

Abstract
Background and Purpose
Non‐alcoholic steatohepatitis (NASH) is the hepatic manifestation of metabolic syndrome and is characterized by steatosis, inflammation and fibrosis. Soluble guanylate cyclase (sGC) stimulation reduces inflammation and fibrosis in experimental models of lung, kidney and heart disease. Here, we tested whether sGC stimulation is also effective in experimental NASH.
Experimental Approach
NASH was induced in mice by feeding a choline‐deficient, l‐amino acid‐defined, high‐fat diet. These mice received either placebo or the sGC stimulator IW‐1973 at two different doses (1 and 3 mg·kg−1·day−1) for 9 weeks. IW‐1973 was also tested in high‐fat diet (HFD)‐induced obese mice. Steatosis, inflammation and fibrosis were assessed by Oil Red O, haematoxylin–eosin, Masson's trichrome, Sirius Red, F4/80 and α‐smooth muscle actin staining. mRNA expression was assessed by quantitative PCR. Levels of IW‐1973, cytokines and cGMP were determined by LC‐MS/MS, Luminex and enzyme immunoassay respectively.
Key Results
Mice with NASH showed reduced cGMP levels and sGC expression, increased steatosis, inflammation, fibrosis, TNF‐α and MCP‐1 levels and up‐regulated collagen types I α1 and α2, MMP2, TGF‐β1 and tissue metallopeptidase inhibitor 1 expression. IW‐1973 restored hepatic cGMP levels and sGC expression resulting in a dose‐dependent reduction of hepatic inflammation and fibrosis. IW‐1973 levels were ≈40‐fold higher in liver tissue than in plasma. IW‐1973 also reduced hepatic steatosis and adipocyte hypertrophy secondary to enhanced autophagy in HFD‐induced obese mice.
Conclusions and Implications
Our data indicate that sGC stimulation prevents hepatic steatosis, inflammation and fibrosis in experimental NASH. These findings warrant further evaluation of IW‐1973 in the clinical setting.
Abbreviations
- ATG
autophagy‐related protein
- BAT
brown adipose tissue
- CDAAH
choline‐deficient, l‐amino acid‐defined, high‐fat
- COL1A1/COL1A2
collagen type I α1/α2
- FAT/CD36
fatty acid translocase
- GUCY1A2
guanylate cyclase 1 soluble subunit α2
- HFD
high‐fat diet
- HSL
hormone‐sensitive lipase
- IL‐1ra
IL‐1 receptor antagonist
- LC3
microtubule‐associated proteins 1A/1B light chain 3
- MCP‐1
monocyte chemotactic protein 1
- NAFLD
non‐alcoholic fatty liver disease
- NAS
non‐alcoholic fatty liver disease activity score
- NASH
non‐alcoholic steatohepatitis
- sGC
soluble GC
- SREBP‐1c
sterol response element‐binding protein 1c
- TAGs
triglycerides
- UCP1
uncoupling protein 1
- VASP
vasodilator‐stimulated phosphoprotein
- WAT
white adipose tissue
- α‐SMA
α‐smooth muscle actin
Introduction
Non‐alcoholic fatty liver disease (NAFLD) is a multifaceted condition that includes pathological manifestations ranging from the accumulation of neutral lipids in the cytoplasm of hepatocytes (steatosis) to the combination of steatosis with inflammation, a stage known as non‐alcoholic steatohepatitis (NASH) (Sanyal, 2005). NASH is the aggressive form of NAFLD and increases the risk for advanced liver disease culminating in hepatic fibrosis, cirrhosis and hepatocarcinoma (Sanyal, 2005). The mechanisms underlying the progression of NASH have not been completely elucidated, but repeated insults to the liver of these patients lead to inflammatory injury, hepatocyte death and the expansion of extracellular matrix (fibrosis) (Friedman, 2004). Therefore, pharmacological interventions that protect liver cells from inflammatory injury and fibrosis could be useful as a therapy for NASH.
Cyclic nucleotides, such as cGMP, play a major role in cell signalling and tissue homeostasis. Pharmacological modulation of the cGMP pathway is a potential target for therapy in complex diseases (Francis et al., 2010; Buglioni and Burnet, 2016). Indeed, small molecules with the ability to activate/stimulate soluble guanylate cyclase (sGC), which produces the second messenger cGMP in response to NO binding, have been reported to be therapeutically efficacious in several conditions, including pulmonary hypertension and chronic thromboembolic pulmonary hypertension (Ghofrani et al., 2013a,b). In preclinical models, drugs modulating the NO–sGC–cGMP pathway have demonstrated potential therapeutic roles in hypertension, atherosclerosis, restenosis and thrombosis (Tulis et al., 2000; Stasch et al., 2002). Also, at the preclinical level, these molecules have been shown to have important reno‐protective and cardiac‐protective actions, ameliorating renal and cardiac fibrosis (Hohenstein et al., 2005; Patrucco et al., 2014; Stasch et al., 2015). Molecules with the ability to impact the NO–sGC–cGMP pathway block inflammatory responses and the conversion of human lung fibroblasts into myofibroblasts (Ahluwalia et al., 2004; Pan et al., 2005; Dunkern et al., 2007). Therefore, pharmacological interventions that affect the NO–sGC–cGMP pathway are good candidates for decreasing inflammation and fibrosis in patients with NASH.
Alterations in the NO–sGC–cGMP pathway resulting in increased tissue cGMP concentrations can be induced by four different interventions, namely, through the use of (i) specific PDE5 and PDE9 inhibitors, (ii) NO donors, (iii) sGC activators and (iv) sGC stimulators (Francis et al., 2010; Buglioni and Burnet, 2016). PDE5 inhibitors, which prevent cGMP hydrolysis, were unable to enhance the exercise capacity of patients with heart failure (Redfield et al., 2013), and NO donors depend on a patient's biometabolism to be activated (Buglioni and Burnet, 2016). In contrast, direct sGC stimulators and activators provide a feasible alternative for the pharmacological manipulation of the NO–sGC–cGMP pathway. Activators of sGC are NO‐ and haem‐independent agonists that act only when the haem group of sGC is oxidized or when sGC is in a haem‐free state. In contrast, stimulators of sGC act directly on the reduced form of this enzyme in a haem‐dependent manner, increasing its sensitivity to NO and thereby amplifying the effects of NO (Buglioni and Burnet, 2016). These characteristics allow sGC stimulators to increase cGMP signalling even when the endogenous NO/cGMP pathway is impaired (Stasch and Hobbs, 2009). In this study, we explored the effects of the sGC stimulator IW‐1973 which is under clinical investigation for the treatment of heart failure with preserved ejection fraction but with the added complications of diabetes. IW‐1973 was selected because of its extensive tissue distribution, high oral bioavailability and long pharmacokinetic half‐life, which is consistent with once daily dosing in humans (Hanrahan et al., 2017). Specifically, in this study, we explored the effects of IW‐1973 in an optimized murine model of NASH induced by the combination of a choline‐deficient l‐amino acid‐defined diet with a high‐fat (CDAAH) diet (Matsumoto et al., 2013). This NASH model is useful for gaining further insights into human liver disease and for the development of effective therapies because it induces hepatic inflammation that rapidly progresses to liver fibrosis. In addition, since adiposity plays a major role in NASH progression (Shoelson et al., 2007), we also explored the effects of IW‐1973 in the classic, long‐term model of dietary obesity induced by feeding a high‐fat diet (HFD).
Methods
Animals and experimental design
Male C57BL/6J mice (Charles River Laboratories, Saint‐Aubin‐lès‐Elbeuf, France) were housed on woodchip bedding in cages with 50–60% humidity and a 12 h light/dark cycle and given free access to food and water. At 6 weeks of age (18–22 g), mice were randomly assigned to four independent groups, which received the following diets for 9 weeks: group 1 were fed a standard pellet chow diet (D12450J; 10% kcal from fat) (n = 10); group 2 were fed the CDAAH diet (A06071302; 60% kcal from fat) (n = 15), as described by Matsumoto et al. (2013); group 3 were fed the CDAAH supplemented with the sGC stimulator IW‐1973 (1 mg·kg−1·day−1) (n = 10); and group 4 were fed the CDAAH plus IW‐1973 (3 mg·kg−1·day−1) (n = 10). Diets were prepared by Research Diets (New Brunswick, NJ, USA). Additional groups of mice were fed the standard pelleted chow (n = 5), a HFD (D12492i; 60% kcal from fat) (n = 10), a HFD supplemented with IW‐1973 (1 mg·kg−1·day−1) (n = 10) or a HFD plus IW‐1973 (3 mg·kg−1·day−1) (n = 10) for 16 weeks. At the end of the intervention period, mice were killed by an overdose of anaesthetic (a mixture of 0.1 mg ketamine.g‐1 body wt and 0.01 mg xylazine.g‐1 body wt). Blood was collected, and plasma was obtained by centrifugation at 800× g for 10 min. Liver, epididymal white adipose tissue (WAT) and scapular brown adipose tissue (BAT) were excised and rinsed in Dulbecco's PBS with calcium and magnesium (DPBS++). Portions of liver and WAT were fixed in 10% formalin and embedded in paraffin. Other portions of liver were placed in OCT immersed in cold 2‐methylbutane on dry ice and kept at −80°C. Portions of liver, WAT and BAT were also snap frozen in liquid nitrogen for further analysis. The administration of the experimental diets had no major impact on animal welfare, apart from a slight decrease in body weight in animals receiving the CDAAH diet. All studies were conducted in accordance with the criteria of the Investigation and Ethics Committee of the Hospital Clínic and the European Community laws governing the use of experimental animals. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Biochemical analyses
Plasma concentrations of glucose, total cholesterol, triglycerides (TAGs), aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were determined by standard laboratory procedures.
Tissue TAG and cholesterol levels
Liver tissue was homogenized in NP40 Substitute Assay Reagent (Cayman Chemicals, Ann Arbor, MI, USA) containing protease inhibitors (Complete Mini; Roche Diagnostics, Basel, Switzerland). Homogenates were centrifuged 10 min at 10 000× g at 4°C, and supernatants were diluted 1:2–1:10 or 1:100–1:400 in assay buffer for TAG and cholesterol measurements respectively. Samples were assayed using triglyceride colorimetric (reference 10010303; Cayman Chemicals) or cholesterol fluorimetric (reference 10007640; Cayman Chemicals) assay kits.
Histological analysis of liver steatosis
Hepatic steatosis was assessed by Oil Red O staining of OCT‐embedded cryosections. Briefly, liver sections were fixed in 60% isopropanol for 10 min and stained with 0.3% Oil Red O in 60% isopropanol for 30 min and subsequently washed with 60% isopropanol. Sections were counterstained with Gill's haematoxylin, washed with 4% acetic acid solution and mounted with aqueous solution. Sections were visualized at 200× magnification in a Nikon Eclipse E600 microscope (Nikon, Kawasaki, Japan). Relative areas of steatosis, expressed as % of Oil Red O staining, were quantified by histomorphometry using a computerized image analysis system (Cell^D; analySIS imaging processing, Münster, Germany). A minimum of 10 independent fields per sample were evaluated.
Histological analysis of inflammation and fibrosis
Liver and WAT samples were fixed in 10% formalin, embedded in paraffin, cut into 5 μm sections and stained with haematoxylin–eosin (H&E) at the Pathology Department of the Hospital Clínic. Liver necroinflammatory injury at 200× magnification was analysed blinded, and the NAFLD activity score (NAS) was calculated by a registered pathologist unaware of the treatments (A. D.). The NAS grading system was described by Liang et al. (2014) as a simple NAFLD scoring system with high reproducibility that is applicable for different rodent models and for all stages of NAFLD aetiology. NAS is calculated as the unweighted sum of the scores for steatosis (including macro‐vesicular and micro‐vesicular steatosis separately and by hepatocellular hypertrophy) and inflammation (scored by analysing the amount of inflammatory foci/field). Liver and WAT sections were also stained with Sirius Red. Briefly, sections were incubated for 10 min in 0.5% thiosemicarbazide and stained in 0.1% Sirius Red F3B in saturated picric acid for 1 h, followed by a wash with a 0.5% acetic acid solution. Fibrosis was also assessed by Masson's trichrome staining at the Pathology Department of the Hospital Clínic. Sections were visualized at 200× magnification in a Nikon Eclipse E600 microscope. Relative areas of fibrosis, expressed as % of Sirius Red or Masson's trichrome staining, were quantified by histomorphometry using a computerized image analysis system (Cell^D; analySIS imaging processing). A minimum of 10 independent fields per sample was evaluated.
Morphometric analysis of adipose tissue sections
Paraffin‐embedded WAT samples were cut into 5 μm sections and stained for H&E and Sirius Red as described above. For image analysis, six photomicrographs per section were randomly obtained under a Nikon Eclipse E600 microscope at 200× magnification. Adipocyte size was calculated from cross‐sectional areas obtained from perimeter tracings using ImageJ software (Macbiophotonics, McMaster University, Hamilton, ON, Canada).
Isolation of hepatocytes, hepatic stellate cells and Kupffer cells
Hepatocytes and non‐parenchymal liver cells were isolated from C57BL/6J mice by a three‐step in situ perfusion procedure using 0.04% collagenase IV through the portal vein, as previously described by López‐Vicario et al. (2015) and Morán‐Salvador et al. (2013).
Differentiation and incubation of 3T3‐L1 adipocytes
Mouse 3T3‐L1 cells were seeded onto 12‐well plates (75 000 cells per well) in DMEM supplemented with 10% (v·v‐1) FBS, 100 U·mL−1 penicillin/streptomycin and 4 mM l‐glutamine in a humidified atmosphere of 5% CO2 at 37°C and allowed to grow to confluence for 2 days. Confluent 3T3‐L1 cells were cultured in adipocyte induction medium containing insulin (5 μg·mL−1), IBMX (0.5 mM), dexamethasone (0.25 μM), penicillin/streptomycin (100 U·mL−1) and l‐glutamine (4 mM) in DMEM supplemented with 10% FBS. After 2 days, the cells were cultured in continuation medium (5 μg·mL−1 insulin) for 3 days and then maintained in DMEM supplemented with 10% FBS until exhibiting an adipocyte phenotype, as assessed in a phase‐contrast microscope. At days 7 and 8 of differentiation, adipocytes were incubated with vehicle (0.005% DMSO) or IW‐1973 at 1, 3 and 10 μM concentrations for 24 h. At the end of the incubation period, adipocytes were scraped into ice‐cold DPBS−/− and resuspended in lysis buffer for protein extraction.
Gene expression analysis by real‐time PCR
Isolation of total RNA was performed using the TRIzol reagent (MRC, Cincinnati, OH, USA). RNA concentration was assessed in a NanoDrop‐1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), and its integrity was tested on a 6000 LabChip in a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). cDNA synthesis from 200 to 1000 ng of total RNA was performed using the High‐Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA). For gene expression quantification by real‐time PCR, validated and pre‐designed TaqMan Gene Expression Assays were used [collagen type I α1 chain (Col1a1; ID: Mm00801666_g1), collagen type I α2 chain (Col1a2; ID: Mm00483888_m1), MMP2 (ID: Mm00439498_m1), TGF‐β1 (ID: Mm00441724_m1), tissue metallopeptidase inhibitor 1 (TIMP‐1; ID: Mm00441818_m1), TNF‐α (ID: Mm00443258_m1), monocyte chemotactic protein 1 (MCP‐1 also known as CCL2; ID: Mm00441242_m1), IL‐6 (ID: Mm00446190_m1), IL‐10 (ID: Mm01288386_m1), IL‐1β (ID: Mm01336189_m1), IL‐1 receptor antagonist (IL‐1ra; ID: Mm00446186_m1), NOD‐like receptor family, pyrin domain containing 3 (NLRP3; ID: Mm00840904_m1), uncoupling protein 1 (UCP1; ID: Mm01244861_m1), cell death activator A (CIDEA; ID: Mm00432554_m1), PPARγ coactivator 1a (PGC‐1α; ID: Mm01298835_m1) and PR domain containing 16 (PRDM16; ID: Mm00712556_m1), NOS3 (also known eNOS; ID: Mm00435217_m1), guanylate cyclase 1 soluble α2 (GUCY1A2; ID: Mm01253540_m1), protein kinase cGMP‐dependent 1 (PKG1; ID: Mm01207548_m1), PKG2 (ID: Mm00435938_m1), vasodilator‐stimulated phosphoprotein (VASP; ID: Mm01299587_m1), PDE5A (ID: Mm00463177_m1), PDE9A (ID: Mm00501039_m1), sterol response element‐binding protein‐1c (SREBP‐1c; ID: Mm00550338_m1), PPARα (ID: Mm00440939_m1) and fatty acid translocase (FAT/CD36; ID: Mm00432403_m1)], using β‐actin (ActB; ID: Mm00607939_s1) as an endogenous control. Real‐time PCR amplifications were performed in 7900HT Fast system (Applied Biosystems). PCR results were analysed with Sequence Detector Software version 2.3 (Applied Biosystems). The amount of target gene, normalized to β‐actin and relative to a calibrator, was determined by the arithmetic equation 2−∆∆Ct described in the comparative Ct method.
Analysis of GUCY1A2 by endpoint RT‐PCR
Endpoint amplification of GUCY1A2 and GAPDH was performed with the following specific oligonucleotides: GUCY1A2, sense 5′‐GAAGTCATCCTCGGCGTATC‐3′ and antisense 5′‐ACCCAGCTTTTTGTTTTTAGTTGA‐3′; GAPDH, sense 5′‐TCCAGTATGACTCCACTC‐3′ and antisense 5′‐ATTTCTCGTGGTTCACAC‐3′. The specificity of primers was confirmed in the GenBank database using the basic local alignment search tool. PCR products were analysed by electrophoresis in 2.0% agarose gels and visualized by SYBR safe staining, using a 100 bp DNA ladder (Invitrogen, Carlsbad, CA, USA).
Analysis of protein expression by Western blot
Total protein was extracted with a lysis buffer containing 50 mM HEPES, 20 mM β‐glycerophosphate, 2 mM EDTA, 1% Igepal, 10% (v·v‐1) glycerol, 1 mM MgCl2, 1 mM CaCl2 and 150 mM NaCl, supplemented with protease inhibitor (Complete Mini; Roche Diagnostics) and phosphatase inhibitor (PhosSTOP; Roche Diagnostics) mixtures. Homogenates were incubated on ice for 15 min with frequent vortexing. Homogenates were centrifuged at 16 000× g for 40 min at 4°C, and supernatants were collected. Hormone‐sensitive lipase (HSL), phospho (Ser563)‐HSL (p‐HSL), microtubule‐associated proteins 1A/1B light chain 3A/3B (LC3‐I/LC3‐II) and p62 protein expression were analysed by Western blot. A total of 50 μg of protein was resuspended in SDS‐containing Laemmli sample buffer and heated for 5 min at 95°C, and proteins were separated for 90 min at 120 V at 4°C in 10% SDS‐PAGE electrophoresis. Transfer was performed by the iBlot Dry Blotting System (Invitrogen) onto PVDF membranes at 20 V over 7 min. Membranes were then soaked for 45 min at room temperature in TBS (20 mM Tris/HCl pH 7.4 and 0.5 M NaCl) containing 0.1% (v·v‐1) Tween 20 (0.1% TBST) and 5% (w·v‐1) non‐fat milk powder. Blots were washed three times for 5 min each with 0.1% TBST and subsequently treated overnight at 4°C with primary rabbit anti‐mouse p‐HSL (4139; dilution 1:1000; Cell Signaling, Danvers, MA, USA), HSL (10006371; dilution 1:1000; Cayman, Ann Arbor, MI, USA), LC3‐I/LC3‐II (12741S; dilution 1/1000; Cell Signaling), p62 (Ab109012; dilution 1:10 000, Abcam, Cambridge, UK), autophagy‐related protein (ATG)7 (8558; dilution 1/1000; Cell Signaling), ATG12 (4180; dilution 1/1000; Cell Signaling), PKG1 (3248; dilution 1:1000; Cell Signaling) and PKG2 (Ab‐126; dilution 1:1000; MyBiosource, San Diego, CA, USA) or β‐actin (5125; dilution 1:1000; Cell Signaling) antibodies in 0.1% TBST. After the blots had been washed three times for 5 min each with 0.1% TBST, membranes were incubated for 1 h at room temperature with an HRP‐linked donkey anti‐rabbit secondary antibody (Ab6885; dilution 1:2000; Abcam) in 0.1% TBST containing 5% BSA. Bands were visualized using the EZ‐ECL chemiluminescence detection kit (Biological Industries, Kibbutz Beit HaEmek, Israel) in a LAS 4000 imaging system (GE Healthcare Life Sciences, Little Chalfont, UK) and quantified using Image GE ImageQuant TL analysis software.
Analysis of cytokine levels
TNF‐α and MCP‐1 levels in liver tissue were assessed in a Luminex 100 system (Luminex Corp., Austin, TX, USA) using a custom‐made MILLIPLEX MAP Mouse High Sensitivity T Cell Magnetic Bead Panel (Merck Millipore, Billerica, MA, USA). Briefly, 25 μL of diluent and 25 μL of liver protein extract were added to each well before the addition of premixed microbeads (25 μL). The plate was incubated overnight at 4°C with shaking, then washed and reincubated with 25 μL of detection antibody for 1 h. The plate was washed again and incubated with 25 μL of streptavidin–phycoerythrin for 30 min. The plate was finally washed twice, and the beads were resuspended with 150 μL of sheath fluid and analysed in the Luminex 100 system. Readouts were detected as mean fluorescence intensity by the instrument, and values were subsequently converted to pg·mL−1 by extrapolation from a set of standards that were run simultaneously in the assay.
Immunohistochemistry
Paraffin‐embedded tissue sections were deparaffinized, rehydrated and pretreated with sodium citrate (0.05%) to unmask the antigen, followed by incubation with H2O2 (0.3%) for 25 min at room temperature in dark conditions, to block endogenous peroxidase activity, and with 2% BSA for 20 min at room temperature, to avoid nonspecific binding of the primary antibody. Thereafter, the liver sections were incubated 1 h at room temperature with a primary rat anti‐mouse F4/80 antibody (dilution 1:100; Bio‐Rad, Hercules, CA, USA) followed by incubation for 15 min at room temperature with a biotinylated rabbit anti‐rat IgG secondary antibody (Abcam) and 30 min with HRP goat anti‐rabbit IgG secondary antibody (dilution 1:200; Abcam). For α‐smooth muscle actin (α‐SMA), sections were incubated with primary rabbit anti‐mouse α‐SMA antibody (dilution 1:250; Abcam) for 1 h with HRP goat anti‐rabbit IgG secondary antibody (dilution 1:200; Abcam) for 30 min. The signal was detected by the Bond Polymer Refine Detection system (Leica Biosystems, Wetzlar, Germany). The sections were visualized at 200×, and positive staining for F4/80 and α‐SMA was quantified by histomorphometry. The results are expressed as a percentage of the F4/80‐positive or α‐SMA‐positive area.
Tissue levels of cAMP and cGMP
Liver tissue was homogenized in 5 vol of 5% trichloroacetic acid (TCA) in order to purify tissue samples from PDEs. Homogenates were centrifuged 10 min at 1500× g at 4°C, and TCA was extracted three times from supernatants using 5 vol of water‐saturated diethyl ether. Extracted samples were heated at 70°C for 5 min to remove residual ether from aqueous layer and stored at 4°C for determination of cAMP and cGMP levels. Standard curves were prepared with the matrix solution of 5% TCA water‐saturated diethyl ether, and standards and samples were acetylated prior to the analysis in competitive enzyme immunoassays (cAMP: reference 581001 and cGMP: reference 581021; Cayman Chemicals).
Liver and plasma levels of IW‐1973
Liver tissue, plasma samples and standards were analysed using a Thermo Vantage Triple Quadrapole LC/MS‐MS (Thermo Fisher Scientific, Waltham, MA, USA) in positive ion‐mode electrospray ionization, linked to an Aquity UPLC (Waters, Milford, MA, USA). Liver tissues were weighed and diluted 30× with 10% acetic acid in 90% water and homogenized using an Omni Prep homogenizer (Omni International, Kennesaw, GA, USA). Liver tissue proteins and plasma samples were precipitated using 50% acetonitrile and 50% methanol containing 20 ng·mL−1 internal standard. The samples were centrifuged at 2670× g at 4°C for 30 min. The supernatant was transferred and dried under nitrogen at 55°C and resuspended in the starting mobile phase. A standard curve from 1–1000 ng·mL−1 was prepared in 30× diluted liver tissue and also in plasma. The samples were loaded onto a Thermo Hypersil Gold 2.1 × 50 mm, 5 μm particle‐size column with a javelin guard (Hypersil Gold 5 μm 10 × 2.1 mm). The mobile phase consisted of aqueous 0.1% formic acid (mobile phase A) and acetonitrile with 0.1% formic acid (mobile phase B). The flow rate was 0.4 mL·min−1. The gradient holds at 80% mobile phase A for 0.5 min, ramps to 5% mobile phase A by 2.5 min and holds for 0.5 min before returning to 80% mobile phase A by 4 min. The total run time was 4 min per sample.
Data and statistical analysis
Statistical analysis of the results was performed with GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA). For multiple comparisons, ANOVA was employed followed by Dunnett's post hoc test [only in those tests where F achieved the necessary level of statistical significance (P < 0.05)]. For single comparisons, Student's unpaired t‐test was used. Results are expressed as mean ± SEM. All measurements were undertaken in at least two technical replicates. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c,d).
Results
We first assessed the status of the NO–sGC–cGMP pathway in CDAAH mice. A schematic diagram of the NO–sGC–cGMP signalling pathway is shown in Figure 1A. As shown in Figure 1B, the gene encoding the key enzyme of this pathway (the sGC GUCY1A2) is constitutively expressed in liver tissue and quiescent hepatic stellate cells (HSC) but not in hepatocytes. GUCY1A2 expression was not detected in activated HSC and Kupffer cells by endpoint PCR. Consistent with impairment of the NO–sGC–cGMP pathway in liver disease, CDAAH mice showed a reduced expression of the genes encoding GUCY1A2 and NOS3 and lower tissue levels of cGMP than chow‐fed control mice (Figure 1C–E). CDAAH mice also displayed reduced mRNA expression of the cGMP‐metabolizing enzymes (i.e. PDE5A and PDE9A) (Figure 1F, G), whereas the expression of PKG1 and PKG2 (Figure 1H, I) and the downstream effector VASP (Supporting Information Figure S1A) was not different between CDAAH mice and controls. CDAAH mice had higher hepatic levels of cAMP compared with chow‐fed controls (Supporting Information Figure S1B). Administration of IW‐1973 to CDAAH mice increased GUCY1A2 and NOS3 expression to similar levels to controls while significantly raising cGMP concentrations in the livers of these animals (Figure 1C–E). Of interest, IW‐1973‐treated mice had a lower expression of cGMP‐metabolizing enzymes (Figure 1F, G). In addition, IW‐1973‐treated mice showed significantly lower mRNA expression of PKG2 (Figure 1I), a finding that was confirmed at the protein level by Western blot analysis (Figure 1J). There were no significant differences in hepatic PKG1 expression (Figure 1H, J) and cAMP levels (Supporting Information Figure S1B) between IW‐1973‐treated and CDAAH mice. Circulating plasma levels of IW‐1973, as determined by LC‐MS/MS, were 11.3 ± 3.7 nM in the group of mice receiving IW‐1973 at a dose of 1 mg·kg−1·day−1 and 30.4 ± 13.8 nM for those receiving IW‐1973 at a dose of 3 mg·kg−1·day−1. The 1 and 3 mg·kg−1·day−1 doses were selected to achieve plasma levels that have no effect on BP in normal mice, and the observed plasma levels of IW‐1973 have no effect on BP in normal mice where plasma levels of approximately 200 nM are required to produce a 4.5 mmHg drop in mean arterial BP measured by telemetry (data not shown). IW‐1973 concentrations in CDAAH mouse livers were significantly higher than in plasma, with levels of 457.4 ± 98.6 and 971.4 ± 190.7 nM for the 1 and 3 mg·kg−1· day−1 respectively.
Figure 1.
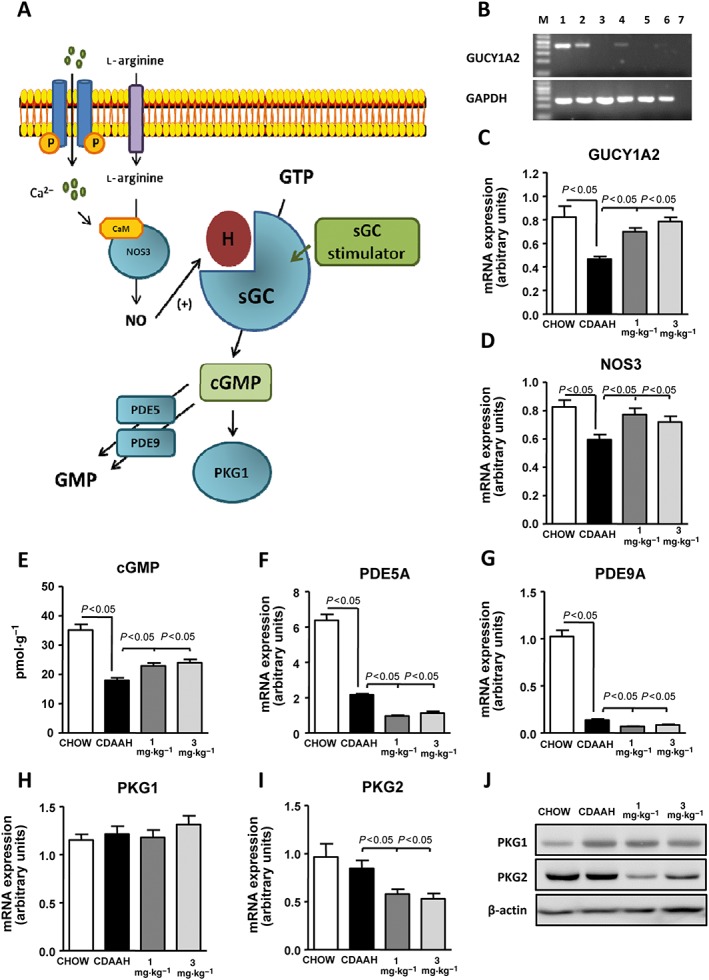
Effects of IW‐1973 on the hepatic cGMP pathway. (A) Schematic diagram summarizing the cGMP signalling pathway and sGC stimulator site of action. CaM, Ca2+‐calmodulin; H, haem group. (B) Representative PCR analysis of GUCY1A2 and GAPDH expression in liver tissue (lane 2), hepatocytes (lane 3), quiescent HSC (lane 4), activated HSC (lane 5) and Kuppfer cells (lane 6). Lane 1 is a positive control (brain tissue), and lane 7 is a blank negative control. (C, D) GUCY1A2 and NOS3 mRNA expression determined by real‐time PCR in liver samples from mice receiving chow diet (n = 10), choline‐deficient, CDAAH diet (n = 15), CDAAH plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or CDAAH plus IW‐1973 at 3 mg·kg−1 (n = 10). (E) Hepatic cGMP levels determined by enzyme immunoassay. (F, G) Hepatic PDE5A and PDE9A mRNA expression determined by real‐time PCR. (H, I) Hepatic PKG1 and PKG2 mRNA expression. (J) Hepatic PKG1 and PKG2 protein expression. Results are expressed as mean ± SEM.
Compared with chow‐fed mice, CDAAH‐fed mice had significantly lower body weights (Figure 2A), significantly higher liver‐to‐body weight ratios (Figure 2B) and lower BAT‐to‐body weight ratios (Supporting Information Figure S2A). No changes in WAT weight were observed (Figure 2C). CDAAH mice showed reduced plasma levels of glucose, total cholesterol and TAG (Supporting Information Figure S2B). Moreover, CDAAH mice exhibited exacerbated hepatic steatosis revealed by extensive Oil Red O staining of neutral lipids (Supporting Information Figure S2C) and hepatic TAG accumulation (Supporting Information Figure S2D). In agreement with enhanced steatosis, CDAAH mice showed down‐regulated expression of the lipolytic PPARα gene accompanied by a marked up‐regulation of FAT/CD36, a gene involved in hepatic fatty acid uptake (Supporting Information Figure S2E). No changes in hepatic cholesterol levels were observed (Supporting Information Figure S2F). Administration of the sGC stimulator IW‐1973 did not alter body weight (Figure 2A) but significantly reduced liver‐ and WAT‐to‐body weight ratios (Figure 2B, C). IW‐1973 also reduced BAT‐to‐body weight ratio (Supporting Information Figure S2A). A modest reduction in plasma glucose levels (Supporting Information Figure S2B) and Oil Red O staining (Supporting Information Figure S2C) was observed in mice receiving IW‐1973. No effects on plasma and hepatic TAG and cholesterol levels were observed (Supporting Information Figure S2B, D, F), whereas PPARα and FAT/CD36 expression was slightly modified after IW‐1973 treatment (Supporting Information Figure S2E).
Figure 2.
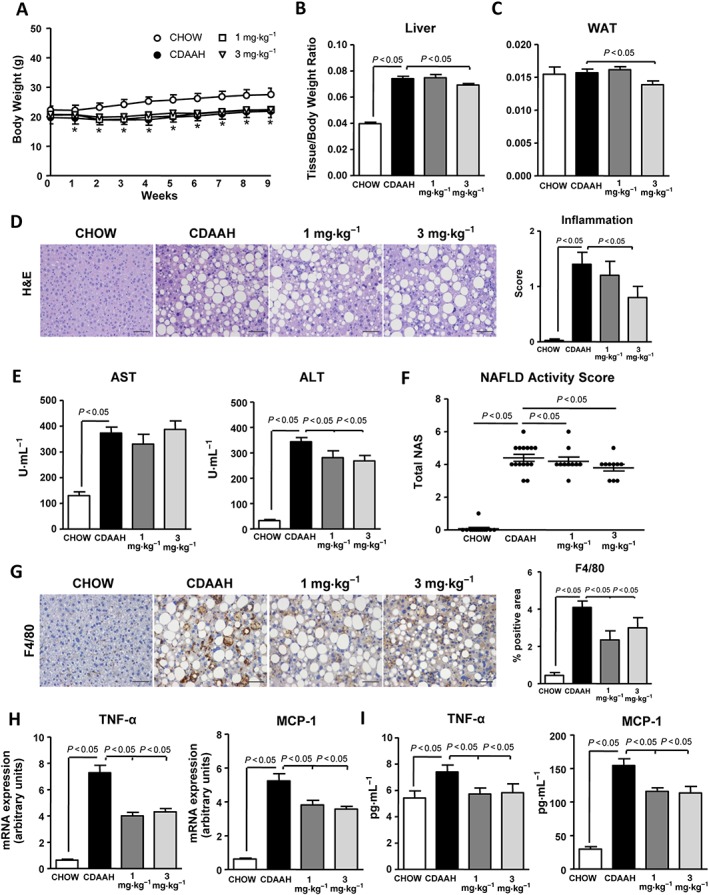
Effects of IW‐1973 on hepatic inflammation. (A) Body weight changes during the 9 weeks of treatment in mice receiving chow diet (n = 10), CDAAH diet (n = 15), CDAAH plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or CDAAH plus IW‐1973 at 3 mg·kg−1 (n = 10). (B, C) Liver and WAT weights, expressed as tissue/body weight ratio. (D) Representative photomicrographs (200× magnification) of liver sections stained with H&E. (Right) Inflammation scores in H&E‐stained liver sections scored by a registered pathologist. (E) Plasma AST and ALT levels. (F) NAS. Inflammation and steatosis were scored by a registered pathologist. NAS was calculated as described in the Methods section. (G) Representative photomicrographs (200× magnification) of liver sections stained with a specific F4/80 antibody used for the assessment of macrophage infiltration. (Right) Histomorphometric analysis of the area stained with the macrophage specific F4/80 antibody. (H) Hepatic TNF‐α and MCP‐1 mRNA expression. (I) Hepatic TNF‐α and MCP‐1 levels. Results are expressed as mean ± SEM. *P < 0.05 for CDAAH versus CHOW. Scale bar = 50 μm.
Consistent with the fact that CDAAH is an established model of inflammatory liver injury, CDAAH mice showed marked hepatic inflammation as compared with controls, as revealed by histological analysis of H&E‐stained liver sections (Figure 2D). CDAAH mice also showed increased plasma AST and ALT levels (Figure 2E). Consistently, CDAAH mice had a higher NAS compared with controls (Figure 2F). CDAAH mice also showed increased area of positive immunostaining for the specific macrophage marker F4/80, indicating greater macrophage infiltration (Figure 2G). Moreover, confirming these histological and immunohistochemical results, CDAAH mice had higher mRNA expression of TNF‐α and MCP‐1 in comparison with chow‐fed control mice (Figure 2H). Increased expression of these inflammatory genes was confirmed at the protein level (Figure 2I). Up‐regulation of IL‐6, IL‐10 and IL‐1ra was also seen in CDAAH mice (Supporting Information Figure S3). Addition of the sGC stimulator IW‐1973 to CDAAH diet was associated with significantly lower hepatic inflammation and plasma ALT levels, reduced NAS and macrophage infiltration and decreased tissue levels of TNF‐α and MCP‐1 (Figure 2D–I). Reduced IL‐6 and IL‐1ra expression was also seen in IW‐1973‐treated mice (Supporting Information Figure S3).
Consistent with the fact that CDAAH is an established model of inflammatory liver injury that rapidly progresses to liver fibrosis, CDAAH mice exhibited increased Masson's trichrome and Sirius Red staining as well as α‐SMA immunostaining indicative of extensive liver fibrosis (Figure 3A, B). Furthermore, the expression of fibrogenic genes, including COL1A1, COL1A2, MMP2, TGF‐β1 and TIMP‐1, was significantly up‐regulated in CDAAH mice (Figure 3C). In CDAAH mice receiving IW‐1973, the areas stained with Masson's trichrome, Sirius Red and α‐SMA were significantly smaller compared with CDAAH‐fed mice (Figure 3A, B). In addition, the expression of all fibrogenic genes was down‐regulated in mice receiving IW‐1973 (Figure 3C).
Figure 3.
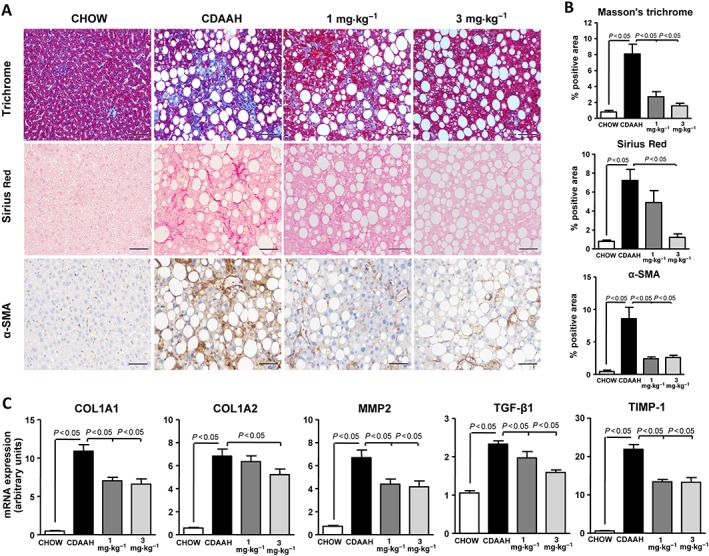
Effects of IW‐1973 on hepatic fibrosis. (A) Representative photomicrographs (200× magnification) of liver sections stained with Masson's trichrome, Sirius Red and a specific α‐SMA antibody used for the assessement of hepatic fibrosis in mice receiving chow diet (n = 10), CDAAH diet (n = 15), CDAAH plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or CDAAH plus IW‐1973 at 3 mg·kg−1 (n = 10). (B) Histomorphometric analysis of the area stained with Masson's trichrome, Sirius Red and α‐SMA. (C) Hepatic COL1A1, COL1A2, MMP2, TGF‐β1 and TIMP‐1 mRNA expression determined by real‐time PCR. Results are expressed as mean ± SEM. Scale bar = 50 μm.
We next explored the effects of IW‐1973 in the classic, long‐term model of obesity‐induced NASH induced by a HFD, which primarily is an established model of hepatic steatosis. As expected, HFD‐induced obese mice exhibited widespread Oil Red O staining, increased hepatic TAG levels and up‐regulated expression of the lipogenic regulator SREBP‐1c and the fatty acid oxidation regulator PPARα (Figure 4A–E). These mice exhibited very mild fibrosis (Figure 4A, B). The addition of the sGC stimulator IW‐1973 produced a significant reduction in hepatic steatosis, hepatic TAG levels and lipogenic gene expression and augmented gene expression of fatty acid oxidation regulators (Figure 4A–E). In contrast, IW‐1973 modestly attenuated Sirius Red and α‐SMA staining (Figure 4A, B). Significant reductions in plasma AST levels were observed in mice receiving IW‐1973 (Supporting Information Figure S4).
Figure 4.
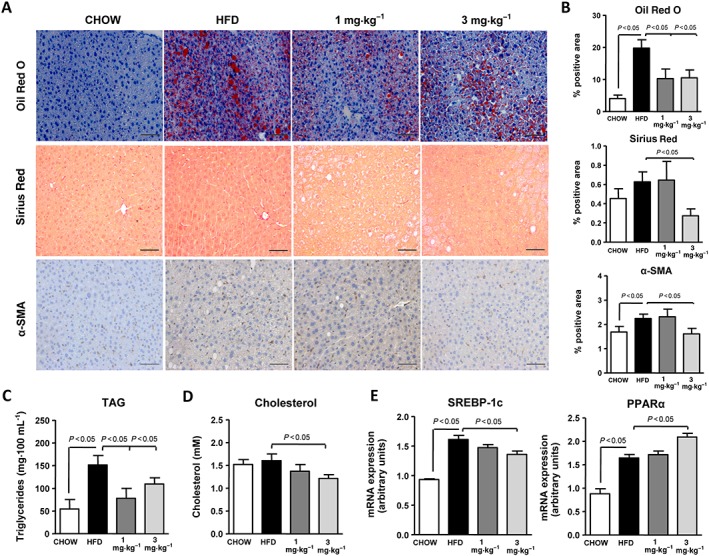
Effects of IW‐1973 on hepatic steatosis and fibrosis in the HFD obesity model. (A) Representative photomicrographs (200× magnification) of liver sections stained with Oil Red O, Sirius Red and α‐SMA antibody in mice receiving chow diet (n = 5), an obesogenic HFD diet (n = 10), HFD plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or HFD plus IW‐1973 at 3 mg·kg−1 (n = 10). (B) Histomorphometric analysis of the area stained with Oil Red O, Sirius Red and α‐SMA. (C) Hepatic TAG levels. (D) Hepatic cholesterol levels. (E) Hepatic SREBP‐1c and PPARα mRNA expression. Results are expressed as mean ± SEM. Scale bar = 50 μm.
WAT plays a crucial role in the development of obesity‐induced NASH (Shoelson et al., 2007). Therefore, we characterized the NO–sGC–cGMP pathway in this insulin‐sensitive tissue, in HFD‐induced obese mice. These mice gained more body weight and had higher WAT‐ and BAT‐to‐body weight ratios compared with controls (Figure 5A, B). As compared with lean chow‐fed control mice, HFD‐induced obese WAT also showed a greater number of crown‐like structures indicating enhanced macrophage infiltration (Figure 5C). Augmented infiltration of macrophages into WAT from HFD‐induced obese mice was confirmed by morphometric analysis of F4/80 immunostaining (Figure 5D). Of interest, morphometric analysis of H&E‐stained WAT sections revealed increased adipocyte area in obese mice (Figure 5C, E). These WAT phenotypic changes in obese mice were associated with significant down‐regulation of NOS3, GUCY1A2, PKG1, PKG2 and PDE5A (Supporting Information Figure S5A–E). VASP expression was not different between the two groups (Supporting Information Figure S5F). Increased tissue remodelling and fibrosis was also a hallmark of obese WAT, as revealed by enhanced Sirius Red staining and TIMP‐1 over‐expression (Supporting Information Figure S5G, H). Of note, TIMP‐1 expression was inversely correlated with the expression of the cGMP‐synthesizing enzyme sGC α subunit gene GUCY1A2 (Supporting Information Figure S5I). In HFD fed mice also treated with the sGC stimulator IW‐1973, the body weight or WAT‐ and BAT‐to‐body weight ratios were not altered compared with controls (Figure 5A, B). However, HFD mice treated with IW‐1973 had fewer crown‐like structures and a lower degree of macrophage infiltration compared with HFD mice receiving placebo (Figure 5C, D). Of interest, WAT from HFD‐induced obese mice treated with IW‐1973 contained adipocytes of reduced size as compared with WAT from placebo‐treated animals (Figure 5E). Smaller adipocyte area was not secondary to greater lipolysis and release of free fatty acids to the circulation, since phosphorylation of the master lipolytic enzyme HSL was not significantly modified by IW‐1973 (Figure 5F). However, IW‐1973 induced an increase in the autophagy flux in WAT, as a higher LC3II/LC3I ratio in conjunction with a lower p62 protein expression was observed in the groups of HFD obese mice receiving IW‐1973 (Figure 5G). Increased autophagy was confirmed in 3T3‐L1 adipocytes incubated with increasing concentrations of IW‐1973 (Figure 5H). Finally, sGC stimulation did not translate into increased WAT thermogenesis, since UCP1, CIDEA, PGC‐1α and PRDM16 expression remained unchanged after IW‐1973 treatment (Supporting Information Figure S6A). Similar effects were observed in BAT (Supporting Information Figure S6B).
Figure 5.
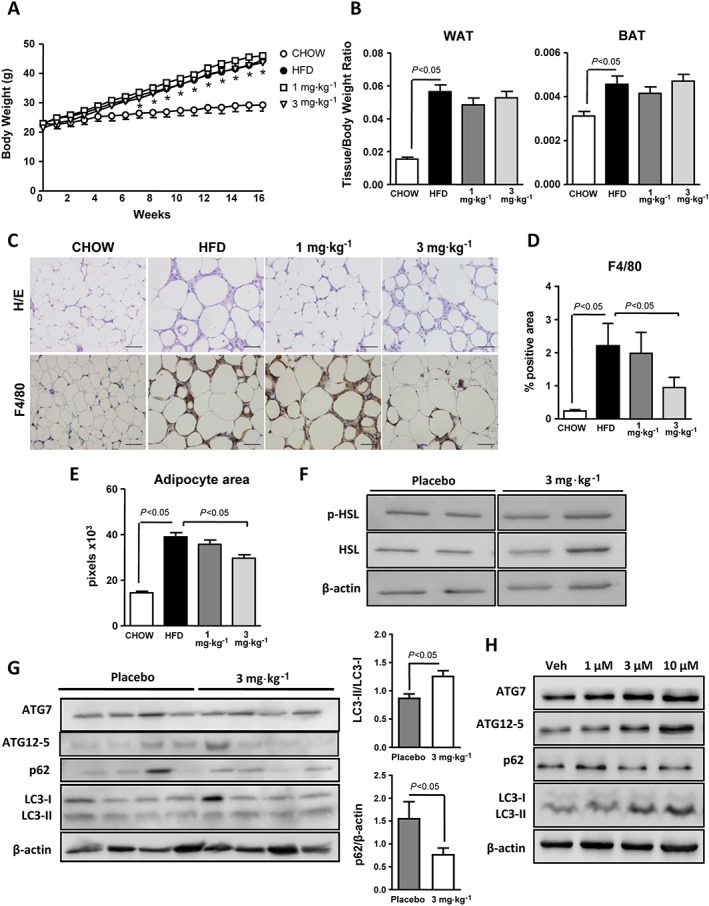
Effects of IW‐1973 on WAT. (A) Body weight progression during the 16 weeks of treatment of the mice receiving chow diet (n = 5), HFD diet (n = 10), HFD plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or HFD plus IW‐1973 at 3 mg·kg−1 (n = 10). (B) WAT and BAT weight to body weight ratios. (C) Representative photomicrographs (200× magnification) of WAT sections stained with H&E and a specific F4/80 antibody. (D) Histomorphometric analysis of the area immunostained with the specific F4/80 macrophage antibody. (E) Adipocyte area assessed by ImageJ morphometric analysis as described in the Methods section. (F) p‐HSL and β‐actin determined in WAT by SDS‐PAGE Western blot. (G) ATG7, ATG12‐5, p62, LC3‐I/ II and β‐actin determined in WAT by SDS‐PAGE Western blot analysis. The densitometric analyses of LC3‐II/LC3‐I ratio and p62 are shown on the right. (H) ATG7, ATG12‐5, p62, LC3‐I/LC3‐II and β‐actin in differentiated 3T3 adipocytes incubated with vehicle (Veh) or increasing concentrations of IW‐1973 (1, 3 and 10 μM). Results are expressed as mean ± SEM. *P < 0.05 for HFD versus CHOW. Blot data subjected to statistical analysis were from n = 5 experiments. Scale bar = 50 μm.
Discussion
Our study provides novel insights into the role of the NO–sGC–cGMP pathway in liver disease. In particular, our study provides evidence at the experimental level that sGC stimulation prevents hepatic steatosis, inflammation and fibrosis, which are the three key features of NASH. One of the major novelties of our study is that it demonstrates for the first time the hepatoprotective actions of a sGC stimulator in two distinct and independent experimental models of NASH. These two models cover the spectrum of liver abnormalities observed in NASH ranging from hepatic steatosis (HFD model) to inflammation progressing to fibrosis (CDAAH model). In the HFD model, a murine model of hepatic steatosis secondary to obesity, the sGC stimulator IW‐1973 had a profound anti‐steatotic effect and a moderate anti‐fibrotic action in the liver accompanied by the promotion of the healthy expansion of adipocytes in the WAT. In the CDAAH model, a murine model of hepatic inflammation and fibrosis, the sGC stimulator IW‐1973 ameliorated hepatic inflammation and exerted a remarkable protection against liver fibrosis in the context of a mild anti‐steatotic action.
As far as we know, the present report is the first study assessing the pharmacological modulation of sGC in the CDAAH model of NASH. The CDAAH is a new model that optimizes the original MCD model, which main drawback is the drastic loss of weight in the animals. In the CDAAH model, this disadvantage is superseded by combining the MCD and HFD models, resulting in a stabilization of the weight loss (Liang et al., 2014). Our data in these mice demonstrated that administration of IW‐1973 restored hepatic GUCY1A2 and NOS3 expression levels and down‐regulated PDE5 and PDE9, leading to the augmentation of hepatic cGMP levels. Augmented cGMP levels in response to IW‐1973 treatment correlated well with direct improvements in hepatic inflammation, resulting in reduced macrophage infiltration, hepatocyte ballooning and the mRNA and protein levels of TNF‐α and MCP‐1. These findings are in agreement with the results communicated by Ahluwalia et al. (2004), which demonstrated that sGC stimulators inhibit IL‐1β‐induced leukocyte rolling and adhesion, secondary to the modulation of adhesion molecules such as L‐selectin and P‐selectin. Furthermore, genetic ablation of cGMP‐dependent PKG1 or VASP causes liver and WAT inflammation (Handa et al., 2011; Lutz et al., 2011).
The most important finding of our study was the remarkable anti‐fibrotic effect exerted by the sGC stimulator IW‐1973 in the liver of CDAAH mice. Indeed, histopathological analysis of liver sections of mice receiving IW‐1973 revealed decreased Masson's trichrome and Sirius Red staining and α‐SMA immunostaining. Moreover, IW‐1973 exerted significantly down‐regulated a battery of genes involved in the fibrogenic/remodelling process such as COL1A1, COL1A2, MMP‐2, TGF‐β1 and TIMP‐1. IW‐1973 is a sGC stimulator with a large volume of distribution in the rat and particularly high levels in the liver (approximately 40‐fold higher levels compared with plasma). The increased liver distribution probably contributes to the antifibrotic effects observed with IW‐1973, which were achieved at doses that correspond to plasma concentrations that have no effect on systemic BP in normal mice. Our findings agree with previous publications demonstrating anti‐remodelling effects of agents modulating the cGMP pathway in in vivo and in vitro models of renal, cardiac, dermal and pulmonary fibrosis (Hohenstein et al., 2005; Dunkern et al., 2007; Cui et al., 2014; Patrucco et al., 2014; Beyer et al., 2015; Wang et al., 2016). Moreover, our findings are consistent with previous publications reporting the anti‐fibrotic actions of NO donors or sGC activators in vitro in renal myofibroblasts (Wang et al., 2016) and in vivo in models of biliary and toxic (i.e. carbon tetrachloride and thioacetamide) liver injury (Moal et al., 2006; Knorr et al., 2008; Xie et al., 2012). The mechanism underlying the anti‐fibrotic actions of agents working on the cGMP pathway is still unclear, but IW‐1973 is probably targeting hepatic stellate cells, the main fibrogenic cell type in the liver. In this regard, Hall et al. (2017) have recently confirmed earlier work showing that hepatic GUCY1 is mainly expressed in hepatic stellate cells but not in hepatocytes. These authors have also shown that hepatic stellate cells and not hepatocytes respond to sGC stimulation by IW‐1973 (Hall et al., 2017). In this study, Hall et al. (2017) have also provided evidence that IW‐1973 is able to block TGF‐β‐induced α‐SMA protein levels in hepatic stellate cells. These findings are consistent with our results showing a significant and dose‐dependent down‐regulation of TGF‐β1 in CDAAH mice receiving IW‐1973, as well as with previous studies indicating that the blockage of the non‐canonical TGF‐β pathway by increased cGMP may be responsible for the anti‐fibrotic effects (Cui et al., 2014, Beyer et al., 2015).
Another major finding of our study was that the administration of the sGC stimulator IW‐1973 blunted hepatic steatosis in the HFD‐induced obesity model. Although the mechanisms underlying this effect are not defined, Ayala et al. (2007) reported that chronic treatment with sildenafil, a selective PDE5 inhibitor, was accompanied by an improvement in energy balance and insulin sensitivity in HFD‐fed mice. In line with the anti‐steatotic actions seen in our study, cardiomyocytes overexpressing a constitutively active sGC showed better fatty acid trafficking and reduced TAG accumulation (Khairallah et al., 2008). An interesting feature of the HFD model is that it provides the optimal setting to investigate the influence of extrahepatic factors, mainly those derived from WAT, on the progression of hepatic steatosis (Heydemann, 2016). Indeed, our study demonstrated that the beneficial effects of the sGC stimulator IW‐1973 were not only limited to the liver but also extended into the WAT depot. WAT plays a critical role in regulating energy homeostasis, and its functional integrity is required for the balanced body metabolism of a healthy organism (Trayhurn, 2005; Shoelson et al., 2007). In obese conditions, the presence of a chronic ‘low‐grade’ state of inflammation in WAT leads to altered WAT endocrine and metabolic functions (Ferrante, 2007, Elks and Francis, 2010). As a consequence of this dysregulation, there is an unbalanced production of pro‐inflammatory adipokines accompanied by an impairment in WAT lipolysis, which results in an overspill of free fatty acids into the liver (Ouchi et al., 2011). Therefore, in recent years, WAT has emerged as a target for the prevention of NASH. The results of the current study demonstrate a significant reduction in macrophage infiltration in response to the administration of the sGC stimulator IW‐1973, thus suggesting that an amelioration of WAT inflammation contributed to the protective actions of this drug on the liver. This finding is consistent with those from a recent study by Sanyal et al. (2017) exploring the mechanisms underlying the anti‐inflammatory effects of sGC stimulators in WAT. These authors performed a comprehensive dissection of the cGMP signalling pathway in this tissue and demonstrated that the intensity of obesity‐induced inflammation correlates well with the amplitude of cGMP dysregulation, through pathways such as TNF‐α‐mediated suppression of sGC and NF‐κB or PKG suppression by JNK signalling (Sanyal et al., 2017).
Another interesting finding of our study was that IW‐1973 treatment was associated with a dose‐dependent reduction in adipocyte area, a finding similar to that reported by Mitschke et al. (2013) in primary murine adipocytes using 8‐pCPT‐cGMP, an analogue of the natural signal molecule cGMP. Along these lines, Hoffmann et al. (2015) also reported a reduction in the size of human adipocytes in response to BAY 41‐8543, a small molecule stimulator of sGC. Overall, these findings are in agreement with the hypothesis by Sun et al. (2011), who suggested that a decrease in adipocyte size is a parameter for healthy expansion of WAT. Since in our study the reduction in the size of adipocytes induced by IW‐1973 was not mediated by changes in HSL, the key enzyme in WAT lipolysis, we explored other mechanisms leading to the reduction in the lipid content such as autophagy. Autophagy is an evolutionary conserved catabolic process by which cellular components are engulfed in an autophagosome and delivered to the lysosome for degradation (Kaur and Debnath, 2015). Autophagy is emerging as a potential regulator of lipid metabolism and obesity (Liu et al., 2016), because this process mediates the lipolysis of TAG and reduces the storage of lipids in adipocytes (Singh et al., 2009; López‐Vicario et al., 2015; Soussi et al., 2015). Finally, it is important to mention that in contrast to previous reports by Mitschke et al. (2013) and Hoffmann et al. (2015), we did not see an increase in WAT browning in IW‐1973‐treated mice, as UCP1 as well as other thermogenic markers such as CIDEA, PGC‐1α and PRDM16 levels remained unchanged in this tissue.
In conclusion, the present study demonstrates at the experimental level that the sGC stimulator IW‐1973 attenuates three hallmarks of NASH: hepatic steatosis, inflammation and fibrosis. In addition, the sGC stimulator IW‐1973 restored WAT homeostasis, which is an insulin‐sensitive tissue that plays a major role in the progression of NASH. Although further studies are needed to clarify the exact mechanisms underlying the beneficial actions of IW‐1973 and the modulation of the NO–sGC–cGMP pathway in the liver, our study provides an important evaluation of the properties of sGC stimulators in NASH. Together, our findings contribute to the potential expansion of the therapeutic armamentarium of liver diseases through the modulation of cGMP levels. Future studies are warranted in patients with NASH to fully confirm the clinical relevance of our findings.
Author contributions
R.F.‐C., J.L.M and J.C. conceived and designed the experiments; R.F.‐C. performed the experiments; E.T. supervised the procedures; A.D., J.A.‐Q. and M.D.‐G. contributed to the acquisition of data; M.C., B.R. and C.L.‐V. contributed to the experiments and procedures; K.H., C.S., R.S., M.C. and J.L.M. participated in critically revising the draft of the article; R.F.‐C. and J.C. wrote the paper.
Conflict of interest
K.H., C.S., R.S., M.C. and J.L.M. are employees of Ironwood Pharmaceuticals.
Part of this work was presented at the International Liver Congress (Amsterdam, The Netherlands, April 19–23, 2017) and was published in an abstract form in J. Hepatol 2017;66:S17.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effects of IW‐1973 on hepatic cyclic nucleotide pathway. (A) VASP mRNA expression as determined by real‐time PCR in liver samples from mice receiving either chow diet (n = 10), choline‐deficient, L‐amino acid‐defined, high‐fat (CDAAH) diet (n = 15), CDAAH plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or CDAAH plus IW‐1973 at 3 mg·kg−1 (n = 10). (B) Hepatic tissue levels of cAMP in these groups of animals as determined by EIA. Results are expressed as mean ± SEM.
Figure S2 Effects of IW‐1973 on plasma biochemistry and steatosis. (A) BAT‐to‐body weight ratio in mice receiving either chow diet (n = 10), CDAAH diet (n = 15), CDAAH plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or CDAAH plus IW‐1973 at 3 mg·kg−1 (n = 10). (B) Plasma glucose, TAG and total cholesterol levels. (C) Representative photomicrographs (200x magnification) of liver sections and histomorphometric analysis of the area stained with Oil red‐O. (D) Hepatic TAG levels. (E) Hepatic PPARα and FAT/CD36 mRNA expression. (F) Hepatic cholesterol levels. Results are expressed as mean ± SEM. Scale bar = 50 μm.
Figure S3 Effects of IW‐1973 on hepatic cytokine expression. Hepatic IL‐6, IL‐10 and IL‐1ra mRNA expression in mice receiving either chow diet (n = 10), CDAAH diet (n = 15), CDAAH plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or CDAAH plus IW‐1973 at 3 mg·kg−1 (n = 10). Results are expressed as mean ± SEM.
Figure S4 Effects of IW‐1973 on plasma transaminase (i.e. AST and ALT) levels in mice receiving either chow diet (n = 5), an obesogenic HFD diet (n = 10), HFD plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or HFD plus IW‐1973 at 3 mg·kg−1 (n = 10). Results are expressed as mean ± SEM.
Figure S5 Changes in WAT cGMP pathway and fibrosis in high‐fat diet (HFD)‐induced obese mice. WAT mRNA expression for NOS3 (A), GUCY1A2 (B), PKG1 (C), PKG2 (D), PDE5a (E) and VASP (F) in mice receiving either chow diet (n = 5), HFD diet (n = 10), HFD plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or HFD plus IW‐1973 at 3 mg·kg−1 (n = 10). (G) Histomorphometric analysis of fibrosis by Sirius red staining. (H) mRNA expression of the fibrosis marker TIMP‐1. (I) Correlation between GUCY1A2 and TIMP‐1 in human obese WAT. Results are expressed as mean ± SEM.
Figure S6 Effects of IW‐1973 on adipose tissue thermogenesis. UCP1, CIDEA, PGC‐1α and PRDM16 mRNA expression in WAT (A) and BAT (B) in mice receiving either chow diet (n = 5), HFD diet (n = 10), HFD plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or HFD plus IW‐1973 at 3 mg·kg−1 (n = 10). Results are expressed as mean ± SEM.
Acknowledgements
This study was supported by grants from Ministerio de Economía y Competitividad (SAF15‐63674‐R and PIE14/00045) under European Regional Development Funds (ERDF). CIBERehd is funded by the Instituto de Salud Carlos III. This study was carried out at the Centre Esther Koplowitz (IDIBAPs), which is a member of the CERCA Programme/Generalitat de Catalunya. The project was also supported by Ironwood Pharmaceuticals. The authors would like to thank Albert Salvatella and Maria Isabel Martínez‐Puchol for their technical help in the completion of the experiments.
Flores‐Costa, R. , Alcaraz‐Quiles, J. , Titos, E. , López‐Vicario, C. , Casulleras, M. , Duran‐Güell, M. , Rius, B. , Diaz, A. , Hall, K. , Shea, C. , Sarno, R. , Currie, M. , Masferrer, J. L. , and Clària, J. (2018) The soluble guanylate cyclase stimulator IW‐1973 prevents inflammation and fibrosis in experimental non‐alcoholic steatohepatitis. British Journal of Pharmacology, 175: 953–967. doi: 10.1111/bph.14137.
References
- Ahluwalia A, Foster P, Scotland RS, McLean PG, Mathur A, Perreti M et al (2004). Antiinflammatory activity of soluble guanylate cyclase: cGMP‐dependent downregulation of P‐selectin expression and leukocyte recruitment. Proc Natl Acad Sci U S A 101: 1386–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala JE, Bracyl DP, Julien BM, Rottman JN, Fueger PT, Wasserman DH (2007). Chronic treatment with sildenafil improves energy balance and insulin action in high fat‐fed conscious mice. Diabetes 56: 1025–1033. [DOI] [PubMed] [Google Scholar]
- Beyer C, Zenzmaier C, Palumbo‐Zerr K, Mancuso R, Distler A, Dees C et al (2015). Stimulation of the soluble guanylate cyclase (sGC) inhibits fibrosis by blocking non‐canonical TGFβ signalling. Ann Rheum Dis 74: 1408–1416. [DOI] [PubMed] [Google Scholar]
- Buglioni A, Burnet JC Jr (2016). New pharmacological strategies to increase cGMP. Annu Rev Med 67: 229–243. [DOI] [PubMed] [Google Scholar]
- Cui W, Maimaitiyiming H, Qi X, Norman H, Zhou Q, Wang X et al (2014). Increasing cGMP‐dependent protein kinase activity attenuates unilateral ureteral obstruction‐induced renal fibrosis. Am J Physiol Renal Physiol 306: 996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkern TR, Feurstein D, Rossi GA, Sabatini F, Hatzelmann A (2007). Inhibition of TGF‐β induced lung fibroblast to myofibroblast conversion by phosphodiesterase inhibiting drugs and activators of soluble guanylyl cyclase. Eur J Pharmacol 572: 12–22. [DOI] [PubMed] [Google Scholar]
- Elks CM, Francis J (2010). Central adiposity, systemic inflammation, and the metabolic syndrome. Curr Hypertens Rep 12: 99–104. [DOI] [PubMed] [Google Scholar]
- Ferrante AW Jr (2007). Obesity‐induced inflammation: a metabolic dialogue in the language of inflammation. J Intern Med 262: 408–414. [DOI] [PubMed] [Google Scholar]
- Francis SH, Busch JL, Corbin JD (2010). cGMP‐dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev 62: 525–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SL (2004). Mechanisms of disease: mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol 1: 98–105. [DOI] [PubMed] [Google Scholar]
- Ghofrani HA, Galiè N, Grimminger F, Grünig E, Humbert M, Jing ZC et al (2013a). Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 369: 330–340. [DOI] [PubMed] [Google Scholar]
- Ghofrani HA, D'Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH et al (2013b). Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 369: 319–329. [DOI] [PubMed] [Google Scholar]
- Hall KC, Jacobson S, Zhang P, Liu G, Sarno R, Catanzano V et al (2017). Inhibition of fibrosis and inflammation by a soluble guanylate cyclase stimulator in models of liver disease. Hepatology 66. [Google Scholar]
- Handa P, Tateya S, Rizzo NO, Cheng AM, Morgan‐Stevenson V, Han CY et al (2011). Contributes to adipose tissue inflammation during high‐fat feeding. Arterioscler Thromb Vasc Biol 31: 2827–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrahan JP, Wakefield J, Wilson P, Mihova M, Chickering JG, Ruff D, et al Multiple‐ascending‐dose study of IW‐1973, a soluble guanylate cyclase stimulator. AHA Council on Hypertension, AHA Council on Kidney in Cardiovascular Disease, American Society of Hypertension Joint Scientific Sessions 2017; September 14–17, 2017; San Francisco, CA.
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydemann A (2016). An overview of murine high fat diet as a model for type 2 diabetes mellitus. J Diabetes Res 2016: 2902351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann LS, Etzrodt J, Willkomm L, Sanyal A, Scheja L, Fischer AWC et al (2015). Stimulation of soluble guanylyl cyclase protects against obesity by recruiting brown adipose tissue. Nat Commun 6: 7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenstein B, Daniel C, Wagner A, Stasch JP, Hugo C (2005). Stimulation of soluble guanylyl cyclase inhibits mesangial cell proliferation and matrix accumulation in experimental glomerulonephritis. Am J Physiol Renal Physiol 288: F685–F693. [DOI] [PubMed] [Google Scholar]
- Kaur J, Debnath J (2015). Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 16: 461–472. [DOI] [PubMed] [Google Scholar]
- Khairallah RJ, Khairallah M, Gélinas R, Bouchard B, Young ME, Allen BG et al (2008). Cyclic GMP signaling in cardiomyocytes modulates fatty acid trafficking and prevents triglyceride accumulation. J Mol Cell Cardiol 45: 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorr A, Hirth‐Dietrich C, Alonso‐Alija C, Hárter M, Hahn M, Keim Y et al (2008). Nitric oxide‐independent activation of soluble guanylate cyclase by BAY 60‐2770 in experimental liver fibrosis. Arzneimittelforschung 58: 71–80. [DOI] [PubMed] [Google Scholar]
- Liang W, Menke AL, Driessen A, Koek GH, Lindeman JH, Stoop R et al (2014). Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS One 9: e115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Takahashi Y, Desai N, Zhang J, Serfass JM, Shi YG et al (2016). Bif‐1 deficiency impairs lipid homeostasis and causes obesity accompanied by insulin resistance. Sci Rep 6: 20453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Vicario C, Alcaraz‐Quiles J, García‐Alonso V, Rius B, Hwang SH, Titos E et al (2015). Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: role for omega‐3 epoxides. Proc Natl Acad Sci U S A 112: 536–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz SZ, Hennige AM, Feil S, Peter A, Gerling A, Machann J et al (2011). Genetic ablation of cGMP‐dependent protein kinase type I causes liver inflammation and fasting hyperglycemia. Diabetes 60: 1566–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Hada N, Sakamaki Y, Uno A, Shiga T, Tanaka C et al (2013). An improved mouse model that rapidly develops fibrosis in non‐alcoholic steatohepatitis. Int J Exp Pathol 94: 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitschke MM, Hoffmann LS, Gnad T, Scholz D, Kruithoff K, Mayer P et al (2013). Increased cGMP promotes healthy expansion and browning of white adipose tissue. FASEB J 27: 1621–1630. [DOI] [PubMed] [Google Scholar]
- Moal F, Veal N, Vuillemin E, Barrière E, Wang J, Fizanne L et al (2006). Hemodynamic and antifibrotic effects of a selective liver nitric oxide donor V‐PYRRO/NO in bile duct ligated rats. World J Gastroenterol 12: 6639–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morán‐Salvador E, Titos E, González‐Périz A, Rius B, García‐Alonso V, López‐Vicario C et al (2013). Cell‐specific knockout mice establish anti‐inflammatory and anti‐fibrogenic properties for PPAR in non‐parenchymal liver cells. J Hepatol 59: 1045–1053. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Parker JL, Lugus JJ, Walsh K (2011). Adipokines in inflammation and metabolic disease. Nat Rev Immunol 11: 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan SL, Guh JH, Peng CY, Chang YL, Cheng FC, Chang JH et al (2005). A potential role of YC‐1 on the inhibition of cytokine release in peripheral blood mononuclear leukocytes and endotoxemic mouse models. Thromb Haemost 93: 940–948. [DOI] [PubMed] [Google Scholar]
- Patrucco E, Domes K, Sbroggió M, Blaich A, Schlossmann J, Desch M et al (2014). Roles of cGMP‐dependent protein kinase I (cGKI) and PDE5 in the regulation of Ang II‐induced cardiac hypertrophy and fibrosis. Proc Natl Acad Sci U S A 111: 12925–12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfield MM, Chen HH, Bourlag BA, Semigran MJ, Lee KL, Lewis G et al (2013). Effect of phosphodiesterase‐5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction, a randomized clinical trial. JAMA 309: 1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A, Naumann J, Hoffmann LS, Chabowska‐Kita A, Ehrlund A, Schlitzer A et al (2017). Interplay between obesity‐induced inflammation and cGMP signaling in white adipose tissue. Cell Rep 18: 225–236. [DOI] [PubMed] [Google Scholar]
- Sanyal AJ (2005). Mechanisms of disease: pathogenesis of nonalcoholic fatty liver disease. Nat Clin Pract Gastroenterol Hepatol 2: 46–53. [DOI] [PubMed] [Google Scholar]
- Shoelson SE, Herrero L, Naaz A (2007). Obesity, inflammation and insulin resistance. Gastroenterology 132: 2169–2180. [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M et al (2009). Autophagy regulates lipid metabolism. Nature 458: 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soussi H, Reggio S, Alili R, Prado C, Mutel S, Pini M et al (2015). DAPK2 downregulation associates with attenuated adipocyte autophagic clearance in human obesity. Diabetes 64: 3452–3463. [DOI] [PubMed] [Google Scholar]
- Stasch JP, Alonso‐Alija C, Apeler H, Dembowsky K, Feurer A, Minuth T et al (2002). Pharmacological actions of a novel NO‐independent guanylyl cyclase stimulator, BAY 41‐8543: in vitro studies. Br J Pharmacol 135: 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasch JP, Hobbs AJ (2009). NO‐independent, haem‐dependent soluble guanylate cyclase stimulators. Hand Exp Pharmacol 191: 278–296. [DOI] [PubMed] [Google Scholar]
- Stasch JP, Schlossmann J, Hocher B (2015). Renal effects of soluble guanylate cyclase stimulators and activators: a review of the preclinical evidence. Curr Opin Pharmacol 21: 95–104. [DOI] [PubMed] [Google Scholar]
- Sun K, Kusminski CM, Scherer PE (2011). Adipose tissue remodeling and obesity. J Clin Invest 121: 2094–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trayhurn P (2005). Endocrine and signalling role of adipose tissue: new perspectives on fat. Acta Physiol Scand 184: 285–293. [DOI] [PubMed] [Google Scholar]
- Tulis DA, Durante W, Peyton KJ, Chapman GB, Evans AJ, Schafer AI (2000). YC‐1, a benzyl indazole derivative, stimulates vascular cGMP and inhibits neointima formation. Biochem Biophys Res Commun 279: 646–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Kemp‐Harper BK, Kocan M, Ang SY, Hewitson TD, Samuel CS (2016). The anti‐fibrotic actions of relaxin are mediated through a NO–sGC–cGMP‐dependent pathway in renal myofibroblasts in vitro and enhanced by the NO donor, diethylamine NONOate. Front Pharmacol 7: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC et al (2012). Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 142: 918–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of IW‐1973 on hepatic cyclic nucleotide pathway. (A) VASP mRNA expression as determined by real‐time PCR in liver samples from mice receiving either chow diet (n = 10), choline‐deficient, L‐amino acid‐defined, high‐fat (CDAAH) diet (n = 15), CDAAH plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or CDAAH plus IW‐1973 at 3 mg·kg−1 (n = 10). (B) Hepatic tissue levels of cAMP in these groups of animals as determined by EIA. Results are expressed as mean ± SEM.
Figure S2 Effects of IW‐1973 on plasma biochemistry and steatosis. (A) BAT‐to‐body weight ratio in mice receiving either chow diet (n = 10), CDAAH diet (n = 15), CDAAH plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or CDAAH plus IW‐1973 at 3 mg·kg−1 (n = 10). (B) Plasma glucose, TAG and total cholesterol levels. (C) Representative photomicrographs (200x magnification) of liver sections and histomorphometric analysis of the area stained with Oil red‐O. (D) Hepatic TAG levels. (E) Hepatic PPARα and FAT/CD36 mRNA expression. (F) Hepatic cholesterol levels. Results are expressed as mean ± SEM. Scale bar = 50 μm.
Figure S3 Effects of IW‐1973 on hepatic cytokine expression. Hepatic IL‐6, IL‐10 and IL‐1ra mRNA expression in mice receiving either chow diet (n = 10), CDAAH diet (n = 15), CDAAH plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or CDAAH plus IW‐1973 at 3 mg·kg−1 (n = 10). Results are expressed as mean ± SEM.
Figure S4 Effects of IW‐1973 on plasma transaminase (i.e. AST and ALT) levels in mice receiving either chow diet (n = 5), an obesogenic HFD diet (n = 10), HFD plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or HFD plus IW‐1973 at 3 mg·kg−1 (n = 10). Results are expressed as mean ± SEM.
Figure S5 Changes in WAT cGMP pathway and fibrosis in high‐fat diet (HFD)‐induced obese mice. WAT mRNA expression for NOS3 (A), GUCY1A2 (B), PKG1 (C), PKG2 (D), PDE5a (E) and VASP (F) in mice receiving either chow diet (n = 5), HFD diet (n = 10), HFD plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or HFD plus IW‐1973 at 3 mg·kg−1 (n = 10). (G) Histomorphometric analysis of fibrosis by Sirius red staining. (H) mRNA expression of the fibrosis marker TIMP‐1. (I) Correlation between GUCY1A2 and TIMP‐1 in human obese WAT. Results are expressed as mean ± SEM.
Figure S6 Effects of IW‐1973 on adipose tissue thermogenesis. UCP1, CIDEA, PGC‐1α and PRDM16 mRNA expression in WAT (A) and BAT (B) in mice receiving either chow diet (n = 5), HFD diet (n = 10), HFD plus sGC stimulator IW‐1973 at 1 mg·kg−1 (n = 10) or HFD plus IW‐1973 at 3 mg·kg−1 (n = 10). Results are expressed as mean ± SEM.