

Abstract
The transition path is the tiny segment of a single molecule trajectory when the free energy barrier between states is crossed and for protein folding contains all of the information about the self-assembly mechanism. As a first step toward obtaining structural information during the transition path from experiments, single molecule FRET spectroscopy has been used to determine average transition path times from a photon-by-photon analysis of fluorescence trajectories. These results, obtained for several different proteins, have already provided new and demanding tests that support both the accuracy of all-atom molecular dynamics simulations and the basic postulates of energy landscape theory of protein folding.
Introduction
There have been several milestones over the past three decades in understanding how proteins fold that have created an active subfield of biological physics and biophysical chemistry. They have come from all three branches of science - experiment, theory and computation. Experimental advances have included the application of linear free energy relations and protein engineering to obtain structural information on the transition state ensemble [1,2] and the introduction of nanosecond laser-triggering to dramatically improve time-resolution in kinetic experiments [3–5]. Theoretical advances have included the statistical mechanical description of folding as diffusion on a low dimensional free energy surface with order parameters as coordinates [6–8], the development of theoretical models based on explicit consideration of only native contacts (i.e., intramolecular contacts present in the folded structure) [9–12] and the application of concepts from polymer physics [13]. The most unexpected advance has been in the area of computation, specifically all-atom molecular dynamics (MD) simulations, in which Newton’s equations of motion are solved for all heavy atoms of the polypeptide and solvent [14,15]. These simulations are now capable of folding a polypeptide from an initially disordered state into a protein with the correct structure for many small proteins [16]. The importance of the MD simulations cannot be overstated. If accurate, with long trajectories to observe a statistically sufficient number of folding/unfolding transitions, the simulations contain everything one would want to know on how a specific protein folds in atomistic detail. These simulations have already provided critical testing grounds for universal theoretical concepts [17] and statistical mechanical models of protein folding [18]. Therefore, new kinds of experiments that further test the accuracy of these simulations are extremely important. One such class of experiments employs single molecule fluorescence spectroscopy, with the ultimate goal of watching the evolution of structure for individual molecules as they self-assemble on transition paths. In this article we describe the first steps toward this goal using single molecule Förster resonance energy transfer (FRET) spectroscopy that have allowed determination of the average duration of transition paths from photon trajectories.
First, what is a transition path? The transition path corresponds to a successful reactant to product crossing of the free energy barrier separating states (free energy minima). For folding of a protein with just two populated states - unfolded and folded - Figure 1 shows that the transition path corresponds to the segment of the molecular trajectory from unfolded to folded for which a position qU close to the unfolded minimum on the reaction coordinate q is crossed and reaches position qF close to the folded minimum without ever re-crossing qU [19]. Consequently, in this case, the transition path contains all of the structural information on how the unfolded polypeptide self-assembles to form the folded, biologically-active structure. Since the transition path is a property of an individual molecule, it can only be observed by watching one molecule at a time. The zeroth order question about transition paths, then, is: what is the average duration of the barrier crossing, i.e. what is the average transition path time? What we have learned from the experimental determination of this time using single molecule FRET is the subject of this article.
Figure 1.

Free-energy surface of protein folding. Protein folding can be described as diffusion on a low-dimensional free energy surface with order parameters as coordinates and has been described successfully as diffusion on a 1D free energy surface, here projected onto the coordinate q. For a two-state protein, the folded and unfolded states are separated by a free energy barrier with the height of . (ωu)2, (ωf)2, and (ω*)2, are curvatures at the bottom of the unfolded and folded state wells and at the top of the barrier, respectively. An unfolded molecule spends the vast majority of time fluctuating in the unfolded well before making a very rapid folding transition over the barrier. The transition path is that part of the molecular trajectory that leaves a position qU on the unfolded side of the barrier and reaches qF on the folded side without re-crossing qU (blue portion of the trajectory). The transition path appears as a near-instantaneous jump in a binned FRET efficiency trajectory (Bottom).
Definition of Transition Path Time
In single molecule experiments it is important to distinguish between the average folding time, tf (the inverse of the rate coefficient for folding, kf) and the average transition path time, tTP. Unlike the average folding and unfolding times, the average transition path time is the same for folding and unfolding regardless of the shape of a free energy barrier. The folding time is the average time that the molecule spends in the unfolded state before a successful barrier crossing to the folded state, which appears as an almost instantaneous jump in the trajectory in Figure 1, while the unfolding time (tu) is the average time that the molecule spends in the folded state before crossing the barrier to the unfolded state. These waiting times are exponentially distributed and their average is the inverse of the corresponding rate coefficients (kf and ku; note that in ensemble experiments, the observed rate is the sum of the two rate coefficients, while in single molecule experiments they are determined individually). The transition path time, on the other hand, is the time actually spent in a successful barrier crossing (the apparent “jump” in Figure 2a).
Figure 2.

Photon (Bottom), binned fluorescence (Middle) and FRET efficiency (Top) trajectories simulated with (a) high (500 ms−1) and (b) low (10 ms−1) photon count rates and the FRET efficiencies of 0.7 and 0.3. The FRET efficiency (E) is the fraction of photons in a bin that are emitted by the acceptor (E = nA/(nA + nD), where nA is the number of acceptor photons and nD is the number of donor photons). The bin time of the FRET efficiency and fluorescence trajectories is 0.5 ms. Purple vertical dashed lines indicate positions of transition between folded and unfolded states. Transition time points are the same in (a) and (b). The photon trajectory in (a) shows photons near the first transition (yellow shaded area in the binned trajectory).
In addition to the large difference in time scales, a major difference between folding times and transition path times is their dependence on the height of the free energy barrier (Figure 1). The folding time is given by the Kramers’ theory [20] as
| (1) |
where D* is the diffusion coefficient at the barrier top, (ω*)2 and (ωu)2 are the curvatures of the free energy surface at the top of the barrier and bottom of the unfolded well, respectively, is the barrier height, β = 1/kBT, kB is the Boltzmann constant, and T is the absolute temperature. With the same one-dimensional (1D) diffusion theory, the average transition path time (for a high parabolic barrier) is [19,21]
| (2) |
where γ (= 0.577 …) is Euler’s constant. These two equations are similar in that both times are inversely proportional to the diffusion coefficient D* and depend on the free energy barrier height . However, the folding time is extremely sensitive to the barrier height (exponentially dependent) while the transition path time is insensitive (logarithmically dependent).
The justification for using equations 1 and 2 for one-dimensional free energy surfaces began with results of early simulations of folding lattice model representations of proteins [22]. These simulations showed that the rates of folding could be accurately reproduced by Kramers’ theory for escape over a one-dimensional free energy barrier with the fraction of native contacts (Q) as the reaction coordinate. Calculation of the barrier height from the relative populations of structures (the potential of mean force) for a given Q, and the decay time for the Q-Q correlation function in the unfolded well as the Kramers pre-exponential factor, resulted in rates within a factor of 2 of the mean first passage times to the folded state observed in the simulations [22]. It is an enormous simplification to project the many degrees of freedom onto this one-dimensional picture, so the results of the lattice simulation were quite surprising. Subsequently, all-atom MD simulations have shown that the one-dimensional free energy surface with an order parameter as reaction coordinate is indeed sufficient to describe both equilibria, i.e. the number and relative free energy of populated minima, and the kinetics [23]. That it is possible to represent the dynamics of a system with so many degrees of freedom as diffusion on a one-dimensional surface may be a consequence of the fraction of native contacts Q being a good reaction coordinate, owing to the dominant role of native contacts in determining folding mechanism [17].
Determination of Transition Path Times from Single Molecule FRET
Förster resonance energy transfer (FRET) [24] has been the most useful single molecule fluorescence method [25–27] for studying protein folding [21,28–59]. Single molecule FRET has also made numerous contributions toward understanding the structure and dynamics of unfolded and intrinsically disorderd proteins (see [60] for recent review). In FRET experiments, a protein molecule is labeled at specific positions with one donor and one acceptor fluorophore. For observing folding/unfolding trajectories, the molecule is immobilized on a coated glass surface. After laser excitation of the donor, emission of both the donor or, if the excitation energy has been transferred, of the acceptor is detected. Since the transfer efficiency (E) depends on the distance (R) between the two fluorophores [E = R06/(R06 + R6), where the constant R0 is the Förster radius], folded and unfolded states are distinguished by the higher FRET efficiency for the folded molecule compared to the unfolded molecule. By performing experiments near the denaturation mid-point, the trajectory contains a roughly equal number of folding and unfolding transitions. If the waiting times are sufficiently long and the photon detection rate is sufficiently high, as in Fig. 2(a), the photons can be binned and the FRET efficiency for each bin is plotted as a function of time. This FRET efficiency trajectory readily yields the individual rate coefficients for folding and unfolding. On the other hand, for waiting times that are too short or photon detection rates are too low, the FRET efficiency trajectory is too noisy to obtain any information about rate coefficients. In this case, the unbinned photon trajectory must be analyzed [37]. According to MD simulations, transition path times are in the nanosecond-microsecond time scale, which poses an enormous challenge for single molecule FRET experiments [16]. Consequently, acquisition of data sufficient to obtain even average transition path times requires photon trajectories with the highest possible detection rate and a robust method of analysis.
Szabo and Gopich have developed a very powerful maximum likelihood method for analyzing such photon trajectories [21,61,62]. The analysis with their method is considerably simplified if a model has already been firmly established in ensemble measurements. Given the number of states contained in the trajectory, the method determines the most likely parameters of the model (FRET efficiencies and rate coefficients) that best reproduce the photon trajectory. For the kinetics of a two-state model, for example, the 4 parameters are the folding and unfolding rate coefficients and the FRET efficiencies for the folded and unfolded states. To obtain average transition path times, a third virtual state is introduced with a FRET efficiency that is assumed to be midway between the folded and unfolded state. The only additional parameter to add to this model is the lifetime of the virtual state, which corresponds to the average transition path time [63]. The likelihood of a two-state model, which implicitly assumes that the transition path time is instantaneous, is then compared with the likelihood of a three-state model with a finite lifetime for the virtual intermediate. If the plot of the difference in log likelihoods for the two- and three-state models versus assumed lifetime for the virtual intermediate shows a statistically-significant difference at a peak in the likelihood-time plot, the value of the assumed lifetime at the peak is the average transition path time. If there is no statistically-significant peak, an upper bound for the average transition path time can be determined.
Figure 3 shows the results for three two-state proteins - the 35-residue all-β WW domain [63], the 56-residue α/β protein GB1 [63], and the 73-residue, designed all-α protein α3D [43,64]. The WW domain is a fast folder (tf = 100 μs), so to slow the rates and increase the transition path times making them more easily measurable, the experiments were performed at a viscosity (η) 10-times higher than water. After correcting for the higher viscosity, the average transition path time in water at 22°C was determined to be 1.6 μs [63]. No viscogen was necessary to resolve the average transition path time for α3D, for which the folding time at 22oC is 3 ms and the average transition path time is 12 μs. There were too few transitions to determine the average transition path time for the slow folding protein GB1 with a folding time of 1 s. However, the analysis gave the initially surprising result that it must be less than 10 μs [63].
Figure 3.

Measurement of average transition path times of folding of the FBP28 WW domain (Left), α3D (Middle), and GB1 (Right). (a) Proteins are labeled with a donor (Alexa 488) and an acceptor (Alexa 594 or Alexa 647) dyes and immobilized on a polyethylene glycol-coated glass surface. (b) Binned donor and acceptor fluorescence trajectories (Top) (bin times of 50 μs for the WW domain and α3D and 100 μs for protein G) and photon trajectories near the folding transition (Bottom). (c) The transition path time is determined by analyzing photon trajectories near transitions using the maximum likelihood method with the three-state model. The average transition path time (tTP) is equal to the lifetime of a virtual intermediate state S (τS), which is determined from the maximum of the difference of the log likelihood, Δln L = ln L(τs) − ln L(0). L(0) is the likelihood for the two-state model, in which transitions are instantaneous (τS = 0). The transition path time of the WW domain is 16 μs (at the viscosity of 10 cP). The transition path time of α3D (12 μs) can be measured without increasing solvent viscosity because its diffusion on the one-dimensional free energy surface is much slower than those of the WW domain and protein GB1. Only the upper bound for the transition path time of 10 μs can be determined for GB1 because no peak is observed. Figure is adapted from refs. [63]. and [43].
Comparison of Transition Path Times with Theory and Simulations
Even though only average transition path times were determined, the results turn out to be quite interesting and important. First is the excellent agreement of tTP determined from the experiments and those observed in the simulations. After correcting for the higher temperature and lower viscosity of the water in the MD simulations, tTP simulations for a WW domain with the same structure, but different sequence, is 1.5 μs compared to 1.6 μs from the experiments [16,63]. In the case of α3D, the measurement of the temperature dependence of tTP allowed extrapolation to the temperature 370 K of the simulations. The experimental value is 1.7 – 2.3 μs, while the simulation tTP is ~1.3 μs after a viscosity (η0.3; see below) correction [43]. From this comparison, one can conclude that the experiments and simulations are mutually consistent.
The transition path times are remarkably similar for the WW domain and protein GB1 even though their folding rates differ by ~104-fold (100 μs vs. 1 s). Equation 2 readily explains this result by showing that tTP is insensitive to the free energy barrier height, which is the primary factor in determining tf in equation 1. Equation 2 was derived with the very same assumptions of Kramers theory, namely that the rate of escape over the barrier top is determined by diffusion of a Brownian particle on a one-dimensional free energy surface. Given the importance of the one-dimensional diffusion picture for describing protein folding kinetics [6], the finding of similar transition path times for the fast and slow folder provides additional support for this theoretical description.
Determination of free energy barrier heights has always been a challenging problem for experiments [65]. α3D presented a unique opportunity. When both the folding and transition path times are known, equations 1 and 2 show that the ratio of these two quantities allows the determination of the free energy barrier height, , if the ratio ω*/ωu is known. ω*/ωu has not yet been determined by experiments, but the success of the MD simulations in reproducing experimental folding mechanisms [66] suggests that the simulated ratio of ~1.3 can be used, which results in a value at the midpoint denaturant concentration [43]. From the denaturant dependence of the folding rate [37], it is predicted that the folding barrier height is close to zero in the absence of denaturant, consistent with the theoretical prediction that free energy barriers in protein folding can become quite small and even disappear [6].
The folding time for the WW domain is proportional to the first power of the viscosity (tf ∝ η), as expected from the results for a β hairpin [67] and for all-β proteins [68], while the dependence of both the folding time and average transition path time for α3D at neutral pH is extremely weak, t ∝ ηα, with α = 0.2 for tf and α = 0.3 for tTP. Subsequent experiments at low pH, where the salt bridges involving aspartates and glutamates are eliminated by protonating the negatively-charged carboxylic acid groups, showed that both tf and tTP markedly decrease [64]. At low pH, tf decreases over 10-fold and tTP also decreases by a similar amount, indicating that the pre-exponential factor in Kramers’ equation 1 is responsible for the decrease in tf rather than the barrier height. The low pH experiments also showed that α, the exponent in the viscosity dependence of the folding time increases from 0.3 to 0.7, the value found for isolated α helices [67]. The exponent less than 1.0 has been explained by dihedral angle rotations occurring faster than the solvent relaxation time [67–69], which is a breakdown of Kramers’ theory in which solvent relaxation is assumed to be instantaneous and therefore α = 1.0.
Extensive MD simulations were performed, which are consistent with the experimental results in that both tTP and tf decrease at low pH in spite of an increase in the free energy barrier height, because the diffusion coefficient D* increases [64]. Moreover, as found in the experiments, the viscosity dependence of both tTP and tf increase at low pH. The experimental and simulation results both point to a rougher underlying energy landscape for α3D at neutral pH compared to the WW domain and protein GB1, confirming a key prediction on the difference in roughness between evolved and designed protein landscapes [70,71].
The pre-exponential factor in Eq. (1) is usually ignored in interpreting rate coefficients because of its much greater sensitivity to ΔG* than to D*. However, it should be pointed out that the pre-exponetial factor provides an estimate of the fastest rate that a particular protein can fold, the so-called protein folding “speed limit” [72]. Moreover, changes in the pre-exponential factor can potentially complicate the interpretation of Φ-values (discussed below). The results for α3D provide a cautionary note.
Significance of Transition Path Experiments for Understanding How Proteins Fold
The answer to the question - how do proteins fold? - depends very much on the discipline of the scientist asking the question. Prior to the discovery of the role of chaperones [73,74], a biologist might argue that, since Anfinsen demonstrated proteins fold to a structure determined only by the amino acid sequence and do so spontaneously, there is no question and nothing more to know. A structural biologist or biochemist will of course want to know more, such as: what is the order of assembly of the various secondary structural elements, such as α helices and β hairpins? A biophysical scientist, on the other hand, seeks universal principles and a theoretical model based on these principles that can quantitatively explain the results of equilibrium and kinetic experiments, as well as those of all-atom molecular dynamics calculations. Only then, is the assembly order of the parts explained or predicted by the model believable.
To appreciate the importance of transition paths for understanding how proteins fold from this biophysical perspective, it is first instructive to point out what has been meant by a protein folding mechanism. The early approach was similar to that of classical organic chemistry, namely determine the number and connectivity of intermediates in kinetic experiments in which the initial condition is the unfolded polypeptide. In this description, the mechanism is a simple sequence (often incorrectly cited as solving the Levinthal paradox[75]): U → I1 → I2 → …N, with off pathway intermediates also permitted [76]. Having identified the number of intermediates, the next step is to characterize their structure. There are two major problems with this scenario. One is that theoretical considerations and MD simulations clearly show that, if viewed at atomistic resolution, no two transition paths are identical. Consequently, all intermediates are enormously large ensembles of structures and folding must proceed via a comparably large ensemble of pathways. For this reason, comprehension of an assembly pathway must involve some level of coarse graining, so the number of pathways in the ensemble is a function of the extent of coarse graining. For example, if the mechanism of the 3-helical villin subdomain is described in terms of the order in which the 3 helices form on the transition path, all 6 possible pathways are populated according to both state-of-the art MD simulations [18,77] and an Ising-like theoretical model [18].
The second problem with the sequential model, U → I1 → I2 → …N, is that many single domain proteins exhibit no detectable intermediates. These so-called two-state proteins exhibit only folded and unfolded states at equilibrium and at all times in kinetic experiments [78]. In this case, Φ-value analysis, in which the effect of a single amino acid replacement on rates and equilibrium constants is commonly used to infer the degree of native structure around the substituted amino acid in the transition state ensemble [1,2]. Although this experiment produces the only detailed structural information on the folding mechanism for two-state proteins, the information is for the average structural environment at a single position along the transition path, i.e. the average degree of native structure in the transition state ensemble at the top of the free energy barrier.
Additional structural information on a two-state folding mechanism can be obtained from NMR experiments [79–81]. These experiments delineate regions of the structure protected from hydrogen-deuterium (HD) exchange in high-lying free energy minima on the folded side of the free energy barrier. To translate the results of these equilibrium experiments into a kinetic mechanism, it is necessary to make the critical assumption that the order of, albeit average, structure formation is determined solely by the relative free energies of the minima. Given the success of a one-dimensional free energy surface description of folding discussed above, the assumption is not unreasonable. However, the order of structure formation is for the segment of the transition path on the folded side of the barrier, with no information on the more important segment between the unfolded state and the top of the free energy barrier, which describes how the protein reaches the barrier top (transition state).
Finally, there is one case where the average properties of a transition path ensemble can be probed in temperature-jump experiments. It is for two-state proteins that fold without a barrier when the temperature is increased - so called “downhill” or “one-state” folders [82–86]. These proteins fold very fast and therefore require probes with high time-resolution, but they do have the potential of yielding considerable average three-dimensional structural information all along the transition path and not just at the top of the barrier, as in Φ-value analysis, or on the folded side of the barrier, as in the HD exchange NMR experiments.
The above discussion points out why we have focused our single molecule FRET studies on measuring properties of transition paths.
Future Directions
Although determining average transition path times is only the first step in the investigation of transition paths in protein folding, these experiments have already played a significant part in motivating numerous interesting theoretical investigations, see, e.g. ref [87–91]. For the slower transition path times found for nucleic acids, advances in single molecule force spectroscopy now permit resolution of the duration of a single barrier crossing, making it possible to obtain a complete distribution of transition path times [92–97]. This is more difficult for single molecule FRET experiments of proteins, where currently only the very longest individual transition path times can be observed (HS Chung and WA Eaton, unpublished results) because of bleaching of the fluorophores at the high illumination intensities that must be employed. So, to observe structure during an individual transition path will require methods that significantly decrease the excited state chemistry that destroys the fluorophores [98,99]. With such improvements it should then be possible to obtain three-dimensional structural information during the transition path from an experiment in which 3 fluorophores are attached to the protein, which can yield 3 distances (Figure 4). Knowing three long-range distances as a function of time during the transition path may place sufficiently severe constraints on polypeptide conformations to provide a demanding test of transition paths calculated from simulations or theoretical models. With technical improvements, it should be fairly straightforward, as shown in Fig. 4, to determine the order of helix formation for helical proteins such as the villin subdomain, which has been predicted by both simulations and a theoretical model [18]. While this can potentially be done for the average pathway by ensemble experiments, observation of the complete distribution, as predicted by simulations and a theoretical model, will require watching folding one molecule at a time.
Figure 4.
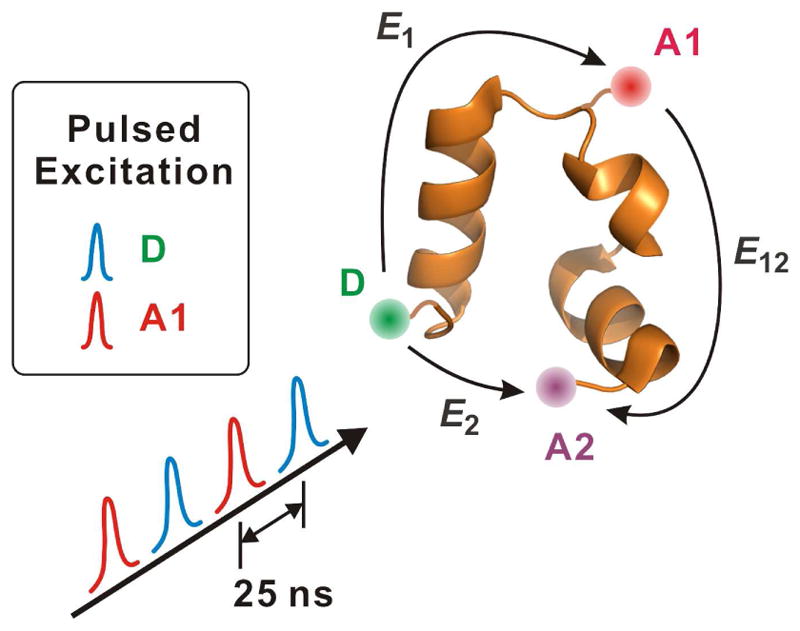
Three-color FRET for observing three-dimensional structure during transition paths. Calling the 3 fluorophores donor (D), acceptor 1 (A1) and acceptor 2 (A2), the idea of the experiment is to alternately excite D and A1 with two different pulsed lasers, which can be done with simple time delays so each dye is excited once every 50 ns. Excitation of D results in transfer of the excitation energy to A1 (E1) and also, albeit less, directly to A2 (E2). However, A1 can transfer excitation energy that it has received from D to A2 (E12). To determine directly the efficiency of transfer from A1 to A2, the second laser excites A1, which can only transfer excitation energy to A2. The alternating excitation therefore allows disentanglement of the results to yield 3 FRET efficiencies, D to A1, D to A2, and A1 to A2, thereby yielding 3 distances [100]. Because the protein is so small, the experiment will require development of fluorophores that have smaller R0’s than those currently employed and are also more resistant to photochemistry.
Highlights.
Single molecule FRET experiments determine average transition path times
Observed times are extremely close to those found in all-atom MD simulations.
Times are very similar for fast- and slow-folding proteins
Longer times for designed protein are due to slower reaction coordinate diffusion
Slower diffusion on reaction coordinate results from non-native interactions
Acknowledgments
We thank Robert Best and Attila Szabo for many helpful discussion and comments on the manuscript. This work was supported by the Intramural Research Program of NIDDK/NIH.
Footnotes
Conflicts of interest
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Recent papers of particular interest have been highlighted as:
* of special interest
** of outstanding interest
- 1.Fersht AR, Matouschek A, Serrano L. The folding of an enzyme. 1. Theory of protein engineering analysis of stability and pathway of protein folding. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 2.Mallam AL, Jackson SE. Use of protein engineering techniques to elucidate protein folding pathways. Progress in Molecular Biology and Translational Science. 2008;84:57–113. doi: 10.1016/S0079-6603(08)00403-0. [DOI] [PubMed] [Google Scholar]
- 3.Jones CM, Henry ER, Hu Y, Chan CK, Luck SD, Bhuyan A, Roder H, Hofrichter J, Eaton WA. Fast events in protein folding initiated by nanosecond laser photolysis. Proc Natl Acad Sci USA. 1993;90:11860–11864. doi: 10.1073/pnas.90.24.11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gruebele M. The fast protein folding problem. Ann Rev Phys Chem. 1999;50:485–516. doi: 10.1146/annurev.physchem.50.1.485. [DOI] [PubMed] [Google Scholar]
- 5.Eaton WA, Munoz V, Hagen SJ, Jas GS, Lapidus LJ, Henry ER, Hofrichter J. Fast kinetics and mechanisms in protein folding. Ann Rev Biophys Biomolec Struct. 2000;29:327–359. doi: 10.1146/annurev.biophys.29.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein-folding - a synthesis. Proteins-Struct Funct Gen. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 7.Onuchic JN, Wolynes PG. Theory of protein folding. Curr Opin Struct Biol. 2004;14:70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Shakhnovich E. Protein folding thermodynamics and dynamics: Where physics, chemistry, and biology meet. Chem Rev. 2006;106:1559–1588. doi: 10.1021/cr040425u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munoz V, Eaton WA. A simple model for calculating the kinetics of protein folding from three-dimensional structures. Proc Natl Acad Sci USA. 1999;96:11311–11316. doi: 10.1073/pnas.96.20.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Portman JJ, Takada S, Wolynes PG. Microscopic theory of protein folding rates. I. Fine structure of the free energy profile and folding routes from a variational approach. J Chem Phys. 2001;114:5069–5081. [Google Scholar]
- 11.Kubelka J, Henry ER, Cellmer T, Hofrichter J, Eaton WA. Chemical, physical, and theoretical kinetics of an ultrafast folding protein. Proc Natl Acad Sci USA. 2008;105:18655–18662. doi: 10.1073/pnas.0808600105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *12.Jacobs WM, Shakhnovich EI. Structure-based prediction of protein-folding transition paths. Biophys J. 2016;111:925–936. doi: 10.1016/j.bpj.2016.06.031. A theoretical model is developed for predicting transition path distributions based on contacts present in the native structure and a principle of substructure cooperativity. The model can be applied to much larger proteins than can be treated by the Ising-like model of refs 9, 11 and 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thirumalai D, O’Brien EP, Morrison G, Hyeon C. Theoretical perspectives on protein folding. Ann Rev Biophys. 2010;39:159–183. doi: 10.1146/annurev-biophys-051309-103835. [DOI] [PubMed] [Google Scholar]
- 14.Lane TJ, Shukla D, Beauchamp KA, Pande VS. To milliseconds and beyond: challenges in the simulation of protein folding. Curr Opin Struct Biol. 2013;23:58–65. doi: 10.1016/j.sbi.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piana S, Klepeis JL, Shaw DE. Assessing the accuracy of physical models used in protein-folding simulations: quantitative evidence from long molecular dynamics simulations. Curr Opin Struct Biol. 2014;24:98–105. doi: 10.1016/j.sbi.2013.12.006. [DOI] [PubMed] [Google Scholar]
- **16.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How fast-folding proteins fold. Science. 2011;334:517–520. doi: 10.1126/science.1208351. This is a landmark paper showing that a special purpose computer for performing all-atom molecular dynamics simulations can fold small, fast-folding proteins to the correct structure. The simulated equilibrium and kinetic properties are sufficiently close to experimental values that the transition paths found in the simulations should serve as a useful guide for designing future experiments to determine folding mechanisms. The simulations have also been important for testing assumptions employed in theoretical models (see comments concerning refs 17, 18 and 23) [DOI] [PubMed] [Google Scholar]
- **17.Best RB, Hummer G, Eaton WA. Native contacts determine protein folding mechanisms in atomistic simulations. Proc Natl Acad Sci USA. 2013;110:17874–17879. doi: 10.1073/pnas.1311599110. Analysis of the simulations in ref 16 uses two measures for comparing the relative importance of native and non-native contacts during the transition path. The analysis shows that, while non-native contacts influence folding rates from their effect on barrier heights and reaction-coordinate diffusion coefficients, they play no significant part in determining folding mechanisms of these small, fast-folding proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **18.Henry ER, Best RB, Eaton WA. Comparing a simple theoretical model for protein folding with all-atom molecular dynamics simulations. Proc Natl Acad Sci USA. 2013;110:17880–17885. doi: 10.1073/pnas.1317105110. The simulations in ref. 16 provide justification for the two key assumptions of an an Ising-like model that only native contacts need to be explicitly considered and that structure grows in just a few regions of the polypeptide. The distribution of transitons paths predicted by the model are remarkably similar to the distribution found in the simulations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hummer G. From transition paths to transition states and rate coefficients. J Chem Phys. 2004;120:516–523. doi: 10.1063/1.1630572. [DOI] [PubMed] [Google Scholar]
- 20.Kramers HA. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica. 1940;VII:284–304. [Google Scholar]
- *21.Chung HS, Gopich IV. Fast single-molecule FRET spectroscopy: theory and experiment. Phys Chem Chem Phys. 2014;16:18644–18657. doi: 10.1039/c4cp02489c. This paper gives an excellent review of practical issues in extracting dynamical parameters from single molecule photon trajectories using maximum likelihood methods. It also presents a derivation of the key equation 2 of the text. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Socci ND, Onuchic JN, Wolynes PG. Diffusive dynamics of the reaction coordinate for protein folding funnels. J Chem Phys. 1996;104:5860–5868. [Google Scholar]
- *23.Zheng WW, Best RB. Reduction of all-atom protein folding dynamics to one-dimensional diffusion. J Phys Chem B. 2015;119:15247–15255. doi: 10.1021/acs.jpcb.5b09741. Analysis of the simulations in ref. 16 shows that rates and transition path times can be accurately described as diffusion on just a single coordinate, the fraction of native contacts (Q) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stryer L, Haugland RP. Energy transfer: a spectroscopic ruler. Proc Natl Acad Sci USA. 1967;58:719–726. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ha T. Single-molecule fluorescence resonance energy transfer. Methods. 2001;25:78–86. doi: 10.1006/meth.2001.1217. [DOI] [PubMed] [Google Scholar]
- 26.Schuler B, Lipman EA, Steinbach PJ, Kumke M, Eaton WA. Polyproline and the “spectroscopic ruler” revisited with single-molecule fluorescence. Proc Natl Acad Sci USA. 2005;102:2754–2759. doi: 10.1073/pnas.0408164102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Best RB, Merchant KA, Gopich IV, Schuler B, Bax A, Eaton WA. Effect of flexibility and cis residues in single-molecule FRET studies of polyproline. Proc Natl Acad Sci USA. 2007;104:18964–18969. doi: 10.1073/pnas.0709567104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lipman EA, Schuler B, Bakajin O, Eaton WA. Single-molecule measurement of protein folding kinetics. Science. 2003;301:1233–1235. doi: 10.1126/science.1085399. [DOI] [PubMed] [Google Scholar]
- 29.Rhoades E, Cohen M, Schuler B, Haran G. Two-state folding observed in individual protein molecules. J Amer Chem Soc. 2004;126:14686–14687. doi: 10.1021/ja046209k. [DOI] [PubMed] [Google Scholar]
- 30.Hertzog DE, Michalet X, Jager M, Kong XX, Santiago JG, Weiss S, Bakajin O. Femtomole mixer for microsecond kinetic studies of protein folding. Anal Chem. 2004;76:7169–7178. doi: 10.1021/ac048661s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orte A, Craggs TD, White SS, Jackson SE, Klenerman D. Evidence of an intermediate and parallel pathways in protein unfolding from single-molecule fluorescence. J Amer Chem Soc. 2008;130:7898–7907. doi: 10.1021/ja709973m. [DOI] [PubMed] [Google Scholar]
- 32.Ting CL, Makarov DE. Two-dimensional fluorescence resonance energy transfer as a probe for protein folding: A theoretical study. J Chem Phys. 2008;128:115102. doi: 10.1063/1.2835611. [DOI] [PubMed] [Google Scholar]
- 33.Huang F, Ying LM, Fersht AR. Direct observation of barrier-limited folding of BBL by single-molecule fluorescence resonance energy transfer. Proc Natl Acad Sci USA. 2009;106:16239–16244. doi: 10.1073/pnas.0909126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allen LR, Paci E. Simulation of fluorescence resonance energy transfer experiments: effect of the dyes on protein folding. J Phys Cond Matt. 2010;22:235103. doi: 10.1088/0953-8984/22/23/235103. [DOI] [PubMed] [Google Scholar]
- 35.Schuetz P, Wuttke R, Schuler B, Caflisch A. Free Energy Surfaces from Single-Distance Information. J Phys Chem B. 2010;114:15227–15235. doi: 10.1021/jp1053698. [DOI] [PubMed] [Google Scholar]
- 36.Ebbinghaus S, Dhar A, McDonald D, Gruebele M. Protein folding stability and dynamics imaged in a living cell. Nature Meth. 2010;7:319–323. doi: 10.1038/nmeth.1435. [DOI] [PubMed] [Google Scholar]
- 37.Chung HS, Gopich IV, McHale K, Cellmer T, Louis JM, Eaton WA. Extracting rate coefficients from single-molecule photon trajectories and FRET efficiency histograms for a fast-folding protein. J Phys Chem A. 2011;115:3642–3656. doi: 10.1021/jp1009669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhi ZY, Liu PC, Wang P, Huang YY, Zhao XS. Domain-specific folding kinetics of staphylococcal nuclease observed through single-molecule FRET in a microfluidic mixer. Chemphyschem. 2011;12:3515–3518. doi: 10.1002/cphc.201100652. [DOI] [PubMed] [Google Scholar]
- 39.Liu JW, Campos LA, Cerminara M, Wang X, Ramanathan R, English DS, Munoz V. Exploring one-state downhill protein folding in single molecules. Proc Natl Acad Sci USA. 2012;109:179–184. doi: 10.1073/pnas.1111164109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben Ishay E, Hazan G, Rahamim G, Amir D, Haas E. An instrument for fast acquisition of fluorescence decay curves at picosecond resolution designed for “double kinetics” experiments: Application to fluorescence resonance excitation energy transfer study of protein folding. Rev Sci Inst. 2012;83:084301. doi: 10.1063/1.4737632. [DOI] [PubMed] [Google Scholar]
- 41.Yang LL, Kao MWP, Chen HL, Lim TS, Fann WS, Chen RPY. Observation of protein folding/unfolding dynamics of ubiquitin trapped in agarose gel by single-molecule FRET. Eur Biophys J Biophys Lett. 2012;41:189–198. doi: 10.1007/s00249-011-0772-6. [DOI] [PubMed] [Google Scholar]
- 42.Chung HS, Cellmer T, Louis JM, Eaton WA. Measuring ultrafast protein folding rates from photon-by-photon analysis of single molecule fluorescence trajectories. Chem Phys. 2013;422:229–237. doi: 10.1016/j.chemphys.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chung HS, Eaton WA. Single-molecule fluorescence probes dynamics of barrier crossing. Nature. 2013;502:685–688. doi: 10.1038/nature12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wunderlich B, Nettels D, Benke S, Clark J, Weidner S, Hofmann H, Pfeil SH, Schuler B. Microfluidic mixer designed for performing single-molecule kinetics with confocal detection on timescales from milliseconds to minutes. Nature Protocols. 2013;8:1459–1474. doi: 10.1038/nprot.2013.082. [DOI] [PubMed] [Google Scholar]
- 45.Kathuria SV, Kayatekin C, Barrea R, Kondrashkina E, Graceffa R, Guo L, Nobrega RP, Chakravarthy S, Matthews CR, Irving TC, et al. Microsecond barrier-limited chain collapse observed by time-Resolved FRET and SAXS. J Mol Biol. 2014;426:1980–1994. doi: 10.1016/j.jmb.2014.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orevi T, Lerner E, Rahamim G, Amir D, Haas E. Ensemble and Ssngle-molecule detected time-resolved FRET methods in studies of protein conformations and dynamics. In: Engelborghs Y, Visser A, editors. Fluorescence Spectroscopy and Microscopy: Methods and Protocols. Vol. 1076. 2014. pp. 113–169. Methods in Molecular Biology. [DOI] [PubMed] [Google Scholar]
- 47.Borgia A, Kemplen KR, Borgia MB, Soranno A, Shammas S, Wunderlich B, Nettels D, Best RB, Clarke J, Schuler B. Transient misfolding dominates multidomain protein folding. Nature Comm. 2015;6:8861. doi: 10.1038/ncomms9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hazan NP, Tomov TE, Tsukanov R, Liber M, Berger Y, Masoud R, Toth K, Langowski J, Nir E. Nucleosome core particle disassembly and assembly kinetics studied using single-molecule fluorescence. Biophys J. 2015;109:1676–1685. doi: 10.1016/j.bpj.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim SE, Lee IB, Hyeon C, Hong SC. Deciphering kinetic information from single-molecule FRET data that show slow transitions. J Phys Chem B. 2015;119:6974–6978. doi: 10.1021/acs.jpcb.5b03991. [DOI] [PubMed] [Google Scholar]
- 50.Ramanathan R, Munoz V. A method for extracting the free energy surface and conformational dynamics of fast-folding proteins from single molecule photon trajectories. J Phys Chem B. 2015;119:7944–7956. doi: 10.1021/acs.jpcb.5b03176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang LG, Zeng Y, Sun QQ, Sun YR, Guo ZH, Qu JNY, Yao SHA. Microsecond protein folding events revealed by time-resolved fluorescence resonance energy transfer in a microfluidic mixer. Anal Chem. 2015;87:5589–5595. doi: 10.1021/acs.analchem.5b00366. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi S, Kamagata K, Oikawa H. Where the complex things are: single molecule and ensemble spectroscopic investigations of protein folding dynamics. Curr Opin Struct Biol. 2016;36:1–9. doi: 10.1016/j.sbi.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 53.Tian PF, Best RB. Structural determinants of misfolding in multidomain proteins. Plos Comp Biol. 2016;12:e1004933. doi: 10.1371/journal.pcbi.1004933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maity H, Reddy G. Folding of protein L with implications for collapse in the denatured state ensemble. J Amer Chem Soc. 2016;138:2609–2616. doi: 10.1021/jacs.5b11300. [DOI] [PubMed] [Google Scholar]
- 55.Holmstrom ED, Nesbitt DJ. Biophysical insights from temperature-dependent single-molecule forster resonance energy transfer. Ann Rev Phys Chem. 2016;67:441–465. doi: 10.1146/annurev-physchem-040215-112544. [DOI] [PubMed] [Google Scholar]
- 56.Haran G. Single-Molecule Fluorescence Spectroscopy of the Folding of a Repeat Protein. 2016. Single-Molecule Fluorescence Spectroscopy of the Folding of a Repeat Protein Introduction; pp. 1–11. Springer Theses-Recognizing Outstanding PhD Research. [DOI] [Google Scholar]
- 57.Stockmar F, Kobitski AY, Nienhaus GU. Fast folding dynamics of an intermediate state in RNase H measured by single-molecule FRET. J Phys Chem B. 2016;120:641–649. doi: 10.1021/acs.jpcb.5b09336. [DOI] [PubMed] [Google Scholar]
- 58.Chung HS, Louis JM, Gopich IV. Analysis of fluorescence lifetime and energy transfer efficiency in single-molecule photon trajectories of fast-folding proteins. J Phys Chem B. 2016;120:680–699. doi: 10.1021/acs.jpcb.5b11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dingfelder F, Wunderlich B, Benke S, Zosel F, Zijlstra N, Nettels D, Schuler B. Rapid microfluidic double-jump mixing device for single-molecule spectroscopy. J Amer Chem Soc. 2017;139:6062–6065. doi: 10.1021/jacs.7b02357. [DOI] [PubMed] [Google Scholar]
- **60.Schuler B, Soranno A, Hofmann H, Nettels D. Single-molecule FRET spectroscopy and the polymer physics of unfolded and intrinsically disordered proteins. Ann Rev Biophys. 2016;45:207–231. doi: 10.1146/annurev-biophys-062215-010915. This paper is an excellent review of the many contributions that single molecule FRET spectroscopy, in combination with the application of polymer physics theory, has made to understanding structure distributions and dynamics of unfolded proteins and intrinsically disordered proteins. The most important contributions have come from the Schuler group. [DOI] [PubMed] [Google Scholar]
- **61.Gopich IV, Szabo A. Decoding the pattern of photon colors in single-molecule FRET. J Phys Chem B. 2009;113:10965–10973. doi: 10.1021/jp903671p. This is the key paper describing the original work by Gopich and Szabo on the development of their maximum likelihood method to extract rates of conformatinoal transitions in single molecule FRET experiments by analyzing the sequence of photon colors and the time interval between their detection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gopich IV. Accuracy of maximum likelihood estimates of a two-state model in single-molecule FRET. J Chem Phys. 2015;142:034110. doi: 10.1063/1.4904381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **63.Chung HS, McHale K, Louis JM, Eaton WA. Single-molecule fluorescence experiments determine protein folding transition path times. Science. 2012;335:981–984. doi: 10.1126/science.1215768. This paper describes the first determination of the average transition path time for a molecular process. The average time is found to differ by less than a factor of 6 for two proteins with folding rates that differ by 10,000-fold. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *64.Chung HS, Piana-Agostinetti S, Shaw DE, Eaton WA. Structural origin of slow diffusion in protein folding. Science. 2015;349:1504–1510. doi: 10.1126/science.aab1369. By employing single molecule FRET and MD simulations, this work identified that the formation of non-native salt bridges during folding transitions causes slow diffusion of a designed protein α3D on a 1D free energy surface. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Godoy-Ruiz R, Henry ER, Kubelka J, Hofrichter J, Munoz V, Sanchez-Ruiz JM, Eaton WA. Estimating free-energy barrier heights for an ultrafast folding protein from calorimetric and kinetic data. J Phys Chem B. 2008;112:5938–5949. doi: 10.1021/jp0757715. [DOI] [PubMed] [Google Scholar]
- 66.Best RB, Hummer G. Microscopic interpretation of folding phi-values using the transition path ensemble. Proc Natl Acad Sci USA. 2016;113:3263–3268. doi: 10.1073/pnas.1520864113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jas GS, Eaton WA, Hofrichter J. Effect of viscosity on the kinetics of alpha-helix and beta-hairpin formation. J Phys Chem B. 2001;105:261–272. [Google Scholar]
- 68.Zheng WW, De Sancho D, Hoppe T, Best RB. Dependence of internal friction on folding mechanism. J Am Chem Soc. 2015;137:3283–3290. doi: 10.1021/ja511609u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Sancho D, Sirur A, Best RB. Molecular origins of internal friction effects on protein-folding rates. Nature Comm. 2014;5:4307. doi: 10.1038/ncomms5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sadqi M, de Alba E, Perez-Jimenez R, Sanchez-Ruiz JM, Munoz V. A designed protein as experimental model of primordial folding. Proc Natl Acad Sci USA. 2009;106:4127–4132. doi: 10.1073/pnas.0812108106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **71.Ferreiro DU, Komives EA, Wolynes PG. Frustration in biomolecules. Quart Rev Biophys. 2014;47:285–363. doi: 10.1017/S0033583514000092. The concept of frustation is described and shown how it contributes to a deeper understanding of protein structure, function, and folding. An example of a quantitative statement of the principle of minimal frustration is that the free energy gap between folded and folded states is sufficiently large that most proteins fold without being trapped in free energy wells corresponding to misfolded states. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kubelka J, Hofrichter J, Eaton WA. The protein folding ‘speed limit’. Curr Opin Struct Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 73.Goloubinoff P, Gatenby AA, Lorimer GH. GroEL heat-shock protein promotes assembly of foreign prokaryotic ribulose biphosphate carboxylase oligomers in escherichia coli. Nature. 1989;337:44–47. doi: 10.1038/337044a0. [DOI] [PubMed] [Google Scholar]
- 74.Cheng MY, Hartl FU, Martin J, Pollock RA, Kalousek F, Neupert W, Hallberg EM, Hallberg RL, Horwich AL. Mictochondrial heat-shock protein HSP60 is essential for assembly of proteins imported into yeasr mitochondria. Nature. 1989;337:620–625. doi: 10.1038/337620a0. [DOI] [PubMed] [Google Scholar]
- 75.Baldwin RL. Clash between energy landscape theory and foldon-dependent protein folding. Proc Natl Acad Sci USA. 2017;114:8442–8443. doi: 10.1073/pnas.1709133114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim PS, Baldwin RL. Intermediates in gthe folding reactions of small proteins. Ann Rev Biochem. 1990;59:631–660. doi: 10.1146/annurev.bi.59.070190.003215. [DOI] [PubMed] [Google Scholar]
- 77.Piana S, Lindorff-Larsen K, Shaw DE. How robust are protein folding simulations with respect to force field parameterization? Biophys J. 2011;100:L47–L49. doi: 10.1016/j.bpj.2011.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jackson SE, Fersht AR. Folding of chymotrypsin inhibitor-2. 1. Evidence for a 2-state transtion. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 79.Bai YW, Sosnick TR, Mayne L, Englander SW. Protein folding intermediates- native state hydrogen exchange. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Englander SW, Mayne L. The case for defined protein folding pathways. Proc Natl Acad Sci USA. 2017;114:8253–8258. doi: 10.1073/pnas.1706196114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *81.Eaton WA, Wolynes PG. Theory, simulations and experiments show that proteins fold by multiple pathways. Proc Natl Acad Sci USA. 2017 doi: 10.1073/pnas.1716444114. (in press). This Letter rebuts the main conclusions of ref. 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sabelko J, Ervin J, Gruebele M. Observation of strange kinetics in protein folding. Proc Natl Acad Sci USA. 1999;96:6031–6036. doi: 10.1073/pnas.96.11.6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eaton WA. Searching for “downhill scenarios” in protein folding. Proc Natl Acad Sci USA. 1999;96:5897–5899. doi: 10.1073/pnas.96.11.5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garcia-Mira MM, Sadqi M, Fischer N, Sanchez-Ruiz JM, Munoz V. Experimental identification of downhill protein folding. Science. 2002;298:2191–2195. doi: 10.1126/science.1077809. [DOI] [PubMed] [Google Scholar]
- 85.Sadqi M, Fushman D, Munoz V. Atom-by-atom analysis of global downhill protein folding. Nature. 2006;442:317–321. doi: 10.1038/nature04859. [DOI] [PubMed] [Google Scholar]
- 86.Munoz V, Cerminara M. When fast is better: protein folding fundamentals and mechanisms from ultrafast approaches. Biochem J. 2016;473:2545–2559. doi: 10.1042/BCJ20160107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chaudhury S, Makarov DE. A harmonic transition state approximation for the duration of reactive events in complex molecular rearrangements. J Chem Phys. 2010;133:034118. doi: 10.1063/1.3459058. [DOI] [PubMed] [Google Scholar]
- 88.Orland H. Generating transition paths by Langevin bridges. J Chem Phys. 2011;134:174114. doi: 10.1063/1.3586036. [DOI] [PubMed] [Google Scholar]
- 89.Kim WK, Netz RR. The mean shape of transition and first-passage paths. J Chem Phys. 2015;143:224108. doi: 10.1063/1.4936408. [DOI] [PubMed] [Google Scholar]
- 90.Daldrop JO, Kim WK, Netz RR. Transition paths are hot. Epl. 2016;113:18004. [Google Scholar]
- 91.Makarov DE. Reconciling transition path time and rate measurements in reactions with large entropic barriers. J Chem Phys. 2017;146:071101. doi: 10.1063/1.4977177. [DOI] [PubMed] [Google Scholar]
- 92.Zhang BW, Jasnow D, Zuckerman DM. Transition-event durations in one-dimensional activated processes. J Chem Phys. 2007;126:074504. doi: 10.1063/1.2434966. [DOI] [PubMed] [Google Scholar]
- **93.Neupane K, Foster DAN, Dee DR, Yu H, Wang F, Woodside MT. Direct observation of transition paths during the folding of proteins and nucleic acids. Science. 2016;352:239–242. doi: 10.1126/science.aad0637. This work represents a major advance in the invesitigation of transition paths in biomolecular folding. By improving the time resolution of optical tweezer experiments, the authors are able to resolve the duration of individual transition paths for folding a DNA hairpin and thereby measure a complete distribution of transition path times. By extracting the free energy profile from their data, together with rates and transition path times, the authors confirm the validity of equation 2 [94] [DOI] [PubMed] [Google Scholar]
- 94.Truex K, Chung HS, Louis JM, Eaton WA. Testing landscape theory for biomolecular processes with single molecule fluorescence spectroscopy. Physical Review Letters. 2015;115:018101. doi: 10.1103/PhysRevLett.115.018101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pollak E. Transition path time distribution and the transition path free energy barrier. Phys Chem Chem Phys. 2016;18:28872–28882. doi: 10.1039/c6cp05052b. [DOI] [PubMed] [Google Scholar]
- 96.Satija R, Das A, Makarov DE. Transition path times reveal memory effects and anomalous diffusion in the dynamics of protein folding. J Chem Phys. 2017;147:152707. doi: 10.1063/1.4993228. [DOI] [PubMed] [Google Scholar]
- 97.Laleman M, carlon E, Orland H. Condensed Matter Preprints. 2017 [Google Scholar]
- 98.Campos LA, Liu JW, Wang X, Ramanathan R, English DS, Munoz V. A photoprotection strategy for microsecond-resolution single-molecule fluorescence spectroscopy. Nature Meth. 2011;8:143–163. doi: 10.1038/nmeth.1553. [DOI] [PubMed] [Google Scholar]
- 99.Zheng QS, Juette MF, Jockusch S, Wasserman MR, Zhou Z, Altman RB, Blanchard SC. Ultra-stable organic fluorophores for single-molecule research. Chem Soc Rev. 2014;43:1044–1056. doi: 10.1039/c3cs60237k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chung HS, Meng F, Kim J-Y, McHale K, Gopich IV, Louis JM. Oligomerization of the tetramerization domain of p53 probed by two- and three-color single-molecule FRET. Proc Natl Acad Sci USA. 2017;114:E6812–E6821. doi: 10.1073/pnas.1700357114. [DOI] [PMC free article] [PubMed] [Google Scholar]