

Abstract
Polycystic liver disease (PLD) is a group of genetic disorders with limited treatment and significant morbidity. Hepatic cysts arise from cholangiocytes exhibiting a hyperproliferative phenotype. Considering that hyperproliferation of many cell types is associated with alterations in autophagy, we hypothesized that autophagy is altered in PLD cholangiocytes, contributes to hepatic cystogenesis, and might represent a potential therapeutic target. We employed Functional Pathway Cluster Analysis (FPCA) and NextGen Sequencing (NGS), transmission electron microscopy, immunofluorescence confocal microscopy, and western blotting to assess autophagy in human and rodent PLD cholangiocytes. A 3-D culture model was utilized to study the effects of molecular and pharmacologic inhibition of autophagy on hepatic cystogenesis in vitro, and the PCK rat, an animal model of PLD, to study the effects of hydroxychloroquine (HCQ), a drug that interferes with the autophagy pathway, on disease progression in vivo. Assessment of the transcriptome of PLD cholangiocytes followed by FPCA revealed that the autophagy-lysosomal pathway is one of the most altered pathways in PLD. Direct evaluation of autophagy in PLD cholangiocytes both in vitro and in vivo showed increased number and size of autophagosomes, lysosomes and autolysosomes, overexpression of autophagy-related proteins (Atg5, Beclin1, Atg7, and LC3), and enhanced autophagic flux associated with activation of the cAMP-PKA-CREB signaling pathway. Molecular and pharmacologic intervention in autophagy with ATG7 siRNA, Bafilomycin A1 and HCQ reduced proliferation of PLD cholangiocytes in vitro and growth of hepatic cysts in 3-D cultures. HCQ also efficiently inhibited hepatic cystogenesis in the PCK rat.
Conclusion
Autophagy is increased in PLD cholangiocytes, contributes to hepatic cystogenesis, and represents a potential therapeutic target for disease treatment.
Keywords: autophagy-lysosomal pathway, Atg proteins, cAMP-PKA-CREB signaling, hydroxychloroquine
Introduction
Autophagy is an evolutionary conserved pathway involved in regulation of cellular homeostasis via degradation and recycling of cytoplasmic constituents in lysosomes. In the course of autophagy, damaged or excess organelles, long-lived proteins, protein aggregates, nucleic acids, lipids and other cytoplasmic materials are transported to autophagosomes that later fuse with lysosomes in which the autophagosomal cargo is degraded and recycled.1 Originally, autophagy was considered a cellular survival mechanism against unfavorable conditions, in particular, starvation. But in recent years an important role for autophagy has emerged in various disorders including diseases of the hepato-biliary system.2-4 Moreover, autophagy is now considered as a druggable target for a number of pathologies.5, 6
In this work, we addressed the role of autophagy in polycystic liver disease (PLD), the hepatic manifestation of autosomal dominant (ADPKD) and autosomal recessive (ARPKD) polycystic kidney diseases. PLD is characterized by fluid-filled hepatic cysts that arise from cholangiocytes and grow progressively over time as a result of structural and functional disturbances in numerous intracellular processes and signaling pathways.7, 8 Considering association of autophagy with rapid cell proliferation2 we speculated that autophagy might play an important role in hepatic cystogenesis, a condition of benign cholangiocyte hyperproliferation.
To our knowledge, studies that implicate autophagy in PLD associated with PKD are limited to a single published work reporting that aberrant activation of the PI3K/mTOR pathway in the PCK rat (an animal model of ARPKD) increased cholangiocyte proliferation and cyst growth, whereas inhibition of this pathway attenuated cholangiocyte proliferation via mechanisms involving apoptosis and/or autophagy.9 However, a direct contribution of autophagy to hepatic cystogenesis, and a role as a potential target for a therapeutic treatment of PLD were not addressed.
Thus, the aims of this work were: (i) to directly assess autophagy in human and rodent cholangiocytes of both forms of PLD (i.e., ADPKD and ARPKD), (ii) to study a possible contribution of autophagy to hepatic cystogenesis, and (iii) to test if autophagy represents a potential therapeutic target for PLD.
Materials and Methods
Animals, cell cultures and reagents
Age- and sex-matched wild type (WT, Harlan Sprague Dawley) and PCK rats (Harlan Sprague Dawley background), WT and Pkd2WS25/- mice and Pkhd1del2/del2 mice (all, C57BL/6 background) were housed at the 12 hours light/dark cycle, maintained on a standard diet and water ad lib. After anesthesia (pentobarbital, 50 mg/kg) livers were removed, fixed and paraffin-embedded. The protocol was approved by the Mayo Institutional Animal Care and Use Committee. Normal (NRC) and PCK rat cholangiocytes, normal human (NHC) and ADPKD cholangiocytes were maintained as we previously described.10 NRC and PCK rat cholangiocytes were stable transfected with LC3-GFP construct (Invitrogen). ADPKD cholangiocytes were transfected with ATG7 or control siRNAs (Invitrogen). Bafilomycin A1 and hydroxychloroquine (HCQ) were purchased from Sigma Aldrich.
RNA preparation and sequencing
Protocol of cholangiocyte RNA-sequencing is described in Supporting Materials and Methods.
Transmission electron microscopy (TEM) of liver sections and cultured cholangiocytes was performed as we previously described.7, 11
Western blotting
Proteins were separated by 4–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and incubated with primary antibodies to Atg5 (Sigma Aldrich), Beclin 1 (Novus), Atg7 (Sigma Aldrich), p62 (Sigma Aldrich), p-CREB, CREB (Cell Signaling) and β-actin (Sigma Aldrich). For LC3 detection, proteins were separated by 4% stacking gel and 20% separating gel followed by incubation with LC3 antibody (Novus). Corresponding secondary horseradish peroxidase-conjugated antibodies (Santa Cruz Biotechnology) were applied and bands visualized with ECL Plus Western Blotting Detection kit (BD Biosciences). Integrated density of protein bands was assessed by ImageJ (NIH) and data presented as arbitrary units compared to β-actin.
Immunofluorescence confocal microscopy
Livers from WT and PCK rats; and healthy humans and patients with ADPKD and ADPLD (provided by the Mayo Clinical Core) were incubated with primary antibodies to Atg5, Atg7, Beclin 1, LC3 and PCNA (Santa Cruz) and then with respective secondary fluorescent antibodies (Molecular Probes). Nuclei were stained with DAPI (4-,6-diamidino-2-phenylindole; Invitrogen). Liver sections and LC3-GFP transfected cultured cholangiocytes were analyzed by ZeissLSM-510 microscope (Carl Zeiss, Thornwood, NY). Fluorescence intensity (arbitrary units) of protein staining was assessed by Adobe Photoshop Elements 10 and expressed as percent change in cystic cholangiocytes compared to respective controls in which fluorescence intensity was considered to be equal 100%. Number of LC3-GFP positive puncta was assessed by ImageJ.
Cholangiocyte proliferation was determined by CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI). Cholangiocytes (2500 cells/well) were grown in regular DMEM/F12 media at 37°C (5% CO2 and 100% humidity) for 48 hours and treated with Bafilomycin A1 and HCQ (both, 100 nM) for additional 24 hours. Alterations in proliferation of cultured cholangiocytes after treatment were expressed as percent change compared to un-treated cholangiocytes in which cell proliferation was considered to be equal 100%. Proliferation of PLD cholangiocytes in vivo was evaluated by the number of PCNA-positive nuclei as we described previously.12
cAMP measurements
Cholangiocytes (10,000/well) were incubated with pasireotide (20 μM), HCQ (100 nM) or both drugs in combination for 15-30 min and cAMP levels were detected using cAMP-Screen® System (Applied Biosystems).
Intrahepatic bile ducts and cholangioids in 3-D cultures
Microdissected from the PCK rat liver intrahepatic cystic bile ducts were grown in three-dimensional matrices as described.7, 11 The cysts were treated with Bafilomycin A1 and HCQ (both, 100 nM) daily. Images were taken at day 1 (i.e., 24 hours after seeding) and at day 5. The circumference of each cyst was measured using ImageJ software (NIH) as described.11
Cholangioids were generated by plating ∼6000 human ADPKD cholangiocytes on the polyHEMA-coated surfaces as we recently described.13 Circumference of cholangioids was measured by Image J and expressed as percent change in ATG7 siRNA-transfected cholangioids compared to cholangioids transfected with scrambled controls (in which circumference was considered to be equal 100%).
Treatment protocol
PCK rats (4-6 weeks old, n=16 females, n=16 males) were divided into four groups: (i) pasireotide-treated (n=4 females, n=4 males), (ii) HCQ-treated (n=4 females, n=4 males), (iii) pasireotide + HCQ-treated (n=4 females, n=4 males), and (iv) un-treated (n=4 females, n=4 males). Pasireotide (20 mg/kg/day) was dissolved in sterile water and administered via osmotic mini-pumps (model 2002, Alzet Osmotic Pumps, Cupertino, CA) implanted subcutaneously on the animal back as previously described.10 Pumps were replaced every 2 weeks; at this time pasireotide concentrations were adjusted to the animal weight. HCQ (15mg/kg bw) was dissolved in DMSO and injected intraperitoneally (IP); doses of HCQ were adjusted to the rat weight every other week. Un-treated group had implanted pumps filled with sterile water and received equal doses of DMSO (IP injections). Rats were sacrificed after 6 weeks of treatment and body and organ weights were assessed. Cystic and fibrotic areas of liver and kidneys were analyzed as previously described.10 Based on our previous experience that percent of liver parenchyma occupied by hepatic cysts do not differ between male and female PCK rats,10, 12, 14 they were combined for estimation of cystic and fibrotic areas.
Statistical analysis
The data are expressed as the MEAN±SD. Statistical analysis was performed by Student's t-test and results were considered statistically significant at P<0.05.
Results
PLD cholangiocytes are characterized by the altered autophagy-lysosomal pathway
Transcriptome profiling of NRC, PCK rat cholangiocytes, NHC and human ADPKD cholangiocytes revealed 1803 genes differentially expressed in the PCK rat cholangiocytes, and 2590 genes differentially expressed in human ADPKD cholangiocytes, compared to NRC and NHC, respectively. The pathway cluster analysis of these genes by the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways indicated 33 altered pathways in the PCK rat cholangiocytes and 30 pathways in human ADPKD cholangiocytes, including the autophagy-lysosomal pathway that was the most altered pathway in the PCK rat (Fig. 1A) and the second most altered pathway in human ADPKD (Fig. 1B) cholangiocytes.
Figure 1. PLD cholangiocytes are characterized by the altered autophagy-lysosomal pathway.
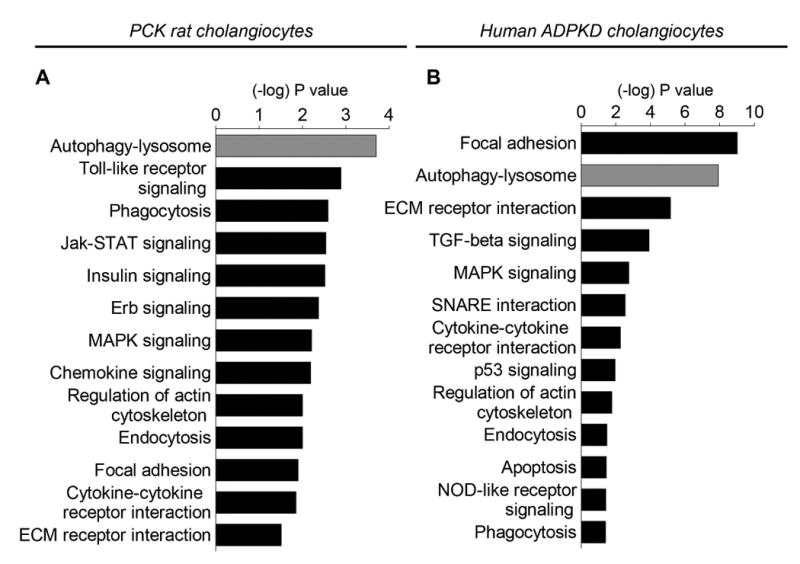
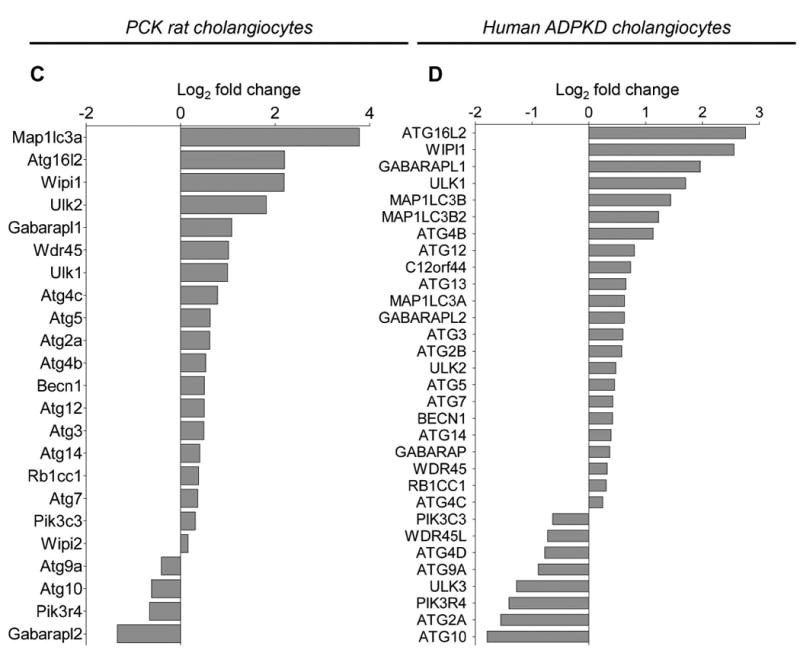
(A) The autophagy-lysosomal pathway is the most altered pathway in PCK rat cholangiocytes and (B) the second most altered pathway in human ADPKD cholangiocytes. The 13 altered pathways are shown. (C) 19 genes out of 26 genes associated with autophagy are up-regulated in PCK rat cholangiocytes, and (D) 23 genes out of 34 genes are up-regulated in human ADPKD cholangiocytes. Changes in gene expression in PCK rat and human ADPKD cholangiocytes compared to NRC and NHC (n=3 for each experimental conditions) were calculated using R package edgeR. FDR 0.05 and log2 fold change >=1 or log2 fold change <=-1 were considered as the cutoff for up and down regulated genes.
We also identified 26 genes associated with autophagy in rat cholangiocytes, and 34 genes in human cholangiocytes. The majority of these genes were up-regulated in PCK rat cholangiocytes [i.e., 19 genes out of 26 genes, (Fig. 1C)], and in human ADPKD cholangiocytes [i.e., 23 genes out of 34 genes, (Fig. 1D)]. In particular, autophagy-related Atg/ATG (i.e., Atg2/ATG2, Atg3/ATG3, Atg4/ATG4, Atg5/ATG5, Becn1/BECN1, Atg7/ATG7, Map1Lc3/MAP1LC3, Agt12/ATG12, Atg14/ATG14, Atg16l2/ATG16L2, Wipi1/WIPI1 and Wdr45/WDR45), genes encoding proteins essential for induction of autophagy (i.e., Ulk1/ULK1, Ulk2/ULK2, Rb1cc1/RB1CC1) and for autophagic flux (i.e., Gabarapl1/GABARAPL1) were up-regulated in both rat and human PLD cholangiocytes.
The number and size of organelles involved in autophagy are increased in PLD cholangiocytes
The number of autophagosomes, lysosomes and autolysosomes, the key intracellular organelles involved in autophagy (Fig. 2A), was 2-3 times greater in cultured PCK rat and human ADPKD cholangiocytes compared to respective controls (Fig. 2B-C). PCK rat cholangiocytes contained per section 3.9±1.1 autophagosomes, 2.7±1.5 lysosomes and 7.5±3.9 autolysosomes compared, correspondingly, to 1.2±1.1 autophagosomes, 1.2±0.4 lysosomes and 4.1±1.9 autolysosomes in NRC (Fig. 2B-C). Human ADPKD cholangiocytes contained per section 3.9±1.9 autophagosomes, 2.1±1.1 lysosomes and 6.4±2.0 autolysosomes compared to 1.8±0.8 autophagosomes, 1.0±0.8 lysosomes and 2.1±1.4 autolysosomes in NHC (Fig. 2B-C).
Figure 2. The number and size of autophagosomes, lysosomes and autolysosomes are increased in PLD cholangiocytes.
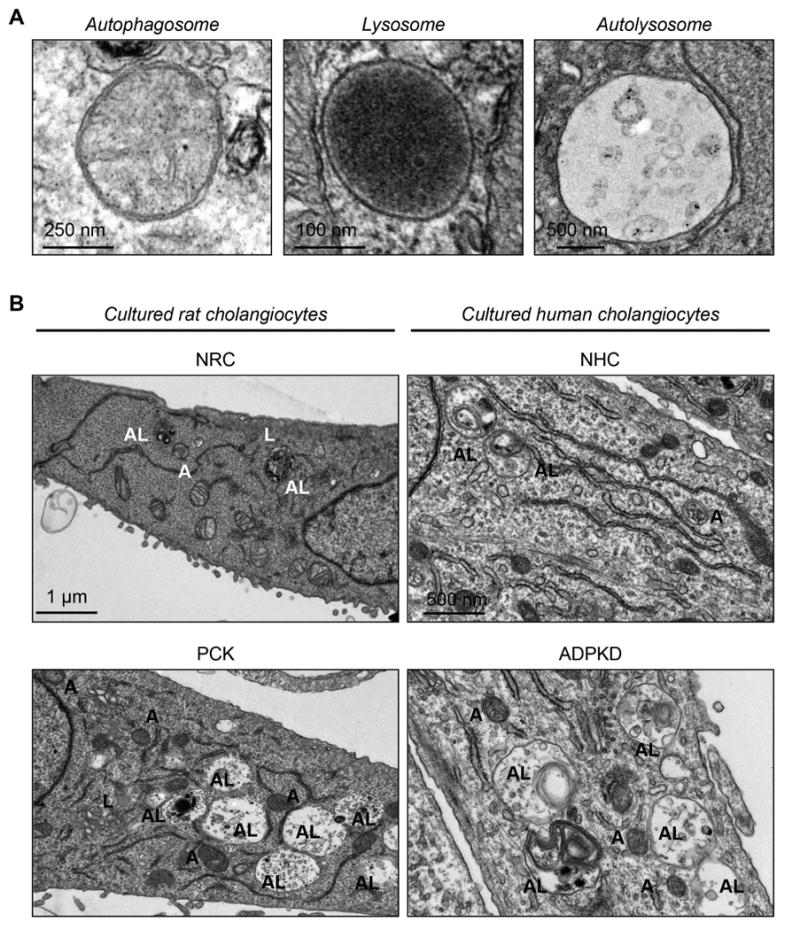
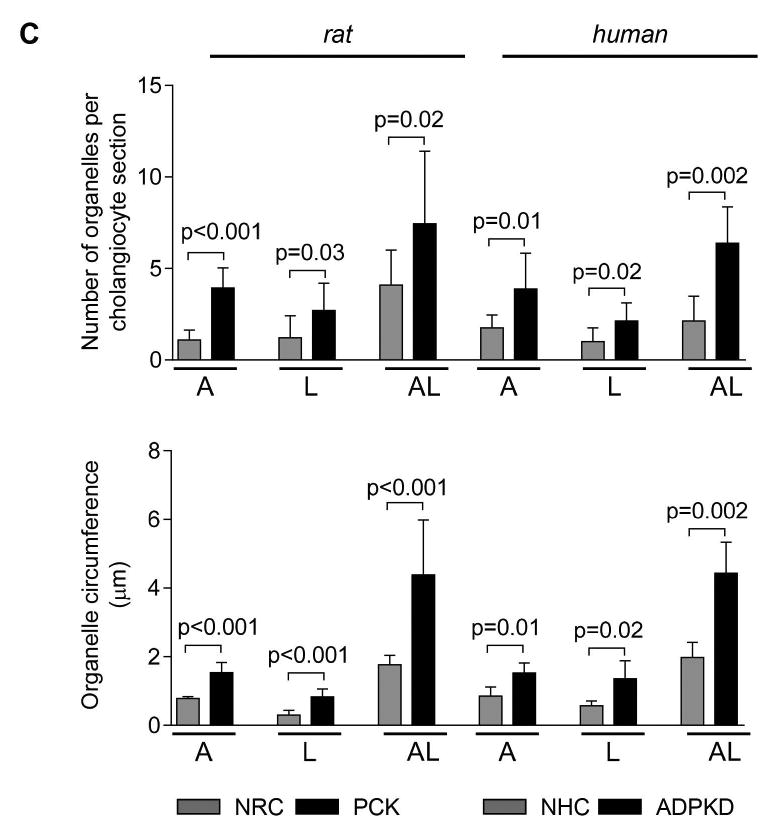
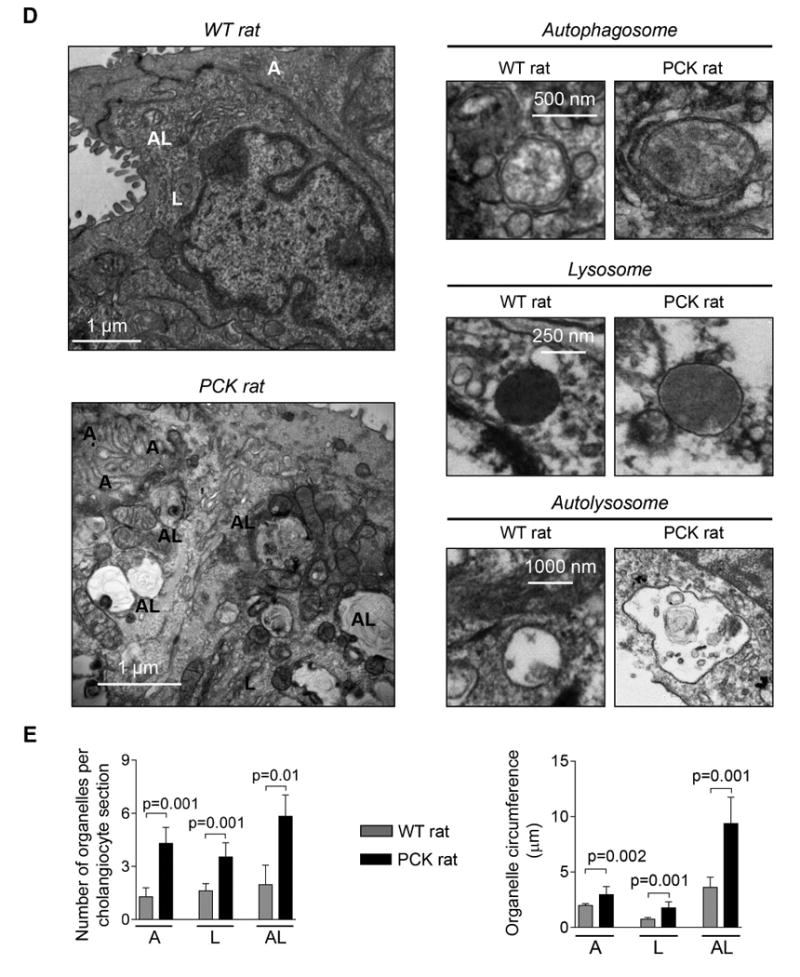
(A) Representative TEM images of rat cholangiocyte autophagosomes, lysosomes and autolysosomes. (B) Representative TEM images and (C) bar graphs demonstrate that the number and size of autophagosomes, lysosomes and autolysosomes are greater in cultured PCK rat and human ADPKD cholangiocytes compared to NRC and NHC, respectively. n=30 cholangiocyte sections for each experimental condition. (D) Representative TEM images and (E) bar graphs show an increase in the number and size of organelles in cholangiocytes lining liver cysts in PCK rats compared to cholangiocytes of WT rats. Five liver sections from each three WT rats and five PCK rats were analyzed. Data are presented as MEAN±SD. Abbreviation: A - autophagosome, L – lysosome, AL – autolysosome, WT – wild type.
Autophagosomes, lysosomes and autolysosomes were also larger in PCK rat and human ADPKD cholangiocytes. In PCK cholangiocytes, the circumference of autophagosomes, lysosomes and autolysosomes was, respectively, 1.53±0.30 μm, 0.83±0.22 μm and 4.39±1.60 μm compared to 0.78±0.06 μm (autophagosomes), 0.29±0.14 μm (lysosomes) and 1.76±0.28 μm (autolysosomes) in NRC (Fig. 2B-C). Similarly, in ADPKD cholangiocytes the circumference of autophagosomes, lysosomes and autolysosomes was, respectively, 1.52±0.29 μm, 1.36±0.53 μm and 4.43±0.91 μm compared to 0.84±0.27 μm (autophagosomes), 0.57±0.14 μm (lysosomes) and 1.98±0.44 μm (autolysosomes) in NHC (Fig. 2B-C).
An increased number and size of these organelles were also observed in vivo. Cholangiocytes of PCK rats contained per section 4.3±0.9 autophagosomes versus 1.3±0.5 in WT rat, 3.5±0.8 lysosomes versus 1.6±0.4 in WT rat, and 5.8±1.2 autolysosomes versus 1.9±1.1 in WT rat (Fig. 2D-E). The circumference of autophagosomes, lysosomes and autolysosomes was, respectively, 2.96±0.73 μm, 1.76±0.54 μm and 9.38±2.38 μm compared to 1.98±0.17 μm (autophagosomes), 0.75±0.14 μm (lysosomes) and 3.62±0.91 μm (autolysosomes) in WT rat (Fig. 2D-E). Comparable changes in the number and size of autophagic organelles were also observed in cholangiocytes of ADPKD patients (Supporting Fig. 1) and Pkd2WS25/- mouse (an animal model of ADPKD) (Supporting Fig. 2).
Expression of autophagy-related proteins is increased in PLD cholangiocytes
Expression of selected Atg proteins (i.e., Atg5, Beclin 1, Atg7 and LC3) in livers of PCK rats and humans with ADPKD and ARPKD was increased 2-3-fold compared to respective controls (Fig. 3A-B). Similar expression of these proteins was also observed in cholangiocytes of Pkd2WS25/- mouse (an animal model of ADPKD) (Supporting Fig. 3). In cultured PCK rat and human ADPKD cholangiocytes, copy numbers of autophagy-related protein transcripts (i.e., Atg5, Beclin 1, Atg7 and LC3; Fig. 3C) and expression of Atg proteins (Fig. 3D-E) were 2-5 fold greater compared to corresponding controls.
Figure 3. Expression of autophagy-related proteins is increased in PLD cholangiocytes.
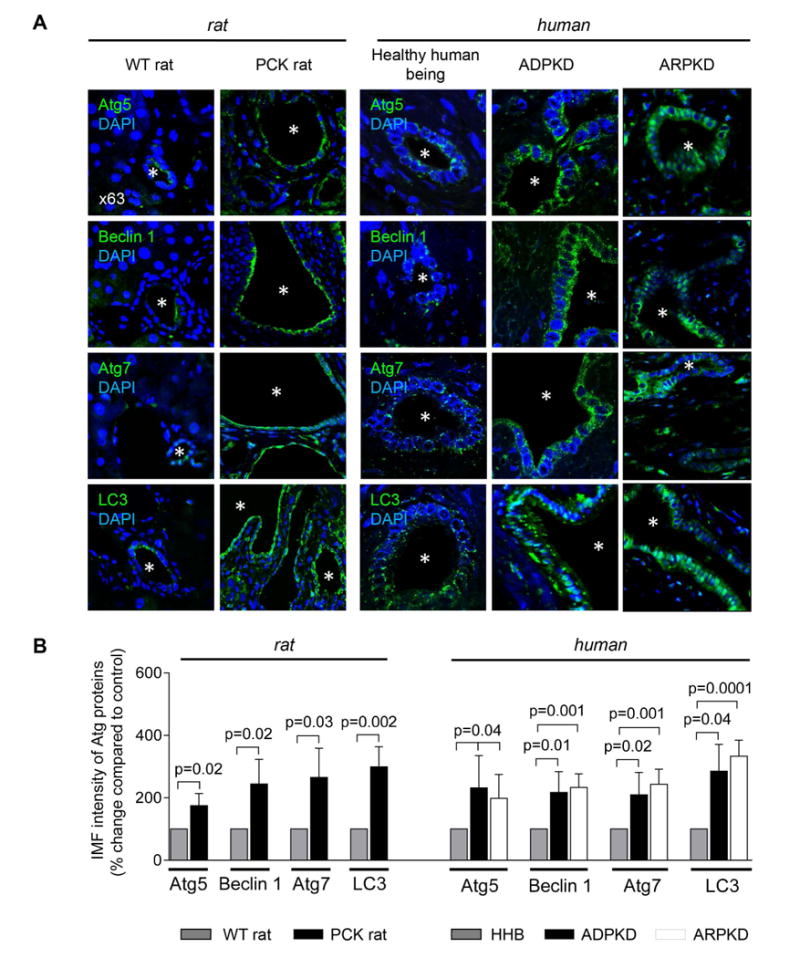
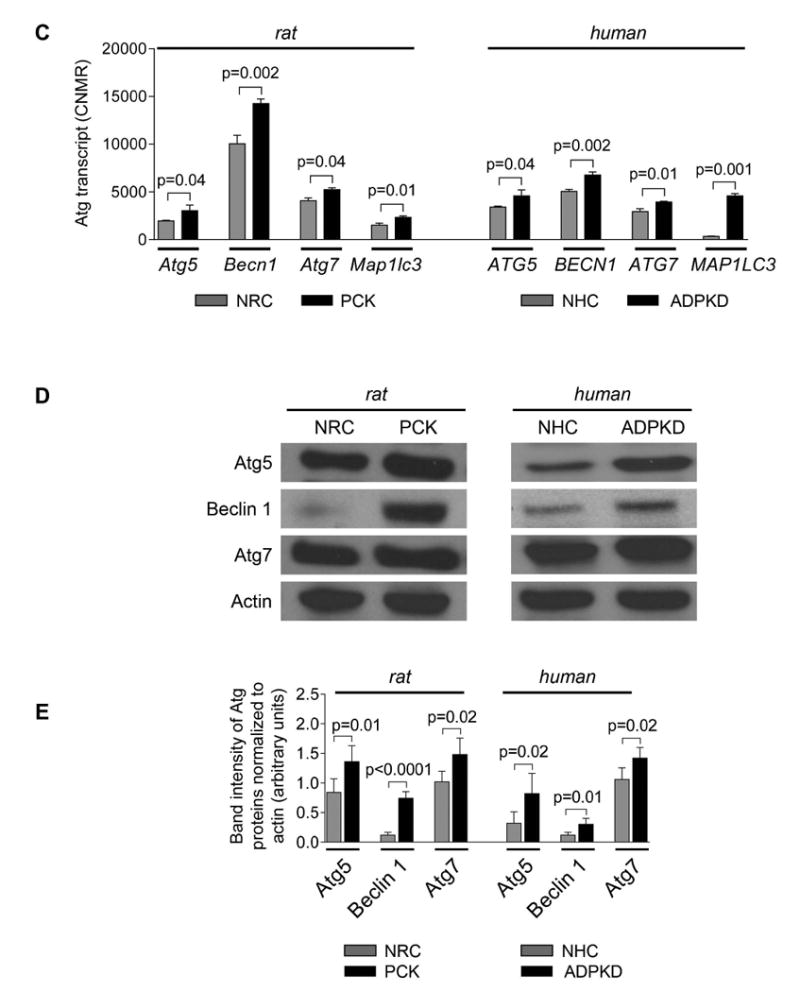
(A) Representative confocal microscopy images of liver sections of wild type rats (n=3), PCK rats (n=5), healthy human beings (n=3), ADPKD (n=3) and ARPKD (n=3) patients and (B) quantitation of relative IMF intensity show increased expression of autophagy-related proteins in PLD cholangiocytes. Five liver sections for each liver were analyzed. Asterisks depict bile duct or cyst lumen. (C) Higher copy numbers of Atg5, Beclin 1, Atg7 and LC3 protein transcripts per million reads (CNMR) were observed in cultured rat and human PLD cholangiocytes compared to controls. (n=3 for each cell line). (D) Representative western blots and (E) quantitation of band density of Atg proteins show that their levels are increased in PLD cholangiocytes. (n=3 for each cell line). Data are presented as MEAN±SD. Abbreviation: WT – wild type, HHB – healthy human beings.
Autophagic flux is increased in PLD cholangiocytes
Expression of the autophagy marker, LC3-II, in PCK rat cholangiocytes was greater than in NRC (Fig. 4A-B) and further increased in the presence of Bafilomycin A1, a drug that inhibits the final steps of autophagy (i.e., prevents autophagosome-lysosome fusion and degradation; Fig. 4A-B) indicating increased autophagic flux (i.e., the entire process of autophagy).
Figure 4. Autophagic flux is enhanced in PLD cholangiocytes.
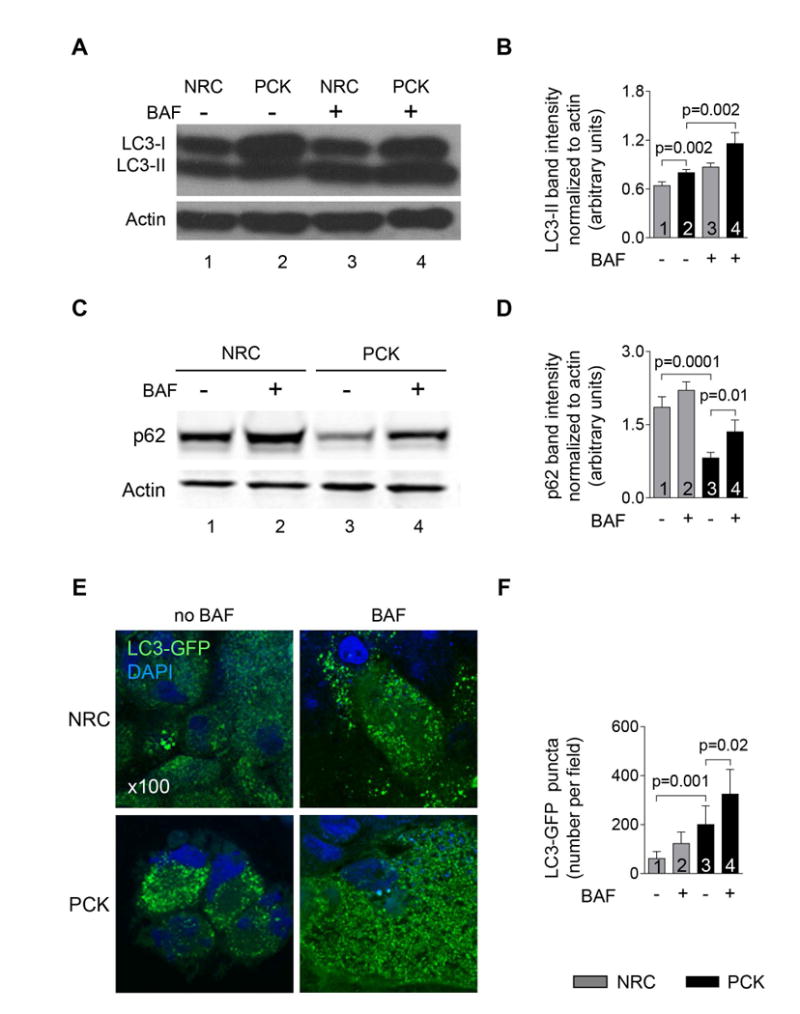
(A&B) Expression of autophagosome marker, LC3-II, was greater in PCK rat cholangiocytes compared to NRC as demonstrated by representative western blots (line 2 versus line 1) and quantitative analysis of LC3-II band intensity (bar 2 versus bar 1). Bafilomycin A1 (100 nM) further increased LC3-II levels in PCK rat cholangiocytes (line 4 versus line 2; bar 4 versus bar 2) demonstrating enhanced autophagic flux. (C&D) Expression of p62/SQSTM1, an autophagy receptor, was lower in PCK rat cholangiocytes compared to NRC (line 3 versus line 1; bar 3 versus bar 1). Bafilomycin A1 (100 nM) increased p62 levels in PCK cholangiocytes (line 4 versus line 3; bar 4 versus bar 3) showing enhanced autophagic flux. (E) Representative immunofluorescence confocal microscopy images of NRC and PCK rat cholangiocytes stable transfected with LC3-GFP construct and (F) quantitation of LC3-GFP-positive puncta demonstrate that the number of LC3-GFP puncta is greater in PCK cholangiocytes compared to NRC (bar 3 versus bar 1). Treatment of PCK rat cholangiocytes with Bafilomycin A1 (100 nM) further increased the number of LC3-GFP-positive puncta (bar 4 versus bar 3) indicating increased autophagic flux. (n=3 for each cell line). Data are presented as MEAN±SD. Abbreviation: BAF – Bafilomycin A1.
The expression of autophagy receptor, p62 (level of which is decreased when autophagic flux is increased15) was reduced in PCK rat cholangiocytes compared to NRC (Fig. 4C-D). The levels of p62 was partially recovered in PCK rat cholangiocytes in the presence of Bafilomycin A1 (Fig. 4C-D) also indicating increased autophagic flux.
An increase in autophagic flux in PLD cholangiocytes was also confirmed in cholangiocytes stable transfected with LC3-GFP construct (Fig. 4E-F). The number of LC3-GFP-positive puncta was greater in PCK rat cholangiocytes compared to NRC. A substantial increase in the number of LC3-GFP puncta in PCK rat cholangiocytes treated with Bafilomycin A1 further indicated increased autophagy.
cAMP signaling contributes to increased autophagy in PLD cholangiocytes
Our bioinformatics analysis revealed that Atg/ATG genes (Atg5/ATG5, Atg7/ATG7, Map1lc3/MAP1LC3) in rat and human cholangiocytes harbor CREB binding sites within their promoters suggesting that over-expression of Atg proteins and increased autophagic flux in PLD cholangiocytes might be associated with the cAMP-PKA-CREB signaling pathway. This hypothesis is supported by three sets of our data. First, we demonstrated that in human ADPKD cholangiocytes treated with an activator of PKA, 6-Phe-cAMP, phosphorylation of CREB (Fig. 5A), the level of LC3 and autophagic flux (Fig. 5B) were increased. The effects of 6-Phe-cAMP on autophagy were CREB-mediated, since they were abolished by pre-treatment of cholangiocytes with the CREB inhibitor, 666-15 (Fig. 5B). Second, expression of Atg5, Atg7 and LC3 in cholangiocytes of PCK rats treated with the somatostatin analogue, paseriotide that lowers cAMP levels,10 was decreased (Fig. 5C). Third, the treatment of cultured human ADPKD cholangiocytes with pasireotide resulted in decreased expression of Atg5 and Atg7 (Fig. 5D) and decreased autophagic flux (Fig. 5 E). The inhibitory effects of pasireotide on Atg5 and Atg7 expression were similar to the effects of the CREB inhibitor, 666-15 (Fig. 5 D).
Figure 5. The cAMP-PKA-CREB signaling pathway contributes to increased autophagy in PLD cholangiocytes.
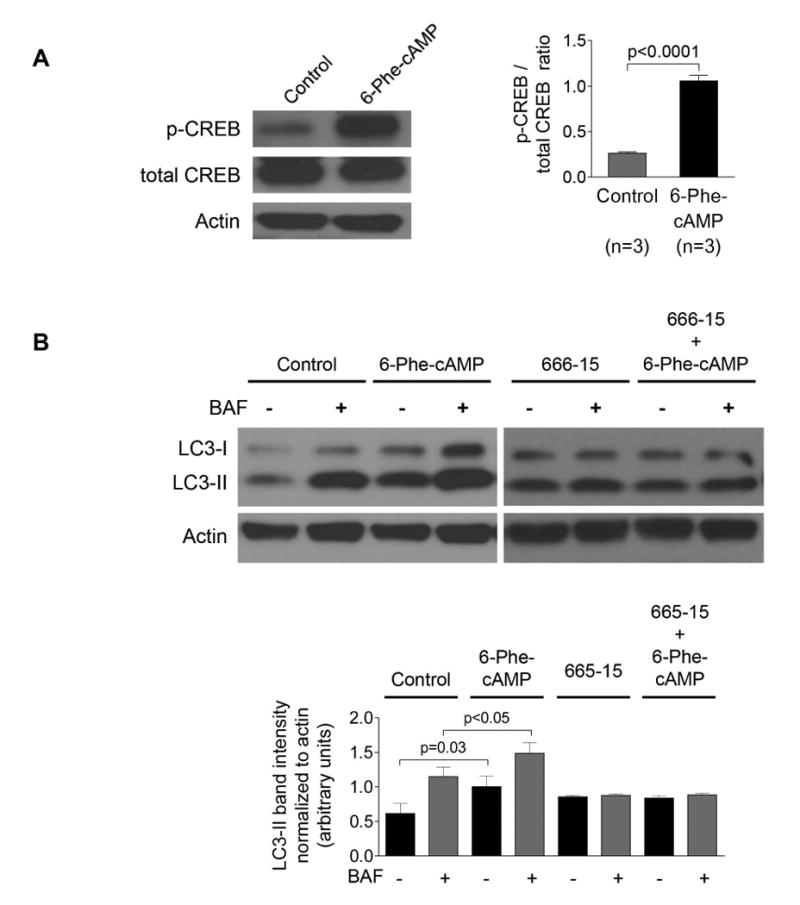
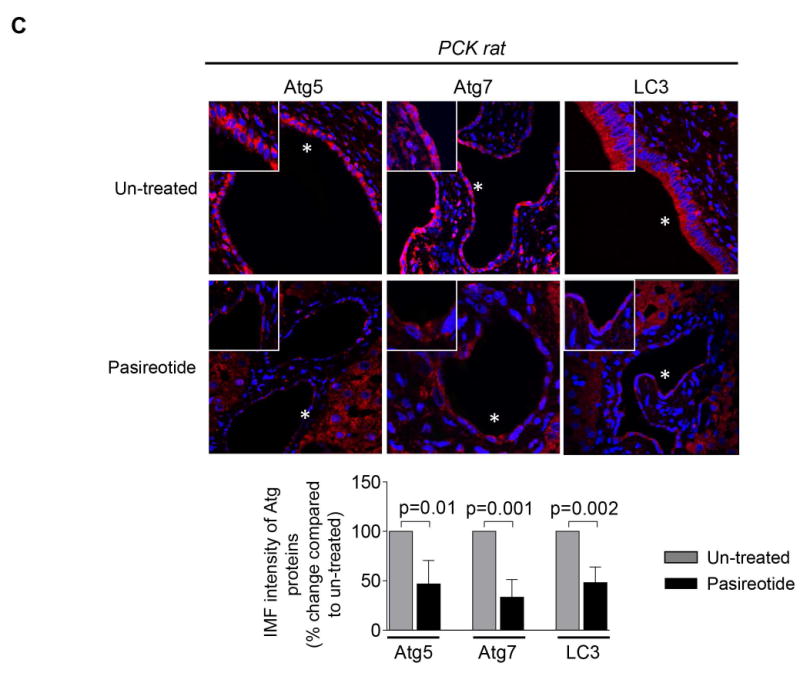
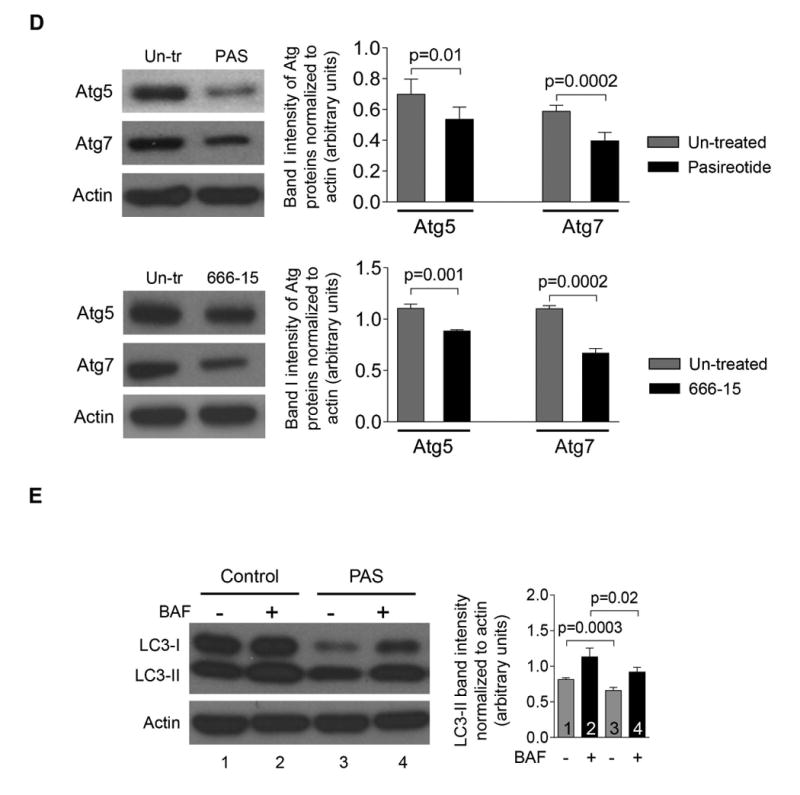
(A) PKA activator, 6-Phe-cAMP, increased phosphorylation of CREB in human ADPKD cholangiocytes by 5-fold. (B) Autophagic flux was increased in 6-Phe-cAMP-treated cholangiocytes in a CREB-mediated manner since the CREB inhibitor, 666-15, abolished these effects. (C) Representative images of liver sections of untreated PCK rats (n=8) and PCK rats treated with a somatostatin analog, paseriotide (n=8). A portion of enlarged hepatic cysts (*) is shown in insets. Paseriotide decreased expression of Atg5, Atg7 and LC3 compared to untreated controls by 2-3-fold. Atg proteins are stained in red; nuclei are stained with DAPI in blue. (D) Pasireotide decreased expression of Atg5 and Atg7 in cultured human ADPKD cholangiocytes by 37% and 25%, respectively. The CREB inhibitor, 666-15, inhibited expression of Atg5 and Atg7 in PLD cholangiocytes with similar magnitude.
Pharmacologic inhibition of autophagy reduced cholangiocyte proliferation and hepatic cyst growth in vitro
PLD cholangiocytes exhibit enhanced proliferative capacity determining hepatic cyst growth.10, 12 The treatment of PCK rat cholangiocytes with inhibitors of autophagic flux, Bafilomycin A1 and HCQ, reduced their proliferation by 50% and 32%, respectively (Fig. 6A). Also, whereas bile ducts isolated from PCK rats expanded in size over time in 3-D culture by ∼75% (Fig. 6B-C), in the presence of Bafilomycin A1 and HCQ, circumferences of cystic bile ducts were increased at day 5 compared to day 1 only by 34% and 25%, respectively (Fig. 6B-C), indicating that autophagy inhibitors attenuated cyst growth in vitro.
Figure 6. Pharmacologic and genetic inhibition of autophagy reduced PLD cholangiocyte proliferation, cyst growth and cholangioid formation of in 3-D cultures.
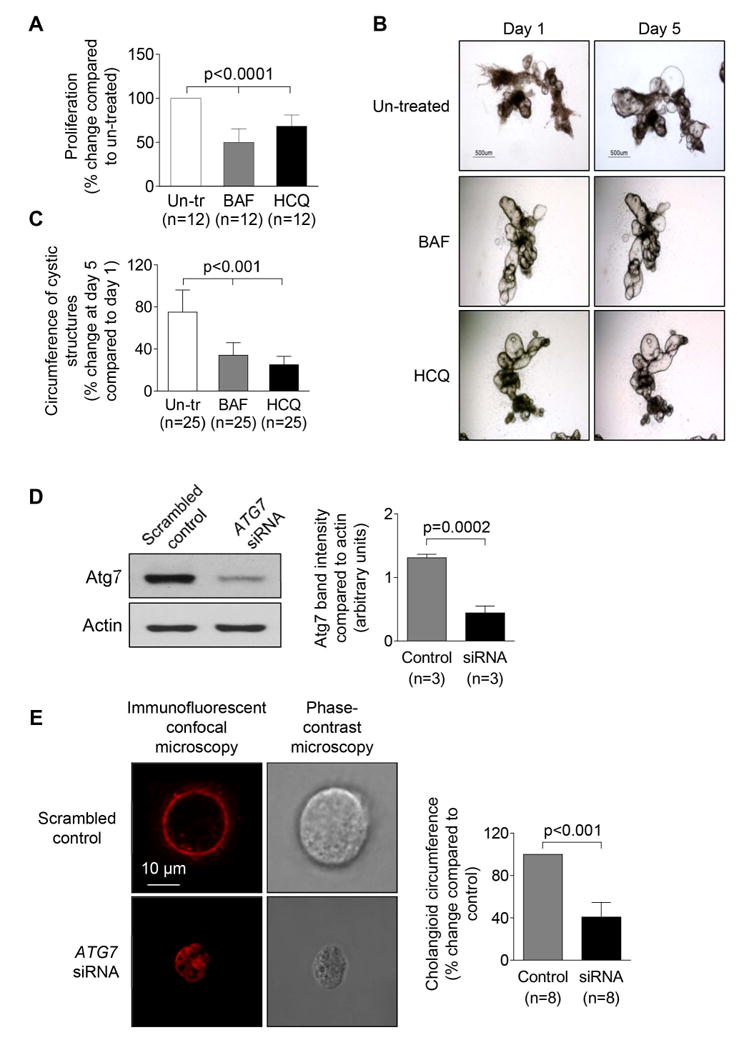
(A) Proliferation of PCK rat cholangiocytes was inhibited by Bafilomycin A1 and HCQ (both, 100 nM) as assessed by MTS absorbance. (B) Representative light microscopy images and (C) bar graphs demonstrate inhibited expansion of dissected PCK rat cystic bile ducts treated with Bafilomycin A1 and HCQ (both, 100 nM). (D) Representative western blots and quantitation of protein band density demonstrate reduced expression of Atg7 in ADPKD cholangiocytes stable transfected with ATG7 siRNA compared to cholangiocytes transfected with scrambled control. (E) Representative confocal and phase-contrast microscopy images of cholangioids formed by human ADPKD cholangiocytes in 3-D culture. ADPKD cholangiocytes transfected with ATG7-siRNA developed smaller cystic structures compared to cholangiocytes transfected with scrambled control siRNA. Data are presented as MEAN±SD. Abbreviation: Un-tr – untreated, BAF – Bafilomycin A1, HCQ – hydroxychloroquine.
Inhibition of ATG7 expression reduced formation of human ADPKD cholangioids in 3-D culture
Transfection of human ADPKD cholangiocytes with ATG7-siRNA reduced expression of Atg7 by 75% (Fig. 6D) and resulted in a decrease of circumference of cholangioids in 3-D culture by 45% (Fig. 6E) demonstrating the important role of autophagy in cyst formation.
HCQ inhibits hepatic cyst growth in PCK rats
To directly address a potential therapeutic application of our in vitro findings, we treated PCK rats with HCQ and found a significant improvement of disease. Liver weights were decreased in male and female rats by 12.9% and 15.2%, respectively (Supporting Table 1). The hepatic cystic and fibrotic areas were decreased in response to treatment from 16.6±1.4% to 12.3±1.1%, and from 10.4±0.8% to 6.4±0.8%, correspondingly (Fig. 7A-B, Supporting Table 1).
Figure 7. HCQ alone and in combination with pasireotide decreases hepatic cyst growth in PCK rats.
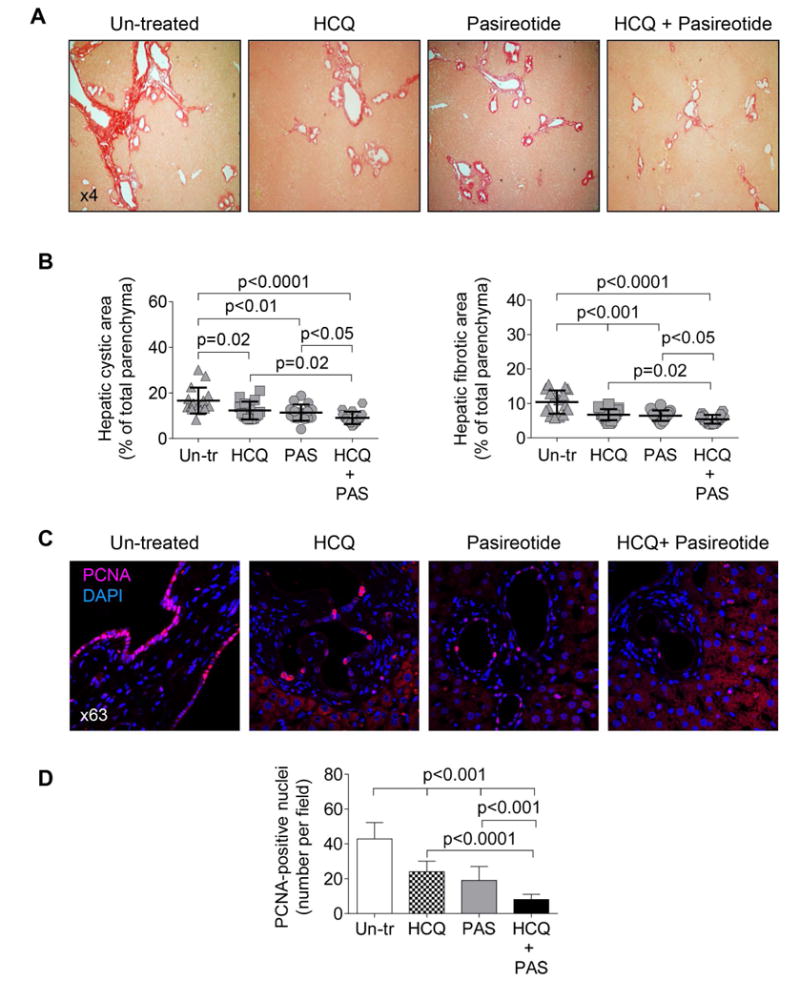
(A) Representative images of picrosirius red stained liver sections of PCK rats which received daily injection of HCQ (15 mg/kg); pasireotide (20 mg/kg/day) introduced via an osmotic pump; a group of rats that simultaneously received HCQ and pasireotide, and a control group that had implanted pumps filled with sterile water and injected with equal doses of DMSO for 6 weeks. Each group of rats consisted of 4 females and 4 males. (B) Scatter plots depict cystic and fibrotic areas of individual liver lobes (two liver lobes from each rat analyzed). (C) Representative images of liver section from un-treated and drug-treated PCK rats stained with PCNA antibody and quantitative analysis show that the number of PCNA-positive nuclei after the treatment with either pasireotide or HCQ alone was decreased; drug combination reduced the number of PCNA-positive nuclei even further. (n=15 microscopic fields for each treatment group). Data are presented as MEAN±SD. Abbreviations: Un-tr- un-treated; PAS – pasireotide; HCQ – hydroxychloroquine.
The beneficial effects of HCQ were also seen in kidneys of PCK rats. Kidney weights were decreased in both male and female rats by 10.1%, and the renal cystic and fibrotic areas were decreased from 36.7±3.4% to 28.9±3.5% and from 3.0±0.3% to 1.9±0.1%, respectively (Supporting Fig. 4; Supporting Table 1).
The therapeutic effects of HCQ on hepatic cystogenesis in PCK rats were similar to effects of somatostatin analogues, octreotide and pasireotide, the only known drugs that are currently used for the treatment of PLD 10, 16 (Fig. 7A-B, Supporting Table 1). Importantly, the efficacy of HCQ and paseriotide administered concurrently was enhanced as reflected by reduction of liver weight in male and female rats by 19.7% and 20.7%, respectively. The hepatic cystic and fibrotic areas were decreased in livers of both males and females from 16.6±1.4% to 9.1±1.6%, and from 10.4±0.8% to 5.4±0.6%, respectively (Fig. 7A-B, Supporting Table 1). Synergistic therapeutic effects of HCQ and pasireotide were also seen in the PCK rat kidneys (Supporting Fig. 4; Supporting Table 1).
After the treatment of PCK rats with either HCQ alone or pasireotide alone, the number of PCNA-positive nuclei in cholangiocytes was decreased, respectively, by 1.8-fold and 2.3-fold. Drug combination reduced the number of PCNA-positive nuclei by 5.1-fold (Fig. 7C-D).
Serum biochemical markers relevant to the functions of the hepatobiliary system were comparable in un-treated and drug-treated PCK rats (Supporting Table 2). This is consistent with our previous published works demonstrating that LFTs remain unchanged in response to treatment with different drugs.10, 12, 17 Presumably, one of the major reasons for this observation is short (i.e., 6 weeks) duration of treatment.
HCQ decreases the levels of cAMP and PCK rat cholangiocyte proliferation
Consistent with our previous reports, 10, 12, 17 cAMP levels were increased in rat and human PLD cholangiocytes compared to healthy controls (Fig. 8A). HCQ alone, pasireotide alone, and combination of these two drugs reduced cAMP levels, respectively, by 25.6%, 31.0% and 43.3% in PCK rat cholangiocytes, and by 24.9%, 26.9% and 36.9% in human ADPKD cholangiocytes (Fig. 8B). HCQ alone and pasireotide alone decreased proliferation of PCK rat cholangiocytes by 33-42% and human ADPKD cholangiocytes by 28-35% (Fig. 8C). Two drugs together had a synergistic effect decreasing proliferation of PCK rat and human ADPKD cholangiocytes by 61% and 67%, respectively (Fig. 8C). Thus, these data demonstrate that therapeutic effects of HCQ on hepatic cystogenesis are presumably associated with inhibition of cAMP levels and cholangiocyte proliferation.
Figure 8. HCQ alone and in combination with pasireotide synergistically decreases cAMP levels and inhibits proliferation of PLD cholangiocytes.
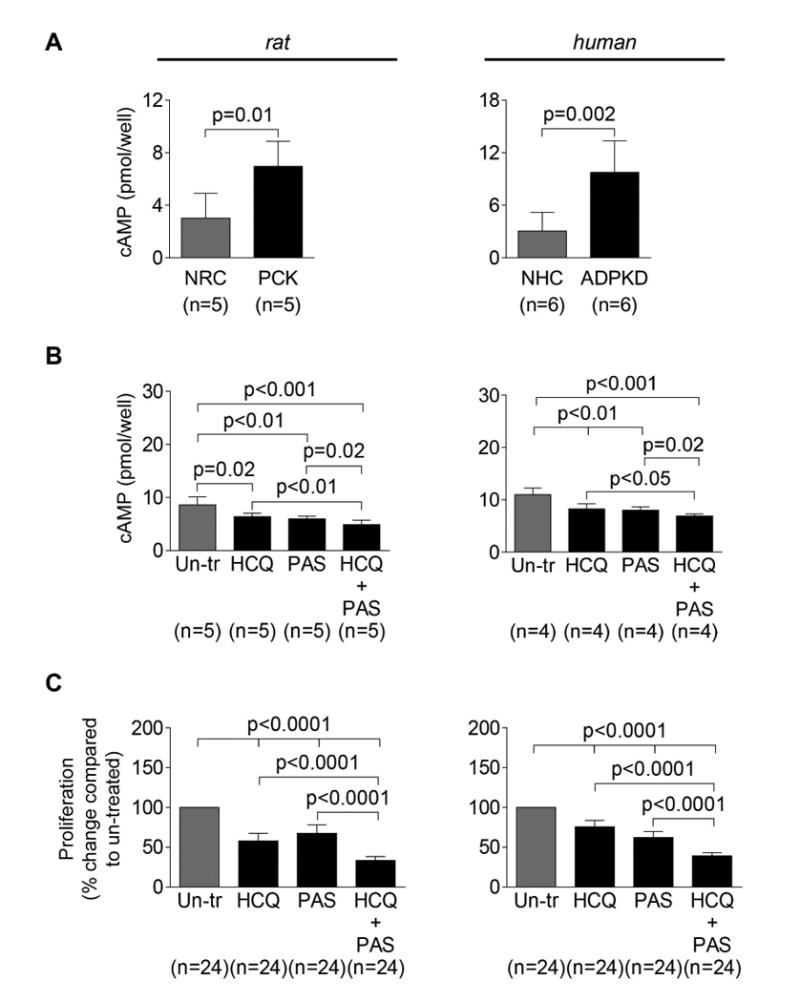
(A) Basal levels of cAMP were increased in PCK rat and ADPKD cholangiocytes compared to respective controls. (B) Levels of cAMP were decreased in PCK rat and human ADPKD cholangiocytes in response to PAS (20 mM) and HCQ (100 nM) alone. Combination of these two drugs resulted in a greater inhibition of cAMP production. (C) Proliferation of both, PCK rat and human ADPKD cholangiocytes was inhibited by PAS (20 mM) and HCQ (100 nM) alone. Combination of these two drugs resulted in a greater inhibition of cholangiocyte proliferation. All data are presented as MEAN±SD. Abbreviation: Un-tr – un-treated.
Discussion
The key findings of this study relate to the pathophysiological role of increased cholangiocyte autophagy in Polycystic Liver Disease. We found that in human and rodent PLD cholangiocytes: (i) the autophagy-lysosomal pathway is altered; (ii) Atg transcripts and transcripts essential for induction of autophagy are up-regulated; (iii) the number and size of autophagosomes, lysosomes and autolysosomes are increased; (iv) selected Atg proteins are overexpressed; (v) autophagic flux is increased; (vi) the cAMP-PKA-CREB signaling pathway contributes to increased autophagy; (vii) intervention in autophagy with ATG7 siRNA and inhibitors of autophagic flux, Bafilomycin A1 and HCQ, reduce PLD cholangiocyte proliferation and hepatic cyst growth in vitro; (viii) HCQ improves disease progression in the PCK rat, an animal model of PLD; and (ix) concurrently HCQ and a somatostatin analogue, pasireotide, reduce hepato-renal cystogenesis in the PCK rat more effectively than each drug alone by decreasing cAMP levels and cholangiocyte proliferation to a greater extent than either single drug.
As mentioned, the potential involvement of autophagy in hepatic cystogenesis was previously proposed,9 and the importance of autophagy for Polycystic Kidney Disease (PKD) was also suggested.18 It has been hypothesized that PKD represents a case of suppressed autophagy because the mTOR pathway, a cellular suppressor of autophagy, is activated in renal cystic epithelial cells.18 Indeed, in zebrafish and mouse models of ADPKD, rapamycin (an inhibitor of the mTOR pathway and an inducer of autophagy) showed positive effects on autophagy in renal epithelial cells.18, 19 It has been also reported in agreement with our observations presented in this study that the number of autophagosomes and the expression of autophagy-related proteins (i.e., Beclin 1 and LC3-II) were increased in cystic renal epithelia of two animal models of PKD: Han:SPRD rats and cpk mice.20 Thus, the experimental evidence on the role of autophagy in PKD remains limited and controversial.
Thus, to our knowledge, our work is the first direct demonstration that autophagy is increased in human and rodent PLD cholangiocytes via mechanisms most probably associated with aberrant cAMP signaling and also contributes to hepatic cystogenesis. Perhaps most importantly, our work demonstrates that, when autophagy is targeted either molecularly or pharmacologically, hepatic cyst growth both in vitro and in vivo is reduced. Moreover, pharmacological targeting of autophagy in an animal model of ARPKD, the PCK rat, is also beneficial for cyst growth in the kidney.
In PLD, hepatic cysts derive from cholangiocytes and grow progressively over time with the involvement of multiple mechanisms tightly associated with the activated cAMP signaling pathway. 7, 8, 10-12 Taking into account the important role of this signaling pathway in regulation of autophagy in different cell types,21, 22 we think that elevated levels of cAMP is the major inducer of autophagy in PLD cholangiocytes, and that mechanisms accounting for increased autophagy in PLD cholangiocytes are associated, at least in part, with the cAMP-PKA-CREB signaling pathway.
Recent studies suggest that cAMP signaling influences autophagy negatively or positively depending on the cell type.21, 23 For example increased levels of cAMP activate PKA that in turn phosphorylates a nuclear transcription factor, CREB, and activated CREB influences autophagy via expression of autophagy-related genes. 24, 25 In particular, in hepatocytes, CREB promotes autophagy under nutrient-deprived conditions via up-regulation of autophagy genes Atg7, Ulk1, and Tfeb.25 Relevant to the possibility that altered autophagy in PLD cholangiocytes is associated with the cAMP-PKA-CREB signaling pathways are four sets of our data. First, in silico bioinformatics analysis revealed that Atg5/ATG5, Becn1/BECN1, Atg7/ATG7, and Map1lc3/MAP1LC3 harbor the CREB binding sites within their promoters. Second, activation of PKA resulted in an increase in CREB phosphorylation, and in autophagic flux in human ADPKD cholangiocytes in culture. Third, expression of Atg5 and Atg7 was reduced in cholangiocytes of PCK rats and in cultured human ADPKD cholangiocytes treated with a somatostatin analogue paseriotide that inhibits the cAMP signaling pathway. Finally, expression of Atg5 and Atg7 was reduced in human ADPKD cholangiocytes treated with a CREB inhibitor, 666-15.
Recently, we demonstrated that a cAMP-linked bile acid receptor, TGR5, contributes to PLD progression,14 and could be considered as one of the major inducers of increased cAMP levels in PLD cholangiocytes, and potentially in cAMP-regulated autophagy. Given the importance of TGR5 activation and cAMP signaling in other chronic cholestatic conditions,26 it is likely that TGR5-cAMP-dependent activation of autophagy may also occur in other liver diseases.
A functional outcome of increased autophagy in PLD is elevated proliferation of cholangiocytes and accelerated hepatic cyst growth. The involvement of autophagy in cell proliferation is known, especially in cancer; however the exact mechanisms of autophagy-dependent cell proliferation remain poorly understood.27 Autophagy can both restrain and promote cell proliferation in a context-dependent manner, (i.e., depending on the cell type, disease stage, expression of Atg proteins, and other conditions).27 For example, Atg5 is essential for T cell proliferation in mice,28 and Beclin1 and Atg7 are essential for stem cell proliferation in C. elegans.29 Our data showing overexpression of Atg5 and Atg7 in hyperproliferative rodent and human PLD cholangiocytes are in agreement with these observations.
Manipulation with autophagy in in vitro and in vivo models of PLD demonstrates strong therapeutic effects. Bafilomycin A1 and HCQ both reduce PLD cholangiocyte proliferation and hepatic cyst growth in 3D culture, and treatment of PCK rats with HCQ reduced hepatic and renal cystogenesis to the same extent as the somatostatin analogues, octreotide and pasireotide, the only drugs used in clinical practice for the treatment of PLD.10, 16 Unfortunately, the chronic treatment of PLD patients with somatostatin analogues is costly ($7,000-$11,000 per month). Thus, a search for new therapeutic options for PLD remains of a great importance. Our data suggest that inhibition of autophagy in PLD cholangiocytes by HCQ, which is an inexpensive FDA approved drug, might be beneficial for PLD patients.
We also found that concurrently HCQ and pasireotide reduce hepato-renal cystogenesis in PCK rats more effectively than each drug alone. The mechanisms of the synergistic effects of these two drugs remain unclear. It has been demonstrated in in vitro and in vivo studies on the rat inner medullary collecting ducts that chloroquine inhibits both basal and arginine vasopressin (AVP)-induced cAMP production.30, 31 Our data show a greater reduction in cholangiocyte cAMP levels when the drugs are used together than when they are used alone. Thus, it appears that both drugs via different mechanisms affect cAMP signaling.
In summary, our study provides the first direct evidence that autophagy is increased in PLD cholangiocytes, contributes to hepatic cystogenesis, and should be considered a novel therapeutic target for the treatment of liver disorders associated with ADPKD and ARPKD.
Supplementary Material
Acknowledgments
The authors thank Dr. Gregory J. Gores, Dr. Frank A. Sinicrope, and Dr. Ryan J. Schulze for the critical reading of the manuscript, a productive discussion, and helpful suggestions.
Financial Support: This work was supported by DK24031 from the NIH, by the Mayo Clinic, the Clinical Core and Optical Microscopy Cores of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567), the Mayo Translational Polycystic Kidney Disease Center and a Mayo Translational PKD Center Pilot and Feasibility Award (NIDDK P30DK090728).
AIM, TVM & NFL supervised project & wrote manuscript. AIM, TVM, MJLP, J(F)D, LL and BQH performed experiments and contributed to discussion.
List of Abbreviations
- PLD
polycystic liver disease
- ADPKD
autosomal dominant polycystic kidney disease
- ARPKD
autosomal recessive polycystic kidney disease
- NGS
NextGen Sequencing
- FPCA
Functional Pathway Cluster Analysis
- HCQ
hydroxychloroquine
Footnotes
Potential conflict of interest: Nothing to report.
References
- 1.Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. (3rd) 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Madrigal-Matute J, Cuervo AM. Regulation of Liver Metabolism by Autophagy. Gastroenterology. 2016;150:328–339. doi: 10.1053/j.gastro.2015.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakanuma Y, Sasaki M, Harada K. Autophagy and senescence in fibrosing cholangiopathies. Journal of hepatology. 2015;62:934–945. doi: 10.1016/j.jhep.2014.11.027. [DOI] [PubMed] [Google Scholar]
- 4.Czaja MJ, Ding WX, Donohue TM, Jr, Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, Perlmutter DH, Randall G, Ray RB, Tsung A, Yin XM. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kroemer G. Autophagy: a druggable process that is deregulated in aging and human disease. The Journal of clinical investigation. 2015;125:1–4. doi: 10.1172/JCI78652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nature reviews Drug discovery. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masyuk TV, Huang BQ, Masyuk AI, Ritman EL, Torres VE, Wang X, Harris PC, Larusso NF. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. The American journal of pathology. 2004;165:1719–1730. doi: 10.1016/S0002-9440(10)63427-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perugorria MJ, Masyuk TV, Marin JJ, Marzioni M, Bujanda L, LaRusso NF, Banales JM. Polycystic liver diseases: advanced insights into the molecular mechanisms. Nature reviews Gastroenterology & hepatology. 2014;11:750–761. doi: 10.1038/nrgastro.2014.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren XS, Sato Y, Harada K, Sasaki M, Furubo S, Song JY, Nakanuma Y. Activation of the PI3K/mTOR pathway is involved in cystic proliferation of cholangiocytes of the PCK rat. PloS one. 2014;9:e87660. doi: 10.1371/journal.pone.0087660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masyuk TV, Radtke BN, Stroope AJ, Banales JM, Gradilone SA, Huang B, Masyuk AI, Hogan MC, Torres VE, Larusso NF. Pasireotide is more effective than octreotide in reducing hepatorenal cystogenesis in rodents with polycystic kidney and liver diseases. Hepatology. 2013;58:409–421. doi: 10.1002/hep.26140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masyuk TV, Lee SO, Radtke BN, Stroope AJ, Huang B, Banales JM, Masyuk AI, Splinter PL, Gradilone SA, Gajdos GB, LaRusso NF. Centrosomal abnormalities characterize human and rodent cystic cholangiocytes and are associated with Cdc25A overexpression. The American journal of pathology. 2014;184:110–121. doi: 10.1016/j.ajpath.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology. 2007;132:1104–1116. doi: 10.1053/j.gastro.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 13.Loarca L, De Assuncao Thiago M, Jalan-Sakrikar N. Development and Characterization of Cholangioids from Normal and Diseased Human Cholangiocytes as an In Vitro Model to Study Primary Sclerosing Cholangitis. Lab Investigation. 2017 doi: 10.1038/labinvest.2017.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masyuk TV, Masyuk AI, Lorenzo Pisarello M, Howard BN, Huang BQ, Lee PY, Fung X, Sergienko E, Ardecky RJ, Chung TDY, Pinkerton AB, LaRusso NF. TGR5 contributes to hepatic cystogenesis in rodents with polycystic liver diseases through cyclic adenosine monophosphate/Galphas signaling. Hepatology. 2017 doi: 10.1002/hep.29284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hogan MC, Masyuk T, Bergstralh E, Li B, Kremers WK, Vaughan LE, Ihrke A, Severson AL, Irazabal MV, Glockner J, LaRusso NF, Torres VE. Efficacy of 4 Years of Octreotide Long-Acting Release Therapy in Patients With Severe Polycystic Liver Disease. Mayo Clinic proceedings. 2015;90:1030–1037. doi: 10.1016/j.mayocp.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masyuk TV, Radtke BN, Stroope AJ, Banales JM, Masyuk AI, Gradilone SA, Gajdos GB, Chandok N, Bakeberg JL, Ward CJ, Ritman EL, Kiyokawa H, LaRusso NF. Inhibition of Cdc25A suppresses hepato-renal cystogenesis in rodent models of polycystic kidney and liver disease. Gastroenterology. 2012;142:622–633 e624. doi: 10.1053/j.gastro.2011.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ravichandran K, Edelstein CL. Polycystic kidney disease: a case of suppressed autophagy? Seminars in nephrology. 2014;34:27–33. doi: 10.1016/j.semnephrol.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 19.Zhu P, Sieben CJ, Xu X, Harris PC, Lin X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Human molecular genetics. 2017;26:158–172. doi: 10.1093/hmg/ddw376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belibi F, Zafar I, Ravichandran K, Segvic AB, Jani A, Ljubanovic DG, Edelstein CL. Hypoxia-inducible factor-1alpha (HIF-1alpha) and autophagy in polycystic kidney disease (PKD) American journal of physiology Renal physiology. 2011;300:F1235–1243. doi: 10.1152/ajprenal.00348.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiological reviews. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 22.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annual review of genetics. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ugland H, Naderi S, Brech A, Collas P, Blomhoff HK. cAMP induces autophagy via a novel pathway involving ERK, cyclin E and Beclin 1. Autophagy. 2011;7:1199–1211. doi: 10.4161/auto.7.10.16649. [DOI] [PubMed] [Google Scholar]
- 24.Namkoong S, Lee KI, Lee JI, Park R, Lee EJ, Jang IS, Park J. The integral membrane protein ITM2A, a transcriptional target of PKA-CREB, regulates autophagic flux via interaction with the vacuolar ATPase. Autophagy. 2015;11:756–768. doi: 10.1080/15548627.2015.1034412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, Ma J, Kemper B, Kemper JK. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature. 2014;516:108–111. doi: 10.1038/nature13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deutschmann K, Reich M, Klindt C, Droge C, Spomer L, Haussinger D, Keitel V. Bile acid receptors in the biliary tree: TGR5 in physiology and disease. Biochim Biophys Acta. 2017 doi: 10.1016/j.bbadis.2017.08.021. [DOI] [PubMed] [Google Scholar]
- 27.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, Kimmelman A, Kumar S, Levine B, Maiuri MC, Martin SJ, Penninger J, Piacentini M, Rubinsztein DC, Simon HU, Simonsen A, Thorburn AM, Velasco G, Ryan KM, Kroemer G. Autophagy in malignant transformation and cancer progression. The EMBO journal. 2015;34:856–880. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. The Journal of experimental medicine. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ames K, Da Cunha DS, Gonzalez B, Konta M, Lin F, Shechter G, Starikov L, Wong S, Bulow HE, Melendez A. A Non-Cell-Autonomous Role of BEC-1/BECN1/Beclin1 in Coordinating Cell-Cycle Progression and Stem Cell Proliferation during Germline Development. Current biology : CB. 2017;27:905–913. doi: 10.1016/j.cub.2017.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Musabayane CT, Wargent ET, Balment RJ. Chloroquine inhibits arginine vasopressin production in isolated rat inner medullary segments induced cAMP collecting duct. Renal failure. 2000;22:27–37. doi: 10.1081/jdi-100100848. [DOI] [PubMed] [Google Scholar]
- 31.von Bergen TN, Blount MA. Chronic use of chloroquine disrupts the urine concentration mechanism by lowering cAMP levels in the inner medulla. American journal of physiology Renal physiology. 2012;303:F900–905. doi: 10.1152/ajprenal.00547.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.