissue homing 18. Neutrophils use the CXC chemokine receptor 2 (CXCR2)‐dependent mechanism [or interleukin‐8 (IL‐8)‐dependent in people] and their ligands to go out from the bone marrow and accumulate to the plaque 18, 22. Increased levels of cholesterol up‐regulate CXCR2 expression in neutrophils thereby promoting their exit from the bone marrow 23. In LDL receptor (LDLR)‐deficient mice (a hypercholesterolaemic model that spontaneously develops atherosclerosis when fed on a fat‐rich diet), knockout of CXCR2 resulted in attenuated atherogenesis indicating the pro‐atherosclerotic role of increased expression of this receptor, especially in neutrophils 24. For senescent neutrophils, the CXCR4/CXCL12 pathway serves as a mechanism to mediate the retrieval of these cell to the bone marrow 25. CXCR4‐inhibiting agents and lipid‐enriched feeding down‐regulate CXCL12 production in the bone marrow and thus led to increased levels of neutrophils and enhanced atherogenesis 18, 26.
Monocytes leaving the bone marrow and recruitment to the lesion are regulated by the C‐C chemokine receptor type 2 (CCR2) 27, 28, 29. In CCR2‐deficient mice, atherosclerosis progression is delayed due to reduced recruitment of monocytes to atherosclerotic lesions 17, 30. In people, increased plasma cholesterol is associated with higher levels of CCL2 that binds to CCR2 23. These findings can suggest that CCR2/CCL2 mechanism plays a role in the mobilization of monocytes to be pro‐atherogenic. Additionally, increased progression to monocytes and neutrophils was detected in the bone marrow in individuals affected by cardiovascular disease.
Other chemokine receptors such as CCR5 and CX3CR1 are also important to the recruitment of monocytes to atherosclerotic plaques as ablation of these receptors and CCL2 prevents monocytosis and dramatically reduces atherosclerosis in apoE‐deficient mice 17. Various monocyte subsets differentially use CCR2, CX3CR1 and CCR5 to accumulate in atherosclerotic lesions. In mice, there are two main subsets producing different chemokine receptor patterns: CCR2+CX3CR1+Ly6Chigh and CCR2−CX3CR1++Ly6Clow monocytes. To enter lesions, CCR2−CX3CR1++Ly6Clow monocytes do not use CX3CR1 but are partially dependent on CCR5. In contrast, CCR2+CX3CR1+Ly6Chigh monocytes employ CCR2, and CCR5 27.
Recently, Quintar with co‐authors employed a new method, long‐term 2‐photon intravital microscopy, to assess the behaviour and frequency of patrolling CX3CR1highLy6Clow monocytes in live CX3CR1+/GFP(green fluorescent protein) mice 31. The numbers of patrolling monocytes became increased by ninefold in CX3CR1‐GFP (green fluorescent protein) mice and by 22‐fold in CX3CR1+/GFP/apoE‐deficient mice fed on cholesterol‐rich diet. In atherosclerosis, the real attachment of patrolling monocytes to the endothelium was observed. These data indicate that patrolling monocytes can be recruited to the atherosclerotic endothelium in a CX3CR1‐independent manner 31. In summary, CCR2, CX3CR1 and CCR5 play independent and additive roles in atherogenesis. Signals mediated via these pathways determine the frequency of monocyte subsets and therefore account for almost all macrophage accumulation into atherosclerotic vessels.
Role of different monocyte subsets in atherosclerosis
Monocyte population is heterogeneous and consists of three subpopulations characterized by the differential expression of the lipopolysaccharide (LPS) receptor (CD14) and the FcγIII receptor (CD16). There are classical CD14++CD16−, intermediate CD14++CD16+ and non‐classical CD14+CD16++ monocytes in humans 32. In non‐inflammatory conditions, classical CD14++CD16− cells are prevalent accounting for up to 90% of all monocytes while the subset of monocytes that highly express CD16 is minor (5–10% of a total monocyte population) 33. However, in inflammation, counts of CD14+CD16++ monocytes rapidly increase 34. These cells show macrophage‐like behaviour because they have the ability to phagocytosis and reactive oxygen species (ROS) production. Compared to classical monocytes, the ability of CD14+CD16++ cells to produce cytokines is greatly decreased 35. Additionally, CD14+CD16++ monocytes express intracellular adhesion molecule‐1 (ICAM‐1), opsonin receptors FcγRII and FcγRI, and high levels of major histocompatibility (MHC) class II molecules 36. Classification of the heterogeneous population of monocytes is the subject of numerous studies and is reviewed elsewhere 37.
Epidemiological and cross‐sectional studies showed association of counts of CD16‐positive (both intermediate and non‐classical) monocytes with various cardiovascular risk characteristics. For example, Schlitt et al. 38 reported positive correlation of increased amounts of CD14+CD16+ cells with coronary atherosclerosis onset and higher serum levels of tumour necrosis factor (TNF)‐α. In patients with unstable angina pectoris, it was found that elevated counts of CD14+CD16+ monocytes positively correlate with coronary fibrous cap thickness 39. In patients with stable angina pectoris, association between CD14+CD16+ monocyte frequencies and advanced vulnerability characteristics of atherosclerotic lesions was also shown 40. Timmerman with co‐authors found that subjects with low‐physical activity had higher levels of inflammatory CD16‐positive monocytes than physically active people 41. In low‐risk individuals, a relationship between CD16‐positive monocytes and both obesity and subclinical atherosclerosis was reported 42.
However, these early studies considered association of a whole population of CD16‐positive monocytes with cardiovascular risk factors but did not discriminate between the impact of CD14++CD16+ and CD14+CD16++ subsets. Finding of the heterogeneity of the population of CD16‐positive monocytes initiated studies aimed to evaluate a distinct role of each subset. Poitou with co‐authors described a major impact of fat mass variations on CD14+D16+ monocyte subset 43. Similarly, an intermediate monocyte subset was found to be an independent predictor of adverse cardiovascular events and sudden death in patients with chronic kidney disease (CKD) 44, 45. The role of elevated CD14++CD16+ cell numbers as a prognostic marker of unfavourable cardiovascular events was also shown in a randomly selected Swedish population 46 and in a cohort study of at‐risk German patients referred for coronary angioplasty 47.
Thus, these findings show that increased amounts of CD16‐positive monocytes and especially the CD14++CD16+ subset are associated with cardiovascular disease and may promote plaque progression and instability. In CKD patients, a greater adhesion ability of CD14+CD16+ monocytes to endothelial cells was observed, because of enhanced expression of chemokines that promote adhesion 48. Accordingly, in in vitro cell model of prolonged lifespan of healthy monocytes, it was found that senescent CD14+CD16+ monocytes possess enhanced ability interact with endothelial cells, increased antigen‐presenting capacity, and higher expression of inflammatory cytokines compared with the CD14++CD16− subset 49. These observations at least in part can explain association of CD14+CD16+ monocytes with increased cardiovascular risk as such pro‐inflammatory monocytes have advanced properties for transendothelial trafficking to the intima media where they can initiate inflammation.
NR4A1‐deficient animal model
Nuclear receptor NR4A1 (also known as Nur77) is a transcriptional factor, that is crucially involved in the development of monocytes especially patrolling Ly6C− monocytes 50. Like in humans, there are three subsets of murine monocytes characterized by high (Ly6Chigh monocytes), intermediate (Ly6Cmiddle monocytes) and low (Ly6Clow monocytes) of Ly6C, an inflammatory monocyte marker. The Ly6Chigh subset exerts pro‐inflammatory properties and phagocytic activity, Ly6Chigh cells are also pro‐inflammatory, while Ly6Clow monocytes have patrolling function and are involved in tissue repair 51.
NR4A1 deficiency was shown to aggravate atherosclerosis progression both in ApoE‐ 52 and LDLR‐deficient mice 53 fed on high‐fat diet. This is accompanied with dramatic reduction of Ly6Clow monocyte counts 53. Furthermore, in NR4A1/LDLR‐deficient mice, macrophages were polarized towards the pro‐inflammatory M1 phenotype characterized by increased expression of pro‐inflammatory markers such as toll‐like receptor 4 (TLR‐4), TNF‐α, inducible NO‐synthase (iNOS), and nuclear factor κB (NF‐κB), decreased expression of Arginase‐1 and enhanced lipid accumulation 53. Thus, these data suggest for the anti‐atherogenic role and anti‐inflammatory role of NR4A1 and Ly6C− monocytes. Loss of protective Ly6Clow monocytes in NR4A1‐deficient mice may therefore be associated with increased macrophage activation and phenotypic pro‐inflammatory M1 polarization of macrophages and lead to advanced atherogenic progression. Ly6Clow monocytes dampen inflammation by releasing IL‐10, an anti‐inflammatory cytokine and differentiate to M2 macrophages after extravasation to the tissue 51. Patrolling Ly6Clow monocytes possess a vasculoprotective function as depletion of this population in NR4A1/apoE‐deficient mice was shown to lead to massive apoptosis of arterial endothelial cells 31.
Factors that associate impaired fat metabolism with the inflammatory responses in atherosclerosis
Liver X receptors
In myeloid cells, especially in mononuclear phagocytes, liver X receptors (LXR‐α and LXR‐β) play the main role in the regulation of the cholesterol reverse transport from monocytes/macrophages, thereby contributing to the lipid efflux and to the liberation of tissues from the ectopic fat deposition 54. LXRs can be activated by oxysterols and by‐products of their biosynthesis 55. The physiological goal of the activation of LXRs is the counteracting to the cholesterol excessive influx that comes from the intestine, liver and bile in order to promote its excretion 56. In addition, LXRs are involved in the immune control, such as down‐regulation of expression of pro‐inflammatory genes including TNF‐α, TLR‐4 and IL‐1β 57, 58, 59.
LXR‐dependent pathways connect the lipid metabolism with inflammation. Both LXR‐α and LXR‐β can be activated by oxysterols and cholesterol biosynthesis metabolites 60. LXRs can suppress inflammation by inhibiting NF‐kB‐dependent signalling pathways via the mechanism of repression 61. LXRs are also involved to the induction of anti‐inflammatory mechanisms in monocytes/macrophages by the regulation of the phagocytosis of apoptotic cells and through up‐regulating expression of arginase that represent an M2‐specific marker 62. The anti‐inflammatory properties of macrophage‐expressed LXRs were shown by the engraftment of the bone marrow from the LXR‐α‐ and LXR‐β‐deficient macrophages to ApoE‐ and LDLR‐deficient mice has already been shown 63.
LXR agonists, such as GW3965, were found to demonstrate anti‐atherosclerotic properties by inducing the plaque regression and influencing lesional composition through decrease of the content of inflammatory cells and the atheroma volume 64, 65, 66. The inhibitory effect of LXR up‐regulation was shown on plaque progression, even when there were no changes in plasma lipid profile. It is likely that LXRs can stimulate the atheroprotective trafficking of lipids from the tissues back to the liver thereby decreasing their accumulation in peripheral organs/tissues 67.
In addition, LXRs participate in the clearance of senescent neutrophils to maintain the neutrophil homoeostasis and prevent excessive amplification of neutrophils. In mice, LXR deficiency is associated with perturbations in the neutrophil homoeostasis that leads to enhanced granulopoiesis 68. LXR mediate the phagocytosis of aged neutrophils through the activation of the proto‐oncogene tyrosine‐protein kinase MER (MERTK) that leads to enhanced phagocytosis by macrophages 69. Treatment with GW3965 and TO901317 (both are agonists of LXRs) resulted in reduced counts of neutrophils in the circulation 70. In addition, in LDLR‐deficient mice, Kappus et al. 71 showed the ability of both LXR agonists to down‐regulate expression of inflammatory genes in macrophages lacking ABCA1/G1. This in turn suggests for existence of anti‐inflammatory signalling pathways that are independent from the mechanism of cholesterol efflux.
High cholesterol and hematopoietic progenitor cells
Hematopoietic stem and multipotential progenitor cells (HSPCs) born in the bone marrow is crucially involved in the production of monocytes and neutrophils. In physiological conditions, neutrophils and monocytes are directly originated from HSPCs that are located in special microenvironments of the bone marrow called stem cell niches. Normally, the production and mobilization of HSPCs are strictly controlled to avoid excessive formation of monocytes/macrophages but to maintain the permanent self‐renewal of these cell populations during the life 72.
In inflammation, these progenitors were shown to be primarily involved in the mobilization of pro‐inflammatory cells 73. Cholesterol metabolism regulates the function of HSPCs and their ability to differentiate to monocytes and other immune cells. In animal models of atherosclerosis, a sufficient extramedullary hematopoiesis was observed. In mice, HSPCs can be mobilized from the bone marrow to the secondary lymphoid organs such as spleen where they can differentiate to monocytes and be involved in atherosclerosis through the recruitment to the plaque 74. Normally, the spleen serves as a source for monocytes but can significantly increase the production of these cells in inflammatory and hypercholesterolaemic conditions 75. Cholesterol‐rich diet permanently increases counts of splenic monocytes indicating the positive role of high‐cholesterol levels on the mobilization of HSPCs from the bone marrow 76.
Moreover, high cholesterol can also induce ‘memory’ effects (through presumably the epigenetic changes) in HSPCs on their direct progeny (Fig. 1). For example, Seijkens with co‐authors reported enhanced ability of bone marrow‐derived HSPCs transplanted from LDLR‐deficient mice to normal mice to induce generation of myeloid cells particularly granulocytes and Ly6Chigh monocytes 77. High cholesterol‐induced HSPCs also induce pro‐inflammatory and pro‐atherosclerotic M1 macrophages that can subsequently produce inflammatory messengers, such as TNF‐α, IL‐6 and CCL2. In addition, enhanced properties of hypercholesterolaemia‐induced HSPCs were shown to then differentiate to pro‐inflammatory monocytes that can accumulate in the plaque and promote the atherosclerotic development 77, 78. However, it should be mentioned that pro‐inflammatory effects of high cholesterol‐induced HSPCs may be mediated not only through epigenetic reprogramming but also through up‐regulation of the IL‐3/macrophage colony‐stimulating factor (GM‐CSF) receptor 79.
Figure 1.
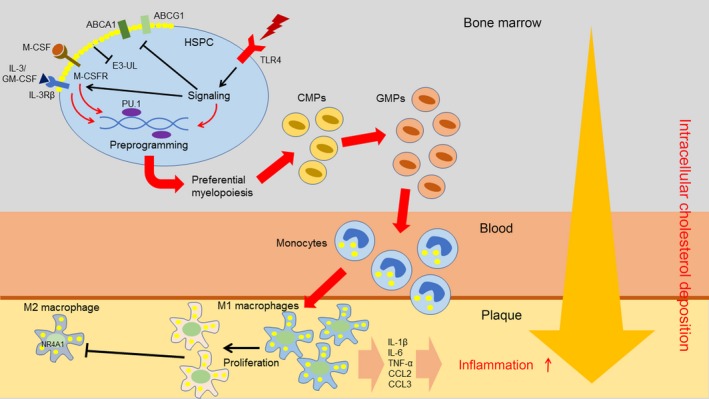
Contribution of high cholesterol‐induced myelocytosis to atherogenesis. High cholesterol up‐regulates expression of inflammatory receptors such as TLR‐4 on the surface of HSPCs. TLR‐4 can be activated by pro‐inflammatory stimuli. TLR‐4‐dependent signalling leads to the inhibition of cholesterol efflux mediated by ABCA1 and ABCG1. Increased membrane cholesterol leads to increased surface expression of myeloid cytokine receptors IL‐3Rβ and M‐CSFR due to failure to activate E3‐ubiquitin ligases (E3‐UL). The receptors mediate enhanced signalling from myeloid cytokines (IL‐3, M‐CSF, GM‐CSF), which in cooperation with cholesterol‐enriched microenvironment promote HSPC reprogramming (mediated by transcription factor PU.1) to produce more myeloid cells. During differentiation to common myeloid progenitors (CMPs) and further to granulocyte‐macrophage progenitors (GMPs), myeloid cells uptake more cholesterol since their reverse cholesterol transport is suppressed. In the circulation, monocytes can accumulate more cholesterol before entering the plaque. In the plaque, lipid‐laden monocytes preferentially differentiate to M1 macrophages that produce a number of pro‐inflammatory cytokines and therefore contribute to inflammation. In these macrophages, expression of transcription factor NR4A1 is suppressed thereby limiting the ability to switch to the anti‐inflammatory M2 phenotype.
Recently, van Kampen with co‐authors showed the ability of the bone marrow‐derived HSPCs from the normal recipient mice to which hypercholesterolaemia‐primed HSPCs were engrafted to induce hypomethylation of CpG regions in the PU.1 and interferon regulatory factor 8 (IRF‐8) genes 80. Both PU.1 and IRF‐8 are transcription factors that control commitment of the myeloid lineage 81. In HSPCs, high cholesterol‐induced epigenetic changes in the promoters of the PU.1 and IRF‐8 genes lead to the transcriptional de‐repression of both genes and their enhanced expression that in turn primes preferential differentiation of HSPCs to the myeloid cells and atherogenesis (Fig. 1) 77. Hematopoietic IRF‐8‐deficiency promotes atherosclerosis in mice 19. Similar with hypercholesterolaemia, hyperglycaemia was shown to up‐regulate myelopoiesis and monocytosis in diabetic mice 82.
ATP‐binding cassette (ABC) transporters such as ABCA1 and ABCG1 are involved to the reverse transport thereby preventing its excessive accumulation in the cell. In LDLR‐deficient mice with genetic deletion of both ABCA1 and ABCG1, increased numbers of leucocytes and enhanced atherosclerosis was observed 83. The mechanism by which ABCA1/ABCG1 deficiency contributes to atherogenesis involves increased proliferation of HSPCs 84. Elevated proliferation of HSPCs correlates with enhanced formation of cholesterol‐rich lipid rafts, up‐regulation of ERK1/2 phosphorylation, RAS activation and increased expression of the IL‐3/GM‐CSF receptor 79. Treatment of ABCA1/ABCG1‐deficient HSPCs with cyclodextrin (a cyclic oligosaccharide that solubilizes cholesterol and therefore abolishes cholesterol‐rich lipid rafts) significantly decreases proliferation of HSPCs 84. Increased deposition of cholesterol in ABCA1/ABCG1‐deficient HSPCs elevates cholesterol content in cell membrane, enhances generation of lipid rafts and increases surface expression of the IL‐3/GM‐CSF receptor that shares a common CD131 subunit. IL‐3/GM‐CSF stimulate Ras/ERK‐dependent signalling, which leads to enhanced proliferation of HSPCs 84 and elevated production of monocytes and neutrophils 85.
In LDLR‐deficient mice transgenic for ApoA1 and fed on high‐fat diet, transplantation of the ABCA1/ABCG1‐deficient bone marrow resulted in significantly decreased atherogenesis compared with non‐transgenic LDLR‐deficient mice HSPCs 84. These observations suggest on the anti‐atherogenic role of ApoA1 and HDL that in part are released through the inhibition of proliferation of HSPCs and subsequent monocytosis and neutrophilia.
ApoE is another anti‐atherogenic lipoprotein that is mainly involved in the clearance of cholesterol‐containing LDL particles and vasculoprotective effects in arteries 86. ApoE was shown to be able to regulate the proliferation of HSPCs 78. Bellosta with co‐authors observed increased propensity of hypercholesterolaemic ApoE‐deficient mice fed on high‐cholesterol diet to develop increased counts of HSPCs, neutrophils and monocytes, while LDLR‐deficient or ApoA1‐deficient mice exhibited only a mild predisposition to monocytosis and neutrophilia. In humans, individuals that have low HDL levels because of genetic mutations do not show increase in numbers of circulating HSPCs and monocytes 87.
The mechanism by which ApoE can down‐regulate HSPC proliferation may involve binding of HSPC‐secreted ApoE to the surface proteoglycans and stimulation of ABCA1/ABCG1, which in turn activates increased reverse cholesterol transport from HSPCs. ABCA1/ABCG1‐deficient HSPCs are especially prone to cholesterol deposition and are sensitive to IL‐3‐ and GM‐CSF‐dependent proliferation‐stimulating effects, which in turn accelerates atherogenesis 10, 18, 87. The treatment of ApoE‐deficient mice with phospholipid/ApoA‐I complexes (that are poor of phospholipids) decreased the overproduction of HSPCs and monocytes/neutrophils 78. Additionally, administration of LXR agonists also had atheroprotective effects via elevating ApoE levels in HSPCs and reducing proliferation of myeloid precursors and monocytes/neutrophils 78.
In summary, cholesterol regulates HSPC proliferation through the mechanism of the reverse cholesterol transport mediated by the activity of ABCA1/ABCG1 and its interaction with ApoE, an acceptor protein for cholesterol. The strong interaction between ApoE and both cholesterol efflux pumps results in the decrease of cholesterol content in the plasma membrane and therefore weaken of IL‐3/GM‐CSF‐dependent mitogenic signals. The ApoE‐dependent proliferative mechanism is independent on HDL or ApoA1 but can be stimulated by the presence of these free cholesterol acceptors or through the activation of LXRs 78.
The repertoire of hematopoietic progenitor cells is biased towards the preferential myeloid production in atherogenesis
Vascular inflammation that chronically persists in atherosclerosis can alter the lineage selection and differentiation of HSPCs towards a preferential generation of pro‐inflammatory cells. In hypercholesterolaemic mice and humans with familial hypercholesterolaemia, an obvious increase in monocyte production was shown along with increased expression of LDL‐handling molecules, such as LDLR, scavenger receptor B1 (SRB1) and low‐density lipoprotein receptor‐related protein 1 (LRP1) 88.
ApoE‐deficient murine macrophages were shown to develop a more pronounced response to TLR‐4 ligands compared with normal cells. In line with that, ABCA1/ABCG1‐deficient macrophages exhibit higher surface expression of TLR‐4 and more sensitive to TLR‐4‐dependent stimuli in comparison with the wild‐type macrophages 71, 89. As TLR‐4 is involved in the recognition of LPS, other bacterial and viral components, TLR‐mediated induction of the pro‐inflammatory response can mediate skewing the preferential differentiation of HSPCs to inflammatory cells observed in acute (such as an infection) and chronic (such as atherosclerosis) inflammatory environment. (Fig. 1). This pathway can exist because TLR‐4 expression was found in HSPCs 90. Furthermore, increased expression of TLR‐4 in HSPCs can cooperate with impaired cholesterol metabolism in HSPCs (in the initiation of the bias to increased generation of myeloid precursors in individuals affected by hypercholesterolaemia, atherosclerosis and other cardiometabolic disorders associated with altered lipid homoeostasis). In inflammation, some new substances, such as oxLDL 91, S100A8/A9 92 and HSP‐70 93 can activate TLR‐4 dependent responses in HSPCs and indeed induce myeloid skewing in HSPC differentiation. However, specifically to atherosclerosis, more studies are required to establish a range of TLR‐4 ligands capable to influence the HSPC differentiation in the atherosclerotic disease. In any case, TLR‐4 activation was shown to dampen the activity of ABCA1/ABCG1 transporters and therefore decrease cholesterol efflux from the cells. In addition, TLR‐4 stimulation increases expression of inflammatory cytokines and chemokines in macrophages thereby contributing to the pro‐inflammatory polarization of macrophages 94. However, it should be definitely shown whether the cooperation between TLR‐4 signalling and impaired lipid metabolism in HSPCs can contribute to the predominant differentiation of HSPCs to the myeloid lineage in atherosclerosis.
Mechanisms of monocyte release from the bone marrow
HSPC‐derived monocytes can retain in the bone marrow and intravasate into the bloodstream in a circadian rhythm‐dependent manner 95. In steady‐state conditions, oscillatory release of monocytes from the blood is controlled by oscillating expression of circadian genes Bmal1, Nrld1 and Dbp in monocytes. For example, the clock gene Bmal1 regulates egress of Ly6Chigh monocytes through controlling expression of chemokines Ccl2 and Ccl8 in monocytes 95. When Bmal1 reaches a nadir at the transition to the active phase, Bmal1‐mediated repression of Ccl2 disappears. This allows Ccl2 levels to rise to peak levels and hence promotes monocyte mobilization to the bloodstream and inflamed peripheral tissues.
In pathological conditions, monocyte emigration from the bone marrow can be promoted by infection or sterile inflammation such as myocardial infarction (MI) and atherosclerosis. Shi et al. 29 showed that challenge with LPS leads to the departure of inflammatory Ly6ChighCCR2+ monocytes from the blood marrow. The departure rate is highly correlated with expression of CCL2, a ligand for CCR2, by mesenchymal stem cells and reticular cells presented in the bone marrow. Accordingly, depletion of CCL2 in these cells abolishes leaving of Ly6ChighCCR2+ monocytes from the bone marrow 29.
In myocardial infarction‐induced heart injury, leucocyte‐expressed β2‐adrenergic receptor (ADRB2)‐dependent signalling was shown to be important for the recruitment of monocytes/macrophages and neutrophils to the damaged heart for induction of cardiac remodelling 96. In Adrb2‐deficient bone marrow, CCR2 expression and responsiveness to CCL2‐mediated migration were abolished. In contrast, ADRB2 activation leads to the up‐regulation of CCR2 expression and migratory responsiveness to CCL2 in the bone marrow. Mechanistically, β‐arrestin2‐biased ADRB2 signalling is required for the regulation of CCR2 expression. Additionally, activator protein 1 (AP‐1), a transcriptional factor, is needed in mediating CCR2 expression in response to ADRB2 stimulation in both murine bone marrow and human monocytes 97. Therefore, ADRB2 expression in immune cells is essential for the initiation of early inflammatory repair response to acute cardiac damage by assisting in infiltration of the infarct by leucocytes.
Local macrophage proliferation is a major source of lesional macrophages
During plaque growth, macrophages progressively accumulate in the diseased arterial wall. Previous concept considered monocyte infiltration as a major source that gives rise to intraplaque macrophages. However, Robbins et al. 98 showed that population of macrophages in advanced plaques of apoE‐deficient mice is predominantly replenished through the mechanism of local macrophage proliferation rather than through macrophage influx. So far, mechanisms that drive local macrophage proliferation are not well understood. GM‐CSF, a primary candidate to serve as a mitogenic signal, is not responsible for macrophage proliferation as this growth factor was shown to mediate proliferation of dendritic cells in the hypercholesterolaemic aortic intima 99. Scavenger receptor A1 (SRA1) abundantly presented on the surface of macrophages was suggested as a key player in macrophage proliferation 100. SRA1 transfers oxLDL into the cell and therefore can contribute to the transformation of macrophages to foam cells in the local lipid‐rich and pro‐inflammatory microenvironment 101. Recent studies showed that hypercholesterolaemia and improper reverse cholesterol transport can cause enhanced proliferation and mobilization of bone marrow‐derived HSPCs, excessive proliferation of splenic myeloid progenitors and monocytes, and increased monocyte influx to the plaque 78, 84, 102. All these cell types can be involved in monocytosis and support enhanced monocyte influx to the plaque. Indeed, intraplaque hypercholesterolaemic microenvironment may represent a key force that drives local amplification of macrophages. However, this possibility should be elucidated.
Contribution of monocytes to plaque progression
Plaque evolution is a complex multistep process. Plaque formation and progression include many consequent events and mechanisms including intimal lipoprotein retention, recruitment of inflammatory cells, foam cell generation and death, VSMC proliferation and migration, extracellular matrix (ECM) synthesis, neovascularization, calcification, arterial wall remodelling, fibrous cup rupture, thrombus formation and other events. Pathogenic mechanisms of atherogenic progression are comprehensively characterized in several recent reviews 103, 104, 105.
Monocytes play an important role in atherogenesis. In the plaque, inflammatory monocytes represent a significant portion of immune cells. Inflammatory monocytes release inflammatory messengers such as TNF‐α, IL‐1β and CCL2 and produce ROS therefore propagating vascular inflammation and promoting oxidative stress 106. In addition to the ability to produce cytokines, mouse inflammatory Ly6C+ monocytes can extravasate through the endothelial barrier to the subendothelial space. The transendothelial trafficking is mediated by interaction of complementary pair CCR2/CCL2 or/and CCR5/CCL5 in a VLA1/VCAM‐1 dependent manner 51. In the arterial intima, Ly6C+ monocytes differentiate to inflammatory and pro‐atherogenic M1 macrophages that actively promote plaque formation and progression through generation of foam cells and release of inflammatory cytokines (TNF‐α, IL‐6), ROS and matrix metalloproteinases (MMPs). By degrading ECM, macrophage‐derived MMPs contribute to the fibrous cup rupture, intraplaque neovascularization and pathological vascular remodelling 107. M1 macrophages express MHC‐I/II molecules and therefore can present antigens to CD4+T cells and initiate an adaptive immune response 108.
Like M1 macrophages, inflammatory monocytes can also present antigens to CD4+ T cells in a CD40‐dependent fashion. CD40/CD40L signalling pair plays a central role in inducing adaptive immunity in atherosclerosis 109. CD40 is expressed on the surface of dendritic cells, macrophages and monocytes, while CD40 ligand (CD40L) is produced by CD4+T cells and platelets. Both CD40 and its ligand are expressed by vascular smooth muscle cells (VSMCs) and endothelial cells 110. In atherosclerosis, oxLDL was suggested as a potential self‐antigen for the activation of CD4+ T cells 111. In human plaques, colocalization of CD40‐CD40L with oxLDL epitopes, SRA1 and CD16 was found thereby indicating a potential antigenic role of oxLDL 112. In apoE‐deficient mice, deletion of CD40 or CD40L leads to attenuation of atherosclerosis, increasing ECM and preferential polarization towards the anti‐inflammatory M2 phenotype 113.
Human CD14++CD16+ monocytes, which are orthologs of murine Ly6C+ monocytes, are involved in the phagocytosis of LDL particles and apoptotic cells. CD14+CD16+ cells (both non‐classical and intermediate) perform oxLDL uptake at activated endothelial cell surfaces as they are more resistant to oxLDL‐induced lipotoxicity 114. Kapinsky with co‐authors showed that the inflammatory CD14++CD16+ subset is more prone to foam cell formation when exposed to enzymatically degraded LDL 115. This may indicate a potential pro‐atherogenic role of this subset. Overall, in atherosclerosis, monocytes mainly serve as a source of macrophages and dendritic cells but are also involved in inflammation, foam cell formation, and activation of T cells.
Factors that regulate macrophage retention or emigration from the plaque
Lesional macrophage counts are dynamically determined by entry of circulating monocytes, and macrophage egress, local proliferation and death 116. Macrophage egress was described in early lesions. However, the emigration rate was shown to decline with plaque progression 117. Macrophage emigration stabilizes plaque phenotype and is associated with atherosclerosis regression.
The balance between retention and emigration signals is likely to be responsible for the content of intraplaque macrophages. These signals are widely unknown but become to be discovered. For example, cholesterol‐induced expression of semaphorin 3E and netrin 1 inhibits migration and stimulates retention of macrophages in the plaque 118, 119. Hypoxia also up‐regulates expression of these molecules 120, 121 and therefore contributes to macrophage retention. LDLR‐deficient mice whose bone marrow lacks netrin 1 develop less advanced atherosclerosis and exhibit enhanced macrophage egress from plaques thereby indicating that netrin 1 is involved in retention of macrophages in lesions 118. In the plaque, netrin 1 is expressed by VSMCs while macrophages express netrin receptor UNC5B. Interaction between these factors underlines molecular mechanism of macrophage retention 122.
Expression of semaphorin 3E and its receptor plexin D1 is up‐regulated in atherosclerotic lesions especially in advanced plaques. Semaphorin 3E is highly expressed by inflammatory M1 macrophages or in macrophages exposed to hypoxia or oxLDL. In a mouse model of atherosclerosis regression, expression of semaphorin 3E is greatly suppressed and associated with reduced content of macrophages in the plaque. Mechanistically, semaphorin 3E inhibits actin polymerization and macrophage motility thereby promoting immobilization of these cells in the plaque 120.
Other molecules, which suppress cell motility (e.g., adhesion molecules) or resolution of inflammation, may also be involved in the regulation of macrophage retention or emigration. Recently, it was reported that the adhesion receptor CD146 regulates the formation of macrophage foam cells and their retention within the lesion 123. Expression of CD146 in macrophages is up‐regulated by oxLDL. This receptor promotes internalization of the scavenger receptor CD36 involved in oxLDL uptake. This leads to increased lipid accumulation in macrophages and reduced migratory ability towards chemokines CCL19 and CCL21 123.
The signals, which stimulate macrophages to leave lesions, remain poorly defined. It seems that the macrophage fate to retain or exit a plaque is determined by a complex pattern of expression of factors that inhibit or promote motility. Further studies are required to define other such factors in animal models of regression.
Neutrophils and their role in atherosclerosis
Neutrophils are crucially involved in atherogenesis. These cells play a sharply pro‐inflammatory role and can seriously damage arterial wall due to the multiple cytotoxic and proteolytic molecules that they carry. In atherosclerosis, inflammatory and metabolic stimuli can significantly increase neutrophil production (i.e., cause neutrophilia) that may have deleterious sequelae for cardiovascular system.
Function and characteristics of neutrophils
Neutrophils belong to the family of polymorphonuclear leucocytes as they have a nucleus divided into several segments. Among leucocytes, these cells are the most frequent (up to 75% of a total population) 124. The lifespan of these cells is relatively short (8–12 hrs in the bloodstream) because neutrophils contain granules with highly toxic compounds released upon activation 125. This is a reason why they are so highly produced by the bone marrow accounting for 55–65% of the hematopoietic potential 126.
In physiological conditions, the primary task of neutrophils is to neutralize and kill various pathogens such as bacteria, fungi and protozoans. Neutrophils possess the capacity to phagocytize bacteria that then are killed by proteases and antimicrobial factors released from multiple cytoplasmic granules to the phagocytosome. In addition, stimulated neutrophils massively release ROS to the phagocytosome that damage phagocytized microorganisms 127. However, the defensive role of neutrophils is more complex because these cells were shown to interact with other leucocytes such natural killer cells, macrophages, dendritic cells and T‐ and B‐lymphocytes 128.
Neutrophils have specific advanced tools such as neutrophil extracellular traps (NETs) to kill pathogens. NETs represent a network of fibres consisted of decondensed chromatin associated with granular protein components 129. Neutrophils throw out NETs in response to pro‐inflammatory stimuli such as LPS, IL‐8. This process is accompanied with the plasma membrane destruction 130. In the NETs, chromatin creates a viscous matrix capable to capture pathogens that will be then eliminated by high concentrations of NET‐associated proteases and antimicrobial peptides [human neutrophil peptide 3 (HNP3), cathelicidin, α‐defensins] 131. NETs generation usually leads to the neutrophil death, but some cells remain mobility and ability to destroy microbes via phagocytosis and degranulation 132. As macrophages cannot neutralize NETs due to their high‐cytotoxic potential, such networks are destructed by exonucleases 130.
Another specific characteristic of neutrophils is a respiratory burst. Neutrophil granules contain three proteins [myeloperoxidase (MPO), NADPH oxidase and lactoferrin] involved in redox reactions and capable to produce ROS. NADPH oxidase is a significant contributor to superoxide anion radical (O2.−) production by activated neutrophils. This enzyme is a multicomponent assembly composed by membrane‐associated cytochrome b558 (includes subunits p22phox and gp91phox) and three cytoplasmic subunits (p47phox, p67phox and Rac1) 133. As NADPH oxidase activation is accompanied with increased ROS production that could damage host tissues, expression and activity of this enzyme is strictly regulated. For example, adenosine inhibits NADPH oxidase by decreasing density of cytochrome b558 components on the plasma membrane through redistribution between intracellular compartments and or/endocytosis 134. Activated neutrophils produce large amounts of the superoxide anion radical that is converted to H2O2, which becomes involved to further reactions to generate ROS 135. Massive ROS production helps to degrade bacteria and other pathogens.
Due to high reactivity and a rich arsenal of cytotoxic agents, neutrophil production and function should be strictly controlled. Neutrophils experience continuous death through apoptosis. Dead or apoptotic neutrophils are then cleared by macrophages 136. Loss of a proper control of neutrophil production or inefficient clearance may lead to neutrophilia and neutrophil overactivity associated with the development of many inflammatory, autoimmune and allergic diseases. The rate of neutrophil infiltration correlated with the severity of disease 137.
Components of neutrophil granules
In the cytoplasm, neutrophils contain multiple granules. There are three types of granules: primary, secondary and tertiary. Primary granules contain MPO, serine proteases (cathepsin G, neutrophil elastase, proteinase 3, neutrophil serine protease 4 and azurocidin 1) and antimicrobial peptides (bactericidal/permeability‐increasing protein, defensins). MPO, cathepsin G and neutrophil elastase are the major protein components of primary granules. MPO produces hypochlorous acid (HOCl) from hydrogen peroxide (H2O2) and chloride anion (Cl−) during neutrophil respiratory burst. This enzyme also oxidizes tyrosine to tyrosyl radical with H2O2 138. HOCl and tyrosyl radicals are cytotoxic agents that kill pathogenic bacteria 139. Cathepsin G is an essential component of NETs that kills microbes 140. By degrading structural ECM proteins, this protease contributes to tissue remodelling in inflamed sites 141 Capodici and Berg 1989. Neutrophil elastase contains two isoforms (2A and 2B) and degrades virulence factors of various Gram‐negative bacteria 142. This serine protease also participates in tissue remodelling by cleaving collagen‐IV and elastin 143.
Secondary granules contain several enzymes [collagenase (also known as MMP‐1), lysozyme, alkaline phosphatase, NADH oxidase), cathelicidin (antimicrobial peptide; also known as Cramp (in mice) or LL37 (in humans)] and lactoferrin, a protein with a wide spectrum of antimicrobial activity. Tertiary granules harbour gelatinases A and B (or MMP‐2 and ‐9, respectively), cathepsin (serine protease) and lipocalin 2, an antimicrobial peptide 7. Thus, neutrophil granules contain a variety of molecules with proteolytic, antimicrobial, cytotoxic and oxidative activity that are released upon degranulation and carry a potent destructive potential.
Neutrophilia and hypercholesterolaemia
In neutrophilia, higher numbers of neutrophils circulate in the blood. Neutrophilia is usually induced as a response to bacterial infection. However, increase in neutrophils in the blood can also result from other factors. As was shown in atherosclerotic models of mouse, rabbit and swine, high cholesterol can cause rise of neutrophils in the circulation 18, 19, 144, 145. In humans, a positive correlation between neutrophil counts, rupture‐prone areas of the plaque and density of intraplaque microvessels was found in patients with carotid atherosclerosis 146. These data indicate that neutrophilia can increase lesional instability and increase risk of plaque rupture. Mechanistically, neutrophil proteases can degrade the plaque fibrous cup and contribute to destabilization of the lesion 147.
Neutrophilia was shown to serve as independent predictor for adverse cardiovascular events in subjects with asymptomatic carotid stenosis 148. Guasti with co‐authors performed systemic analysis of studies in which a prognostic value of neutrophil counts for unfavourable cardiovascular events was evaluated on a total population of 34,000 individuals with acute coronary syndromes and/or after cardiac revascularization 16. A significance of neutrophilia (if evaluated concomitantly with total leucocyte numbers and levels of C‐reactive protein) as an independent predictor for cardiovascular outcomes was shown 16.
Overall, in hypercholesterolaemic animals, neutrophilia and monocytosis can be induced concomitantly through the common mechanism involving increased surface expression of the IL‐3/GM‐CSF receptor due to cholesterol accumulation in cell membrane of HSCs. This in turn stimulates their proliferative potential 84. By studying children with familial hypercholesterolaemia, Tolani et al. 88 demonstrated that similar mechanism of induction of cholesterol‐induced HCS proliferation also exists in humans.
Mediators of neutrophil recruitment to lesions
In the bone marrow, commitment of the neutrophil‐specific lineage is controlled by G‐CSF and IL‐23. In myeloid progenitors, down‐regulation of the reverse cholesterol transport stimulates production of neutrophils. However, neutrophils hold in the bone marrow because of the interaction of CXCL12 with its receptor CXCR4 149. Interruption of the CXCL12/CXCR4 axis or elevated plasma CXCL1/CXCL2 concentrations induces mobilization of neutrophils from the bone marrow that leads to neutrophilia (Fig. 2). CXCL1/CXCL2 bind to CXCR1 located on the surface of neutrophils and stimulates entry of these cells to the bloodstream 150. Chemotactic stimuli such as macrophage‐derived CCL3 and platelet‐derived CCL5 stimulate neutrophils via CCR1, CCR3 and CCR5 and promote firm attachment of neutrophils to the inflamed endothelium 151. Sensing of neutrophil‐derived CCR5 induces sialylation of this receptor mediated by the α2,3‐sialyltransferase IV (St3Gal4). This enzyme is also essential for CCL5‐mediated neutrophil recruitment and extravasation. St3Gal4‐deficient murine neutrophils exhibited decreased binding of Ccl5 and aberrant Ccl5‐mediated integrin activation. Accordingly, Ccl5‐dependent immobilization of neutrophils on the endothelial surface was almost abolished. Ccl5‐triggered neutrophil transmigration across the endothelial barrier was drastically reduced. As a result, St3Gal4/apoE‐deficient mice develop smaller and less inflammatory lesions 151.
Figure 2.
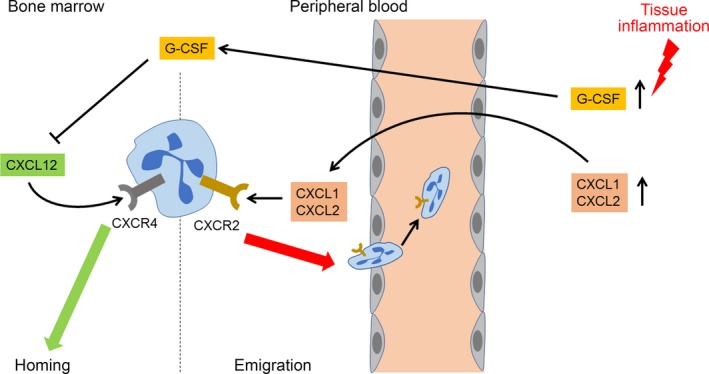
Mobilization of neutrophils from the bone marrow in inflammatory conditions. Chemokine receptor CXCR4 is responsible for retention of neutrophils in the bone marrow while CXCR2 triggers neutrophil emigration from the bone to the circulation. The CXCR4/CXCL12 axis promotes neutrophil homing in the bone marrow. In tissue inflammation, levels of G‐CSF, CXCL1 and CXCL2 are up‐regulated. G‐CSF mobilizes neutrophils from the bone marrow by increasing the ratio of CXCR4 to CXCR2 ligands in the bone marrow. Both CXCL1 and CXCL2 are CXCR2 ligands that stimulate neutrophil entrance to the bloodstream. Increased G‐CSF and CXCL1 are produced by the bone marrow and local inflamed tissue.
In line with this, transplantation of the bone marrow deficient for CCL3 (a ligand for CCR1, CCR4 and CCR5) to LDLR‐deficient mice resulted in diminished aortic atherosclerosis progression likely due to prevention of intraplaque accumulation CCL3‐deficient neutrophils. Presumably, CCL3 can provide a chemotactic gradient essential for supporting neutrophil movement to the plaque 152.
CXCL1 also essential for neutrophil recruitment. In a mouse model of shear stress‐induced atherogenesis, implementation of Evasin‐3, a natural protein inhibitor of both chemokines, resulted in specific reduction of inflammation driven by neutrophils, less production of MMP‐9 and increased collagen deposits in the plaque 153. Concomitant administration of Evasin‐3 (inhibits CXC chemokines) and Evasin‐4 (inhibits CC chemokines) had beneficial effects on post‐infarction heart failure in mice by decreasing cardiac inflammation, oxidative stress and neutrophil infiltration as a result of blocking CXCL1 and CCL2 154. Similarly, in apoE‐deficient mice, treatment with a nicotinamide phosphoribosyltransferase inhibitor (FK866) diminished neutrophil and MMP‐9 content in the plaque and increased collagen content by decrease of systemic CXCL1 155.
In addition to chemokines, other proteins can regulate neutrophil recruitment. Fatty acid amide hydrolase (FAAH)/apoE‐deficient mice had smaller lesions enriched by MMP‐9 and neutrophils. Interestingly, in apoE‐deficient mice, FAAH inhibition led to increased recruitment of neutrophils but not monocytes 156. Hoyer with co‐authors observed no change in plaque size but confirmed elevated production of MMP‐9 and higher neutrophil counts in the lesions of FAAH/apoE‐deficient mice 157. FAAH catalyses degradation of anandamide, an endocannabinoid, which presumably can control mobility of leucocytes through the cannabinoid receptor type 2 (CB2R) 158. Stimulation of CB2R with a specific agonist JWH‐133 caused significant decrease neutrophil counts and MMP‐9 production in mouse aortic root plaques; a less profound reduction was observed in carotid lesions 159. In a murine model of ischaemia/reperfusion, CBR2 possessed cardioprotective functions by limiting oxidative stress and neutrophil infiltration in the infarcted myocardium 160. It was also shown that CBR2 exerts a protective function against stroke by blocking neutrophil movement to ischaemic brain regions 161. The inhibitory mechanism is consisted in CBR2‐mediated suppression of CXCL2‐dependent activation of MAP kinase p38, that is required for neutrophil migration towards CXCL2 161. Another cannabinoid receptor GPR55, which is expressed in human neutrophils, was shown to cooperate with CBR2 by suppressing neutrophil migration towards 2‐arachidonoylglycerol, a CBR2 agonist, while down‐regulating neutrophil degranulation and ROS generation 162. Thus, anti‐inflammatory effects of endocannabinoids at least in part could be explained by inhibition of neutrophil recruitment to inflamed tissues but also by blockage of neutrophil effector function 163.
Krüppel‐like factor 2 (KLF2), a transcription factor, is another regulator of neutrophil recruitment. LDLR‐deficient mice lacking KLF2 displayed increased attachment of neutrophils to endothelial cells, accumulation of neutrophils and monocytes and up‐regulation of MPO activity in aortic roots 164. KLF2‐dependent suppression of recruitment and extravasation of inflammatory cells including neutrophils appears to be attributed to its protective effects on endothelial cells by maintaining endothelial barrier integrity 165 and inhibition of expression of adhesion molecules such as E‐selectin and VCAM‐1 166. As known, KLF2 expression is regulated by mechanical forces. Steady laminar flow induces KLF2 expression while perturbed flow down‐regulates expression of this factor 167. Perturbed flow presented in atheroprone arterial regions causes pro‐inflammatory activation of the local endothelium associated with production of inflammatory messengers and adhesion molecules. This attracts inflammatory cells such monocytes and neutrophils to inflamed sites 168. Recently, Franck with co‐authors reported that disturbed bloodflow can stimulate neutrophil recruitment and accumulation, arterial denudation due to the endothelial death 169. Endothelial apoptosis and detachment are mediated by TLR2. Finally, this can induce superficial erosion and thrombosis especially in VSMC‐rich plaques 170.
Pro‐inflammatory cytokines such as TNF‐α promote neutrophil recruitment 171. IL‐17A also exhibit stimulatory effects on neutrophils by generating a pro‐inflammatory neutrophil subset 172 and promoting neutrophil recruitment 173.
Neutrophil granule components in atherosclerosis
In degranulation, activated neutrophils release large amounts of diverse cytotoxic and destructive factors capable to induce dramatic damage of vascular tissue. For example, neutrophil MPO contributes to ROS formation that in turn can be involved in oxidative modification of LDL and activation of endothelial cells thereby promoting a pro‐atherogenic environment 174. MPO also mediates irreversible protein modification via nitrosylation and the formation of 3‐chlorotyrosine and dityrosine crosslinks in the protein molecule 175, 176, 177. Jerke with co‐authors observed direct MPO transfer from neutrophils to endothelial cells mediated by β2‐integrin upon cell–cell contact followed by MPO‐mediated endothelial dysfunction 178. Granule‐derived proteases such as MMP‐1, MMP‐2, MMP‐9, neutrophil elastase and cathepsin participate in ECM degradation and therefore can contribute to vascular remodelling, fibrous cap thinning and atherothrombosis 179, 180, 181.
It should be stressed that neutrophils, monocytes and macrophage cooperate in induction and propagation of atherosclerotic inflammation. Neutrophils attract and guide inflammatory monocytes to plaques. Depletion of neutrophils results in decreased monocyte/macrophage counts within aortic lysates from mice 18. Neutrophils release a number of proteins such as azurocidin, cathepsin G, α‐defensins and LL37 (Cramp in mouse) that exhibit direct chemotactic activity on monocytes 182. Cramp/apoE‐deficient mice develop less advanced plaques with reduced monocyte/macrophage content. Cramp is deposited by neutrophils on the surface of inflamed endothelium. In this place, cathelicidins mediate adhesion of neutrophils and classical monocytes 183. The mechanism by which Cramp assists in the monocyte recruitment involves release of this peptide by extravasated neutrophils and reverse trafficking across the endothelium to the lumen where Cramp stimulates formyl‐peptide receptor 2–mediated β 1/2 integrin activation and triggers monocyte recruitment 184. In humans, LL37 promotes recruitment of neutrophils, monocytes and T cells in a similar fashion employing formyl‐peptide receptor‐like 1 (FPRL1) 185.
Antimicrobial peptides α‐defensins exhibit pro‐atherogenic activity by enhancing monocyte recruitment, platelet activation and generation of foam cells 186. Azurocidin 1 (also known as cationic antimicrobial protein CAP37) stimulates monocyte recruitment through stimulation of surface expression of adhesion molecules (E‐selectin, ICAM1 and VCAM‐1) in endothelial cells 187, 188.
Lipocalin‐2 (also known as neutrophil gelatinase‐associated lipocalin; NGAL) is a small antimicrobial protein. In cardiovascular pathology, serum NGAL concentrations are elevated (particularly in subjects with carotid atherosclerosis and ischaemic cerebrovascular disease 189, 190, 191, 192 and serve as a marker of neutrophil activation and inflammation in preclinical atherosclerosis 193, 194. Furthermore, NGAL was shown to have a value as a prognostic marker for adverse cardiovascular events in patients with ST‐segment elevation myocardial infarction 195, acute myocardial infarction 196 and at‐risk subjects randomly selected from a general population 197, 198.
S100 calcium‐binding protein A9 (S100A9) may represent another potential mediator of neutrophil‐driven atherogenesis. This protein is constitutively expressed by neutrophils and other myeloid cells. S100A9 forms heterodimeric complex with S100A8 199. In the aorta, S100A9/apoE‐deficient mice had plaques of less size thereby suggesting for the pro‐atherogenic action of this protein 200. In patients with type 1 diabetes, serum levels of S100A9 were increased and correlated with higher neutrophil levels and coronary artery disease 82. The S100A9/8 complex promotes myelopoiesis and neutrophil migration as blockage of S100A9/8 results in reduced motility of neutrophils 201, 202. In infection, S100A9 is responsible for mobilization and recruitment of myeloid cells. However, in pathological conditions such diabetes or atherosclerosis, function of this protein may be compromised 203.
NETs in atherosclerosis
NETs were detected in the luminal regions of human and murine lesions 204. Recently, Borissoff with co‐authors showed correlation between increased circulating NET levels and atherothrombosis 205. The presence of self‐DNA complexes with neutrophil granular proteins (that compose NETs) in the circulation activates plasmacytoid DCs (pDCs) and induces pro‐atherogenic pDC‐driven type I interferon (IFN) response 206. In ApoE‐deficient mice, pDCs were shown to aggravate atherosclerosis‐associated inflammation 207. Furthermore, Villanueva et al. 208 observed cytotoxic effects of NETs against ECs in inflammation. In ApoE‐deficient mice, Warnatsch et al. 209 showed that cholesterol crystals induce generation of NETs, which in turn activate Th17 cells and macrophage‐driven IL‐1β release. Platelet‐derived chemokines stimulate NET production 210. NETs were shown to stimulate local pro‐coagulant properties by thrombin generation via platelet‐dependent mechanisms 211. On the other hand, NETs could reciprocally enhance platelet activation and thereby promote atherothrombosis as demonstrated in a murine model of deep vein thrombosis 212.
Conclusion
Impaired cholesterol efflux is crucially involved in ectopic fat accumulation in the arterial wall and the development of atherosclerotic disease. High‐blood cholesterol significantly promotes atherogenesis in obese and dislipidaemic people through several mechanisms, including induction of HSPC proliferation and mobilization from the bone marrow, and activation of preferential differentiation of HSPCs to myeloid precursors, which leads to the increase of pro‐inflammatory immune cells population 213. Although the overall beneficial effect of lipid‐lowering therapy towards the anti‐inflammation, cardioprotection and vasculoprotection is well established, it is difficult to estimate or predict the total outcome due to the multiplicity of molecular mechanisms and physiological functions targeted by statins and other lipid‐lowering agents.
Therapeutic approaches to exploit the anti‐atherogenic potential of HDL, for example with help of ApoA1 mimetic peptides or attempts to increase HDL levels in the circulation had promising effects in animals 214. However, in humans, clinical studies with the use of therapeutic agents designed to raise HDL failed to reach significant reduction of the cardiovascular risk 215, 216, 217.
Topics considered in this review may provide a more specific focus on the regulation of HSPC proliferation and differentiation through targeting reverse cholesterol transport pathways to prevent monocytosis/neutrophilia and reduce atherosclerotic inflammation. In experimental mouse models of atherosclerosis, activation of LXR‐dependent pathways in HSPCs was shown to inhibit their proliferation and pathological myelopoiesis, suppress foam cell formation, an attenuate inflammation and plaque progression. Due to significant curative actions shown by LXR agonists in experimental models of atherogenesis, these agents have a good therapeutic potential to be used for targeting cholesterol efflux mechanisms.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
Funding source: This work was supported by Russian Science Foundation (Grant # 14‐15‐00112).
References
- 1. Spence JD. Recent advances in pathogenesis, assessment, and treatment of atherosclerosis. F1000Res. 2016; 5 pii: F1000 Faculty Rev‐1880. https://doi.org/10.12688/f1000research.8459.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soehnlein O, Swirski FK. Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol Metab. 2013; 24: 129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nissen SE, Tuzcu EM, Schoenhagen P, et al Reversal of atherosclerosis with aggressive lipid lowering (REVERSAL) investigators. Statin therapy, LDL cholesterol, C‐reactive protein, and coronary artery disease. N Engl J Med. 2005; 352: 29–38. [DOI] [PubMed] [Google Scholar]
- 4. Alexander C, Day CE. Distribution of serum lipoproteins of selected vertebrates. Comp Biochem Physiol B. 1973; 46: 295–312. [DOI] [PubMed] [Google Scholar]
- 5. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003; 19: 71–82. [DOI] [PubMed] [Google Scholar]
- 6. Chistiakov DA, Sobenin IA, Orekhov AN, et al Dendritic cells in atherosclerotic inflammation: the complexity of functions and the peculiarities of pathophysiological effects. Front Physiol. 2014; 5: 196 https://doi.org/10.3389/fphys.2014.00196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chistiakov DA, Bobryshev YV, Orekhov AN. Neutrophil's weapons in atherosclerosis. Exp Mol Pathol. 2015; 99: 663–71. [DOI] [PubMed] [Google Scholar]
- 8. Chistiakov DA, Orekhov AN, Bobryshev YV. Immune‐inflammatory responses in atherosclerosis: role of an adaptive immunity mainly driven by T and B cells. Immunobiology. 2016; 221: 1014–33. [DOI] [PubMed] [Google Scholar]
- 9. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. 2014; 5: 514 https://doi.org/10.3389/fimmu.2014.00514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Swirski FK, Libby P, Aikawa E, et al Ly‐6Chi monocytes dominate hypercholesterolemia‐associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007; 117: 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soehnlein O, Drechsler M, Hristov M, et al Functional alterations of myeloid cell subsets in hyperlipidaemia: relevance for atherosclerosis. J Cell Mol Med. 2009; 13: 4293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011; 473: 317–25. [DOI] [PubMed] [Google Scholar]
- 13. Friedman GD, Klatsky AL, Siegelaub AB. The leukocyte count as a predictor of myocardial infarction. N Engl J Med. 1974; 290: 1275–8. [DOI] [PubMed] [Google Scholar]
- 14. Olivares R, Ducimetière P, Claude JR. Monocyte count: a risk factor for coronary heart disease? Am J Epidemiol. 1993; 137: 49–53. [DOI] [PubMed] [Google Scholar]
- 15. Patiño R, Ibarra J, Rodriguez A, et al Circulating monocytes in patients with diabetes mellitus, arterial disease, and increased CD14 expression. Am J Cardiol. 2000; 85: 1288–91. [DOI] [PubMed] [Google Scholar]
- 16. Guasti L, Dentali F, Castiglioni L, et al Neutrophils and clinical outcomes in patients with acute coronary syndromes and/or cardiac revascularisation. A systematic review on more than 34,000 subjects. Thromb Haemost. 2011; 106: 591–9. [DOI] [PubMed] [Google Scholar]
- 17. Combadière C, Potteaux S, Rodero M, et al Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008; 117: 1649–57. [DOI] [PubMed] [Google Scholar]
- 18. Drechsler M, Megens RT, van Zandvoort M, et al Hyperlipidemia‐triggered neutrophilia promotes early atherosclerosis. Circulation. 2010; 122: 1837–45. [DOI] [PubMed] [Google Scholar]
- 19. Döring Y, Soehnlein O, Drechsler M, et al Hematopoietic interferon regulatory factor 8‐deficiency accelerates atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2012; 32: 1613–23. [DOI] [PubMed] [Google Scholar]
- 20. Rothe G, Gabriel H, Kovacs E, et al Peripheral blood mononuclear phagocyte subpopulations as cellular markers in hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1996; 16: 1437–47. [DOI] [PubMed] [Google Scholar]
- 21. Tani S, Matsumoto M, Anazawa T, et al Development of a model for prediction of coronary atherosclerotic regression: evaluation of high‐density lipoprotein cholesterol level and peripheral blood monocyte count. Heart Vessels. 2012; 27: 143–50. [DOI] [PubMed] [Google Scholar]
- 22. Eash KJ, Greenbaum AM, Gopalan PK, et al CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010; 120: 2423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim CS, Park HS, Kawada T, et al Circulating levels of MCP‐1 and IL‐8 are elevated in human obese subjects and associated with obesity‐related parameters. Int J Obes. 2006; 30: 1347–55. [DOI] [PubMed] [Google Scholar]
- 24. Boisvert WA, Santiago R, Curtiss LK, et al A leukocyte homologue of the IL‐8 receptor CXCR‐2 mediates the accumulation of macrophages in atherosclerotic lesions of LDL receptor‐deficient mice. J Clin Invest. 1998; 101: 353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heidt T, Sager HB, Courties G, et al Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014; 20: 754–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zernecke A, Bot I, Djalali‐Talab Y, et al Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res. 2008; 102: 209–17. [DOI] [PubMed] [Google Scholar]
- 27. Tacke F, Alvarez D, Kaplan TJ, et al Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007; 117: 185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Engel DR, Maurer J, Tittel AP, et al CCR2 mediates homeostatic and inflammatory release of Gr1(high) monocytes from the bone marrow, but is dispensable for bladder infiltration in bacterial urinary tract infection. J Immunol. 2008; 181: 5579–86. [DOI] [PubMed] [Google Scholar]
- 29. Shi C, Jia T, Mendez‐Ferrer S, et al Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll‐like receptor ligands. Immunity. 2011; 34: 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saederup N, Chan L, Lira SA, et al Fractalkine deficiency markedly reduces macrophage accumulation and atherosclerotic lesion formation in CCR2‐/‐ mice: evidence for independent chemokine functions in atherogenesis. Circulation. 2008; 117: 1642–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quintar A, McArdle S, Wolf D, et al Endothelial protective monocyte patrolling in large arteries intensified by Western diet and atherosclerosis. Circ Res. 2017; 120: 1789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Idzkowska E, Eljaszewicz A, Miklasz P, et al The role of different monocyte subsets in the pathogenesis of atherosclerosis and acute coronary syndromes. Scand J Immunol. 2015; 82: 163–73. [DOI] [PubMed] [Google Scholar]
- 33. Ziegler‐Heitbrock HW, Ströbel M, Fingerle G, et al Small (CD14 + /CD16 + ) monocytes and regular monocytes in human blood. Pathobiology. 1991; 59: 127–30. [DOI] [PubMed] [Google Scholar]
- 34. Fingerle G, Pforte A, Passlick B, et al The novel subset of CD14 + /CD16 + blood monocytes is expanded in sepsis patients. Blood. 1993; 82: 3170–6. [PubMed] [Google Scholar]
- 35. Ziegler‐Heitbrock HW, Ströbel M, Kieper D, et al Differential expression of cytokines in human blood monocyte subpopulations. Blood. 1992; 79: 503–11. [PubMed] [Google Scholar]
- 36. Ziegler‐Heitbrock HW, Fingerle G, Ströbel M, et al The novel subset of CD14 + /CD16 + blood monocytes exhibits features of tissue macrophages. Eur J Immunol. 1993; 23: 2053–8. [DOI] [PubMed] [Google Scholar]
- 37. Ziegler‐Heitbrock L, Ancuta P, Crowe S, et al Nomenclature of monocytes and dendritic cells in blood. Blood. 2010; 116: e74–80. [DOI] [PubMed] [Google Scholar]
- 38. Schlitt A, Heine GH, Blankenberg S, et al CD14 + CD16 + monocytes in coronary artery disease and their relationship to serum TNF‐alpha levels. Thromb Haemost. 2004; 92: 419–24. [DOI] [PubMed] [Google Scholar]
- 39. Imanishi T, Ikejima H, Tsujioka H, et al Association of monocyte subset counts with coronary fibrous cap thickness in patients with unstable angina pectoris. Atherosclerosis. 2010; 212: 628–35. [DOI] [PubMed] [Google Scholar]
- 40. Kashiwagi M, Imanishi T, Tsujioka H, et al Association of monocyte subsets with vulnerability characteristics of coronary plaques as assessed by 64‐slice multidetector computed tomography in patients with stable angina pectoris. Atherosclerosis. 2010; 212: 171–6. [DOI] [PubMed] [Google Scholar]
- 41. Timmerman KL, Flynn MG, Coen PM, et al Exercise training‐induced lowering of inflammatory (CD14 + CD16 + ) monocytes: a role in the anti‐inflammatory influence of exercise? J Leukoc Biol. 2008; 84: 1271–8. [DOI] [PubMed] [Google Scholar]
- 42. Rogacev KS, Ulrich C, Blömer L, et al Monocyte heterogeneity in obesity and subclinical atherosclerosis. Eur Heart J. 2010; 31: 369–76. [DOI] [PubMed] [Google Scholar]
- 43. Poitou C, Dalmas E, Renovato M, et al CD14dimCD16 + and CD14 + CD16 + monocytes in obesity and during weight loss: relationships with fat mass and subclinical atherosclerosis. Arterioscler Thromb Vasc Biol. 2011; 31: 2322–30. [DOI] [PubMed] [Google Scholar]
- 44. Rogacev KS, Seiler S, Zawada AM, et al CD14 + +CD16 + monocytes and cardiovascular outcome in patients with chronic kidney disease. Eur Heart J. 2011; 32: 84–92. [DOI] [PubMed] [Google Scholar]
- 45. Heine GH, Ortiz A, Massy ZA, et al Monocyte subpopulations and cardiovascular risk in chronic kidney disease. Nat Rev Nephrol. 2012; 8: 362–9. [DOI] [PubMed] [Google Scholar]
- 46. Berg KE, Ljungcrantz I, Andersson L, et al Elevated CD14 + +CD16‐ monocytes predict cardiovascular events. Circ Cardiovasc Genet. 2012; 5: 122–31. [DOI] [PubMed] [Google Scholar]
- 47. Rogacev KS, Cremers B, Zawada AM, et al CD14 + +CD16 + monocytes independently predict cardiovascular events: a cohort study of 951 patients referred for elective coronary angiography. J Am Coll Cardiol. 2012; 60: 1512–20. [DOI] [PubMed] [Google Scholar]
- 48. Ramírez R, Carracedo J, Merino A, et al CD14 + CD16 + monocytes from chronic kidney disease patients exhibit increased adhesion ability to endothelial cells. Contrib Nephrol. 2011; 171: 57–61. [DOI] [PubMed] [Google Scholar]
- 49. Merino A, Buendia P, Martin‐Malo A, et al Senescent CD14 + CD16 + monocytes exhibit proinflammatory and proatherosclerotic activity. J Immunol. 2011; 186: 1809–15. [DOI] [PubMed] [Google Scholar]
- 50. Hamers AA, Hanna RN, Nowyhed H, et al NR4A nuclear receptors in immunity and atherosclerosis. Curr Opin Lipidol. 2013; 24: 381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang J, Zhang L, Yu C, et al Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res. 2014; 2: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hu YW, Zhang P, Yang JY, et al Nur77 decreases atherosclerosis progression in apoE(‐/‐) mice fed a high‐fat/high‐cholesterol diet. PLoS One. 2014; 9: e87313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hanna RN, Shaked I, Hubbeling HG, et al NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res. 2012; 110: 416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid‐activated nuclear receptors. Nature. 2008; 454: 470–7. [DOI] [PubMed] [Google Scholar]
- 55. Janowski BA, Willy PJ, Devi TR, et al An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996; 383: 728–31. [DOI] [PubMed] [Google Scholar]
- 56. Zhao C, Dahlman‐Wright K. Liver X receptor in cholesterol metabolism. J Endocrinol. 2010; 204: 233–40. [DOI] [PubMed] [Google Scholar]
- 57. Joseph SB1, Castrillo A, Laffitte BA, et al Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003; 9: 213–9. [DOI] [PubMed] [Google Scholar]
- 58. Castrillo A, Joseph SB, Marathe C, et al Liver X receptor‐dependent repression of matrix metalloproteinase‐9 expression in macrophages. J Biol Chem. 2003; 278: 10443–9. [DOI] [PubMed] [Google Scholar]
- 59. Ogawa D, Stone JF, Takata Y, et al Liver x receptor agonists inhibit cytokine‐induced osteopontin expression in macrophages through interference with activator protein‐1 signaling pathways. Circ Res. 2005; 96: e59–67. [DOI] [PubMed] [Google Scholar]
- 60. Töröcsik D, Szanto A, Nagy L. Oxysterol signaling links cholesterol metabolism and inflammation via the liver X receptor in macrophages. Mol Aspects Med. 2009; 30: 134–52. [DOI] [PubMed] [Google Scholar]
- 61. Ghisletti S, Huang W, Jepsen K, et al Cooperative NCoR/SMRT interactions establish a corepressor‐based strategy for integration of inflammatory and anti‐inflammatory signaling pathways. Genes Dev. 2009; 23: 681–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marathe C, Bradley MN, Hong C, et al The arginase II gene is an anti‐inflammatory target of liver X receptor in macrophages. J Biol Chem. 2006; 281: 32197–206. [DOI] [PubMed] [Google Scholar]
- 63. Tangirala RK, Bischoff ED, Joseph SB, et al Identification of macrophage liver X receptors as inhibitors of atherosclerosis. Proc Natl Acad Sci USA. 2002; 99: 11896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Terasaka N, Hiroshima A, Koieyama T, et al T‐0901317, a synthetic liver X receptor ligand, inhibits development of atherosclerosis in LDL receptor‐deficient mice. FEBS Lett. 2003; 536: 6–11. [DOI] [PubMed] [Google Scholar]
- 65. Levin N, Bischoff ED, Daige CL, et al Macrophage liver X receptor is required for antiatherogenic activity of LXR agonists. Arterioscler Thromb Vasc Biol. 2005; 25: 135–42. [DOI] [PubMed] [Google Scholar]
- 66. DiBlasio‐Smith EA, Arai M, Quinet EM, et al Discovery and implementation of transcriptional biomarkers of synthetic LXR agonists in peripheral blood cells. J Transl Med. 2008; 6: 59 https://doi.org/10.1186/1479-5876-6-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee SD, Tontonoz P. Liver X receptors at the intersection of lipid metabolism and atherogenesis. Atherosclerosis. 2015; 242: 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hong C, Kidani Y, A‐Gonzalez N, et al Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J Clin Invest. 2012; 122: 337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zahuczky G1, Kristóf E, Majai G, et al Differentiation and glucocorticoid regulated apopto‐phagocytic gene expression patterns in human macrophages. Role of Mertk in enhanced phagocytosis. PLoS One. 2011; 6: e21349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Korf H, Vander Beken S, Romano M, et al Liver X receptors contribute to the protective immune response against Mycobacterium tuberculosis in mice. J Clin Invest. 2009; 119: 1626–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kappus MS, Murphy AJ, Abramowicz S, et al Activation of liver X receptor decreases atherosclerosis in Ldlr⁻/⁻ mice in the absence of ATP‐binding cassette transporters A1 and G1 in myeloid cells. Arterioscler Thromb Vasc Biol. 2014; 34: 279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mercier FE, Ragu C, Scadden DT. The bone marrow at the crossroads of blood and immunity. Nat Rev Immunol. 2011; 12: 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lambert C, Wu Y, Aanei C. Bone marrow immunity and myelodysplasia. Front Oncol. 2016; 6: 172 https://doi.org/10.3389/fonc.2016.00172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Robbins CS, Chudnovskiy A, Rauch PJ, et al Extramedullary hematopoiesis generates Ly‐6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012; 125: 364–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tall AR, Yvan‐Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015; 15: 104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dutta P, Courties G, Wei Y, et al Myocardial infarction accelerates atherosclerosis. Nature. 2012; 487: 325–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Seijkens T, Hoeksema MA, Beckers L, et al Hypercholesterolemia‐induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis. FASEB J. 2014; 28: 2202–13. [DOI] [PubMed] [Google Scholar]
- 78. Murphy AJ, Akhtari M, Tolani S, et al ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011; 121: 4138–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang M, Subramanian M, Abramowicz S, et al Interleukin‐3/granulocyte macrophage colony‐stimulating factor receptor promotes stem cell expansion, monocytosis, and atheroma macrophage burden in mice with hematopoietic ApoE deficiency. Arterioscler Thromb Vasc Biol. 2014; 34: 976–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. van Kampen E, Jaminon A, van Berkel TJ, et al Diet‐induced (epigenetic) changes in bone marrow augment atherosclerosis. J Leukoc Biol. 2014; 96: 833–41. [DOI] [PubMed] [Google Scholar]
- 81. Álvarez‐Errico D, Vento‐Tormo R, Sieweke M, et al Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol. 2015; 15: 7–17. [DOI] [PubMed] [Google Scholar]
- 82. Nagareddy PR, Murphy AJ, Stirzaker RA, et al Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013; 17: 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Out R, Hoekstra M, Habets K, et al Combined deletion of macrophage ABCA1 and ABCG1 leads to massive lipid accumulation in tissue macrophages and distinct atherosclerosis at relatively low plasma cholesterol levels. Arterioscler Thromb Vasc Biol. 2008; 28: 258–64. [DOI] [PubMed] [Google Scholar]
- 84. Yvan‐Charvet L, Pagler T, Gautier EL, et al ATP‐binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010; 328: 1689–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Westerterp M, Murphy AJ, Wang M, et al Deficiency of ATP‐binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res. 2013; 112: 1456–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Getz GS, Reardon CA. Use of mouse models in atherosclerosis research. Methods Mol Biol. 2015; 1339: 1–16. [DOI] [PubMed] [Google Scholar]
- 87. Bellosta S, Mahley RW, Sanan DA, et al Macrophage‐specific expression of human apolipoprotein E reduces atherosclerosis in hypercholesterolemic apolipoprotein E‐null mice. J Clin Invest. 1995; 96: 2170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tolani S, Pagler TA, Murphy AJ, et al Hypercholesterolemia and reduced HDL‐C promote hematopoietic stem cell proliferation and monocytosis: studies in mice and FH children. Atherosclerosis. 2013; 229: 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yvan‐Charvet L, Welch C, Pagler TA, et al Increased inflammatory gene expression in ABC transporter‐deficient macrophages: free cholesterol accumulation, increased signaling via toll‐like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008; 118: 1837–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nagai Y, Garrett KP, Ohta S, et al Toll‐like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006; 24: 801–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yang K, Wang X, Liu Z, et al Oxidized low‐density lipoprotein promotes macrophage lipid accumulation via the toll‐like receptor 4‐Src pathway. Circ J. 2015; 79: 2509–16. [DOI] [PubMed] [Google Scholar]
- 92. Oduro KA Jr, Liu F, Tan Q, et al Myeloid skewing in murine autoimmune arthritis occurs in hematopoietic stem and primitive progenitor cells. Blood. 2012; 120: 2203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dybdahl B, Wahba A, Lien E, et al Inflammatory response after open heart surgery: release of heat‐shock protein 70 and signaling through toll‐like receptor‐4. Circulation. 2002; 105: 685–90. [DOI] [PubMed] [Google Scholar]
- 94. Yvan‐Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol. 2010; 30: 139–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nguyen KD, Fentress SJ, Qiu Y, et al Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science. 2013; 341: 1483–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Grisanti LA, Gumpert AM, Traynham CJ, et al Leukocyte‐expressed β2‐adrenergic receptors are essential for survival after acute myocardial injury. Circulation. 2016; 134: 153–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Grisanti LA, Traynham CJ, Repas AA, et al β2‐Adrenergic receptor‐dependent chemokine receptor 2 expression regulates leukocyte recruitment to the heart following acute injury. Proc Natl Acad Sci USA. 2016; 113: 15126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Robbins CS, Hilgendorf I, Weber GF, et al Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013; 19: 1166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhu SN, Chen M, Jongstra‐Bilen J, et al GM‐CSF regulates intimal cell proliferation in nascent atherosclerotic lesions. J Exp Med. 2009; 206: 2141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sakai M, Miyazaki A, Hakamata H, et al The scavenger receptor serves as a route for internalization of lysophosphatidylcholine in oxidized low density lipoprotein‐induced macrophage proliferation. J Biol Chem. 1996; 271: 27346–52. [DOI] [PubMed] [Google Scholar]
- 101. Chistiakov DA, Melnichenko AA, Orekhov AN, et al How do macrophages sense modified low‐density lipoproteins? Int J Cardiol. 2017; 230: 232–40. [DOI] [PubMed] [Google Scholar]
- 102. Westerterp M, Gourion‐Arsiquaud S, Murphy AJ, et al Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell. 2012; 11: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sakakura K, Nakano M, Otsuka F, et al Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ. 2013; 22: 399–411. [DOI] [PubMed] [Google Scholar]
- 104. Bentzon JF, Otsuka F, Virmani R, et al Mechanisms of plaque formation and rupture. Circ Res. 2014; 114: 1852–66. [DOI] [PubMed] [Google Scholar]
- 105. Otsuka F, Yasuda S, Noguchi T, et al Pathology of coronary atherosclerosis and thrombosis. Cardiovasc Diagn Ther. 2016; 6: 396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhang D, Fang P, Jiang X, et al Severe hyperhomocysteinemia promotes bone marrow‐derived and resident inflammatory monocyte differentiation and atherosclerosis in LDLr/CBS‐deficient mice. Circ Res. 2012; 111: 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Toutouzas K, Synetos A, Nikolaou C, et al Matrix metalloproteinases and vulnerable atheromatous plaque. Curr Top Med Chem. 2012; 12: 1166–80. [DOI] [PubMed] [Google Scholar]
- 108. Koltsova EK, Garcia Z, Chodaczek G, et al Dynamic T cell‐APC interactions sustain chronic inflammation in atherosclerosis. J Clin Invest. 2012; 122: 3114–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Mach F, Schönbeck U, Sukhova GK, et al Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature. 1998; 394: 200–3. [DOI] [PubMed] [Google Scholar]
- 110. Jansen MF, Hollander MR, van Royen N, et al CD40 in coronary artery disease: a matter of macrophages? Basic Res Cardiol. 2016; 111: 38 https://doi.org/10.1007/s00395-016-0554-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tedgui A, Bernard C. Cytokines, immune‐inflammatory response and atherosclerosis. Eur Cytokine Netw. 1994; 5: 263–70. [PubMed] [Google Scholar]
- 112. Häkkinen T, Karkola K, Ylä‐Herttuala S. Macrophages, smooth muscle cells, endothelial cells, and T‐cells express CD40 and CD40L in fatty streaks and more advanced human atherosclerotic lesions. Colocalization with epitopes of oxidized low‐density lipoprotein, scavenger receptor, and CD16 (Fc gammaRIII). Virchows Arch. 2000; 437: 396–405. [DOI] [PubMed] [Google Scholar]
- 113. Lutgens E, Lievens D, Beckers L, et al Deficient CD40‐TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med. 2010; 207: 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Mosig S, Rennert K, Krause S, et al Different functions of monocyte subsets in familial hypercholesterolemia: potential function of CD14 + CD16 + monocytes in detoxification of oxidized LDL. FASEB J. 2009; 23: 866–74. [DOI] [PubMed] [Google Scholar]
- 115. Kapinsky M, Torzewski M, Büchler C, et al Enzymatically degraded LDL preferentially binds to CD14(high) CD16(+) monocytes and induces foam cell formation mediated only in part by the class B scavenger‐receptor CD36. Arterioscler Thromb Vasc Biol. 2001; 21: 1004–10. [DOI] [PubMed] [Google Scholar]
- 116. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013; 13: 709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gerrity RG, Naito HK. Lipid clearance from fatty streak lesions by foam cell migration. Artery. 1980; 8: 215–9. [PubMed] [Google Scholar]
- 118. van Gils JM, Derby MC, Fernandes LR, et al The neuroimmune guidance cue netrin‐1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat Immunol. 2012; 13: 136–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. van Gils JM, Ramkhelawon B, Fernandes L, et al Endothelial expression of guidance cues in vessel wall homeostasis dysregulation under proatherosclerotic conditions. Arterioscler Thromb Vasc Biol. 2013; 33: 911–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wanschel A, Seibert T, Hewing B, et al Neuroimmune guidance cue Semaphorin 3E is expressed in atherosclerotic plaques and regulates macrophage retention. Arterioscler Thromb Vasc Biol. 2013; 33: 886–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ramkhelawon B, Yang Y, van Gils JM, et al Hypoxia induces netrin‐1 and Unc5b in atherosclerotic plaques: mechanism for macrophage retention and survival. Arterioscler Thromb Vasc Biol. 2013; 33: 1180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Oksala N, Pärssinen J, Seppälä I, et al Association of neuroimmune guidance cue netrin‐1 and its chemorepulsive receptor UNC5B with atherosclerotic plaque expression signatures and stability in human(s): Tampere Vascular Study (TVS). Circ Cardiovasc Genet. 2013; 6: 579–87. [DOI] [PubMed] [Google Scholar]
- 123. Luo Y, Duan H, Qian Y, et al Macrophagic CD146 promotes foam cell formation and retention during atherosclerosis. Cell Res. 2017; 27: 352–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Witko‐Sarsat V, Rieu P, Descamps‐Latscha B, et al Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest. 2000; 80: 617–53. [DOI] [PubMed] [Google Scholar]
- 125. Dancey JT, Deubelbeiss KA, Harker LA, et al Neutrophil kinetics in man. J Clin Invest. 1976; 58: 705–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Edwards SW. The development and structure of mature neutrophils In: Edwards SW, editor. Biochemistry and physiology of the neutrophil. New York, NY: Cambridge University Press; 2005. pp. 33–76. [Google Scholar]
- 127. Dupré‐Crochet S, Erard M, Nüβe O. ROS production in phagocytes: why, when, and where? J Leukoc Biol. 2013; 94: 657–70. [DOI] [PubMed] [Google Scholar]
- 128. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013; 13: 159–75. [DOI] [PubMed] [Google Scholar]
- 129. Brinkmann V, Reichard U, Goosmann C, et al Neutrophil extracellular traps kill bacteria. Science. 2004; 303: 1532–5. [DOI] [PubMed] [Google Scholar]
- 130. Fuchs TA, Abed U, Goosmann C, et al Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007; 176: 231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009; 30: 513–21. [DOI] [PubMed] [Google Scholar]
- 132. Yipp BG, Petri B, Salina D, et al Infection‐induced NETosis is a dynamic process involving neutrophil multitasking in vivo . Nat Med. 2012; 18: 1386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hogan D, Wheeler RT. The complex roles of NADPH oxidases in fungal infection. Cell Microbiol. 2014; 16: 1156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Swain SD, Siemsen DW, Nelson LK, et al Inhibition of the neutrophil NADPH oxidase by adenosine is associated with increased movement of flavocytochrome b between subcellular fractions. Inflammation. 2003; 27: 45–58. [DOI] [PubMed] [Google Scholar]
- 135. Ciz M, Denev P, Kratchanova M, et al Flavonoids inhibit the respiratory burst of neutrophils in mammals. Oxid Med Cell Longev. 2012; 2012: 181295 https://doi.org/10.1155/2012/181295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ren Y, Stuart L, Lindberg FP, et al Nonphlogistic clearance of late apoptotic neutrophils by macrophages: efficient phagocytosis independent of beta 2 integrins. J Immunol. 2001; 166: 4743–50. [DOI] [PubMed] [Google Scholar]
- 137. Pimentel TA, Sampaio AL, D'Acquisto F, et al An essential role for mast cells as modulators of neutrophils influx in collagen‐induced arthritis in the mouse. Lab Invest. 2011; 91: 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Teng N, Maghzal GJ, Talib J, et al The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox Rep. 2017; 22: 51–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Schwartz J, Leidal KG, Femling JK, et al Neutrophil bleaching of GFP‐expressing staphylococci: probing the intraphagosomal fate of individual bacteria. J Immunol. 2009; 183: 2632–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Delgado‐Rizo V, Martínez‐Guzmán MA, Iñiguez‐Gutierrez L, et al Neutrophil extracellular traps and its implications in inflammation: an Overview. Front Immunol. 2017; 8: 81 https://doi.org/10.3389/fimmu.2017.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Capodici C, Berg RA. Cathepsin G degrades denatured collagen. Inflammation. 1989; 13: 137–45. [DOI] [PubMed] [Google Scholar]
- 142. Belaaouaj A, Kim KS, Shapiro SD. Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science. 2000; 289: 1185–8. [DOI] [PubMed] [Google Scholar]
- 143. Watanabe H, Hattori S, Katsuda S, et al Human neutrophil elastase: degradation of basement membrane components and immunolocalization in the tissue. J Biochem. 1990; 108: 753–9. [DOI] [PubMed] [Google Scholar]
- 144. Averill LE, Meagher RC, Gerrity RG. Enhanced monocyte progenitor cell proliferation in bone marrow of hyperlipemic swine. Am J Pathol. 1989; 135: 369–77. [PMC free article] [PubMed] [Google Scholar]
- 145. Feldman DL, Mogelesky TC, Liptak BF, et al Leukocytosis in rabbits with diet‐induced atherosclerosis. Arterioscler Thromb. 1991; 11: 985–94. [DOI] [PubMed] [Google Scholar]
- 146. Ionita MG, van den Borne P, Catanzariti LM, et al High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture‐prone lesions. Arterioscler Thromb Vasc Biol. 2010; 30: 1842–8. [DOI] [PubMed] [Google Scholar]
- 147. Moreno JA, Ortega‐Gómez A, Delbosc S, et al In vitro and in vivo evidence for the role of elastase shedding of CD163 in human atherothrombosis. Eur Heart J. 2012; 33: 252–63. [DOI] [PubMed] [Google Scholar]
- 148. Mayer FJ, Gruenberger D, Schillinger M, et al Prognostic value of neutrophils in patients with asymptomatic carotid artery disease. Atherosclerosis. 2013; 231: 274–80. [DOI] [PubMed] [Google Scholar]
- 149. von Vietinghoff S, Ley K. Homeostatic regulation of blood neutrophil counts. J Immunol. 2008; 181: 5183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Köhler A, De Filippo K, Hasenberg M, et al G‐CSF‐mediated thrombopoietin release triggers neutrophil motility and mobilization from bone marrow via induction of Cxcr2 ligands. Blood. 2011; 117: 4349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Döring Y, Drechsler M, Soehnlein O, et al Neutrophils in atherosclerosis: from mice to man. Arterioscler Thromb Vasc Biol. 2015; 35: 288–95. [DOI] [PubMed] [Google Scholar]
- 152. de Jager SC, Bot I, Kraaijeveld AO, et al Leukocyte‐specific CCL3 deficiency inhibits atherosclerotic lesion development by affecting neutrophil accumulation. Arterioscler Thromb Vasc Biol. 2013; 33: e75–83. [DOI] [PubMed] [Google Scholar]
- 153. Copin JC, da Silva RF, Fraga‐Silva RA, et al Treatment with Evasin‐3 reduces atherosclerotic vulnerability for ischemic stroke, but not brain injury in mice. J Cereb Blood Flow Metab. 2013; 33: 490–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Braunersreuther V, Montecucco F, Pelli G, et al Treatment with the CC chemokine‐binding protein Evasin‐4 improves post‐infarction myocardial injury and survival in mice. Thromb Haemost. 2013; 110: 807–25. [DOI] [PubMed] [Google Scholar]
- 155. Nencioni A, da Silva RF, Fraga‐Silva RA, et al Nicotinamide phosphoribosyltransferase inhibition reduces intraplaque CXCL1 production and associated neutrophil infiltration in atherosclerotic mice. Thromb Haemost. 2014; 111: 308–22. [DOI] [PubMed] [Google Scholar]
- 156. Lenglet S, Thomas A, Soehnlein O, et al Fatty acid amide hydrolase deficiency enhances intraplaque neutrophil recruitment in atherosclerotic mice. Arterioscler Thromb Vasc Biol. 2013; 33: 215–23. [DOI] [PubMed] [Google Scholar]
- 157. Hoyer FF, Khoury M, Slomka H, et al Inhibition of endocannabinoid‐degrading enzyme fatty acid amide hydrolase increases atherosclerotic plaque vulnerability in mice. J Mol Cell Cardiol. 2014; 66: 126–32. [DOI] [PubMed] [Google Scholar]
- 158. Cabral GA, Ferreira GA, Jamerson MJ. Endocannabinoids and the immune system in health and disease. Handb Exp Pharmacol. 2015; 231: 185–211. [DOI] [PubMed] [Google Scholar]
- 159. Montecucco F, Di Marzo V, da Silva RF, et al The activation of the cannabinoid receptor type 2 reduces neutrophilic protease‐mediated vulnerability in atherosclerotic plaques. Eur Heart J. 2012; 33: 846–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Montecucco F, Lenglet S, Braunersreuther V, et al CB(2) cannabinoid receptor activation is cardioprotective in a mouse model of ischemia/reperfusion. J Mol Cell Cardiol. 2009; 46: 612–20. [DOI] [PubMed] [Google Scholar]
- 161. Murikinati S1, Jüttler E, Keinert T, et al Activation of cannabinoid 2 receptors protects against cerebral ischemia by inhibiting neutrophil recruitment. FASEB J. 2010; 24: 788–98. [DOI] [PubMed] [Google Scholar]
- 162. Balenga NA, Aflaki E, Kargl J, et al GPR55 regulates cannabinoid 2 receptor‐mediated responses in human neutrophils. Cell Res. 2011; 21: 1452–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Malfitano AM, Basu S, Maresz K, et al What we know and do not know about the cannabinoid receptor 2 (CB2). Semin Immunol. 2014; 26: 369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Lingrel JB, Pilcher‐Roberts R, Basford JE, et al Myeloid‐specific Krüppel‐like factor 2 inactivation increases macrophage and neutrophil adhesion and promotes atherosclerosis. Circ Res. 2012; 110: 1294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Sangwung P, Zhou G, Nayak L, et al KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight. 2017; 2: e91700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. SenBanerjee S, Lin Z, Atkins GB, et al KLF2 is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004; 199: 1305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Wang W, Ha CH, Jhun BS, et al Fluid shear stress stimulates phosphorylation‐dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood. 2010; 115: 2971–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Chistiakov DA, Orekhov AN, Bobryshev YV. Effects of shear stress on endothelial cells: go with the flow. Acta Physiol (Oxf). 2017b; 219: 382–408. [DOI] [PubMed] [Google Scholar]
- 169. Franck G, Mawson T, Sausen G, et al Flow perturbation mediates neutrophil recruitment and potentiates endothelial injury via TLR2 in mice ‐ implications for superficial erosion. Circ Res. 2017; 121: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Quillard T, Araújo HA, Franck G, et al TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur Heart J. 2015; 36: 1394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Chen PJ, Wang YL, Kuo LM, et al Honokiol suppresses TNF‐α‐induced neutrophil adhesion on cerebral endothelial cells by disrupting polyubiquitination and degradation of IκBα. Sci Rep. 2016; 6: 26554 https://doi.org/10.1038/srep26554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Taylor PR, Roy S, Leal SM Jr, et al Activation of neutrophils by autocrine IL‐17A‐IL‐17RC interactions during fungal infection is regulated by IL‐6, IL‐23, RORγt and dectin‐2. Nat Immunol. 2014; 15: 143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Butcher MJ1, Gjurich BN, Phillips T, et al The IL‐17A/IL‐17RA axis plays a proatherogenic role via the regulation of aortic myeloid cell recruitment. Circ Res. 2012; 110: 675–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Malle E, Marsche G, Arnhold J, et al Modification of low‐density lipoprotein by myeloperoxidase‐derived oxidants and reagent hypochlorous acid. Biochim Biophys Acta. 2006; 1761: 392–415. [DOI] [PubMed] [Google Scholar]
- 175. Tien M. Myeloperoxidase‐catalyzed oxidation of tyrosine. Arch Biochem Biophys. 1999; 367: 61–6. [DOI] [PubMed] [Google Scholar]
- 176. Wu W, Chen Y, Hazen SL. Eosinophil peroxidase nitrates protein tyrosyl residues. Implications for oxidative damage by nitrating intermediates in eosinophilic inflammatory disorders. J Biol Chem. 1999; 274: 25933–44. [DOI] [PubMed] [Google Scholar]
- 177. Freeman TA, Parvizi J, Della Valle CJ, et al Reactive oxygen and nitrogen species induce protein and DNA modifications driving arthrofibrosis following total knee arthroplasty. Fibrogen Tissue Rep. 2009; 2: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Jerke U, Rolle S, Purfürst B, et al β2 integrin‐mediated cell‐cell contact transfers active myeloperoxidase from neutrophils to endothelial cells. J Biol Chem. 2013; 288: 12910–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Dorweiler B, Torzewski M, Dahm M, et al Subendothelial infiltration of neutrophil granulocytes and liberation of matrix‐destabilizing enzymes in an experimental model of human neo‐intima. Thromb Haemost. 2008; 99: 373–81. [DOI] [PubMed] [Google Scholar]
- 180. Henriksen PA, Sallenave JM. Human neutrophil elastase: mediator and therapeutic target in atherosclerosis. Int J Biochem Cell Biol. 2008; 40: 1095–100. [DOI] [PubMed] [Google Scholar]
- 181. Li X, de Boer OJ, Ploegmaker H, et al Granulocytes in coronary thrombus evolution after myocardial infarction–time‐dependent changes in expression of matrix metalloproteinases. Cardiovasc Pathol. 2016; 25: 40–6. [DOI] [PubMed] [Google Scholar]
- 182. Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. 2012; 110: 875–88. [DOI] [PubMed] [Google Scholar]
- 183. Döring Y, Drechsler M, Wantha S, et al Lack of neutrophil‐derived CRAMP reduces atherosclerosis in mice. Circ Res. 2012b; 110: 1052–6. [DOI] [PubMed] [Google Scholar]
- 184. Wantha S, Alard JE, Megens RT, et al Neutrophil‐derived cathelicidin promotes adhesion of classical monocytes. Circ Res. 2013; 112: 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Yang D, Chen Q, Schmidt AP, et al LL‐37, the neutrophil granule‐ and epithelial cell‐derived cathelicidin, utilizes formyl peptide receptor‐like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. 2000; 192: 1069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Quinn KL, Henriques M, Tabuchi A, et al Human neutrophil peptides mediate endothelial‐monocyte interaction, foam cell formation, and platelet activation. Arterioscler Thromb Vasc Biol. 2011; 31: 2070–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Lee TD, Gonzalez ML, Kumar P, et al CAP37, a neutrophil‐derived inflammatory mediator, augments leukocyte adhesion to endothelial monolayers. Microvasc Res. 2003; 66: 38–48. [DOI] [PubMed] [Google Scholar]
- 188. Watorek W. Azurocidin – inactive serine proteinase homolog acting as a multifunctional inflammatory mediator. Acta Biochim Pol. 2003; 50: 743–52. [PubMed] [Google Scholar]
- 189. Ramos‐Mozo P, Madrigal‐Matute J, Vega de Ceniga M, et al Increased plasma levels of NGAL, a marker of neutrophil activation, in patients with abdominal aortic aneurysm. Atherosclerosis. 2012; 220: 552–6. [DOI] [PubMed] [Google Scholar]
- 190. Cheng JM, Akkerhuis KM, Meilhac O, et al Circulating osteoglycin and NGAL/MMP9 complex concentrations predict 1‐year major adverse cardiovascular events after coronary angiography. Arterioscler Thromb Vasc Biol. 2014; 34: 1078–84. [DOI] [PubMed] [Google Scholar]
- 191. Soylu K, Nar G, Aksan G, et al Serum neutrophil gelatinase‐associated lipocalin levels and aortic stiffness in noncritical coronary artery disease. Cardiorenal Med. 2014; 4: 147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Soylu K, Aksan G, Nar G, et al Serum neutrophil gelatinase‐associated lipocalin levels are correlated with the complexity and the severity of atherosclerosis in acute coronary syndrome. Anatol J Cardiol. 2015; 15: 450–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Elneihoum AM, Falke P, Hedblad B, et al Leukocyte activation in atherosclerosis: correlation with risk factors. Atherosclerosis. 1997; 131: 79–84. [DOI] [PubMed] [Google Scholar]
- 194. Anwaar I, Gottsäter A, Hedblad B, et al Endothelial derived vasoactive factors and leukocyte derived inflammatory mediators in subjects with asymptomatic atherosclerosis. Angiology. 1998; 49: 957–66. [DOI] [PubMed] [Google Scholar]
- 195. Lindberg S, Pedersen SH, Mogelvang R, et al Prognostic utility of neutrophil gelatinase‐associated lipocalin in predicting mortality and cardiovascular events in patients with ST‐segment elevation myocardial infarction treated with primary percutaneous coronary intervention. J Am Coll Cardiol. 2012; 60: 339–45. [DOI] [PubMed] [Google Scholar]
- 196. Sahinarslan A, Kocaman SA, Bas D, et al Plasma neutrophil gelatinase‐associated lipocalin levels in acute myocardial infarction and stable coronary artery disease. Coron Artery Dis. 2011; 22: 333–8. [DOI] [PubMed] [Google Scholar]
- 197. Daniels LB, Barrett‐Connor E, Clopton P, et al Plasma neutrophil gelatinase‐associated lipocalin is independently associated with cardiovascular disease and mortality in community‐dwelling older adults: the Rancho Bernardo Study. J Am Coll Cardiol. 2012; 59: 1101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Lindberg S, Jensen JS, Mogelvang R, et al Plasma neutrophil gelatinase‐associated lipocalinin in the general population: association with inflammation and prognosis. Arterioscler Thromb Vasc Biol. 2014; 34: 2135–42. [DOI] [PubMed] [Google Scholar]
- 199. Teigelkamp S, Bhardwaj RS, Roth J, et al Calcium‐dependent complex assembly of the myeloic differentiation proteins MRP‐8 and MRP‐14. J Biol Chem. 1991; 266: 13462–7. [PubMed] [Google Scholar]
- 200. Croce K, Gao H, Wang Y, et al Myeloid‐related protein‐8/14 is critical for the biological response to vascular injury. Circulation. 2009; 120: 427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Vandal K, Rouleau P, Boivin A, et al Blockade of S100A8 and S100A9 suppresses neutrophil migration in response to lipopolysaccharide. J Immunol. 2003; 171: 2602–9. [DOI] [PubMed] [Google Scholar]
- 202. Averill MM, Barnhart S, Becker L, et al S100A9 differentially modifies phenotypic states of neutrophils, macrophages, and dendritic cells: implications for atherosclerosis and adipose tissue inflammation. Circulation. 2011; 123: 1216–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Pereira HA. Cationic antimicrobial protein of Mr 37 kDa: a multifunctional inflammatory protein. Chin Med J. 2001; 114: 9–13. [PubMed] [Google Scholar]
- 204. Megens RT, Vijayan S, Lievens D, et al Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb Haemost. 2012; 107: 597–8. [DOI] [PubMed] [Google Scholar]
- 205. Borissoff JI, Joosen IA, Versteylen MO, et al Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol. 2013; 33: 2032–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Döring Y, Manthey HD, Drechsler M, et al Auto‐antigenic protein‐DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation. 2012c; 125: 1673–83. [DOI] [PubMed] [Google Scholar]
- 207. Macritchie N, Grassia G, Sabir SR, et al Plasmacytoid dendritic cells play a key role in promoting atherosclerosis in apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol. 2012; 32: 2569–79. [DOI] [PubMed] [Google Scholar]
- 208. Villanueva E, Yalavarthi S, Berthier CC, et al Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011; 187: 538–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Warnatsch A, Ioannou M, Wang Q, et al Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015; 349: 316–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Rossaint J, Herter JM, Van Aken H, et al Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap‐mediated sterile inflammation. Blood. 2014; 123: 2573–84. [DOI] [PubMed] [Google Scholar]
- 211. Gould TJ, Vu TT, Swystun LL, et al Neutrophil extracellular traps promote thrombin generation through platelet‐dependent and platelet‐independent mechanisms. Arterioscler Thromb Vasc Biol. 2014; 34: 1977–84. [DOI] [PubMed] [Google Scholar]
- 212. Brill A, Fuchs TA, Savchenko AS, et al Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012; 10: 136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Tall AR, Yvan‐Charvet L, Westerterp M, et al Cholesterol efflux: a novel regulator of myelopoiesis and atherogenesis. Arterioscler Thromb Vasc Biol. 2012; 32: 2547–52. [DOI] [PubMed] [Google Scholar]
- 214. Degoma EM, Rader DJ. Novel HDL‐directed pharmacotherapeutic strategies. Nat Rev Cardiol. 2011; 8: 266–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Schwartz GG, Olsson AG, Ballantyne CM, et al Rationale and design of the dal‐OUTCOMES trial: efficacy and safety of dalcetrapib in patients with recent acute coronary syndrome. Am Heart J. 2009; 158: e3. [DOI] [PubMed] [Google Scholar]
- 216. Robinson JG. Dalcetrapib: a review of Phase II data. Expert Opin Investig Drugs. 2010; 19: 795–805. [DOI] [PubMed] [Google Scholar]
- 217. AIM‐HIGH Investigators , Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes‐Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011; 365: 2255–67. [DOI] [PubMed] [Google Scholar]