

Abstract
Alcohol is a well-established risk factor for hepatocellular carcinoma, but the mechanisms by which it promotes liver cancer are not well understood. Several studies have shown that cellular protein arginine methylation is inhibited by alcohol. Arginine methylation is controlled by the reciprocal activity of protein arginine methyltransferases, primarily PRMT1, and a demethylase JMJD6. The aim of this study was to explore the role of arginine methylation changes in alcohol pathogenesis. We found that PRMT1 activity is inhibited in livers of mice fed with alcohol compared to pair-fed mice. Using hepatocyte specific PRMT1 knockout mice we identified that loss of PRMT1 results in enhanced hepatocyte proliferation and a 33% increase in liver size. This increased hepatocyte proliferation was associated with reduced expression of Hnf4α, an important regulator of liver tumorigenesis. We found that PRMT1 regulates Hnf4α expression directly through arginine methylation at the Hnf4α promoter. In the absence of PRMT1, JMJD6 can demethylate the Hnf4α promoter and suppress its expression. We were able to restore Hnf4α expression and abolish the increase in hepatocyte proliferation by knockdown of JMJD6 in PRMT1 knockout mice. Knockdown of JMJD6 in alcohol fed mice similarly increased Hnf4α expression. We then examined whether loss of arginine methylation might play a role in alcohol-associated liver cancers. We examined 25 human HCC specimens and found a strong correlation (R=0.8, P<0.01) between arginine methylation levels and Hnf4α expression in these specimens suggesting that above mechanism is relevant in patients.
Conclusion
Taken together these data suggest that PRMT1 inhibition such as induced by alcohol may result in epigenetic changes leading to loss of Hnf4α. This effect may contribute to alcohol's ability to promote liver tumors.
Keywords: histone arginine methylation, alcohol, HCC, alcoholic liver disease, asymmetric dimethyl arginine
Protein arginine methylation is a common posttranslational modification that plays a role in multiple pathways, including cell cycle control, RNA processing and DNA replication. The protein arginine methyltransferase PRMT1 is responsible for about 85% of total cellular arginine methylation (1) and catalyzes arginine mono- and dimethylation using S-adenosyl methionine (SAM) as a methyl donor. PRMT1 methylates histone H4 at arginine 3, generating H4R3me2a, a transcriptional activation mark (1-5), thus contributing to the histone code. As a transcriptional coactivator, PRMT1 is recruited to promoters by a number of different transcription factors (3, 6, 7).
Abnormal function of PRMT1 is closely associated with several types of cancer and cardiovascular disease. Arginine methylation impacts gene transcription and splicing as well as upstream signal transduction (4). Until recently, arginine methylation was thought to be an irreversible modification due to the absence of demethylation enzymes. But PRMT1 is involved in processes such as cell cycle control, where responses are of short duration and signal turnover is rapid. This suggests a possible dynamic regulation of this modification. Protein arginine demethylases have not been well described, but one of the Jumonji family proteins, JMJD6, has been reported to have arginine demethylase activity (8, 9). JMJD6 is a dioxygenase that can act as both an arginine demethylase and a lysyl-hydroxylase. It is required during embryogenesis and is a key regulator of hematopoietic differentiation through its targets U2AF2/U2AF65 (10). We and others have recently shown that JMJD6 can demethylate PRMT1 targets (8, 9, 11).
About 40% of individuals who chronically consume alcohol develop fatty liver, which can advance to alcoholic hepatitis, cirrhosis, and cancer. Alcohol interacts with other causes of liver disease, including hepatitis B and C, and conditions, such as diabetes and obesity, to increase the risk for developing HCC, either synergistically or additively (12-14). A number of studies have been performed to define the changes induced by alcohol consumption that can lead to progression of liver disease and cancer development. Mouse models suggest that chronic EtOH consumption stimulates hepatocyte proliferation through activation of the Wnt/β-catenin signaling pathway thus promoting tumorigenesis following an initiating insult in the liver (15, 16). Others suggest that increased neutrophil and macrophage activation induced by chronic alcohol exposure contributes to hepatocyte damage and tumor development (16-18). We have recently shown that PRMT1 activity and cellular arginine methylation are inhibited in models of liver disease induced by alcohol and HCV infection (19).
This study examines the role of PRMT1 in alcohol pathogenesis in the liver. This study explores one possible mechanism by which alcohol promotes tumor development in the liver. We found that alcohol inhibits arginine methylation in the liver. Arginine methylation levels in hepatocytes are regulated by PRMT1 and JMJD6 levels which in turn control HNF4α activity and expression. Alcohol induced PRMT1 inhibition results in reduced HNF4α expression which can contribute to alcohol pathogenesis and cancer progression.
Experimental Procedures
Mice
C57BL/6NTac-Prmt1tm1a(EUCOMM)Wtsi /WtsiCnbc mice were obtained from EUCOMM (EUCOMM project: 40181) and bred with Flp recombinase mice to get homozygous Prmt1 floxed breeders as decribed before (20). These mice were next crossed with Albumin-Cre mice to generate mice lacking Prmt1 in hepatocytes. For experiments, PRMT1fl/fl Cre/wt mice were used together with PRMT fl/fl wt/wt littermates as a control.
All mice were housed in a temperature-controlled, specific pathogen-free environment with 12-hour light-dark cycles and fed regular mouse chow and water ad libitum. All animal handling procedures were approved by the Institutional Animal Care and Use Committees at the University of Kansas Medical Center (Kansas City, KS).
Antibodies used
Primary antibodies
Anti-PRMT1 (F339), Acetyl-Histone H3 (Lys27) (D5E4), anti-CyclinD and anti-β-catenin antibodies were from Cell Signaling. Anti-Lamin B (C20), anti-β-actin were from Santa Cruz. Rabbit Anti-PRMT1 antibody (against aa 300-361), were from Abcam. Anti-asymmetric-dimethyl-arginine antibodies and Anti-H4R3me2a antibodies were from ActiveMotif. Monoclonal anti-HNF4α (H1415) antibodies were from R&D Systems Inc. Mouse anti- β-actin, mouse Monoclonal Anti-PRMT1 clone 171 (against aa 1-361), Anti-JMJD6 (SAB3500233) antibodies were from Sigma-Aldrich, Saint Louis, MO. Anti-GAPDH was from Ambion.
Secondary antibodies
IRDye 800CW goat anti-mouse IgG and IRDye 680RD goat anti-rabbit IgG were from Li-COR. General HRP-conjugated secondary antibodies were from Southern Biotechnology Associates (Birmingham, AL).
Cell culture
Huh7.5 cells (21) (obtained from Dr. Charles Rice) were maintained in Dulbecco's Modified Eagle's Medium (Invitrogen, Carlsbad, CA) containing 10% FBS, 50U mL-1 penicillin and 50 mg mL-1 streptomycin. AMI-1 was obtained from EMD4 Biosciences and used at 10 μM for 16-24h prior to harvest. Cells were transfected using Lipofectamine LTX transfection reagent (Invitrogen) according to the manufacturer's protocol.
Vectors
p6352 MSCV-CMV-CMV-Flag-HA-JMJD6 was provided by Peter Howley via Addgene (Addgene plasmid 31358). pCMV6-PRMT1 was from Origene. AAV8.TBG.PI.Null.bGH (AV-8-PV0148) and AAV8.TBG.PI.Cre.rBG (AV-8-PV1091) were a gift from Dr Lisa Zhang and were from Penn Vector Core, Philadelphia, PA. AAV8-GFP-U6-m-JMJD6-shRNA (shAAV-262522) and AAV8-GFP-U6-scrmb-shRNA were from Vector BioLabs, Malvern, PA.
Human specimens
De-identified human liver specimens from liver explants were obtained from the Liver Center Tissue Bank at the University of Kansas Medical Center. All studies using human tissue samples were approved by the Human Subjects Committee of the University of Kansas Medical Center.
Real Time PCR
RNA was extracted from cultured cells using the RNeasy Mini Kit (Qiagen). cDNA was generated using the RNA reverse transcription kit (Applied Biosystems, Cat.No 4368814). Quantitative real time RT-PCR was performed in a CFX96 Real time system (Bio-Rad) using specific sense and antisense primers combined with iQ SYBR Green Supermix (Bio-Rad) for 40 amplification cycles: 5 s at 95 °C, 10 s at 57 °C, 30 s at 72 °C.
Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation was performed as described before (20). Mouse liver tissue was homogenized, cross-linked by the addition of 1% formaldehyde for 10 minutes. Cells were lysed with [10mM Tris-HCl (pH 8.0), 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40]. Nuclei were collected by centrifugation, resuspended in [1% SDS, 5 mmol/L EDTA, 50 mmol/L Tris-HCl (pH 8.0)] and sonicated to generate chromatin to an average length of ∼100 to 500 bp. Next, samples in (1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl of pH 8.1, 150 mM NaCl), were immunoprecipitated overnight at 4°C with 4 μg ChIP-grade antibody. 20 μl of magnetic beads (Dynabeads M-280, Invitrogen) were used to purify immunocomplexes. Following purification, cross-links were reverted by incubation at 65°C for 6 h. Samples were purified with Qiagen kit. For ChIP re-ChIP analysis, DNA-protein complexes were eluted in 25 μL of TE buffer containing 10 mM DTT for 30 min at 37°C. Eluates were diluted 1:20 with ChIP Dilution buffer and used for second immunoprecipitation.
Western Blots
Protein extracts (15 μg) were subjected to 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE), electrophoretically transferred to nitrocellulose membranes (Amersham Hybond ECL, GE Healthcare), and blocked in 3% BSA/PBS at RT for 1 hour. Primary antibodies were incubated overnight at manufacturer recommended concentrations. Immunoblots were detected with the ECL Plus Western Blotting Detection System (Amersham Biosciences, Piscataway, NJ) or using near-infrared fluorescence with the ODYSSEY Fc, Dual-Mode Imaging system (Li-COR). Expression levels were evaluated by quantification of relative density of each band normalized to that of the corresponding β-actin or GAPDH band density.
Immunohistochemistry
Immunostaining on formalin-fixed sections was performed by deparaffinization and rehydration, followed by antigen retrieval by heating in a pressure cooker (121°C) for 5 minutes in 10 mM sodium citrate, pH 6.0. Peroxidase activity was blocked by incubation in 3% hydrogen peroxide for 10 minutes. Sections were rinsed three times in PBS/PBS-T (0.1% Tween-20) and incubated in Dako Protein Block (Dako) at room temperature for 1 hour. After removal of blocking solution, slides were placed into a humidified chamber and incubated overnight with an antibody, diluted 1:300 in Dako Protein Block at 4°C. Antigen was detected using the SignalStain Boost IHC detection reagent (catalogue # 8114; Cell Signaling Technology, Beverly, MA), developed with diaminobenzidene (Dako, Carpinteria, CA), counterstained with hematoxylin (Sigma-Aldrich), and mounted. Signal intensity and percent PCNA positive cells were analyzed by Aperio ImageScope 12.1.
Statistics
Results are expressed as mean ± SD. The Student t test, paired t test, Pearson's correlation, or one-way ANOVA with Bonferroni post hoc test was used for statistical analyses. p value < 0.05 was considered significant.
Results
Hepatocyte specific PRMT1 deletion results in an increase of hepatocyte proliferation
Our previous work has shown that PRMT1 activity, but not protein level, is reduced by alcohol exposure in Huh 7.5 cells (19). We confirmed the inhibition using an in vivo model of Lieber-DeCarli alcohol feeding for three weeks in wild type mice. Since PRMT1 protein expression is not affected by alcohol we measured PRMT1 activity by immunohistochemical staining for PRMT1 dependent asymmetric dimethyl arginine (ADMA) (4). The anti-ADMA antibody broadly recognizes di-methylated proteins of various molecular sizes (Figure S1A). We found that alcohol feeding dramatically reduced ADMA levels in alcohol fed mice compared to mice fed control liquid diet (Figure 1A). Additionally we confirmed the reduction of PRMT1 activity by analyzing methylation of a well described target protein, SAM68 (Figure S1B)
Figure 1. Hepatocyte specific PRMT1 deletion results in an increase of hepatocyte proliferation.
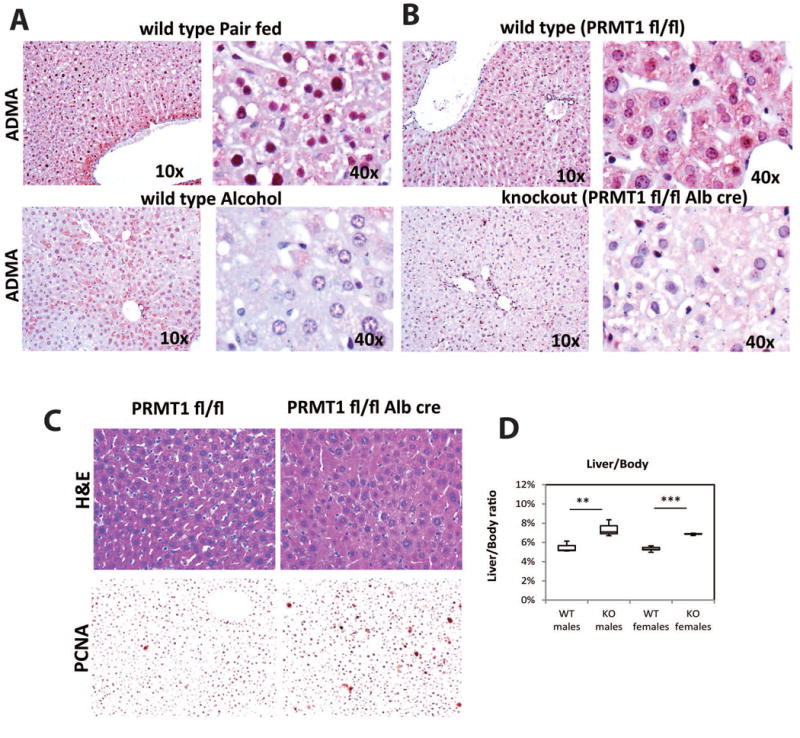
A. Representative images of immunohistochemical staining of asymmetric di-methyl arginine levels (ADMA) in the liver sections of wild type mice fed control liquid diet (pair fed) or alcohol diet (alcohol) for 3 weeks. B. ADMA staining as in A. of liver sections from PRMT1 flox/flox mice and PRMT1 flox/flox Albumin-Cre littermates. C. H&E staining of liver sections from PRMT1 flox/flox mice and PRMT1 flox/flox Albumin-Cre mice. Representative images of immunohistochemical staining of PCNA positive cells in those livers are shown below. D. Liver/body weight ratio from PRMT1 flox/flox mice (WT) and PRMT1 flox/flox Albumin-Cre (KO). **p < 0.01, ***p < 0.001, n=3 per group for males, n=5 per group for females.
To study how loss of PRMT1 activity can contribute to alcohol pathogenesis we created hepatocyte specific PRMT1 knockout mice by breeding PRMT1 fl/fl mice with albumin-Cre mice. Similar knockout mice have been described before to have a defect in PRMT1 dependent FOXO1 activity (22). We confirmed that knockout mice show a loss of ADMA staining in hepatocytes compared to their wild type littermates (Figure 1B). The decrease in ADMA was similar to that seen in alcohol fed mice (compare Figure 1A and 1B). Next we analyzed the livers by H&E staining and found that PRMT1 knockout mice developed ‘nuclear atypia’, but otherwise appeared normal (Figure 1C). Nuclear atypia is characteristic of pre-neoplastic changes and malignancy and often a sign of dedifferentiation and higher proliferation (23-25). This phenotype correlated with increased levels of hepatocyte proliferation in the knockout mice (Figure 1C) and an increase in liver size (Figure 1D). The phenotype was not gender specific; we observed similar liver weight changes in both male and female mice (Figure 1D). Interestingly we found that PRMT1 knockout mice had high levels of serum ALT without apparent liver injury; we found that this was due to increased expression of the ALT gene gpt in the livers of those mice (Figure S2A, B).
PRMT1 deletion results in an increase in β-catenin expression and inhibition of HNF4α expression
To study the mechanism of PRMT1 dependent regulation of hepatocyte proliferation we performed PCR array analysis of proliferation related gene expression in whole liver mRNA from wild type and PRMT1 knockout mice (Figure S3). Examples of genes that are differentially regulated in PRMT1 knockout livers are presented in Figure 2A. The genes that were upregulated or down regulated more than 1.8 fold were used for Ingenuity Pathway Analysis (IPA). We found that HNF4α is predicted to be inhibited in PRMT1 knockout mice and β-catenin is predicted to be activated (Figure S3B).
Figure 2. PRMT1 deletion results in activation of β-catenin signaling and inhibition of HNF4α expression.
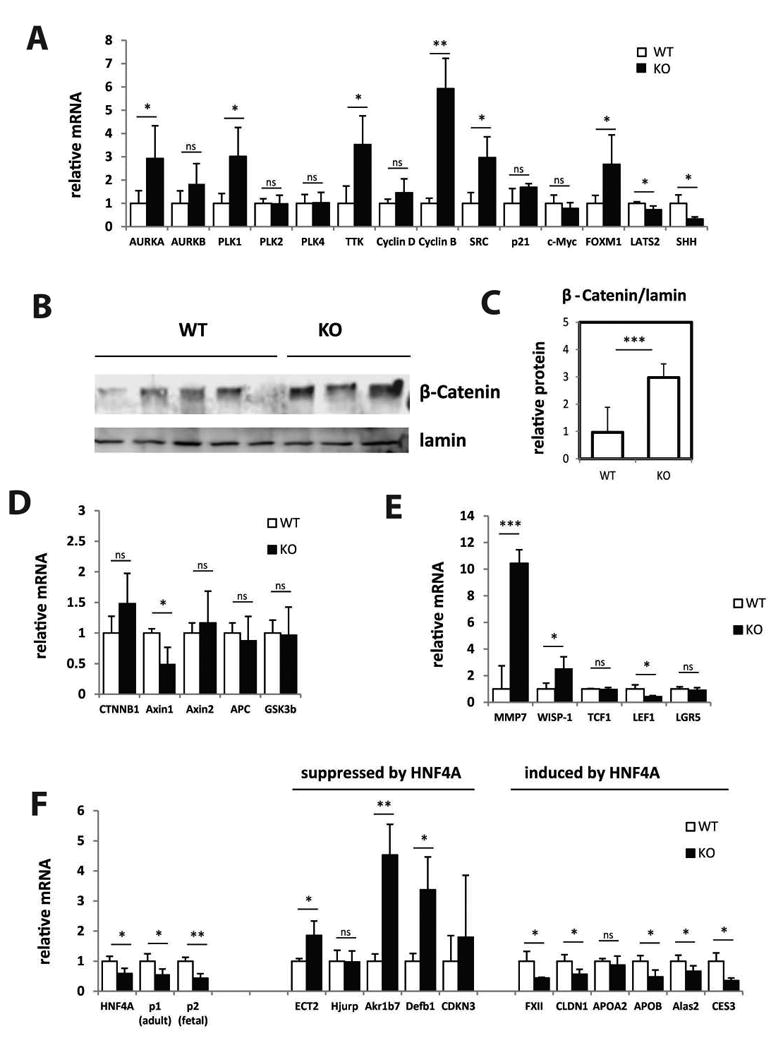
Livers from PRMT1 flox/flox mice (WT) and PRMT1 flox/flox Albumin-Cre (KO) littermates were analyzed for gene expression changes. A. Relative mRNA levels in the livers of WT and KO mice. Data are presented as mean ± SD. n=3 per group. **p < 0.01, *p < 0.05. B, Western blot analysis of β-catenin protein levels in the nuclear extracts from livers of WT and KO mice. C. Densitometry analysis of β-catenin relative protein expression. Data are presented as mean ± SD. N=3-5 per group. ***p < 0.001. D-F. Relative mRNA levels in the livers of WT and KO mice. Data are presented as mean ± SD. n=3 per group. ***p < 0.001, **p < 0.01, *p < 0.05.
To confirm these results we performed series of experiments to analyze those pathways apparently regulated by PRMT1. We found that β-catenin protein levels were increased 3-fold in PRMT1 knockout livers (Figure 2B, C). However, we found no change in β-catenin mRNA levels (Figure 2D). These data suggest that β-catenin stability was increased. β-catenin is a part of the β-catenin/APC/Axin complex and binding by APC/Axin results in β-catenin phosphorylation and degradation. Lack of complex components or increased GSK3B kinase activity can result in β-catenin accumulation (26). We analyzed the expression of those genes and found that only Axin1 mRNA was decreased in PRMT1 knockout compared to wild type livers (Figure 2D). Previous studies showed that PRMT1 regulates β-catenin signaling through expression of Axin1 (27). Our data suggest that in hepatocytes PRMT1 also regulates Axin1 gene expression. Interestingly we found that only a subset of downstream β-catenin target genes were upregulated in PRMT1 knockout mouse liver, while others were not changed or inhibited (Figure 2E). These data suggest that activation of the β-catenin pathway may not be responsible for the increase in hepatocyte proliferation.
Next we analyzed the direct effect of PRMT1 deletion on HNF4α. Previously it has been shown that PRMT1 can bind and methylate HNF4α as well as its target gene promoters, and regulate HNF4α activity (3). We found that HNF4α mRNA expression itself is also regulated by PRMT1 (Figure 2F), possibly, because HNF4α is its own target gene. We also analyzed the expression of HNF4α target genes (28) and confirmed that genes that are suppressed by HNF4α are upregulated in PRMT1 knockout and genes that are induced by HNF4α are inhibited by PRMT1 knockout (Figure 2F). These data suggest that PRMT1 regulates expression of both HNF4α and its downstream target genes.
PRMT1 regulates HNF4α and Axin1 gene expression through promoter histone arginine methylation
Next we examined the mechanism of PRMT1 regulation of HNF4α and Axin1 gene expression. Previously we showed that PRMT1 regulates expression through H4R3me2a histone methylation of gene promoters (2, 4). H4R3me2a promotes recruitment of acetyltransferases and transcriptional activation of the target genes (6). PRMT1 knockout livers have dramatically lower global levels of H4R3me2a compared to wild type litter mates (Figure 3A). We analyzed histone arginine methylation of APC, Axin1, Axin2 and HNF4α and found that PRMT1 knockout livers have significantly lower levels of methylation at Axin1 and HNF4α promoters (Figure 3B, C). Furthermore, binding of HNF4α to its target promoters including its own promoter is reduced in knockout livers (Figure 3D).
Figure 3. PRMT1 regulates HNF4α and Axin1 gene expression through promoter histone arginine methylation.
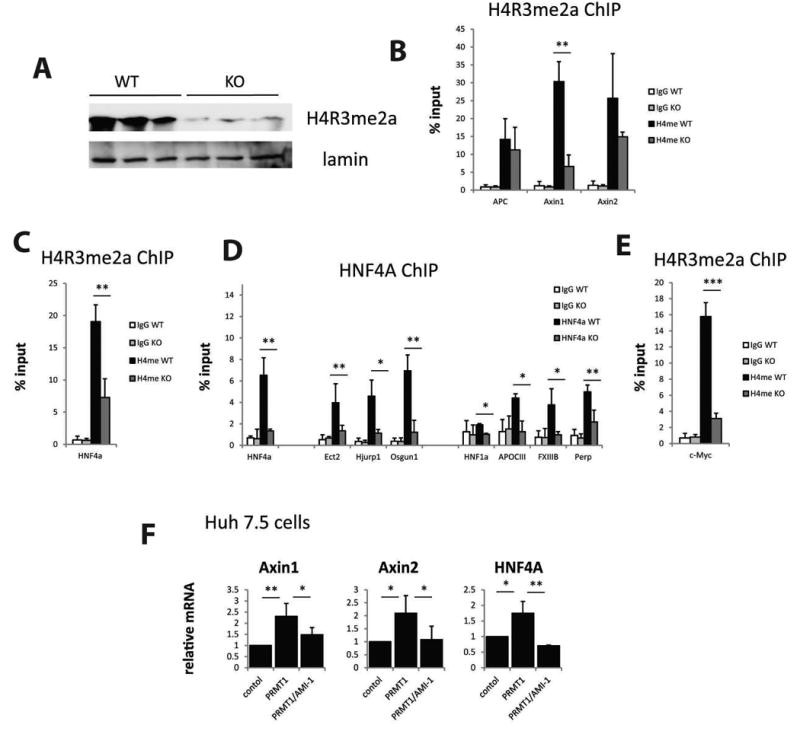
Livers from PRMT1 flox/flox mice (WT) and PRMT1 flox/flox Albumin-Cre (KO) littermates were analyzed for histone methylation changes. A. Levels of PRMT1-dependent histone methylation mark H4R3me2a in the livers of WT and KO mice. B-C, E. Chromatin immunoprecipitation (ChIP) analysis of H4R3me2a levels at gene promoters using anti-H4R3me2a antibody or IgG as a negative control. Average % input for each ChIP is presented. Data are presented as mean ± SD. N=3-4, ***p < 0.001, **p < 0.01. D. Chromatin immunoprecipitation using anti-HNF4α antibody or IgG as a negative control from the livers of WT or KO mice. Data are presented as mean % input ± SD. N=3, *p < 0.05, **p < 0.01. E. Huh 7.5 cells were transfected with wild type PRMT1 in the presence or absence of PRMT1 inhibitor AMI-1. Relative mRNA levels are presented as mean ± SD, N=3, *p < 0.05, **p < 0.01.
Previous studies indicated that HNF4α inhibition or β-catenin activation results in an increase of c-Myc expression, which promotes proliferation (15, 26, 28). However we did not see an increase in c-Myc mRNA in the PRMT1 knockout (Figure 2A). These data suggest that the increase in proliferation observed in PRMT1 knockout mice is c-Myc independent. It also raises the question of why HNF4α inhibition in the context of loss of PRMT1 fails to activate c-Myc expression. This lack of c-Myc activation is likely explained by the direct regulation of c-Myc promoter activity by PRMT1 dependent promoter methylation (2). As shown in Figure 3E, c-Myc promoter methylation is dramatically decreased by the PRMT1 knockout.
Finally, we confirmed that PRMT1 regulates gene expression of Axin1 and HNF4α in vitro using Huh 7.5 cells expressing wild type PRMT1 (Figure 3F). We found that PRMT1 overexpression can induce expression of those genes and it requires PRMT1 activity, since treatment with the PRMT1 inhibitor AMI-1 prevents this induction.
PRMT1 dependent histone methylation is inhibited in alcohol fed mice
To confirm that loss of PRMT1 activity is relevant in human disease we analyzed H4R3me2a levels in the livers of donors or patients with non-alcohol steatohepatitis (NASH), alcoholic liver disease (ALD) and primary biliary cirrhosis (PBC) (Figure 4A). H4R3me2a levels vary between individuals, but only the ALD group showed significantly lower levels compared to normal liver donors.
Figure 4. PRMT1 dependent histone methylation is inhibited in alcohol fed mice.
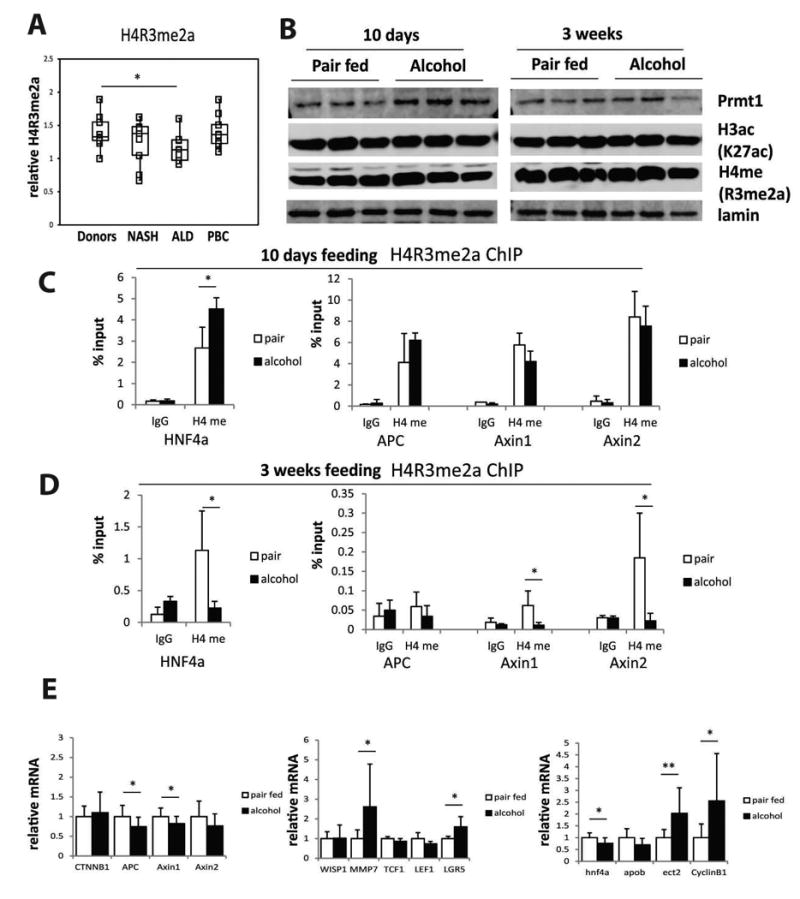
A. Levels of PRMT1-dependent histone methylation mark H4R3me2a in the livers of donors or patients with alcohol liver disease (ALD), non-alcoholic steatohepatitis (NASH), or primary biliary cirrhosis (PBC). N= 9-10 per group, *p < 0.05. B. Western blot analysis of PRMT1 protein expression and H3K27ac and H4R3me2a levels in the livers of wild type mice fed control liquid diet (pair fed) or alcohol diet (alcohol) for 10 days or 3 weeks. C-D. Chromatin immunoprecipitation (ChIP) analysis of H4R3me2a levels at gene promoters using anti-H4R3me2a antibody or IgG as a negative control. Average % input for each ChIP is presented. Data are presented as mean ± SD. N=3, *p < 0.05. E. Relative mRNA levels in the livers of wild type mice fed control liquid diet (pair fed) or alcohol diet (alcohol) for 3 weeks. Data are presented as mean ± SD. n=10 per group. **p < 0.01, *p < 0.05.
To confirm the presence of this mechanism using a mouse model of alcohol feeding, we measured H4R3me2a levels in the livers of mice fed alcohol or control liquid diet for 10 days or 3 weeks (Figure 4B). We did not observe any change in the total level of H4R3me2a in these animals. Interestingly PRMT1 protein level increased at 10 days alcohol feeding compared to control animals. These data suggest that inhibition of PRMT1 activity is not profound enough and/or the duration of inhibition is not long enough to suppress total histone methylation. We thus hypothesized that alcohol might affect methylation more selectively at specific promoters. We found that at 3 weeks but not 10 days of alcohol feeding, specific histone arginine methylation at the promoters of Axin1, Axin2 and HNF4α was reduced (Figure 4C, D). Additionally we found that ethanol feeding produced small but significant decreases in Axin1, APC and HNF4α mRNA, and increases in a subset of β-catenin target gene expression. The decrease in HNF4α mRNA was associated with the predicted changes in its target genes Apob and Ect2 (Figure 4E). These effects of alcohol were similar to those seen in PRMT1 knockout animals in comparison to wild type litter mates (Fig. 2D-F). We found no significant change in β-catenin, c-Myc and JMJD6 protein levels at these conditions (Figure S4).
AAV-Cre mediated PRMT1 deletion in adult mice results in inhibition of HNF4α expression and an increase in hepatocyte proliferation
Since Albumin Cre-genetic deletion of genes in hepatocytes can have off target effects such as Cre expression in biliary cells or expression during development, we confirmed our observations using a second model of PRMT1 deletion in hepatocytes, AAV8-TBG-Cre or AAV-control virus injection into adult PRMT1 fl/fl mice (Figure 5). Figure 5A shows that this treatment eliminated detectible ADMA staining in nearly 100% of hepatocytes. At 3 weeks post AAV injection we observed a dramatic increase in PCNA positive cells in the liver (Figure 5A) and an increase in liver/body weight ratio from 5.2% to 6.2% (Figure 5B). We also detected an increase in proliferation related genes in the knockout animals (Figure 5C). Interestingly, PRMT1 knockout reduced the expression of Axin1 (Figure 5D), but β-catenin level was not increased and its downstream target gene expression was not activated and was even decreased (Figure 5D, E). These data suggests that PRMT1 might have other targets in the β-catenin pathway apart from Axin1 (27, 29). Previous studies found that PRMT1 can positively regulate β-catenin pathway via arginine methylation of G3BP1 (30).
Figure 5. AAV-Cre mediated PRMT1 deletion in adult mice results in inhibition of HNF4α expression and an increase in hepatocyte proliferation.
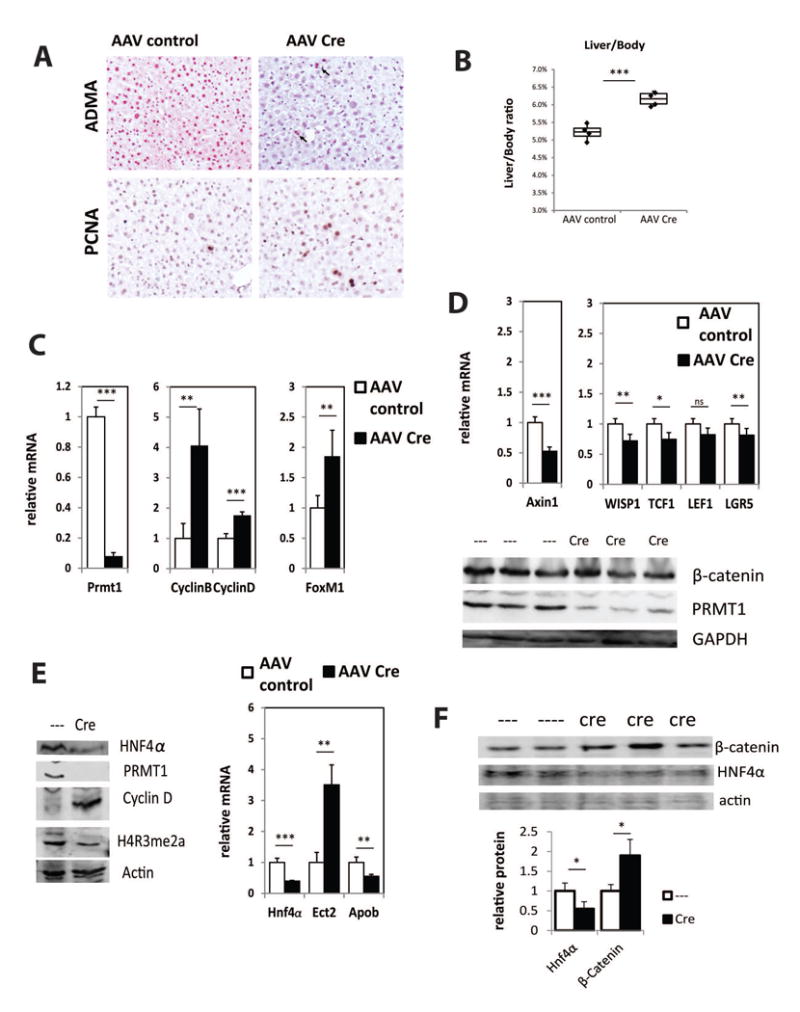
A. Representative images of immunohistochemical staining of asymmetric di-methyl arginine levels (ADMA) in the liver sections of PRMT1 flox/flox mice after 3 weeks post injection of AAV8-TBG-Cre or AAV-control virus 10̂11 gc/mouse. Arrows indicate the hepatocytes that did not receive Cre recombinase and have wild type levels of ADMA. Representative images of immunohistochemical staining of PCNA positive cells in these livers are shown below. B. Liver/body weight ratio from PRMT1 flox/flox mice after 3 weeks post injection of AAV8-TBG-Cre or AAV-control virus 10̂11 gc/mouse. ***p < 0.001, n=4 per group. C-E. Relative mRNA levels and western blot analysis of protein expression in the livers of mice as in B. Data are presented as mean ± SD. N=4 per group, ***p < 0.001, **p < 0.01. F. Western blot analysis of protein expression in the livers of PRMT1 flox/flox mice after 12 weeks post injection of AAV8-TBG-Cre or AAV-control virus 10̂11 gc/mouse. Densitometry analysis of β-catenin and HNF4α relative protein expression. Data are presented as mean ± SD. N=4-5 per group. *p < 0.05.
Next we examined HNF4α expression and found that PRMT1 knockout in adults resulted in a decrease in protein and mRNA levels of HNF4α, decrease in promoter arginine methylation, as well as loss of HNF4α mediated activation of Apob or suppression of Ect2 and Cyclin D (Figure 5F, S5A,B). This data is consistent with previous reports suggesting that reduced expression of Hnf4α is sufficient to induce an increase in hepatocyte proliferation (28). Taken together these data suggest that PRMT1 regulates hepatocyte proliferation mainly through HNF4α regulation.
To determine if differences between the albumin-cre and the AAV-Cre models is simply due to time of exposure to PRMT1 knock out, we injected AAV-Cre to induce PRMT1 knockout for 12 weeks (Figure 5F, S5B). We found a decrease in protein and mRNA levels of HNF4α as well as accumulation of β-catenin protein, similar to the situation in the Alb-Cre mice, but without the increase in expression of the downstream gene WISP1seen in the Alb-Cree knockout (Figure S5B vs Figure 2).
JMJD6 knockdown can restore ADMA and HNF4α levels in PRMT1 knockout mice
Until recently arginine methylation was thought to be a stable modification which can be removed only through degradation and synthesis of new unmethylated proteins. However, recent studies from our lab and others have found that JMJD6 can demethylate some of the PRMT1 targets (8, 9, 11). Apart from being a demethylase, JMJD6 also has other functions that make it hard to evaluate its role in vitro and in vivo. Here we decided to study whether JMJD6 has the opposing role in regulating PRMT1 targets that are important for hepatocyte proliferation (Figure 6). First, we analyzed how JMJD6 overexpression affects HNF4α and Axin1 expression in vitro in Huh 7.5 cells (Figure 6A). We found that JMJD6 suppresses expression of both genes in vitro, suggesting that it can have an opposite effect to that of PRMT1.
Figure 6. JMJD6 knockdown can restore ADMA and HNF4α levels in PRMT1 knockout mice.
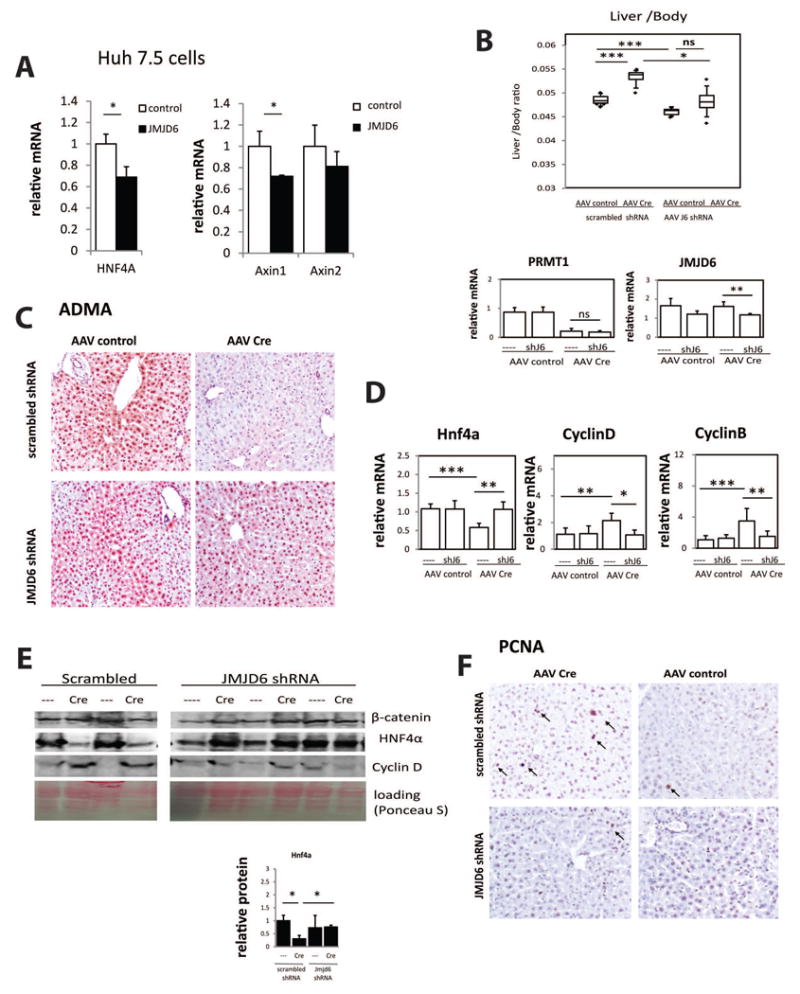
A. Huh 7.5 cells were transfected with wild type JMJD6. Relative mRNA levels are presented as mean ± SD, N=3, *p < 0.05. B. Liver/body weight ratio from PRMT1 flox/flox mice after 3 weeks post injection of AAV8-TBG-Cre or AAV-control virus 10̂11 gc/mouse followed by second injection of AAV8-GFP-U6-m-JMJD6-shRNA or AAV8-GFP-U6-scrmb-shRNA 2 × 10̂11 gc/mouse. *p < 0.05, ***p < 0.001, n=4-6 per group. Relative mRNA levels of PRMT1 and JMJD6 in livers of these mice is presented below. Data are presented as mean ± SD, **p < 0.01. C. Representative images of immunohistochemical staining of asymmetric di-methyl arginine levels (ADMA) in the liver sections of mice as in B. D. Relative mRNA levels in livers of mice as in B. Data are presented as mean ± SD, *p < 0.05, **p < 0.01., ***p < 0.001, n=4-6 per group. E. Western blot analysis of protein expression levels in the livers of mice as in B. Densitometry analysis of HNF4α relative protein expression is presented below. Data are presented as mean ± SD. N=3 per group. *p < 0.05.F. Representative images of immunohistochemical staining of PCNA positive cells in livers of mice as in B. PCNA positive hepatocytes are indicated by arrows.
Next we analyzed the role of JMJD6 in vivo. PRMT1 fl/fl mice received injection of AAV8-TBG-cre or control AAV. Three days later mice received a second injection of AAV8-JMJD6 shRNA or AAV8 scrambled control. Three weeks post injection mice were analyzed. We found that in the JMJD6 shRNA group PRMT1 knockout did not result in a significant increase in liver weight compared to wild type control (Figure 6B). We confirmed that this was not due to inefficient recombination, since both knockout groups had a similar level of PRMT1expression (Figure 6B, S6A,B). JMJD6 knockdown partially restored ADMA levels in PRMT1 knockout mice (Figure 6C). JMJD6 knockdown also reduced Cyclin B and Cyclin D expression and increased HNF4α expression in the livers of PRMT1 knockout mice (Figure 6D,E). Similarly we saw a decrease in the number of PCNA positive cells in the livers of PRMT1 knockout mice that received JMJD6 shRNA compared to mice that received scrambled shRNA (Figure 6F). Taken together these data suggest that JMJD6 knockdown can restore HNF4α levels in PRMT1 knockout hepatocytes and prevent an increase in hepatocyte proliferation. Interestingly, JMJD6 knockdown did not restore ALT levels in those mice suggesting that gpt expression is not regulated by JMJD6 (Figure S6C).
JMJD6 knockdown can restore ADMA and HNF4α levels in alcohol fed mice
Next we examined whether JMJD6 knockdown can restore arginine methylation in alcohol fed mice. Wild type mice were fed alcohol or control liquid diet for 3 weeks and on the first day of alcohol mice received either AAV-JMJD6 shRNA or AAV-scrambled (Figure 7, S7). We found that JMJD6 knock down can increase ADMA levels in livers of alcohol fed mice (Figure 7A, B). In addition, JMJD6 knock down prevented HNF4α promoter demethylation induced by alcohol and efficiently restored HNF4α expression (Figure 7C, D, S6A). These data suggest that the reciprocal activities of PRMT1 and JMJD6 on ADMA regulate HNF4α expression in alcohol fed mice. This is consistent with data presented in Figures 5 and 6. However we found no effect of JMJD6 knock down on expression of proliferation associated genes (Fig. 7D), hepatocyte proliferation markers (Figure 7E), or liver size (Figure S7B) suggesting that alcohol increases hepatocyte proliferation independent of HNF4α inhibition.
Figure 7. JMJD6 knockdown can restore ADMA and HNF4α levels in alcohol fed mice.
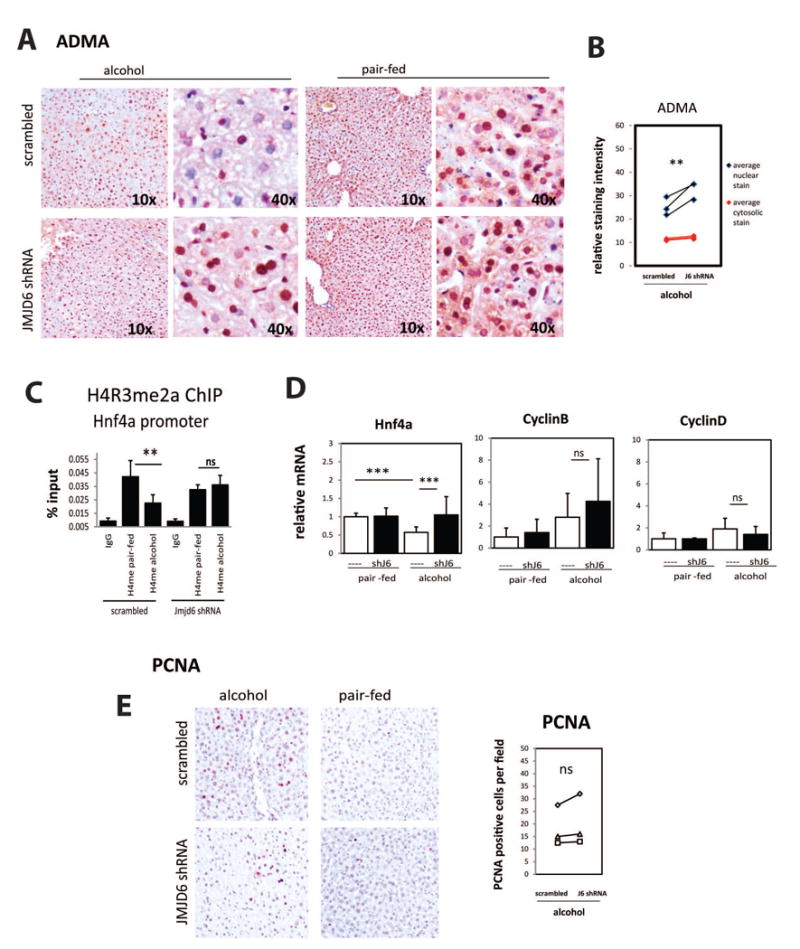
Wild type mice were injected 2 × 10̂11 gc/mouse AAV8-GFP-U6-m-JMJD6-shRNA or AAV8-GFP-U6-scrmb-shRNA and fed control liquid diet (pair fed) or alcohol diet (alcohol) for 3 weeks. A. Representative images of immunohistochemical staining of asymmetric di-methyl arginine levels (ADMA) in the liver sections of these mice. B. Relative staining intensity of ADMA in livers of alcohol fed littermates that received AAV8- JMJD6-shRNA or AAV8-scrmb-shRNA measured using Aperio Image Scope. **p < 0.01, n=3. C. Chromatin immunoprecipitation (ChIP) analysis of H4R3me2a levels atHNF4α gene promoter using anti-H4R3me2a antibody or IgG as a negative control. Average % input for each ChIP is presented. Data are presented as mean ± SD. N=3, **p < 0.01. D. Relative mRNA levels in livers of these mice. Data are presented as mean ± SD, ***p < 0.001, n=4-6 per group. E. Representative images of immunohistochemical staining of PCNA positive cells in livers of these mice.
ADMA levels correlate with HNF4α expression in human HCC
To confirm that PRMT1 and JMJD6 dependent ADMA regulation of HNF4α expression is relevant to human disease, we measured ADMA levels in the nucleus and HNF4α expression in HCC sections from liver explants of HCC patients undergoing liver transplantation. We found a significant correlation between those two parameters (Figure 8A, B). We also measured β-catenin expression in those tissues and found no correlation with ADMA (Figure 8A).
Figure 8. ADMA levels correlate with HNF4α expression in human HCC.
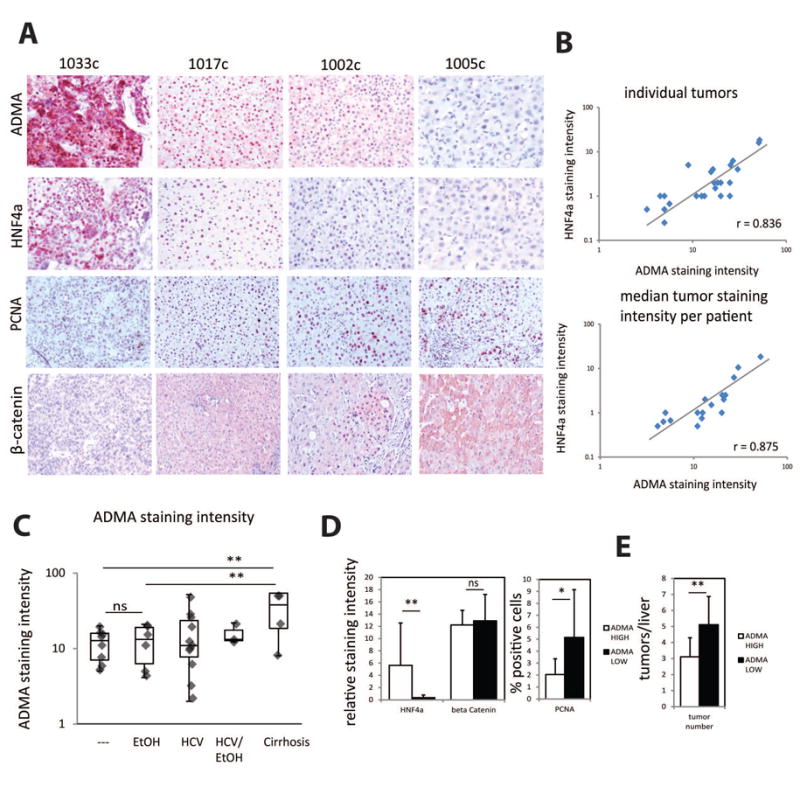
A. Examples of immunohistochemical staining of asymmetric di-methyl arginine levels (ADMA), HNF4α, PCNA and β-catenin levels in the sections of HCC specimen from patients. B. Correlation of ADMA nuclear staining intensity and HNF4α staining intensity in sections from individual tumors or median staining intensity for each patient. C. ADMA nuclear staining intensity in in sections from patients with cirrhosis (n=4), HCC not associated with HCV of alcohol (---, n=7), HCC associated with alcohol (EtOH, n=6), HCV HCC (n= 11), HCV and alcohol HCC (n=3). **p < 0.01. D. Relative staining intensity of HNF4α and β-catenin in HCC sections with low ADMA and high ADMA staining N=10 per group. Percent PCNA positive cells in HCC sections as above. *p < 0.05, **p < 0.01. E. Number of tumors in livers of patients with low and high ADMA staining. N=10 per group, **p < 0.01.
We found that in HCC samples ADMA levels were significantly lower compared to livers with liver cirrhosis. However there was no difference in ADMA levels between different etiologies of HCC (Figure 8C). Interestingly PRMT1 protein levels were not decreased in HCC (Figure S8) and were significantly higher than in donor livers. These data suggests that PRMT1 activity is altered in HCC similar to the situation in alcohol fed mice (Figure 1A).
We found that low ADMA also correlates with higher proliferation in human HCC (Figure 8D). We did not find any correlation with the tumor size, possibly since most of the HCCs were treated with chemoembolization therapy one or multiple times prior to transplant. However the number of tumors at the time of transplant was significantly higher in the group with low ADMA compared to high ADMA group (Figure 8E).
Discussion
Hepatocellular carcinoma (HCC) is the third most common cause of cancer related death (31, 32). Unlike other cancers with known risk factors, causes of HCC are not completely understood. Most HCC patients have a history of chronic liver disease and cirrhosis (12). Commonly, the HCC is associated with hepatitis B and C and chronic alcohol drinking (13). HCC development is a multistep process that involves genetic and epigenetic modifications in the liver that lead to malignant transformation of hepatocytes (33).
Alcohol promotes liver tumors by multiple mechanisms (15-18). Here we describe a novel mechanism of alcohol pathogenesis that can contribute to tumor development. We found that alcohol can dramatically reduce cellular arginine methylation levels. By studying the impact loss of arginine methylation can have on cellular processes we found that two enzymes: the methyltransferase PRMT1 and the demethylase JMJD6 can positively (PRMT1) and negatively (JMJD6) regulate levels of cellular asymmetric dimethyl arginine and histone arginine methylation at promoters of target genes. Specifically they regulate expression of HNF4α in hepatocytes. Loss of PRMT1 results in dramatic downregulation of HNF4α in the liver and an increase in hepatocyte proliferation. JMJD6 knockdown on the other hand can restore HNF4α levels in PRMT1 knockout hepatocytes and prevent the increase in proliferation.
Interestingly we found that genetic ablation of PRMT1 in hepatocytes resulted in upregulation of β-catenin, however we found no increase in downstream target gene expression. Similar results were found in the conditional knockout model (AAV-Cre) after 12 weeks post AAV injections. Previous studies have also described a dependence of steady-state beta catenin level on PRMT1 with PRMT1 knock down causing an increase through effects of Axin1 and G3BP1 (27, 30). While our data confirms that PRMT1 knockdown increases beta catenin level, we fail to see a subsequent increase in downstream target expression. This suggests that PRMT1 knockout has a second effect that prevents increased expression of these targets.
Alcohol feeding duplicates some but not all of the PRMT1 knockout effects. Similar to PRMT1 knockout alcohol caused reduced promoter arginine methylation and gene expression of HNF4α and Axin1. Downstream HNF4α signaling was similarly inhibited in both alcohol and PRMT1 knockout models. Alcohol feeding resulted in upregulation of a subset of β-catenin target genes, which was not present in the PRMT1 conditional knockout model. PRMT1 knockout resulted in dramatic increase in hepatocyte proliferation. Alcohol feeding also resulted in higher hepatocyte proliferation, but the increase in proliferation was not due to reduced ADMA levels, since restoring ADMA with JMJD6 knockdown did not prevent it.
On the other hand, restoring ADMA levels by JMJD6 knock down was sufficient to induce HNF4α expression in alcohol fed mice to the control levels. Additionally we found that there is a strong correlation between ADMA and HNF4α as well as proliferation in human HCC tissue specimen, suggesting that this mechanism is relevant in patients.
PRMT1 and JMJD6 in this setting play opposing roles in control of gene expression and proliferation. However it is likely that not every target of PRMT1 is a target of JMJD6 as well. This study does not investigate the role of other methyltransferases such as CARM1 and PRMT5, which are known to have different substrate specificity, whether or not those enzymes are affected by alcohol and whether or not JMJD6 can demethylate their targets. This important aspect will require further investigation.
We found that PRMT1 and JMJD6 regulate HNF4α. HNF4α is an important regulator of hepatic function. Apart from its role in controlling hepatocyte differentiation, recent studies identified it as a regulator of hepatocyte proliferation (28, 34). HNF4α is downregulated in majority of HCC (35-37). Recent studies have demonstrated that inhibition or loss of HNF4α plays a key role in tumorigenesis in the liver and colon (35, 38).
PRMT1 also regulates other transcription factors that are involved in regulation of cell cycle progression and proliferation, such as FOXOs (22), p53 (6), c-Myc (2, 39), as well as upstream signaling molecules involved in proliferation, EGFR (40). We analyzed PRMT1 knockout mouse gene expression and used IPA analysis to find the main upstream regulators affected by PRMT1. For the top upstream regulators we confirmed whether documented target genes were activated or inhibited by PRMT1 as predicted. Only the HNF4α target gene expression pattern correlated with its inactivation and increased hepatocyte proliferation phenotype.
Taken together, the presented data suggest that the alcohol effect on hepatocyte ADMA levels can result in the suppression of HNF4α expression and contribute to alcohol associated cancer development. We found that JMJD6 knock down is an efficient way to restore HNF4α levels in alcohol fed mice. Further studies are necessary to evaluate the efficiency of targeting JMJD6 in alcohol induced cancer models. Previous studies indicate that JMJD6 is upregulated in several types of cancers and some studies suggest that it promotes cancer cell proliferation (41). Future development of specific JMJD6 inhibitors might be a promising strategy in cancer therapy for HCC and other types of cancer.
Supplementary Material
Acknowledgments
The human specimens used in this study were derived from samples provided by the University of Kansas Liver Center Tissue Bank. We thank Drs. Jameson Forster and Bashar Abdulkarim for their assistance in obtaining these specimens. We thank Dr Lisa Zhang for providing AAV vectors.
Grant support: This study was supported by grants AA012863 from the National Institute on Alcoholism and Alcohol Abuse, COBRE Pilot grant NCRR/NIH P20 RR021940 and 2016 Pinnacle Award from AASLD. This work was supported in part by grants from the National Institute of General Medical Sciences (P20GM103549 & P30GM118247) of the National Institutes of Health
Abbreviations
- JMJD6
Jumonji C domain-containing protein 6
- SAM
S-adenosylmethionine
- PRMT1
protein arginine methyl transferase 1
- HCV
hepatitis C virus
- NASH
nonalcoholic steatohepatitis
- PBC
Primary biliary cirrhosis
- ALD
alcoholic liver disease
- HCC
hepatocellular carcinoma
- ADMA
asymmetric dimethyl arginine
- HNF4α
Hepatocyte nuclear factor 4 alpha
- PCNA
Proliferating cell nuclear antigen
- IP
immunoprecipitation
Footnotes
Disclosures: The authors have no financial, professional or personal conflicts of interest to disclose.
References
- 1.Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, Clarke S, et al. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J Biol Chem. 2000;275:7723–7730. doi: 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y, McBride KM, Hensley S, Lu Y, Chedin F, Bedford MT. Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol Cell. 2014;53:484–497. doi: 10.1016/j.molcel.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrero MJ, Malik S. Two functional modes of a nuclear receptor-recruited arginine methyltransferase in transcriptional activation. Mol Cell. 2006;24:233–243. doi: 10.1016/j.molcel.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chittka A. Dynamic distribution of histone H4 arginine 3 methylation marks in the developing murine cortex. PLoS One. 2010;5:e13807. doi: 10.1371/journal.pone.0013807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Hassa PO, Covic M, Bedford MT, Hottiger MO. Protein arginine methyltransferase 1 coactivates NF-kappaB-dependent gene expression synergistically with CARM1 and PARP1. J Mol Biol. 2008;377:668–678. doi: 10.1016/j.jmb.2008.01.044. [DOI] [PubMed] [Google Scholar]
- 8.Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a histone arginine demethylase. Science. 2007;318:444–447. doi: 10.1126/science.1145801. [DOI] [PubMed] [Google Scholar]
- 9.Poulard C, Rambaud J, Hussein N, Corbo L, Le Romancer M. JMJD6 regulates ERalpha methylation on arginine. PLoS One. 2014;9:e87982. doi: 10.1371/journal.pone.0087982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boeckel JN, Guarani V, Koyanagi M, Roexe T, Lengeling A, Schermuly RT, Gellert P, et al. Jumonji domain-containing protein 6 (Jmjd6) is required for angiogenic sprouting and regulates splicing of VEGF-receptor 1. Proc Natl Acad Sci U S A. 2011;108:3276–3281. doi: 10.1073/pnas.1008098108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tikhanovich I, Kuravi S, Artigues A, Villar MT, Dorko K, Nawabi A, Roberts B, et al. Dynamic Arginine Methylation of Tumor Necrosis Factor (TNF) Receptor-associated Factor 6 Regulates Toll-like Receptor Signaling. J Biol Chem. 2015;290:22236–22249. doi: 10.1074/jbc.M115.653543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanyal AJ, Yoon SK, Lencioni R. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist. 2010;15(Suppl 4):14–22. doi: 10.1634/theoncologist.2010-S4-14. [DOI] [PubMed] [Google Scholar]
- 13.Altekruse SF, Henley SJ, Cucinelli JE, McGlynn KA. Changing hepatocellular carcinoma incidence and liver cancer mortality rates in the United States. Am J Gastroenterol. 2014;109:542–553. doi: 10.1038/ajg.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joshi K, Kohli A, Manch R, Gish R. Alcoholic Liver Disease: High Risk or Low Risk for Developing Hepatocellular Carcinoma? Clin Liver Dis. 2016;20:563–580. doi: 10.1016/j.cld.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 15.Mercer KE, Hennings L, Sharma N, Lai K, Cleves MA, Wynne RA, Badger TM, et al. Alcohol consumption promotes diethylnitrosamine-induced hepatocarcinogenesis in male mice through activation of the Wnt/beta-catenin signaling pathway. Cancer Prev Res (Phila) 2014;7:675–685. doi: 10.1158/1940-6207.CAPR-13-0444-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ambade A, Satishchandran A, Gyongyosi B, Lowe P, Szabo G. Adult mouse model of early hepatocellular carcinoma promoted by alcoholic liver disease. World J Gastroenterol. 2016;22:4091–4108. doi: 10.3748/wjg.v22.i16.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol. 2015;12:387–400. doi: 10.1038/nrgastro.2015.94. [DOI] [PubMed] [Google Scholar]
- 19.Tikhanovich I, Kuravi S, Campbell RV, Kharbanda KK, Artigues A, Villar MT, Weinman SA. Regulation of FOXO3 by phosphorylation and methylation in hepatitis C virus infection and alcohol exposure. Hepatology. 2014;59:58–70. doi: 10.1002/hep.26618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tikhanovich I, Zhao J, Olson J, Adams A, Taylor R, Bridges B, Marshall L, et al. Protein Arginine Methyltransferase 1 modulates innate immune responses through regulation of peroxisome proliferator-activated receptor gamma-dependent macrophage differentiation. J Biol Chem. 2017 doi: 10.1074/jbc.M117.778761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones CT, Catanese MT, Law LM, Khetani SR, Syder AJ, Ploss A, Oh TS, et al. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat Biotechnol. 2010;28:167–171. doi: 10.1038/nbt.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi D, Oh KJ, Han HS, Yoon YS, Jung CY, Kim ST, Koo SH. Protein arginine methyltransferase 1 regulates hepatic glucose production in a FoxO1-dependent manner. Hepatology. 2012;56:1546–1556. doi: 10.1002/hep.25809. [DOI] [PubMed] [Google Scholar]
- 23.Hou YY, Lu SH, Zhou Y, Xu JF, Ji Y, Hou J, Qi WD, et al. Predictive values of clinical and pathological parameters for malignancy of gastrointestinal stromal tumors. Histol Histopathol. 2009;24:737–747. doi: 10.14670/HH-24.737. [DOI] [PubMed] [Google Scholar]
- 24.Terada T, Nakanuma Y. Cell proliferative activity in adenomatous hyperplasia of the liver and small hepatocellular carcinoma. An immunohistochemical study demonstrating proliferating cell nuclear antigen. Cancer. 1992;70:591–598. doi: 10.1002/1097-0142(19920801)70:3<591::aid-cncr2820700309>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 25.Wanless IR. Liver biopsy in the diagnosis of hepatocellular carcinoma. Clin Liver Dis. 2005;9:281–285. vii. doi: 10.1016/j.cld.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 26.Pez F, Lopez A, Kim M, Wands JR, Caron de Fromentel C, Merle P. Wnt signaling and hepatocarcinogenesis: molecular targets for the development of innovative anticancer drugs. J Hepatol. 2013;59:1107–1117. doi: 10.1016/j.jhep.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Cha B, Kim W, Kim YK, Hwang BN, Park SY, Yoon JW, Park WS, et al. Methylation by protein arginine methyltransferase 1 increases stability of Axin, a negative regulator of Wnt signaling. Oncogene. 2011;30:2379–2389. doi: 10.1038/onc.2010.610. [DOI] [PubMed] [Google Scholar]
- 28.Walesky C, Gunewardena S, Terwilliger EF, Edwards G, Borude P, Apte U. Hepatocyte-specific deletion of hepatocyte nuclear factor-4alpha in adult mice results in increased hepatocyte proliferation. Am J Physiol Gastrointest Liver Physiol. 2013;304:G26–37. doi: 10.1152/ajpgi.00064.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baldwin RM, Morettin A, Paris G, Goulet I, Cote J. Alternatively spliced protein arginine methyltransferase 1 isoform PRMT1v2 promotes the survival and invasiveness of breast cancer cells. Cell Cycle. 2012;11:4597–4612. doi: 10.4161/cc.22871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bikkavilli RK, Malbon CC. Arginine methylation of G3BP1 in response to Wnt3a regulates beta-catenin mRNA. J Cell Sci. 2011;124:2310–2320. doi: 10.1242/jcs.084046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 32.Soerjomataram I, Lortet-Tieulent J, Parkin DM, Ferlay J, Mathers C, Forman D, Bray F. Global burden of cancer in 2008: a systematic analysis of disability-adjusted life-years in 12 world regions. Lancet. 2012;380:1840–1850. doi: 10.1016/S0140-6736(12)60919-2. [DOI] [PubMed] [Google Scholar]
- 33.Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27:55–76. doi: 10.1055/s-2006-960171. [DOI] [PubMed] [Google Scholar]
- 34.Bonzo JA, Ferry CH, Matsubara T, Kim JH, Gonzalez FJ. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. J Biol Chem. 2012;287:7345–7356. doi: 10.1074/jbc.M111.334599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walesky C, Apte U. Role of hepatocyte nuclear factor 4alpha (HNF4alpha) in cell proliferation and cancer. Gene Expr. 2015;16:101–108. doi: 10.3727/105221615X14181438356292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramesh V, Ganesan K. Integrative functional genomic delineation of the cascades of transcriptional changes involved in hepatocellular carcinoma progression. Int J Cancer. 2016;139:1586–1597. doi: 10.1002/ijc.30195. [DOI] [PubMed] [Google Scholar]
- 37.Desert R, Rohart F, Canal F, Sicard M, Desille M, Renaud S, Turlin B, et al. Human Hepatocellular Carcinomas with a Periportal Phenotype Have the Lowest Potential for Early Recurrence after Curative Resection. Hepatology. 2017 doi: 10.1002/hep.29254. [DOI] [PubMed] [Google Scholar]
- 38.Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, Gurumurthy S, et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature. 2014;513:110–114. doi: 10.1038/nature13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eberhardt A, Hansen JN, Koster J, Lotta LT, Jr, Wang S, Livingstone E, Qian K, et al. Protein arginine methyltransferase 1 is a novel regulator of MYCN in neuroblastoma. Oncotarget. 2016;7:63629–63639. doi: 10.18632/oncotarget.11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao HW, Hsu JM, Xia W, Wang HL, Wang YN, Chang WC, Arold ST, et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J Clin Invest. 2015;125:4529–4543. doi: 10.1172/JCI82826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aprelikova O, Chen K, El Touny LH, Brignatz-Guittard C, Han J, Qiu T, Yang HH, et al. The epigenetic modifier JMJD6 is amplified in mammary tumors and cooperates with c-Myc to enhance cellular transformation, tumor progression, and metastasis. Clin Epigenetics. 2016;8:38. doi: 10.1186/s13148-016-0205-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.