

Abstract
Intestinal inflammation is the central pathological feature of colitis and the inflammatory bowel diseases. These syndromes arise from unidentified environmental factors. We found that recurrent non-lethal gastric infections of Gram-negative Salmonella enterica Typhimurium (ST), a major source of human food poisoning, caused inflammation of murine intestinal tissue, predominantly the colon, which persisted following pathogen clearance and irreversibly escalated in severity with repeated infections. ST progressively disabled a host mechanism of protection by inducing endogenous neuraminidase activity thereby accelerating the molecular aging and clearance of intestinal alkaline phosphatase (IAP). Disease was linked to a Tlr4-dependent mechanism of IAP desialylation and deficiency with resulting accumulation of lipopolysaccharide-phosphate (LPS-P). Oral administration of IAP or the marketed anti-viral neuraminidase inhibitor Zanamivir were therapeutic by maintaining IAP dephosphorylation of LPS-P.
Introduction
Inflammation of the intestinal tract is the defining feature of colitis and the human inflammatory bowel diseases (IBDs) including Crohn’s disease and ulcerative colitis (UC). In these syndromes, chronic inflammation disrupts intestinal homeostasis and provokes immune-mediated tissue damage (1–3). Disease origins remain mysterious and involve one or more environmental factors as genome sequence variation may play a secondary and sometimes minor role (4). Among multiple human monozygotic twin comparisons, the genetic contribution to the origin of UC is approximately 20%, while in Crohn’s disease genetics can play a larger role (5). Among possible environmental origins of disease, pathogenic infection has been studied as a factor in precipitating intestinal inflammation (6). Interestingly, bacterial infections have been linked to seasonal increases in hospital admissions involving intestinal inflammation and IBD (7).
Small inoculum bacterial infections that are brief and self–limited are likely to be the most common of infections, and they may frequently go unreported, potentially leading to an underappreciation of the numbers of infections among individuals. We considered that there may be cumulative effects of repeated small inoculum and sub-clinical infections, which if correct might be detected in a model of human food poisoning. We designed such a study using recurrent low-titer non-lethal gastrointestinal infection by the bacterium Salmonella enterica Typhimurium (ST), a common human pathogen. Non-typhoidal Salmonella (NTS) causes a greater human disease burden than any other foodborne bacterial pathogen in the U.S. causing more than a million illnesses annually (8, 9). Globally, NTS causes 93.8 million cases and 155,000 deaths each year (10), and is responsible for up to 50% of bacteremias among young children in developing countries (11, 12).
Results
ST infection elicits intestinal inflammation by diminishing host IAP levels
Beginning at 8 weeks of age, wild-type C57BL/6J mice were infected by gastrointubation with 2 × 103 ST colony–forming units (cfu) every 4 weeks for 6 consecutive months. Following infection, ST was detected transiently and predominantly in the small intestine and some lymphoid tissues. The pathogen was cleared by the host to undetectable levels by 21 days post-infection monitoring that further noted the absence of overt symptoms or mortality among the animals (Fig. S1, A and B). The onset of disease required more than a single infection with this low titer while multiple disease signs were evident among the majority of animals prior to the fourth infection. Phenotypes consistent with the onset of intestinal dysfunction included weight loss, reduced colon length, altered stool consistency (diarrhea), and the presence of fecal blood (Fig. 1, A–D and Fig. S1C). By 20 weeks of age and prior to the fourth infection, disease signs were present among the majority of animals and further included an epithelial barrier defect (Fig. 1E). At 32–48 weeks, rectal prolapse was observed among some animals undergoing recurrent infections (Fig. 1F). The frequency of disease signs escalated with successive recurrent infections and persisted for at least 5 months following the cessation of infections.
Fig. 1. Recurrent ST infection diminishes the abundance and protective role of IAP.
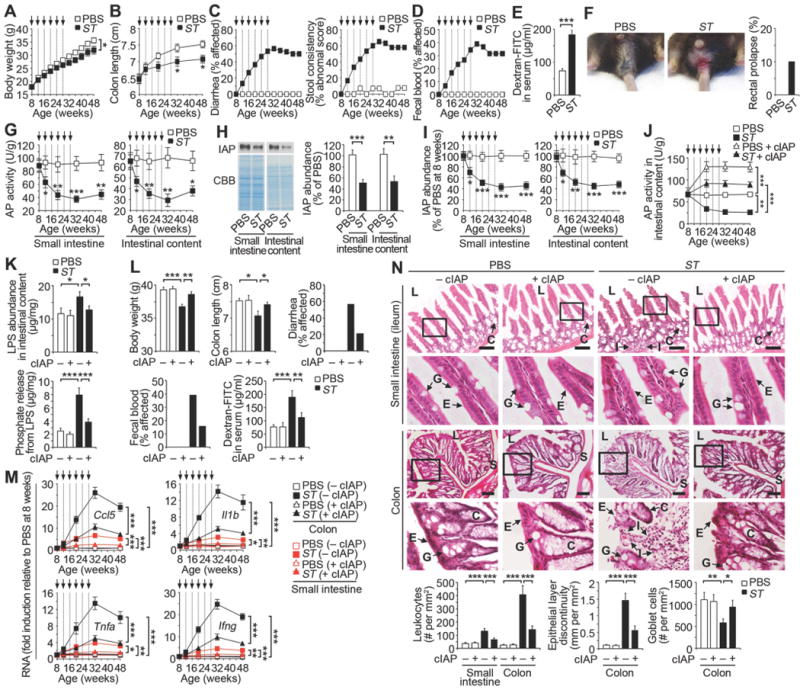
Wild-type mice were analyzed during a course of recurrent ST infection (2 × 103 cfu) or uninfected (PBS) at indicated time points (arrows). (A) Body weight (ST, n = 20; PBS, n = 19). (B) Colon length (n = 40 per condition). (C) Diarrhea and stool consistency (ST, n = 19; PBS, n = 13). (D) Fecal blood (ST, n = 19; PBS, n = 13). (E) Intestinal epithelial barrier function (n = 8 per condition) at 20 weeks of age prior to the fourth infection. (F) Rectal prolapse (ST, n = 30; PBS, n = 20) at 32–48 weeks of age, or 4–20 weeks following last ST infection (representative image). (G) AP activity (n = 40 per condition). (H) Immunoblot blot analysis of IAP at 20 weeks of age prior to the fourth infection (n = 8 per condition). (I) Relative IAP abundance (n = 40 per condition). (J) AP activity +/− calf IAP (cIAP) (n = 40 per condition). (K) LPS abundance and phosphate released from LPS (n = 8 per condition) at 20 weeks of age. (L) Body weight (n = 10 per condition), colon length (n = 8 per condition), diarrhea (ST, n = 23; PBS, n = 14; ST + cIAP, n = 19; PBS + cIAP, n = 15), fecal blood (ST, n = 23; PBS, n = 14; ST + cIAP, n = 19; PBS + cIAP, n = 15) at 48 weeks of age (20 weeks following last ST infection), and intestinal epithelial barrier function (n = 8 per condition) at 20 weeks of age prior to the fourth infection. (M) Cytokine mRNA expression (n = 30 per condition). (N) H & E-stained intestinal tissues at 48 weeks of age (20 weeks following last ST infection). L, intestinal lumen; E, epithelial layer; C, crypt; G, goblet cell; S, submucosa; I, infiltration of leukocytes. Graphs are representative of 16 fields of view (4 mice per condition). All scale bars: 100 µm. Error bars, means ± SEM. ***P < 0.001; **P < 0.01; *P < 0.05; Student’s t test (A), (B), (E), (G), (H), and (I) or one-way ANOVA with Tukey’s multiple comparisons test (J) to (N).
Reductions of alkaline phosphatase (AP) activity and intestinal alkaline phosphatase (IAP) abundance were detected in both the small intestine and intestinal contents, whereas levels of duodenal tissue mRNA that encode IAP were unchanged (Fig. 1, G–I and Fig. S2A). Mammalian IAP is produced exclusively by enterocytes of the duodenum of the small intestine and is released from the cell surface into the lumen where it can dephosphorylate and thereby detoxify the lipopolysaccharide (LPS) endotoxin of Gram-negative bacteria (13–16). We found that oral supplementation with calf intestinal alkaline phosphatase (cIAP) maintained normal AP activity levels in the intestinal tract among animals receiving recurrent ST infections (Fig. 1J). Analysis of LPS isolated from the intestinal tract at 20 weeks of age prior to the fourth infection and following ST clearance revealed a four-fold increase in endogenous LPS-phosphate levels in the context of a 50% increase in total LPS, both of which were maintained close to normal levels in mice receiving cIAP treatment (Fig. 1K). Augmentation of AP activity by cIAP treatment also protected against the development of disease signs encompassing weight loss, colon length, diarrhea, fecal blood, and epithelial barrier function (Fig. 1L).
Inflammatory cytokines associated with intestinal tissue inflammation include chemokine ligand 5 (Ccl5), interleukin-1β (Il1b), tumor necrosis factor-α (Tnfa), and interferon-γ (Ifng). Recurrent ST infection progressively increased inflammatory cytokine mRNA expression levels predominantly in the colon, unless cIAP treatment was provided (Fig. 1M). Histopathological changes were also predominantly seen in the colon including the infiltration of leukocytes into the lamina propria that included neutrophils, monocytes, and T cells, as well as erosion of the epithelial barrier, and reduced goblet cell numbers, whereas a much lesser effect was observed in the small intestine and in addition only in the ileum (Fig. 1N, Fig. S2b, and Fig. S3). Thus, recurrent non-lethal gastrointestinal infection of ST diminished the expression of host IAP activity that normally confers host protection against intestinal inflammation and tissue damage predominantly in the colon. Studies were further undertaken to identify the mechanisms that regulate IAP function.
IAP deficiency is linked to an accelerated rate of de-sialylation and endocytic localization
IAP is synthesized as a glycosylphosphatidylinositol (GPI)-linked glycoprotein residing on the enterocyte cell surface of the duodenum until it is released into the intestinal lumen by phospholipase activity (13, 17). IAP production was investigated by pulse–chase experiments of ex vivo primary enterocyte cultures derived from tissue of the small intestine. Normal rates of IAP synthesis and appearance at the cell surface were observed among all enterocyte cultures regardless of previously cleared infections (Fig. 2A). In contrast, a significant decrease in IAP cell surface half-life with reduced IAP abundance in the culture media were measured among enterocyte samples from mice that had cleared multiple infections (Fig. 2, B and C). Co-localization studies revealed increased co-localization of IAP with markers of early endosomes and lysosomes coincident with reduced cell surface IAP abundance (Fig. 2, D and E).
Fig. 2. Mechanism of IAP regulation during recurrent ST infection.
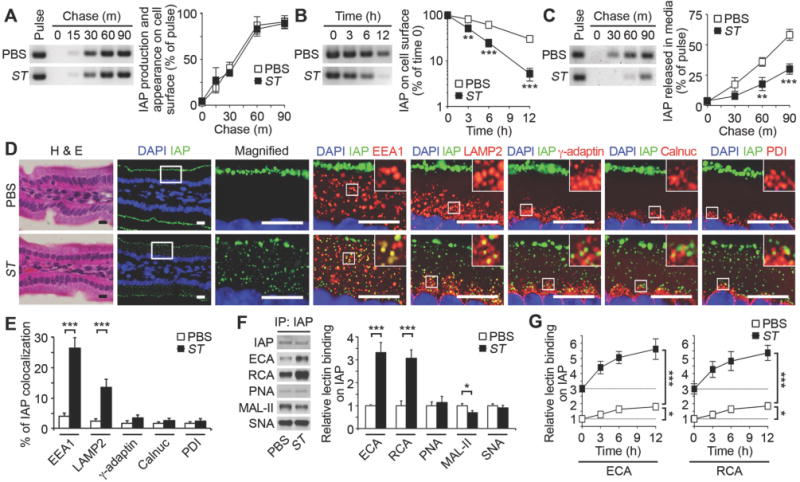
(A–C) Pulse–chase analyses of IAP synthesis, trafficking and cell-surface half-life among cultured primary enterocytes isolated from wild-type mice at 20 weeks of age (prior to fourth ST infection). (D and E) In situ localization and intracellular co-localization of IAP in duodenum sections stained with H & E or with fluorescent antibodies to IAP (green) and intracellular compartment proteins (red) including early endosomes (EEA1), lysosomes (LAMP2), trans-Golgi (γ-adaptin), cis-Golgi (Calnuc), or the endoplasmic reticulum (PDI), depicting the percentage of IAP co-localization (yellow). Graphs are representative of ten fields of view (four mice per condition). Scale bars: 10 µm. (F) Lectin blot of IAP from small intestine. (G) Lectin binding of IAP on cultured enterocytes following cell-surface biotinylation. (A–G) Wild-type mice at 20 weeks of age prior to fourth infection. (A–C and G) n = 6 per condition. (F) n = 8 per condition. Error bars, means ± SEM. ***P < 0.001; **P < 0.01; *P < 0.05; Student’s t test (A), (B), (C), (E), and (F) or one-way ANOVA with Tukey’s multiple comparisons test (G).
IAP is also glycosylated during its synthesis in the secretory pathway. We next analyzed the glycan linkages attached to enterocyte IAP using analytical lectins including the α2–3 sialic acid-specific lectin Maackia amurensis lectin II (MAL-II) and the galactose-specific lectins Erythrina cristagalli agglutinin (ECA) and Ricinus communis agglutinin (RCA). These lectins have been validated biochemically and genetically although their binding does not provide full structure resolution of the various types of glycan structures that can present these linkages. A significant reduction of terminal sialic acid linkages coincident with exposure of underlying galactose linkages were measured in the context of recurrent infection (Fig. 2F). No comparative changes to other specific glycan linkages were detected including the sialylation state of Core 1 O-glycans, or changes in abundance of α2–6-linked sialic acids, using Peanut Agglutinin (PNA) and Sambucus nigra (SNA) lectins, respectively. In the absence of infection, we observed a progressive de-sialylation of the glycans attached to nascent IAP on the enterocyte cell surface, indicative of a feature of its molecular aging. This basal rate of IAP de-sialylation was significantly increased by recurrent ST infection and was concurrent with increased IAP internalization and degradation in enterocytes (Fig. 2G). Although IAP deficiency appeared to be the predominant factor in disease onset as indicated by the effects of cIAP treatment, multiple enterocyte cell surface glycoproteins were observed to be de-sialylated and internalized from the cells surface of endocytes including sucrase-isomaltase, dipeptidyl peptidase 4, and lactase (Fig. S4). These results suggest that the presence of one or more sialyltransferases establishes the normal half-lives (and consequent abundance) of enterocyte cell surface glycoproteins including IAP.
ST3Gal6 maintains IAP sialylation in protecting against intestinal inflammation
The ST3Gal6 sialyltransferase generates α2–3 sialic acid linkages on glycoproteins and is highly expressed in the intestinal tract (18, 19). In mice lacking a functional St3gal6 gene, we detected a significant reduction in AP activity and IAP abundance, whereas mRNA encoding IAP was unchanged (Fig. 3A, 3B and Fig. S5A). Although LPS levels in the intestinal tract were similar at 8 weeks of age, a three-fold increase of LPS-phosphate abundance was measured consistent with reduced AP activity (Fig. 3C). Absent ST3Gal6 function, IAP sialic acid linkages were also deficient with increased galactose exposure (Fig. 3D) similar to findings in wild-type mice experiencing recurrent ST infection. Glycan alterations were further detected histologically among small intestinal epithelial cells (Fig. S5B). Diminished IAP sialylation in ST3Gal6 deficiency was linked to reduced IAP cell surface residency and increased IAP co-localization with markers of endosomes and lysosomes (Fig. 3, E–I). Thus, IAP sialylation by ST3Gal6 functions to maintain IAP half-life and abundance in the intestinal lumen. The impact of IAP deficiency caused by the absence of ST3Gal6 was further investigated.
Fig. 3. Mechanism of IAP regulation by ST3Gal6 sialylation.
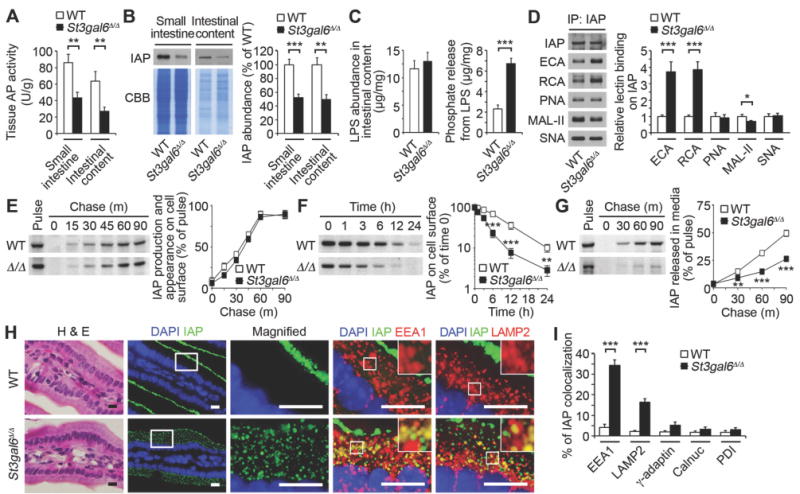
(A) AP activity. (B) IAP protein abundance. (C) LPS abundance and phosphate released from LPS of intestinal content. (D) Lectin blot of IAP from small intestine. (E–G) Pulse–chase of IAP synthesis and trafficking and IAP cell-surface half-life among cultured primary enterocytes isolated from uninfected ST3Gal6-deficient mice and wild-type littermates at 8–10 weeks of age. (H and I) In situ localization and intracellular co-localization of IAP in duodenum, depicting the percentage of IAP co-localization (yellow). Graphs are representative of ten fields of view (4 mice per genotype). Scale bars, 10 µm. (A–I) ST3Gal6-deficient mice and wild-type littermates at 8–10 weeks of age, uninfected. (A–D) n = 8 per condition. (E–G) n = 6 per condition. Error bars, means ± SEM. ***P < 0.001; **P < 0.01; *P < 0.05; Student’s t test (A) to (G) and (I).
Mice aging in the absence of ST3Gal6 were compared with wild-type littermates in the presence and absence of recurrent ST infection and cIAP therapy. Spontaneous phenotypes detected in uninfected mice lacking ST3Gal6 included reduced body weight, reduced colon length, diarrhea, the presence of fecal blood, and epithelial barrier dysfunction, all of which were exacerbated by recurrent ST infection. The addition of cIAP to drinking water normalized AP activity levels during adult life and reduced or eliminated disease signs (Fig. 4, A–C). Similarly, increased LPS-phosphate levels were closely associated with reduced IAP and the onset of disease signs, whereas total LPS increased only modestly in the intestinal contents of infected mice (Fig. 4D). LPS is generated predominantly in the colon by commensal microbes that may serve as markers or effectors of intestinal inflammatory disease (20).
Fig. 4. ST3Gal6 sialylation of IAP in preventing intestinal inflammation.
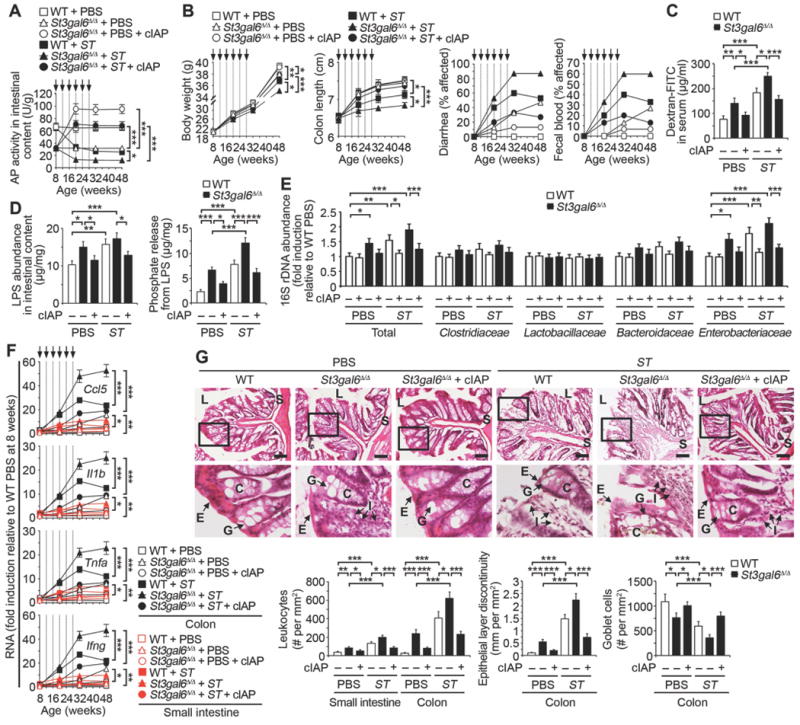
Indicated genotypes following ST re-infection (arrows) were analyzed in absence or presence of cIAP. (A) AP activity (n = 32 per condition). (B) Body weight (n = 10 per condition), colon length (n = 32 per condition), diarrhea (n = 30 per condition), and fecal blood (n = 30 per condition). (C) Intestinal epithelial barrier function. (D) LPS abundance and phosphate released from LPS of intestinal content. (E) Commensal microbiome 16S rDNA in intestinal content (n = 10 per condition). (F) Inflammatory cytokine RNA in colon and small intestine (n = 24 per condition). (G) H & E-stained colon sections. Graphs are representative of ten fields of view (four mice per condition). Scale bars: 100 µm. (C and D) Data were acquired from mice 20 weeks of age prior to fourth infection. (E–G) Data were acquired from mice 32 weeks of age and 4 weeks following the last infection. (C and D) n = 8 per condition. Error bars, means ± SEM. ***P < 0.001; **P < 0.01; *P < 0.05; one-way ANOVA with Tukey’s multiple comparisons test (A) to (G).
A survey of microbiota using 16S rDNA probes revealed a 50% increase of intestinal bacterial load attributed primarily to the Gram-negative Enterobacteriaceae, consistent with the magnitude of increase of total LPS. These findings were present among both uninfected St3gal6-null mice and wild-type littermates subjected to recurrent ST infection infection and ST3Gal6 deficiency (Fig. 4E). The temporal acquisition of these microbiota changes observed at 32 weeks were found to emerge progressively among wild-type mice receiving periodic ST infections, and were coincident with the emergence and increase in the severity of disease signs Fig. S6). These findings are also consistent with reports of altered commensal microbiota populations that can contribute intestinal inflammation and which often include elevated levels of Enterobacteriaceae (3, 21).
ST3Gal6 deficiency spontaneously increased inflammatory cytokines in the intestinal tissues of uninfected animals, and this was further exacerbated by recurrent ST infection (Fig. 4F). Similarly, histopathological findings in the absence of ST3Gal6 correlated with inflammatory marker expression and included leukocyte infiltration, epithelial layer discontinuity, and reduced goblet cell numbers, which were also increased in severity by recurrent ST infection (Fig. 4G). These results demonstrated that the ST3Gal6 sialyltransferase is essential to support normal IAP expression and host protection against spontaneous intestinal inflammation. The cause of IAP de-sialylation following ST infection was unaccounted for but implicated as a significant trigger of pathogenesis.
Disease onset with neuraminidase induction is Tlr4-dependent and recapitulated by LPS
Neuraminidase (Neu) enzymes, also known as sialidases, hydrolyze sialic acids attached to glycan polymers and are encoded in the genomes of diverse organisms including bacteria, mice, and humans. However, the genome of the ST isolate used in our studies does not encode a neuraminidase (22), indicating a host source of the induced Neu activity. Four neuraminidase genes have been identified in mammalian genomes (Neu1–4), with Neu1 and Neu3 enzymes expressed in multiple compartments including the cell surface and in blood circulation (23, 24). An increase of neuraminidase (Neu) activity occurred in the small intestine due to ST infection. Using Toll-like receptor 4 (Tlr4)-null mice, this induction of host Neu activity was dependent upon Tlr4 function (Fig. 5A). Among mammalian Neu isozymes, only Neu3 abundance and Neu3 RNA expression correlated with Tlr4-dependent induction of Neu activity (Fig. 5, B and C). Absence of Neu3 induction due to Tlr4 deficiency resulted in normal IAP expression at the enterocyte cell surface (Fig. 5D). This was coincident with normal levels of sialic acid linkages on IAP and among apical glycoproteins of the small intestinal epithelium (Fig. S5, C and D). AP activity and IAP abundance also remained normal in Tlr4 deficiency and LPS-phosphate levels did not increase significantly following ST infections (Fig. 5, E–G). The induction of inflammatory cytokines caused by recurrent ST infection was also blocked by Tlr4 deficiency, and disease signs were reduced or eliminated with the maintenance of epithelial barrier function (Fig. 5, H and I).
Fig. 5. Host neuraminidase induction by Tlr4 during recurrent ST infection.
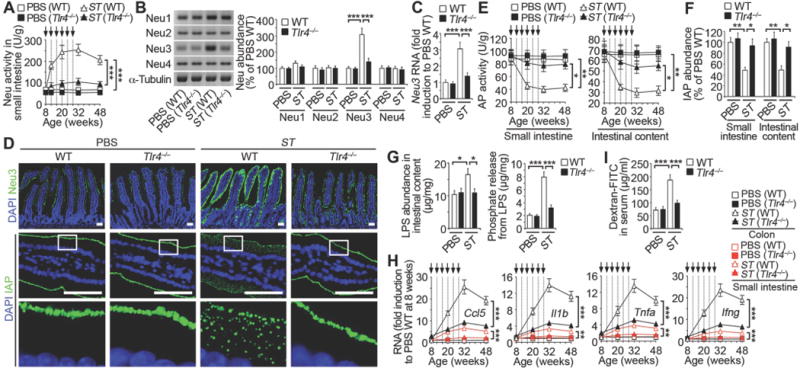
Wild-type and Tlr4-deficient mice were analyzed following ST re-infection or LPS administration (arrows). (A) Neuraminidase (Neu) activity (n = 30 per condition). (B) Neu1, 2, 3 and 4 protein abundance in small intestine. (C) Neu3 mRNA expression in small intestine. (D) In situ localization of Neu3 and IAP in duodenum. Images are representative of ten fields of view (4 mice per condition). Scale bars: 50 µm. (E) AP activity (n = 24 per condition). (F) IAP protein abundance. (G) LPS abundance and phosphate released from LPS of intestinal content. (H) Inflammatory cytokine RNA abundance (n = 16 per condition). (I) Intestinal epithelial barrier function. (B, C, F, G, and I) n = 6 per condition. (B, C, F, G, and I) Animals were 20 weeks of age and prior to the fourth infection. Error bars, means ± SEM. ***P < 0.001; **P < 0.01; *P < 0.05; one-way ANOVA with Tukey’s multiple comparisons test (A) to (I).
Tlr4 is activated by binding to its LPS ligand (25, 26). This bacterial sensor of the LPS endotoxin is expressed on all segments of the intestinal tract and is especially prominent in the small intestine (27, 28), where it functions in pro-inflammatory signaling to engage immune cells in the onset and development of disease (29, 30). We investigated whether LPS was itself responsible for Neu induction, IAP deficiency, and concurrent elevations of inflammatory cytokines. Dose-response analyses using commercially obtained LPS were undertaken to determine minimal dosage and timing for further study (Fig. S7, A and B). We found that gastrointubation of LPS induced Neu activity, Neu3 abundance, and Neu3 RNA levels with reductions in AP activity and IAP abundance, all of which were dependent upon Tlr4 function (Fig. 6, A–F). The induction of Neu activity by LPS resulted in Tlr4-dependent reductions of sialic acid linkages resulting in galactose exposure on isolated IAP and among apical cell surface glycoproteins of the small intestinal epithelium (Fig. S7, C and D). LPS increased the internalization and co-localization of IAP with endocytic markers coincident with reduced IAP expression at the cell surface (Fig. S7E). Normal IAP levels were retained following LPS administration in Tlr4 deficiency with relatively low abundance of LPS-phosphate (Fig. 6G). LPS–induced inflammatory cytokine induction was also blocked by Tlr4 deficiency with the retention of epithelial barrier function (Fig. 6, H and I). Thus, LPS/Tlr4 signaling resulting from recurrent low-titer ST infection is linked to the induction of host Neu3 and increased Neu activity, resulting in IAP de-sialylation and its subsequent pathogenic deficiency. Thus, Neu inhibition may be therapeutic in the context of recurrent ST infection.
Fig. 6. Host neuraminidase induction by Tlr4 and LPS.
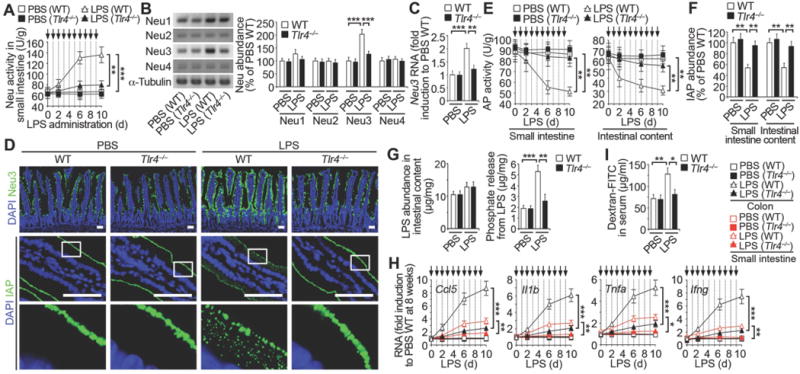
(A) Neu activity in mice at 8 weeks of age prior to repeated LPS administrations (arrows). (B and C) Neu protein abundance and Neu3 RNA expression in small intestine. (D) In situ localization of Neu3 and IAP in duodenum sections, representative of ten fields of view (four mice per condition). Scale bars: 50 µm. (E) AP activity prior to repeated LPS administrations (arrows). (F) IAP protein abundance. (G–I) Phosphate released from LPS of intestinal content, inflammatory cytokine RNA abundance and intestinal epithelial barrier function. (A and E) n = 24 per condition. (B, C, F, G, and I) n = 6 per condition. (H) n = 16 per condition. (B, C, F, G, and I) Mice on day 6 following LPS administration. Error bars, means ± SEM. ***P < 0.001; **P < 0.01; *P < 0.05; one-way ANOVA with Tukey’s multiple comparisons test (A) to (I).
Therapeutic effect of Zanamivir with maintenance of IAP abundance and function
The marketed antiviral drug Zanamivir inhibits influenza neuraminidase activity (31), and has been used in research on mammalian Neu isozymes (24, 32, 33). At pharmacological dosages, Zanamivir inhibits Neu2, Neu3, and Neu4, but not Neu1 (32, 33). We found that oral treatment with Zanamivir maintained normal Neu activity levels in animals experiencing recurrent ST infections (Fig. 7A). Zanamivir did not block the physical induction of Neu3, which persisted for at least 20 weeks following the last ST infection (Fig. 7B). However, the inhibition of Neu activity by Zanamivir maintained host AP activity, IAP abundance, and normal lectin-binding patterns among apical glycoproteins of the small intestinal epithelium (Fig. 7C, Fig. 7D, and Fig. S8). The maintenance of IAP expression and activity was linked to the retention of relatively low LPS-phosphate levels that were otherwise elevated by ST infection (Fig. 7E). Zanamivir inhibited IAP de-sialylation coincident with normal IAP expression at the enterocyte cell surface (Fig. 7, F and G). Zanamivir also inhibited the induction of inflammatory cytokines and reduced the appearance of disease markers of intestinal inflammation including alterations of commensal microbiota, and barrier dysfunction (Fig. 7, H–J and Fig. S9). In comparisons with the acute and chronic model of chemically induced colitis using dextran sodium sulfate (DSS) (34, 35), Zanamivir had no effect, whereas ST3Gal6 deficiency exacerbated disease signs, consistent with the presence of different mechanisms of inflammation that may be responsive to IAP treatment (Fig. S10). Our findings together indicate a disease mechanism of environmental and pathogen origin that encompasses the different locations and functions of the small intestine and colon (Fig. 8).
Fig. 7. Effects of neuraminidase inhibitor Zanamivir on intestinal inflammation.
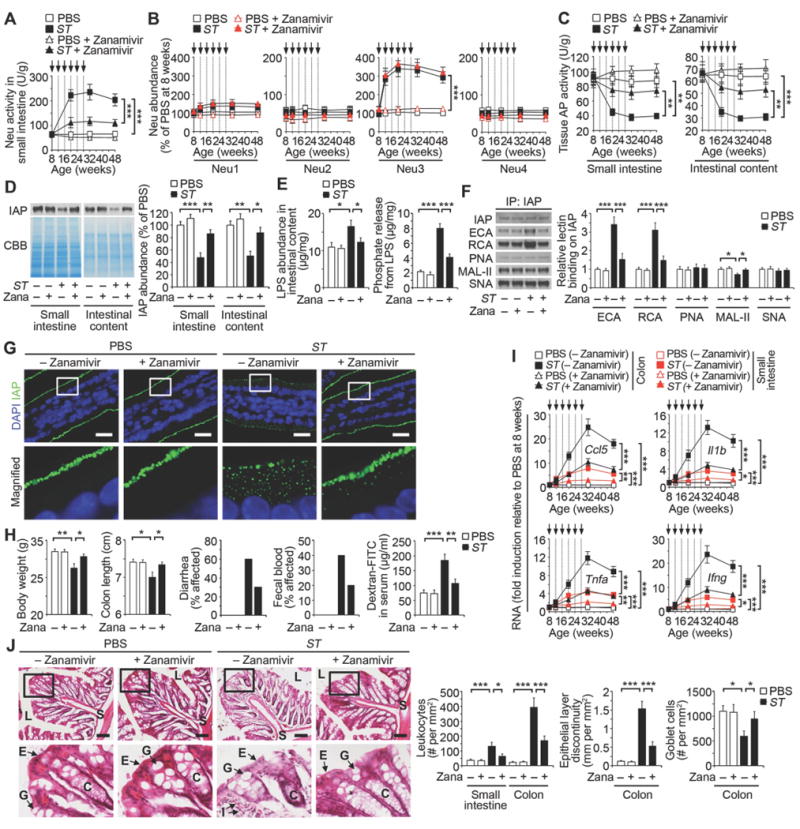
Wild-type mice were analyzed at indicated ages prior to ST re-infection (arrows) in absence or presence of Zanamivir (Zana) (0.5 mg/ml) provided in drinking water immediately after first infection. (A) Neu activity. (B) Neu1, 2, 3 and 4 protein abundance in small intestine. (C) AP activity. (D) IAP protein abundance. (E) LPS abundance and phosphate released from LPS of intestinal content. (F) Lectin blotting of IAP protein from small intestine. (G) In situ localization of IAP in duodenum, representative of ten fields of view (four mice per condition). Scale bars, 20 µm. (H) Body weight (n = 10 per condition), colon length (n = 8 per condition), diarrhea (n = 10 per condition) and fecal blood (n = 10 per condition) at 32 weeks of age and intestinal epithelial barrier function (n = 8 per condition) at 20 weeks of age. (I) Inflammatory cytokine RNA abundance. (J) H & E-stained colon sections at 32 weeks of age. Graphs are representative of ten fields of view (four mice per condition). Scale bars: 100 µm. (D–G) Mice at 20 weeks of age. (A and C) n = 32 per condition. (B and I) n = 30 per condition. (D–F) n = 6 per condition. Error bars, means ± SEM. ***P < 0.001; **P < 0.01; *P < 0.05; one-way ANOVA with Tukey’s multiple comparisons test (A) to (J).
Fig. 8. Model of intestinal inflammation due to recurrent Gram-negative ST infection.
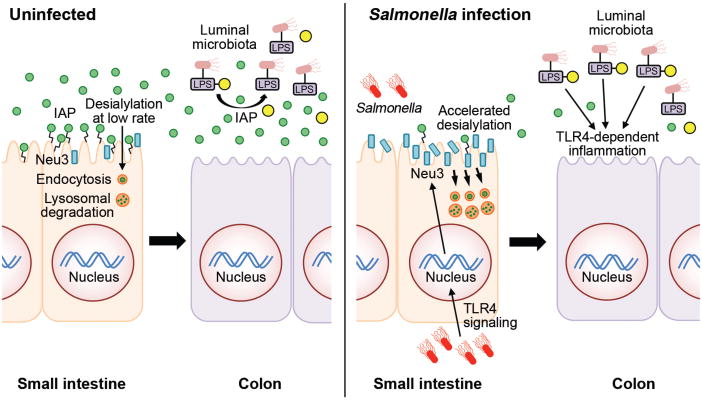
In the absence of infection of the small intestine, the anti-inflammatory GPI–linked IAP glycoprotein (green circles) is highly expressed on the enterocyte cell surface. IAP is eventually released into the lumen and travels through the intestinal tract to the colon where it detoxifies LPS-phosphate produced by Gram-negative and commensal bacteria via dephosphorylation (yellow circles). Nascent IAP at the enterocyte cell surface undergoes a low rate of desialylation linked to the rate of internalization and degradation involving a normal mechanism of IAP aging and turnover. Enterocytes of the small intestine respond to LPS-phosphate and ST infection by activating Tlr4 function, which induces host Neu3 neuraminidase (blue bars) on the enterocyte surface. Increased neuraminidase activity accelerates the rate of IAP de-sialylation and internalization (orange circles), reducing IAP abundance and resulting in increased levels of LPS-phosphate in the colon where Tlr4 activation elicits inflammation and disease.
Discussion
An increasingly severe colitis developed from recurrent low-titer non-lethal transient gastric infections of the Gram-negative pathogen ST. In this mouse model of repeated human food poisoning, the host rapidly cleared the pathogen. Nevertheless, these subsequent recurrent infections progressively disabled a mechanism in the host that normally protects against spontaneous intestinal inflammation. This anti-inflammatory mechanism operates primarily in the colon but is dependent upon IAP production and release from duodenal enterocytes of the small intestine. ST infection targeted this protective mechanism by activating host Tlr4 function in the duodenum and thereby inducing host Neu activity with elevated Neu3 expression at the luminal surface of the enterocyte. Neu induction accelerated the rate of nascent IAP desialylation on the enterocyte cell surface, reducing IAP half-life, causing IAP internalization and degradation, and resulting in a downstream IAP deficiency in the colon. IAP deficiency was linked to deficient dephosphorylation of LPS endotoxin molecules produced by commensal microbiota. This Tlr4-dependent disease manifested primarily in the colon with signs more similar to UC than to Crohn’s disease, and was linked to increases of the pro-inflammatory Tlr4 ligand, LPS-phosphate. Similarly, the genetic disruption of ST3Gal6-dependent sialic acid linkage formation during IAP synthesis caused IAP deficiency resulting in a spontaneous colitis that increased in severity with age and which was exacerbated by recurrent ST infections. In both cases, diminished glycoprotein sialylation among enterocytes resulted in reduced IAP half-life and deficiency with markedly elevated LPS-phosphate abundance in the colon. Consistent with a Tlr4-dependent mechanism, LPS administration alone recapitulated Neu3 induction and IAP deficiency, bypassing the requirement for IAP deficiency to increase colonic LPS-phosphate levels in provoking inflammation.
Intestinal inflammation failed to resolve following the discontinuation of periodic recurrent infections and persisted for months afterwards as a lasting outcome. The degree of this persistence may be determined in part by initial infection titers and the time between recurrent infections, and may result from multiple mechanisms. One possibility is the generation of epigenetic modifications to inflammatory gene promoters regulated by Tlr4 function that result in the persistence of inflammatory cytokine expression and may explain observations of the slow resolution of inflammatory processes (36, 37). The enterocyte Neu3 allele is perhaps similarly regulated in this way as its expression remained induced long after periodic ST infections were discontinued. It is also possible that the escalating inflammation resulting from increased recruitment and activation of innate and adaptive immune cells reaches a point wherein the degree of immunological activation and damage to the epithelium is not easily reversed or attenuated. Disease persistence was also coincident with microbiota alterations that we found emerged concurrently with disease signs and endured following the discontinuation of recurrent infections. Interestingly, acute enteric infections with Yersinia can trigger gut microbiota dysbiosis and chronic inflammation after pathogen clearance in Tlr1-deficient mice (38). The microbiota alterations we observed predominantly involved Enterobacteriaceae, which are frequently imbalanced in studies of intestinal inflammatory disease (21,38).
Neuraminidases function in a variety of processes including pathogen virulence, glycan catabolism, and biological signaling (39–41). Neu function may differ among isozymes and origins, such as indicated from caecal sources in contributing to DSS-induced colitis (34). The Neu3 gene of mammals includes presumptive binding sites for transcriptional factors Stat-3, Rreb-1, MyoD, Ik-2, Pax-2, Aml1a, Hoxa9, and Meis-1, and may employ alternate promoters controlled by Sp1/Sp3 transcription factors (42). Indeed, Stat-3 and Sp1 transcription factors are activated by LPS (43, 44). Transcriptional activation of Neu3 may further underlie Neu3 induction in cancers of colon, renal, and prostate tissues, and mice lacking Neu3 exhibited fewer colitis-associated colonic tumors (45–47). Our studies of an ST isolate that lacks an annotated and demonstrable neuraminidase gene have further linked the degree of Neu3 RNA induction to elevation of Neu activity, supporting the view that host Neu3 is involved in the desialylation of the IAP glycoprotein thereby causing IAP internalization and deficiency. However it remains possible that the Neu activity we have measured comes from an unannotated Neu enzyme or the endogenous microbiota, neither of which can be resolved until Neu3-deficient mice are further investigated. Neu3 activity is primarily active towards gangliosides, however studies reported significant but lesser activity towards glycoprotein substrates (48). It is also possible that Neu3 could somehow act indirectly via desialylation of its canonical ganglioside substrates. Nevertheless, Neu3 has been reported to desialylate the epidermal growth factor receptor (EFGR) glycoprotein (49). Moreover, data similar to our findings herein have further implicated Neu3 in the desialylation of circulating glycoproteins in the blood linked to a mechanism determining the various half-lives of plasma proteins (24).
Regulation of enterocyte IAP trafficking by sialylation extends this recently discovered mechanism of secreted protein aging and turnover to include the determination of protein half-lives at the cell surface. Although ST infection resulted in the desialylation and internalization of multiple enterocyte glycoproteins expressed at the cell surface, the disease phenotype was largely due to IAP deficiency. This represents an example of a specific glycan linkage that is commonly found on secreted and cell surface proteins as having a biological purpose more restrictively associated with one or few such glycoproteins (50–52). This can be explained in part by the presence of multiple sialyltransferases operating in the intestinal tract that are responsible for the sialylation of different subsets of bioactive glycoproteins and that function in different biological processes, such as findings involving ST3Gal6 deficiency herein compared with other studies of ST3Gal4 deficiency (34). Additional factors that could influence IAP expression and disease onset include mutations of glycan acceptor sites of IAP and transcriptional or mutagenic modifications to relevant glycosyltransferase and glycosidase genes, each of which can contribute to glycoprotein function (53). In the control of IAP half-life, increased internalization and degradation of desialylated IAP infers that a sialic acid-binding lectin analogous to the mammalian siglecs of leukocytes (54) may normally bind nascent sialylated IAP on the enterocyte surface to inhibit premature IAP endocytosis thereby allowing subsequent release into the lumen. Alternatively, the exposure of underlying galactose may unmask cryptic ligands for galactose-binding lectins such as the galectins that provoke glycoprotein endocytosis (50, 55).
Diminished alkaline phosphatase activity has been described in patients with colitis and celiac disease and oral alkaline phosphatase supplementation is under study to treat inflammatory diseases including the IBDs (56–59). Moreover, Neu3 protein abundance and activity is increased in human IBD patients (60). In animal studies, IAP deficiency contributes to colitis and allowed greater bacterial passage from the intestinal lumen to the mesenteric lymph nodes (16, 35, 56, 61). We have found that IAP is highly regulated and that ST infection disables IAP function in host protection while progressively eroding microbial barriers by successive rounds of what might otherwise be considered as unproductive infections. Environmental Gram-negative pathogens that access and infect the small intestine may have a similar strategy towards misappropriating host Tlr4 function to diminish IAP activity and increase colonic LPS-phosphate levels thereby provoking intestinal inflammation. This further emphasizes the dual nature of host Tlr4 function, which may be either advantageous or disadvantageous depending upon the context of exposure and perhaps the severity of infection. The link identified herein between host Tlr4 function and Neu3 induction appears to favor the pathogen as it is currently unclear why Tlr4 signaling is linked to the induction of enterocyte Neu3 expression. This likely reflects contextual features of infection and possible advantageous host responses that remain to be discovered. IAP augmentation and Neu inhibition nevertheless represent candidate therapies for intestinal inflammation resulting from recurrent infections of the ST pathogen in a model of repeated human food poisoning that escalates the host production of LPS endotoxin and Tlr4 ligands.
Supplementary Material
One Sentence Summary.
A leading cause of human food poisoning disrupts a mechanism of host protection in causing chronic intestinal inflammation.
Acknowledgments
This research was funded by NIH grants HL125352 (J.D.M. and V.N.) and HL131474 (J.D.M., M.J.M., and V.N.). Additional support was provided by the Wille Family Foundation (J.D.M) and by SFB914 from the German Research Foundation (M.S.). The data presented in this study are in the main text and in the Supplementary Materials.
Footnotes
References
- 1.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;7351:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sansonetti PJ. War and peace at mucosal surfaces. Nat Rev Immunol. 2004;4:953–964. doi: 10.1038/nri1499. [DOI] [PubMed] [Google Scholar]
- 4.Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62:1505–1510. doi: 10.1136/gutjnl-2012-303954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halfvarson J. Genetics in twins with Crohn’s disease: less pronounced than previously believed? Inflamm Bowel Dis. 2011;17:6–12. doi: 10.1002/ibd.21295. [DOI] [PubMed] [Google Scholar]
- 6.Eckmann L. Animal models of inflammatory bowel disease: lessons from enteric infections. Ann N Y Acad Sci. 2006;1072:28–38. doi: 10.1196/annals.1326.008. [DOI] [PubMed] [Google Scholar]
- 7.Sonnenberg A. Seasonal variation of enteric infections and inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:955–959. doi: 10.1002/ibd.20408. [DOI] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention (CDC) Vital signs: incidence and trends of infection with pathogens transmitted commonly through food-foodborne diseases active surveillance network, 10 U.S. sites, 1996–2010. MMWR Morb Mortal Wkly Rep. 2011;60:749–755. [PubMed] [Google Scholar]
- 9.Scallan E, et al. Foodborne illness acquired in the United States-major pathogens. Emerg Infect Dis. 2011;17:7–15. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Majowicz SE, et al. The global burden of nontyphoidal Salmonella gastroenteritis. Clin Infect Dis. 2010;50:882–889. doi: 10.10861/650733. [DOI] [PubMed] [Google Scholar]
- 11.de Jong HK, Parry CM, van der Poll T, Wiersinga WJ. Host-pathogen interaction in invasive Salmonellosis. PLoS Pathog. 2012;10:e1002933. doi: 10.1371/journal.ppat.1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feasey NA, Dougan G, Kingsley RA, Heyderman RS, Gordon MA. Invasive non-typhoidal salmonella disease: an emerging and neglected tropical disease in Africa. Lancet. 2012;379:2489–2499. doi: 10.1016/S0140-67361161752-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hinnebusch BF, et al. Enterocyte differentiation marker intestinal alkaline phosphatase is a target gene of the gut-enriched Kruppel-like factor. Am J Physiol Gastrointest Liver Physiol. 2004;286:G23–G30. doi: 10.1152/ajpgi.00203.2003. [DOI] [PubMed] [Google Scholar]
- 14.Poelstra K, Bakker WW, Klok PA, Hardonk MJ, Meijer DK. A physiologic function for alkaline phosphatase: endotoxin detoxification. Lab Invest. 1997;76:319–227. [PubMed] [Google Scholar]
- 15.Koyama I, Matsunaga T, Harada T, Hokari S, Komoda T. Alkaline phosphatases reduce toxicity of lipopolysaccharides in vivo and in vitro through dephosphorylation. Clin Biochem. 2002;35:455–461. doi: 10.1016/S0009-91200200330-2. [DOI] [PubMed] [Google Scholar]
- 16.Bates JM, Akerlund J, Mittge E, Guillemin K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe. 2007;2:371–382. doi: 10.1016/j.chom.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sussman NL, et al. Intestinal alkaline phosphatase is secreted bidirectionally from villous enterocytes. Am J Physiol. 1989;257:G14–G23. doi: 10.1152/ajpgi.1989.257.1.G14. [DOI] [PubMed] [Google Scholar]
- 18.Ellies LG, et al. Sialyltransferase specificity in selectin ligand formation. Blood. 2002;100:3618–3625. doi: 10.1182/blood-2002-04-1007. [DOI] [PubMed] [Google Scholar]
- 19.Yang WH, Nussbaum C, Grewal PK, Marth JD, Sperandio M. Coordinated roles of ST3Gal-VI and ST3Gal-IV sialyltransferases in the synthesis of selectin ligands. Blood. 2012;120:1015–1026. doi: 10.1182/blood-2012-04-424366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13:321–335. doi: 10.1038/nri3430. [DOI] [PubMed] [Google Scholar]
- 21.Nagalingam NA, Lynch SV. Role of the microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2012;18:968–984. doi: 10.1002/ibd21866. [DOI] [PubMed] [Google Scholar]
- 22.Figueroa-Bossi N, Uzzau S, Maloriol D, Bossi L. Variable assortment of prophages provides a transferable repertoire of pathogenic determinants in Salmonella. Mol Microbiol. 2001;39:260–271. doi: 10.1046/j.1365-2958.2001.02234.x. [DOI] [PubMed] [Google Scholar]
- 23.Monti E, et al. Sialidases in vertebrates: a family of enzymes tailored for several cell functions. Adv Carbohydr Chem Biochem. 2010;64:403–479. doi: 10.1016/S0065-23181064007-3. [DOI] [PubMed] [Google Scholar]
- 24.Yang WH, et al. An intrinsic mechanism of secreted protein aging and turnover. Proc Natl Acad Sci USA. 2015;112:13657–13662. doi: 10.1073/pnas.1515464112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 26.Beutler B. Endotoxin, toll-like receptor 4, and the afferent limb of innate immunity. Curr Opin Microbiol. 2000;3:23–28. doi: 10.1016/S1369-52749900046-6. [DOI] [PubMed] [Google Scholar]
- 27.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;10:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 28.Huhta H, et al. The Expression of Toll-like Receptors in Normal Human and Murine Gastrointestinal Organs and the Effect of Microbiome and Cancer. J Histochem Cytochem. 2016;64:470–482. doi: 10.1369/0022155416656154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dheer R, et al. Intestinal Epithelial Toll-Like Receptor 4 Signaling Affects Epithelial Function and Colonic Microbiota and Promotes a Risk for Transmissible Colitis. Infect Immun. 2016;84:798–810. doi: 10.1128/IAI.01374-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leaphart CL, et al. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol. 2007;179:4808–4820. doi: 10.4049/jimmunol.179.7.4808. [DOI] [PubMed] [Google Scholar]
- 31.Moscona A. Neuraminidase inhibitors for influenza. N Engl J Med. 2005;353:1363–1373. doi: 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]
- 32.Hata K, et al. Limited inhibitory effects of oseltamivir and zanamivir on human sialidases. Antimicrob Agents Chemother. 2008;52:3484–3491. doi: 10.1128/AAC.00344-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stamatos NM, et al. LPS-induced cytokine production in human dendritic cells is regulated by sialidase activity. J Leukoc Biol. 2010;88:1227–1239. doi: 10.1189/jlb.1209776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang YL, Chassard C, Hausmann M, von Itzstein M, Hennet T. Sialic acid catabolism drives intestinal inflammation and microbial dysbiosis in mice. Nat Commun. 2015;6:8141. doi: 10.1038/ncomms9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramasamy S, et al. Intestinal alkaline phosphatase has beneficial effects in mouse models of chronic colitis. Inflamm Bowel Dis. 2011;17:532–542. doi: 10.1002/ibd.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dolowschiak T, et al. IFN-γ Hinders Recovery from Mucosal Inflammation during Antibiotic Therapy for Salmonella Gut Infection. Cell Host Microbe. 2016;20:238–249. doi: 10.1016/j.chom.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 37.Foster SL, Medzhitov R. Gene-specific control of the TLR-induced inflammatory response. Clin Immunol. 2009;130:7–15. doi: 10.1016/j.clim.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamdar K, et al. Genetic and metabolic signals during acute enteric bacterial infection alter the microbiota and drive progression to chronic inflammatory disease. Cell Host Microbe. 2016;19:21–31. doi: 10.1016/j.chom.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manco S, et al. Pneumococcal neuraminidases A and B both have essential roles during infection of the respiratory tract and sepsis. Infect Immun. 2006;74:4014–4020. doi: 10.1128/IAI.01237-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwerdtfeger SM, Melzig MF. Sialidases in biological systems. Pharmazie. 2010;65:551–561. doi: 10.1002/chin.201047267. [DOI] [PubMed] [Google Scholar]
- 41.Varki A, Gagneux P. Multifarious roles of sialic acids in immunity. Ann N Y Acad Sci. 2012;1253:16–36. doi: 10.1111/j.1749-6632.2012.06517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamaguchi K, et al. Regulation of plasma-membrane-associated sialidase NEU3 gene by Sp1/Sp3 transcription factors. Biochem J. 2010;430:107–117. doi: 10.1042/BJ20100350. [DOI] [PubMed] [Google Scholar]
- 43.Ma W, et al. The p38 mitogen-activated kinase pathway regulates the human interleukin-10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide-stimulated human macrophages. J Biol Chem. 2001;276:13664–13674. doi: 10.1074/jbc.M011157200. [DOI] [PubMed] [Google Scholar]
- 44.Carl VS, Gautam JK, Comeau LD, Smith MF., Jr Role of endogenous IL-10 in LPS-induced STAT3 activation and IL-1 receptor antagonist gene expression. J Leukoc Biol. 2004;76:735–742. doi: 10.1189/jlb.1003526. [DOI] [PubMed] [Google Scholar]
- 45.Kakugawa Y, et al. Up-regulation of plasma membrane-associated ganglioside sialidase (Neu3) in human colon cancer and its involvement in apoptosis suppression. Proc Natl Acad Sci USA. 2002;99:10718–10723. doi: 10.1073/pnas.152597199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawamura S, et al. Plasma membrane-associated sialidase (NEU3) regulates progression of prostate cancer to androgen-independent growth through modulation of androgen receptor signaling. Cell Death Differ. 2012;19:170–179. doi: 10.1038/cdd.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamaguchi K, et al. Reduced susceptibility to colitis-associated colon carcinogenesis in mice lacking plasma membrane-associated sialidase. PLoS One. 2012;7:e41132. doi: 10.1371/journal.pone.0041132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wada T, et al. Cloning, expression, and chromosomal mapping of a human ganglioside sialidase. Biochem Biophys Res Comm. 1999;261:21–27. doi: 10.1006/bbrc.1999.0973. doi: 1006/bbrc.1999.0973. [DOI] [PubMed] [Google Scholar]
- 49.Mozzi A, et al. NEU3 activity enhances EGFR activation without affecting EGFR expression and acts on its sialylation levels. Glycobiology. 2015;23:1499–1509. doi: 10.1093/jglycob/cwv026. [DOI] [PubMed] [Google Scholar]
- 50.Ohtsubo K, et al. Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell. 2005;123:1307–1321. doi: 10.1016/j.cell.2005.09.041. [DOI] [PubMed] [Google Scholar]
- 51.Weinhold B, et al. Genetic ablation of polysialic acid causes severe neurodevelopmental defects rescued by deletion of the neural cell adhesion molecule. J Biol Chem. 2005;280:42971–42977. doi: 10.1074/jbc.M511097200. [DOI] [PubMed] [Google Scholar]
- 52.Grewal PK, et al. ST6Gal-I restrains CD22-dependent antigen receptor endocytosis and Shp-1 recruitment in normal and pathogenic immune signaling. Mol Cell Biol. 2006;26:4970–4981. doi: 10.1128/MCB.00308-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 54.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 55.Liu FT, Rabinovich GA. Galectins: regulators of acute and chronic inflammation. Ann N Y Acad Sci. 2010;1183:158–182. doi: 10.1111/j.1749-6632.2009.05131.x. [DOI] [PubMed] [Google Scholar]
- 56.Tuin A, et al. Role of alkaline phosphatase in colitis in man and rats. Gut. 2009;58:379–387. doi: 10.1136/gut.2007.128868. [DOI] [PubMed] [Google Scholar]
- 57.Lukas M, et al. Exogenous alkaline phosphatase for the treatment of patients with moderate to severe ulcerative colitis. Inflamm Bowel Dis. 2010;16:1180–1186. doi: 10.1002/ibd.21161. [DOI] [PubMed] [Google Scholar]
- 58.Molnar K, et al. Intestinal alkaline phosphatase in the colonic mucosa of children with inflammatory bowel disease. World J Gastroenterol. 2012;18:3254–3259. doi: 10.3748/wjg.v18.i25.3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alam SN, et al. Intestinal alkaline phosphatase prevents antibiotic-induced susceptibility to enteric pathogens. Ann Surg. 2014;259:715–722. doi: 10.1097/SLA.0b013e31828fae14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miklavcic JJ, et al. Increased catabolism and decreased unsaturation of ganglioside in patients with inflammatory bowel disease. World J Gastroenterol. 2015;21:10080–10090. doi: 10.3748/wjg.v21.i35.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goldberg RF, et al. Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc Natl Acad Sci USA. 2008;105:3551–3556. doi: 10.1073/pnas.0712140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heithoff DM, et al. Intraspecies variation in the emergence of hyperinfectious bacterial strains in nature. PLoS Pathog. 2012;8:e1002647. doi: 10.1371/journal.ppat.1002647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu J, et al. Loss of intestinal core 1-derived O-glycans causes spontaneous colitis in mice. J Clin Invest. 2011;121:1657–1666. doi: 10.1172/JCI45538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Narisawa S, et al. Accelerated fat absorption in intestinal alkaline phosphatase knockout mice. Mol Cell Biol. 2003;23:7525–7530. doi: 10.1128/MCB.23.21.7525-7530.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ohtsubo K, Chen MZ, Olefsky JM, Marth JD. Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat Med. 2011;17:1067–1075. doi: 10.1038/nm.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ellies LG, et al. Sialyltransferase ST3Gal-IV operates as a dominant modifier of hemostasis by concealing asialoglycoprotein receptor ligands. Proc Natl Acad Sci USA. 2002;99:10042–10047. doi: 10.1073/pnas.142005099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davis MR, Jr, Goldberg JB. Purification and visualization of lipopolysaccharide from Gram-negative bacteria by hot aqueous-phenol extraction. J Vis Exp. 2012;63:3916. doi: 10.3791/3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee CH, Tsai CM. Quantification of bacterial lipopolysaccharides by the purpald assay: measuring formaldehyde generated from 2-keto-3-deoxyoctonate and heptose at the inner core by periodate oxidation. Anal Biochem. 1999;267:161–168. doi: 10.1006/abio.1998.2961. [DOI] [PubMed] [Google Scholar]
- 69.Foot NJ, et al. Ndfip1-deficient mice have impaired DMT1 regulation and iron homeostasis. Blood. 2011;117:638–646. doi: 10.1182/blood-2010-07-295287. [DOI] [PubMed] [Google Scholar]
- 70.Fuhrer A, et al. Milk sialyllactose influences colitis in mice through selective intestinal bacterial colonization. J Exp Med. 2010;207:2843–2854. doi: 10.1084/jem.20101098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zaki MH, et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.