

Abstract
Nicotinamide adenine dinucleotide (NAD+) is an essential metabolite participating in cellular redox chemistry and signaling, and the complex regulation of NAD+ metabolism is not yet fully understood. To investigate this, we established a NAD+-intermediate specific reporter system to identify factors required for salvage of metabolically linked nicotinamide (NAM) and nicotinic acid (NA). Mutants lacking components of the NatB complex, NAT3 and MDM20, appeared as hits in this screen. NatB is an Nα-terminal acetyltransferase responsible for acetylation of the N terminus of specific Met-retained peptides. In NatB mutants, increased NA/NAM levels were concomitant with decreased NAD+. We identified the vacuolar pool of nicotinamide riboside (NR) as the source of this increased NA/NAM. This NR pool is increased by nitrogen starvation, suggesting NAD+ and related metabolites may be trafficked to the vacuole for recycling. Supporting this, increased NA/NAM release in NatB mutants was abolished by deleting the autophagy protein ATG14. We next examined Tpm1 (tropomyosin), whose function is regulated by NatB-mediated acetylation, and Tpm1 overexpression (TPM1-oe) was shown to restore some NatB mutant defects. Interestingly, although TPM1-oe largely suppressed NA/NAM release in NatB mutants, it did not restore NAD+ levels. We showed that decreased nicotinamide mononucleotide adenylyltransferase (Nma1/Nma2) levels probably caused the NAD+ defects, and NMA1-oe was sufficient to restore NAD+. NatB-mediated N-terminal acetylation of Nma1 and Nma2 appears essential for maintaining NAD+ levels. In summary, our results support a connection between NatB-mediated protein acetylation and NAD+ homeostasis. Our findings may contribute to understanding the molecular basis and regulation of NAD+ metabolism.
Keywords: cell metabolism, metabolic regulation, metabolism, NAD biosynthesis, yeast genetics
Introduction
NAD+ and its reduced form NADH are essential metabolites mediating redox reactions in cellular metabolism. NAD+ is a substrate in protein modifications, such as sirtuin (Sir2 family proteins)-mediated protein deacetylation, and ADP-ribosylation by poly(ADP-ribose) polymerases. These protein modifications contribute to regulation of chromatin structure, DNA repair, circadian rhythm, metabolic responses, and life span (1–4). Aberrant NAD+ metabolism is associated with a number of diseases, including diabetes, cancer, and neuron degeneration (2, 3, 5–8). Administration of NAD+ precursors, such as nicotinamide mononucleotide (NMN),2 nicotinamide (NAM), and nicotinamide riboside (NR), has been shown to ameliorate deficiencies related to aberrant NAD+ metabolism in yeast, mouse, and human cells (3, 5–12). However, molecular mechanisms that underlie these beneficial effects remain unclear.
NAD+ biosynthesis is highly conserved between yeast and vertebrates. In yeast, NAD+ is synthesized de novo from tryptophan or salvaged from intermediates, such as NA, NAM, and NR (Fig. 1A). The NAD+ levels maintained by these pathways converge at several different points and share cellular pools of ATP, phosphoribosyl pyrophosphate, and glutamine, while adding to total pools of ribose, AMP, phosphate, formate, alanine, glutamate, and others. NAD+ is a NADP+ precursor, which, like NAD+, is carefully balanced with its reduced form to maintain a favorable redox state. Some of these molecules contribute to other biosynthesis pathways or have signaling functions. Therefore, the cell must maintain these metabolites and their flux in a controlled manner. We do not fully understand all of the mechanisms by which the cell can sense and tune these metabolites, but some known NAD+ homeostasis-regulatory mechanisms include transcriptional control, feedback inhibition, and enzyme or metabolite compartmentalization (1, 13–18). The complex and dynamic flexibility of NAD+ precursors makes studying NAD+ metabolism complicated. For example, NAM can both replenish NAD+ pools and inhibit the activity of NAD+-consuming enzymes. In addition, little is known about the signaling pathways that regulate NAD+ precursor homeostasis, in part due to the lack of sensitive and specific genetic screening systems to identify these NAD+ homeostasis factors.
Figure 1.

Identification of yeast mutants with altered NA and NAM homeostasis. A, a simplified model of the Saccharomyces cerevisiae NAD+ synthesis pathway. NAD+ can be synthesized de novo from Trp and by salvaging NAD+ intermediates through the NA/NAM and NR cycles. NaMN, nicotinic acid mononucleotide. NaAD, deamido NAD+. Abbreviations of protein names are shown in parentheses. B, deletions of NPT1 and PNC1 confer increased NA/NAM and NAM release, respectively. NA/NAM-dependent recipient cells were plated onto niacin-free SD plates as a lawn. Next, WT, npt1Δ, and pnc1Δ feeder cells were spotted on top of the lawn and allowed to grow for 3–4 days at 30 °C. Recipient cells on the top panel (bna6Δnrk1Δnrt1Δ) can utilize both NA and NAM, whereas recipient cells on the bottom panel (bna6Δnrk1Δnrt1Δpnc1Δ) can only utilize NA. The number and size of the satellite colonies surrounding the feeder spots positively correlate with the amount of NA/NAM released from the feeders. C, overview of the genetic screen to identify mutants with increased NA/NAM release. Haploid single deletion mutants (feeder cells) were spotted onto a lawn of NA/NAM-dependent bna6Δnrk1Δnrt1Δ recipient cells, whose growth is dependent on NA/NAM released from the feeder cells in a dose-dependent manner. D, deletions of NAT3 and MDM20 confer increased NAM release. Various feeder strains were spotted onto a lawn of bna6Δnrk1Δnrt1Δ (top) and bna6Δnrk1Δnrt1Δpnc1Δ (bottom) recipient cells. For clarity, inverse images are shown. E, the nat3Δ and mdm20Δ mutants show significant increases in both released and intracellular NA/NAM levels. NA/NAM levels were determined in both growth media (released) and cell extracts (intracellular). Data shown are representative of three independent experiments. Feeding of collected lysate to recipient cells was conducted in triplicate. Error bars, S.D. The p values are calculated using Student's t test (*, p < 0.05).
We have previously shown that NR-mediated NAD+ synthesis plays important roles in the maintenance of NAD+ pools and calorie restriction-induced life span (19). To further understand the regulation of NAD+ homeostasis, we developed an NR-specific reporter-based bioassay to screen for mutants with altered NR release. This screen system was based on our observations that yeast cells release and transport NR back into the cell (19), a phenomenon that is also conserved in human cells (20). Our studies have identified novel NAD+ homeostasis factors, including the phosphate-responsive signaling (PHO) pathway (17), the SPS amino acid-sensing pathway (21), the conserved vacuolar NR transporter Fun26 (1, 17), and several NAD+ metabolic enzymes (22).
In this study, we developed a reporter system targeting the NA/NAM branch of NAD+ metabolism. The hypothesis is that cells defective in NA/NAM salvage (Fig. 1A) would show altered NA/NAM release. The mdm20Δ and nat3Δ mutants were among the top hits that showed increased NA/NAM release. Nat3 and Mdm20 are the catalytic and auxiliary subunits of NatB, respectively (23, 24). NatB belongs to a family of N-terminal acetyltransferases (NATs) that add an acetyl group from acetyl-CoA to the α-amino group on the first amino acid of a protein. In yeast, there are four functional NATs, and each has distinct substrate specificity (23, 25, 26). About 20% of proteins are potential substrates of NatB in yeast and humans (27). N-terminal acetylation plays important roles in regulating protein stability, complex formation, and subcellular localization and has been implicated in various cellular processes and human diseases (27). Our studies are the first to link NatB to NAD+ homeostasis. Here, we characterized the NatB mutants as well as the NatB downstream targets to provide a molecular basis underlying NatB-mediated regulation of NAD+ homeostasis.
Results
A cell-based reporter assay to study NA/NAM homeostasis factors
We first determined whether cells indeed release more NA and/or NAM when NA/NAM salvage is blocked. A cross-feeding assay is established using bna6Δnrk1Δnrt1Δ mutants as “recipient cells” (depend on NA or NAM for growth) and mutants of interest as “feeder cells.” In this system, recipient cells cannot grow on niacin-free SD (NA/NAM-free), but when feeder cells are placed in proximity, feeder cell–released NA or NAM supports recipient cell growth by “cross-feeding.” As a result, this assay determines relative levels of total NA and NAM released by feeder cells. As shown in Fig. 1B (top), recipient cells were spread onto niacin-free SD plates as a lawn. Next, npt1Δ (blocks NA salvage) and pnc1Δ (blocks NAM salvage) feeder cells were spotted on top of the lawn and allowed to grow. Recipient cells near the npt1Δ and pnc1Δ feeder spots appeared as satellite colonies after 3 days, and the number and size of the satellite colonies positively correlate with the amount of NA/NAM released from the feeders. To separate NAM release from NA/NAM release, PNC1 was deleted in the recipient cell strain to block its ability to utilize NAM. Based on the biochemical function of Npt1 and Pnc1 (Fig. 1A), we expect that npt1Δ cells release more NA (and perhaps NAM) and pnc1Δ cells release more NAM. Indeed, as shown in Fig. 1B (bottom), pnc1Δ feeder cells failed to support the growth of the new recipient strain (bna6Δnrk1Δnrt1Δpnc1Δ, which cannot utilize NAM). In contrast, npt1Δ cells still supported the growth of bna6Δnrk1Δnrt1Δpnc1Δ. These results confirmed that mutants with altered NA/NAM homeostasis could be identified using this system.
Components of the NatB complex are novel NA/NAM homeostasis factors
Next, we used the haploid yeast deletion collection as feeder cells to identify mutants with increased NA/NAM release (Fig. 1C). Mutants were assigned a score of 2–5 (WT = 1). A higher score represents more NA/NAM release. To eliminate false-positives, mutants with a score of ≥3 (81 mutants) were reexamined. A total of ∼70 mutants passed the secondary screen (Table S1). Known NAD+ homeostasis factors were identified, including npt1Δ and pnc1Δ. The mdm20Δ and nat3Δ mutants were among the top hits. Nat3 and Mdm20 are subunits of NatB, a member of the NATs that adds an acetyl group from acetyl-CoA to the α-amino group on the first amino acid of a protein (23, 24). Deletion of either NatB subunit results in stress sensitivity growth defects (23, 24, 28, 29). To verify that observed phenotypes are due to the featured mutations and not to secondary cryptic mutations in the deletion collection, we reconstructed all deletion mutants used in this study. We confirmed both nat3Δ and mdm20Δ indeed released more NA/NAM (Fig. 1D, top). Similar to pnc1Δ mutant (Fig. 1B), both nat3Δ and mdm20Δ released mostly NAM, not NA (Fig. 1D, bottom). Fig. 1E showed that increased NA/NAM release in nat3Δ and mdm20Δ cells correlates with increased intracellular NA/NAM levels determined by a quantitative liquid assay.
NR metabolism contributes to increased NAM release in NatB mutants
To understand how NatB complex affects NAM/NAD+ homeostasis, we first tested whether Pnc1 level or activity was reduced in NatB mutants because Pnc1 is a major NAM metabolic factor (nicotinamidase), and the pnc1Δ mutant showed strong NAM release (Fig. 1B). Although Pnc1 is not a predicted NatB substrate, Pnc1 may be indirectly regulated by NatB. Our results showed that Pnc1 activity (Fig. 2B) and protein expression (Fig. 2A) were only slightly decreased in nat3Δ cells. Interestingly, although pnc1Δ cells show WT level of NAD+ under normal (NA-rich) growth conditions (19), the levels of NAD+ were significantly reduced in NatB mutants (Fig. 2C). These studies suggest that additional factors probably contribute to the altered NAM/NAD+ homeostasis observed in NatB mutants. Increased production of NR has been associated with increased activity of the Pi-sensing PHO signaling pathway (17). We therefore determined whether NatB mutants showed altered PHO activity by measuring the activity of rAPase (Pho5, a periplasmic phosphatase activated by PHO signaling). As shown in Fig. 2D (left), PHO activity was moderately increased in the nat3Δ mutant. As controls, deleting PHO5 largely blocked the increase of rATPase activity. The residual rATPase activity observed in nat3Δpho5Δ cells probably came from Pho5-like phosphatases Pho11 and Pho12. Interestingly, observed PHO activation in nat3Δ cells appeared independent of the conventional PHO transcription factor Pho4-Pho2 complex, because deleting PHO4 did not significantly reduce the PHO activation in nat3Δ cells (Fig. 2D, left). Consistent with this result, deleting PHO4 did not prevent NA/NAM release in nat3Δ cells (Fig. 2D, right panel). Next, we determined whether and how NR contributes to increased NAM in NatB mutants. Intracellular NAM may come from NR via the reactions of cytoplasmic nucleosidases Urh1 and Pnp1 (Fig. 2E, top) (30). As shown in Fig. 2E (bottom), deleting URH1 and PNP1 largely diminished NA/NAM release in NatB mutants. Moreover, deleting the vacuolar phosphatase PHO8 (Fig. 2E, top) (17) diminished NA/NAM release (Fig. 2E, bottom), whereas deleting the cytoplasmic nucleotidases ISN1 and SDT1 (Fig. 2E, top) had a lesser effect (Fig. 2E, bottom). These results suggest that increased NAM in NatB mutants probably results from vacuolar NR, which is converted to NAM by cytoplasmic nucleosidases Urh1 and Pnp1. Vacuolar NR may exit the vacuole through the Fun26 transporter (human lysosomal hENT homolog) into the cytoplasm (17), where it is converted to NAM by Urh1 and Pnp1 (Fig. 2E, top). Supporting this, deleting FUN26 indeed blocked NAM release in the NatB mutant (Fig. 2E, bottom). Moreover, nat3Δ cells carrying deletions of URH1 and PNP1 (which converts NR to NAM) showed increased intracellular NR levels (Fig. 2F, middle left) and reduced NA/NAM levels (Fig. 2F, middle right). Deleting PHO8 (which produces NR in the vacuole) blocked the increase of both NR and NA/NAM in nat3Δ cells (Fig. 2F, bottom panels). All of these results support vacuolar NR as the main source of increased NA/NAM in nat3Δ cells.
Figure 2.

Characterizations of NAD+ homeostasis factors in NatB mutants. A, Pnc1 protein levels in WT and nat3Δ cells. Pnc1 level is slightly decreased in nat3Δ mutant during late log phase (13 h). B, relative Pnc1 deamidase activities determined in WT and nat3Δ cell lysates. Pnc1 activity is slightly decreased in nat3Δ cells during late log phase (13 h). C, measurements of intracellular NAD+ levels. Deleting NAT3 significantly decreases NAD+ levels. D, relative repressible acid phosphatase (rAPase) activities in WT and nat3Δ cells. Increased rAPase activity in nat3Δ cells indicates increased PHO signaling activity (left). Deleting PHO4 does not abolish rAPase activity, whereas deleting PHO5 largely diminishes the rAPase activity (left). Deleting PHO4 does not block NA/NAM release in nat3Δ cells (right). Data shown are representative of three independent experiments. E, NR salvage contributes to increased NA/NAM production in NatB mutants. Intracellular compartmentalization of NR salvage factors (top). Deleting cytosolic nucleosidases URH1 and PNP1 (which convert NR to NAM) diminishes NA/NAM release in nat3Δ and mdm20Δ cells (bottom). Deleting vacuolar NR-producing factor Pho8 also diminishes NA/NAM release, whereas deleting the cytosolic NR-producing factors Isn1 and Sdt1 has a lesser effect (bottom). Deleting vacuolar transporter FUN26 also abolishes NA/NAM release in nat3Δ cells. This suggests that increased NAM in NatB mutants comes from vacuolar NR, which is converted to NAM by Urh1 and Pnp1 (top). F, intracellular NR (top left) and NA/NAM (top right) levels are increased in nat3Δ mutant. Deleting URH1 and PNP1 diminishes NA/NAM production (middle right) with a commitment increase of intracellular NR level (middle left). Deleting PHO8 diminishes both NR and NA/NAM levels (bottom panels). For B, C, D, and F, the error bars denote S.D. values of triplicated samples. The p values are calculated using Student's t test (*, p < 0.05).
Blocking NA/NAM production and release is not sufficient to restore NAD+ levels in the NatB mutants
So far, our results suggest that in NatB mutants, increased NA/NAM release is probably due to increased NAM production, which originates from vacuolar NR. It remains unclear how NAD+ and its intermediates are transported into the vacuole for turnover and storage. Because most cellular NAD+ and NADH are bound to proteins, NAD+ intermediates may enter the vacuole via vesicular trafficking and/or autophagy. Because autophagy is a critical regulator of organelle homeostasis and recycling of macromolecules (31, 32), we therefore asked whether inducing autophagy in yeast cells would indeed increase intracellular NR and NA/NAM levels. As shown in Fig. 3A, both NR (left) and NA/NAM (right) levels were increased by nitrogen starvation, a condition known to induce autophagy (31, 33, 34). These increases were largely dependent on Atg14, a key regulator of autophagic membrane tethering and fusion (31). These data suggest that autophagy may contribute to increased NA/NAM in NatB mutants. Supporting this, we showed that deleting ATG14 largely blocked NA/NAM release in the nat3Δ mutant (Fig. 3B). Next, we tested whether deleting ATG14 was sufficient to restore NAD+ levels in the NatB mutants. As shown in Fig. 3C (left), deleting ATG14 did not rescue the low NAD+ levels in nat3Δ cells. Similarly, deleting other factors contributing to NR and NAM production (Figs. 1A and 2E) in nat3Δ cells also failed to restore the NAD+ level back to wildtype (Fig. 3C, right). Most reported NatB mutant phenotypes were attributed to defective N-terminal acetylation of tropomyosins (Tpm1 and Tpm2 in yeast) (23, 28, 29). Tropomyosins are actin filament–binding proteins that stabilize actin cables (29, 35–38). Overexpression of TPM1 (TPM1-oe) and expression of a gain-of-function allele of TPM1 (TPM1-5) were shown to restore actin cable formation in NatB mutants (23, 24, 28, 29). Interestingly, both TPM1-oe and TPM1-5 largely blocked NA/NAM release in the nat3Δ mutant (Fig. 3D), suggesting a role for the actin cytoskeleton in NAM homeostasis regulation. However, TPM1-oe and TPM1-5 still failed to restore the NAD+ levels in nat3Δ cells (Fig. 3E). These studies indicate that the low NAD+ phenotype of nat3Δ cells cannot be rescued simply by decreasing NAM production and/or release and that additional factors are probably involved.
Figure 3.

Blocking NA/NAM production or release is not sufficient to restore NAD+ levels in the NatB mutants. A, nitrogen starvation (N-starv) increases NR (left) and NA/NAM (right) production, which are diminished by deleting ATG14, an autophagy-specific phosphatidylinositol 3-kinase subunit. B, increased NA/NAM levels in nat3Δ cells are diminished by deleting ATG14. C, blocking NA/NAM production by deleting ATG14, FUN26, PHO8, URH1, and PNP1 is not sufficient to restore NAD+ levels in nat3Δ cells. D, TPM1 overexpression (TPM1-oe) and expression of a gain-of-function allele of TPM1 (TPM1-5) diminish NA/NAM release in nat3Δ cells. E, TPM1-oe and expression of TPM1-5 are not sufficient to restore NAD+ levels in nat3Δ cells. Data shown are representative of three independent experiments. For A, C, and E, the error bars denote S.D. values of triplicated samples. The p values are calculated using Student's t test (*, p < 0.05).
Nma1 and Nma2 are responsible for the low NAD+ levels in NatB mutants
To gain further insight, we tested whether supplementing particular NAD+ precursors to nat3Δ cells can restore the NAD+ levels. Interestingly, all NAD+ precursors tested (NA, NAM, NR, and QA) failed to restore NAD+ levels (Fig. 4A). This suggests that nat3Δ cells may be defective in a step of NAD+ biosynthesis that is common to the utilization of QA, NA, and NR. Nma1 and Nma2 are dual functional nicotinamide mononucleotide adenylyltransferases (NMNATs), which utilize both NMN and nicotinic acid mononucleotide (NaMN) as substrates and are required for all three NAD+ biosynthesis pathways (Fig. 1A). N-terminal acetyltransferases have specific targets that are largely determined by the sequence of the first two amino acids. NatB acetylates proteins with a Met-retained residue, followed by Asp, Glu, Gln, or Asn as the second residue (25, 26, 39, 40). Nma1 and Nma2 have a Met-Asp N terminus, but neither protein has been identified as a NatB target. Similar to nat3Δ mutant, nma1Δ cells showed decreased NAD+ levels, which could not be rescued by supplementing NR (Fig. 4B). As for the controls, NR efficiently restored the NAD+ level in the npt1Δ mutant, which has functional NR salvage (Fig. 4B). The similarities between nma1Δ and nat3Δ mutants suggested that Nma1 and Nma2 activities are probably reduced in nat3Δ cells. Because N-terminal acetylation may affect the turnover of target proteins, we first determined whether reduced Nma1/Nma2 activities were due to decreased Nma1/Nma2 protein levels. As shown in Fig. 4C, Nma1 and Nma2 protein levels were indeed decreased in nat3Δ cells (Fig. 4C). In addition to Nma1 and Nma2, NAD+ homeostasis factors Bna2, Bna5, and Hst1 are also potential NatB targets. Bna2 and Bna5 are biosynthesis enzymes in the de novo pathway (Fig. 1A), and Hst1 represses the expression of BNA genes (13). Because blocking the de novo pathway does not affect NAD+ levels under standard growth conditions (NA-rich media), the low NAD+ phenotype of nat3Δ mutant is unlikely to be due to reduced de novo activities. To determine de novo activities in nat3Δ cells, we developed a cell-based readout for QA production. In this assay, more QA release indicates increased de novo activities, and vice versa. As expected, hst1Δ released more QA (Fig. 4D, right) due to derepression of BNA genes, and deletions of BNA genes completely blocked QA release (data not shown). Similar to hst1Δ mutant, nat3Δ mutant also released more QA, and double deletions of NAT3 and HST1 did not further increase QA release (Fig. 4D). In comparison, hst1Δ cells did not release more NA/NAM (Fig. 4D, left), indicating that increased NA/NAM in the nat3Δ mutant is not simply due to increased de novo activities. This is in line with our findings that in nat3Δ cells, increased NA/NAM arises from NR (Fig. 2E). Overall, these results demonstrate that the low NAD+ phenotype of nat3Δ cells is mainly due to reduced protein levels of Nma1 and Nma2. To confirm this, we examined whether overexpression of NMA1 is sufficient to reduce NA/NAM release. As shown in Fig. 4E (left), NMA1-oe completely blocked NA/NAM release in nat3Δ cells. Moreover, NMA1-oe also reduced QA release in nat3Δ cells (Fig. 4E, right). The reduction in QA and NA/NAM release is probably due to more efficient NA/NAM and QA assimilation by NMA1-oe. Supporting this, NMA1-oe was sufficient to restore the NAD+ levels in nat3Δ cells back to WT levels (Fig. 4F). Interestingly, NMA1-oe also increased NAD+ levels in WT cells. Next, we examined whether Nma1 and Nma2 are N-terminal acetylated by NatB. Immunoprecipitated Nma1-HA protein complexes obtained from WT and nat3Δ cells were treated with chymotrypsin and then analyzed by tandem mass spectrometry. Both Nma1 and Nma2 N-terminal peptides were identified in WT and nat3Δ cells. As shown in Fig. 4G, among identified specific Nma1 and Nma2 N-terminal peptides from WT cells, all of them (66 for Nma1, 25 for Nma2) were acetylated on the first methionine. Conversely, of the identified Nma1 and Nma2 N-terminal peptides from nat3Δ cells (20 for Nma1, 10 for Nma2), only one Nma1 peptide showed acetylation (Fig. 4G). These results indicate that Nma1 and Nma2 are indeed N-terminally acetylated in a NatB-dependent manner.
Figure 4.

NMNATs are responsible for the NAD+ decrease in NatB mutants. A, supplementing NAD+ precursors (NA, NAM, NR, and QA) does not restore NAD+ levels in the nat3Δ mutants. B, nat3Δ and nma1Δ mutants share similar low NAD+ levels, which cannot be rescued by NR supplementation. As a control, NR is able to rescue the low NAD+ level in npt1Δ cells. C, the levels of NMNATs proteins (Nma1 and Nma2) are decreased in nat3Δ cells. D, both nat3Δ and hst1Δ cells show increased QA release, which is indicative of increased de novo activities (right). Unlike nat3Δ cells, hst1Δ cells do not show increased NA/NAM release (left). These findings suggest that increased NA/NAM in nat3Δ cells is not simply due to increased de novo activity. The npt1Δnrk1Δbna4Δ mutant was used as the recipient cells for determining QA release (QA-dependent growth). E, NMA1 overexpression (Nma1-oe) diminishes both NA/NAM and QA release in nat3Δ cells. F, NMA1-oe restores the NAD+ levels in nat3Δ cells. Data shown are representative of three independent experiments. G, among specific Nma1 and Nma2 N-terminal peptides identified from WT cells, 100% are acetylated at the first methionine. Acetylations are mostly absent among specific Nma1 and Nma2 N-terminal peptides identified in nat3Δ mutant, indicating that Nma1 and Nma2 are N-terminally acetylated in a NAT3 (NatB)-dependent manner. For A, B, F, and G, the error bars denote S.D. values of duplicated samples. The p values are calculated using Student's t test (*, p < 0.05).
Discussion
In this study, we characterized the NatB complex as a novel NAD+ homeostasis factor. Mutants lacking components of the NatB complex, NAT3 and MDM20, produce and release an excess amount of NA and NAM. Our studies showed that two pathways downstream of NatB contribute to NAD+ homeostasis (Fig. 5). In one, NatB is important for proper NAD+ biosynthesis by regulating Nma1/Nma2. NatB mutants have low NAD+ levels (Fig. 2C), and all NAD+ precursors examined failed to restore the NAD+ levels (Fig. 4, A and B). This suggests that a NAD+ biosynthesis factor(s) required for utilization of all NAD+ precursors is defected in NatB mutants. Nma1 and Nma2 are such targets because they are the only factors required for all three NAD+ biosynthesis pathways (de novo, NA/NAM salvage, and NR salvage) (Fig. 1A), and the N-terminal amino acid sequences are a match for NatB acetylation. In addition, nma1Δ and nat3Δ mutants showed similar NAD+ utilization defects (Fig. 4B). Although Nma2 is present in both mutants, Nma2 is known to play a minor role in NAD+ metabolism. Supporting this model, decreased Nma1 protein level was observed in the nat3Δ mutant (Fig. 4C), and overexpressing NMA1 was sufficient to restore the NAD+ levels (Fig. 4F). Our studies showed that Nma1 and Nma2 are indeed acetylated at the N terminus in a NatB-dependent manner (Fig. 4G). Further studies are required to understand the mechanisms of NatB-mediated regulation of Nma1/Nma2, but the decreased level of both proteins in nat3Δ suggests that the nonacetylated counterparts may be degraded at a higher rate by the N-end rule pathway. The N-end rule pathway recognizes N-terminal degradation signals and marks proteins for degradation by specific E3 ligases. This pathway can be broken up into three branches called the Arg/N-rule pathway, the Pro/N-rule pathway, and the Ac/N-rule pathway (42, 43). The Ac/N-rule branch recognizes N-acetylated proteins. However, in the absence of acetylation, the proteins may be stabilized or targeted by other N-end rule branches, which can lead to different rates of degradation (42, 44, 45). On the other hand, N-acetylated proteins may also bury their acetylated N termini within physically associated proteins (46–48). This provides two important functions. First, it shields the N terminus from Ac/N-rule E3 ligases. Second, it stabilizes the protein complex. The absence of a normally present acetylated N terminus can drastically decrease the affinity of a protein to complex with its partners (24, 47), leaving it more dissociated and prone to targeted degradation (42). Therefore, it is possible that in addition to protein stabilization, N-terminal acetylation may also be important for Nma1 and Nma2 protein-protein interactions. Current research suggests that Nma1 and Nma2 may function as tetramers (49–51).
Figure 5.
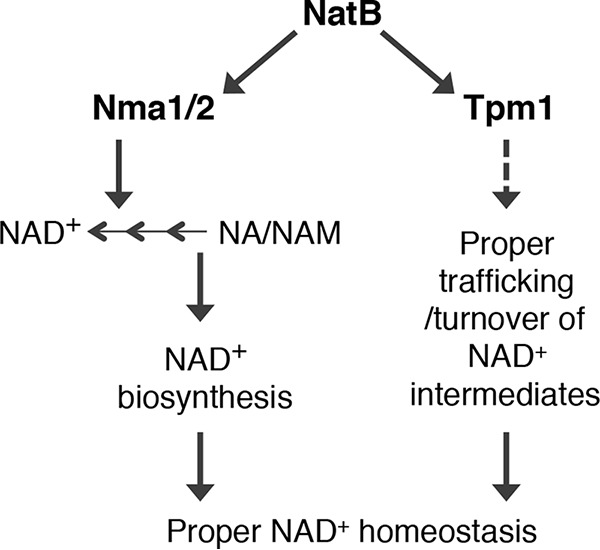
A proposed model depicting factors contributing to altered NAD+ homeostasis in NatB mutants. We identified Nma1 and Nma2 as the acetylation targets of NatB. NatB regulates NAD+ biosynthesis and NAD+ intermediate trafficking via Nma1/Nma2 and Tpm1, respectively. Dashed line, the mechanism of this pathway remains largely unclear.
In the other pathway, NatB- and Tpm1-mediated proper vesicular trafficking appears to specifically affect NA/NAM homeostasis (Fig. 5). TPM1 encodes one of two tropomyosin proteins in yeast, which bind to actin filaments and stabilize actin cables. Acetylation of TPM1 by NatB greatly improves its affinity for actin filaments (24). During cell division and growth, actin cables provide tracks for organelle segregation and endocytic trafficking (35, 38, 52, 53). Interestingly, although overexpression of TPM1 and expression of a dominant TPM1-5 completely blocked NA/NAM release (Fig. 3D), they failed to restore the NAD+ levels in the NatB mutants. This is in line with the model that Nma1 and Nma2 abnormalities are responsible for observed NAD+ defects in NatB mutants. NMA1 overexpression, however, was sufficient to block NA/NAM release (Fig. 4E) and restore NAD+ level in NatB mutants (Fig. 4F). These results show that the two NatB-controlled NAD+ homeostasis pathways are interconnected. How may TPM1 and actin cytoskeleton affect NAD+ homeostasis? It is possible that NAD+ and related precursors (free and protein-bound) are trafficked by means of vesicles, which would require Tpm1 and actin cables to mediate their directed movement (35, 37). Deleting TPM1 alone abolishes the appearance of actin cables, whereas deleting TPM2 does not (36). Neither tpm1Δ nor tpm2Δ single mutant (tpm1Δtpm2Δ double mutant is lethal) appeared in our screen for high NA/NAM release mutants. It is probably because Tpm1 and Tpm2 have some functional redundancy (36, 54), and they can substitute for each other when one is absent. During asymmetrical cell division, targeted and increased vesicular transport to daughter may concomitantly release NA/NAM and other NAD+ intermediates. It is possible that unacetylated Tpm1 fails to efficiently assist the formation and delivery of endocytic vesicles, leading to increased NA/NAM release observed in the NatB mutants.
Our studies showed that increased NA/NAM in NatB mutants came from the NR pool (Fig. 2E). Increased NA/NAM release in NatB mutants was abolished by deleting cytosolic nucleosidases (URH1/PNP1), which convert NR to NAM. Increased NR mostly originates from the vacuolar pool because deleting vacuolar NR-producing phosphatase (PHO8) abolished NA/NAM increase, whereas deleting cytosolic NR-producing nucleotidases (ISN1/SDT1) had a lesser effect (Fig. 2E, bottom). Vacuolar NR may exit the vacuole through Fun26 transporter (human lysosomal hENT homolog) and enter the cytoplasm (17), where it is converted to NAM by nucleosidases. Indeed, deleting FUN26 blocked NAM release in the NatB mutant (Fig. 2E, bottom). It remains unclear how NAD+ and its intermediates are transported into the vacuole. Our studies suggest that autophagy may also play a role. Autophagy is a critical regulator of organelle homeostasis and recycling of macromolecules (55). It has been reported that NatB mutants failed to properly segregate mitochondria during cell division (mitochondrial inheritance defects) (23, 24, 28), which may lead to increased mitochondrial turnover by autophagy. Supporting this, many mutants we identified in our screen affect mitochondrial function (Table S1). In addition, we showed that intracellular levels of both NA/NAM and NR were increased by starvation conditions known to induce autophagy (Fig. 3A). Finally, deleting ATG14, a key regulator of autophagic membrane tethering and fusion (56), blocked NAM release in the nat3Δ mutant (Fig. 3B). Although TPM1-oe shared similar phenotypes with ATG14 deletion, it is unclear whether Tpm1-mediated vesicular trafficking and autophagy function in the same pathway to regulate NA/NAM homeostasis. Further evidence is required to detail the functional link between these factors.
It remains unclear why NatB mutants release more NA/NAM and not NR. Our studies show that the NR pools in NatB mutants are quickly converted to NAM by cytoplasmic Urh1 and Pnp1 (Fig. 2E). In line with this, the NatB mutants were previously identified in a different screen as “low-NR” mutants (17). Although Urh1 and Pnp1 are not predicted direct targets of NatB (23, 25, 26), their activities and localization could be indirectly affected by NatB mutations. Some NatB mutant phenotypes may also be due to aberrant energy homeostasis (57). Supporting this, the Glc7 phosphatase, which inhibits Snf1 (yeast AMPK) activity, is acetylated by NatB, and Snf1 activation was observed in nat3Δ cells (57). In addition, Snf1 activation was associated with Pnp1 activity in starvation-induced ribonucleotide salvage (58). Another NAM metabolic factor that may contribute to increased NA/NAM release is Pnc1. Although Pnc1 levels and activities are only slightly decreased in NatB mutants (Fig. 2, A and B), its localization may be altered in NatB mutants. Pnc1 has been shown to localize in peroxisomes in normal conditions. In response to nutrient starvation and stress, Pnc1 expression is induced and becomes ubiquitous (15, 59). In addition, Pnc1 has been shown to be released extracellularly (60), and it is most likely retained in the periplasmic space (61). It is possible that during asymmetrical cell division, increased vesicular transport to daughter may concomitantly increase NAM release. Pnc1 may be released to convert NAM to NA to support NAD+ biosynthesis and/or eliminate NAM toxicity (62–64). In mammals, eNAMPT is also released extracellularly to help metabolize NAM (65); however, yeast does not have NAMPT.
In summary, our studies have uncovered novel NAD+ homeostasis factors. It would be informative to further investigate the regulation of NAD+ homeostasis by NatB in future studies. These studies may also provide insights into the regulation of NAD+ metabolism and the molecular basis of disorders associated with aberrant NAD+ metabolism.
Experimental procedures
Yeast strains, growth media, and plasmids
Yeast strain BY4742 MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 acquired from Open Biosystems (66) was used for this study. Standard media, such as yeast extract-peptone-dextrose (YPD), synthetic minimal (SD), and synthetic complete (SC) media, were made as described (67). Niacin-free SD and SC were made by using niacin-free yeast nitrogen base (Sunrise Science Products). Niacin (vitamin B3) comprises both NA and NAM. All gene deletions were generated by replacing wildtype genes with reusable loxP-Kanr-loxP cassettes as described (68). Multiple deletions were carried out by removing the Kanr marker using a galactose-inducible Cre recombinase (68). PNC1, NMA1, and NMA2 were tagged by the HA or EGFP epitope tag in the genome using the pFA6a-3HA-HIS3MX or pFA6a-link-yoEGFP-CaURA3 plasmid as described previously (69, 70). The NMA1 and TPM1 overexpression constructs, pADH1-NMA1 and pADH1-TPM1, were made in the integrative pPP81 (LEU2) vector as described (71). The resulting constructs were verified by DNA sequencing. TPM1-5 in pRS316 was a gift from Dr. B. Polevoda. TPM1-5 encodes an extended 7 amino acids at the N terminus of TPM1 (23).
NA/NAM cross-feeding spot assays
The bna6Δnrk1Δnrt1Δ mutant (which utilizes both NA and NAM) and bna6Δnrk1Δnrt1Δpnc1Δ mutant (which only utilizes NA) were used as “recipient cells,” and yeast strains of interest were used as “feeder cells.” First, recipient cells were plated onto niacin-free SD (NA/NAM-free) as a lawn (∼104 cells/cm2). Next, 2 μl of each feeder cell strain (∼104 cells; cell suspension was made in sterile water at A600 of 2) was spotted onto the lawn of recipient cells. Plates were then incubated at 30 °C for 3 days. The extent of the recipient cell growth indicates the levels of NA/NAM released by feeder cells.
Genetic screen using the yeast deletion collection
The haploid yeast deletion collection (∼4500 strains) established in the BY4742 background was acquired from Open Biosystems (72). To screen for mutants with increased NA/NAM release, 2 μl of each strain was directly taken from the frozen stock and then spotted onto niacin-free SD plates spread with the bna6Δnrk1Δnrt1Δ recipient cells at a density of ∼104 cells/cm2. After incubation at 30 °C for 3 days, we scored the cross-feeding activity (which indicates the level of NA/NAM release) of each mutant by comparing the diameter of the cross-feeding zones with that of the wildtype. Mutants were assigned a score of 2–5 (WT = 1). A higher score represents more NA/NAM release. To eliminate false-positives, mutants with a score of ≥3 (81 mutants) were reexamined. Cells from the frozen stock were first recovered in fresh media for 2 days. 104 cells of each strain were then spotted and scored as described above. Both the bna6Δnrk1Δnrt1Δ mutant (which utilizes both NA and NAM) and bna6Δnrk1Δnrt1Δpnc1Δ mutant (which only utilizes NA) were used as “recipient cells.” A total of 63 mutants passed the secondary screen, and interestingly, most mutants release more NAM. Known NAD+ homeostasis factors were identified, including npt1Δ and pnc1Δ (Table S1).
Measurement(s) of NAD+, NADH, NR, NA, and NAM
Total intracellular levels of NAD+ and NADH were determined using enzymatic cycling reactions as described (71). NR levels were determined by a liquid-based cross-feeding bioassay (17, 19). Relative NA/NAM levels were determined similarly as NR with some modifications. To prepare cell extracts for intracellular NA/NAM determination, ∼2.5 × 109 (∼250 A600 units) of cells (donors of interest) grown to late log phase (∼16-h growth from an A600 of 0.1) were lysed by bead-beating (Biospec Products) in 800 μl of ice-cold 50 mm ammonium acetate solution. After filter sterilization, 50–200 μl of clear extract was used to supplement 8-ml cultures of recipient cells (the NA/NAM-dependent bna6Δnrk1Δnrt1Δ mutant) with starting A600 of 0.05 in niacin-free SC. To determine extracellular NA/NAM levels, supernatant of donor cell culture was collected and filter-sterilized, and then 4 ml was added to 4 ml of recipient cell culture with total starting A600 of 0.05 in 2× niacin-free SC. A control culture of recipient cells in niacin-free SC without supplementation was included in all experiments. After incubation at 30 °C for 24 h, growth of the recipient cells (A600) was measured and normalized to the cell number of each donor strain. A600 readings were then converted to NA/NAM concentrations using the standard curve established as described previously (19).
Deamidase activity assay
About 300 A600 unit cells grown overnight in YPD were harvested, and cell lysate was obtained by beads beating in breaking buffer (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, Roche protease inhibitors). Cell lysate containing 70 μg of total cellular proteins was added to 300 μl of deamidase reaction mix (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm MgCl2) (15, 73) using 8 mm NAD+, NMN, NmR, or Nam as substrates, followed by incubation at 30 °C for 45 min. 100 μl of the deamidase reaction mix was then added to 1 ml of ammonia assay mix (3.4 mm α-ketoglutarate, 0.23 mm NADPH, 50 mm phosphate buffer, pH 7.4, 10 units of glutamate dehydrogenase) followed by reaction at room temperature for 15 min (15, 73). The amount of ammonia was calculated by the decrease in absorbance at 340 nm using standard curve derived from the ammonia standard solutions (Sigma).
Repressible acid phosphatase (rAPase) activity assay
The rAPase liquid assay was performed as described (74) on cells grown to late log phase in SDC. In brief, ∼2.5 × 107 cells were harvested, washed, and resuspended in 150 μl of water. 600 μl of substrate solution (5.6 mg/ml p-nitrophenylphosphate in 0.1 m sodium acetate, pH 4) was added to cell suspension, and the mixture was incubated at 30 °C for 15 min. The reaction was stopped by adding 600 μl of ice-cold 10% trichloroacetic acid. 600 μl of this final mixture was then added to 600 μl of saturated Na2CO3 to allow color (neon yellow) development. Cells were pelleted to acquire the supernatant for A420 determination. The rAPase activities were determined by normalizing A420 readings to total cell number (A600).
Nitrogen starvation
We followed previous nitrogen starvation regimens that were shown to induce autophagy in yeast cells with some modifications (31, 33, 34). 8 ml of cells grown in SC overnight were harvested by centrifugation, washed twice before inoculating into 250 ml of SC −niacin at A600 of 0.2. SC −niacin was used to reduce the interference of extracellular niacin (NA/NAM) while determining intracellular NA/NAM. After 6 h at 30 °C, 2 × 100 ml of cells were collected and washed twice in water. One tube of washed cells were subject to lysate collection (control, 0 h) for NA/NAM and NR measurements. The other tube of washed cells were inoculated into 250 ml of SC −niacin −nitrogen (without ammonium sulfate and amino acids) and allowed to grow for 4 h (N-starvation, 4 h) before lysate collection for NA/NAM and NR measurements. SC −niacin and SC −niacin −nitrogen were made with special yeast nitrogen base from Sunrise Science Products.
Protein extraction and Western blot analysis
A total of 100 ml of cells were grown in SC or SD to mid-logarithmic phase (A600 of 1–2), concentrated by centrifugation. Total protein extract from cell lysate was obtained by bead-beating in lysis buffer (50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 1% Nonidet P-40, and Roche cOmplete Mini protease inhibitor mixture). 30 or 70 μg of total protein was loaded in each lane. After electrophoresis, the protein was transferred to nitrocellulose membrane (Whatman). The membranes were then washed and blotted with either anti-HA antibody (Pierce) or anti-actin antibody (Abcam). Protein was visualized using anti-mouse (GE Healthcare) or rabbit (Pierce) antibody conjugate to the horseradish peroxidase and the ECL reagents (GE Healthcare). The chemiluminescent image was analyzed using the Alpha Innotech imaging system and software provided by the manufacturer.
Immunoprecipitation and analysis of N-terminal acetylation of Nma1 and Nma2
About 500 ml of WT and nat3Δ cells carrying HA-tagged Nma1 grown to A600 of 1–2 were collected by centrifugation and washed twice in PBS. Cell lysate was obtained by bead-beating in immunoprecipitation lysis buffer (25 mm Tris-HCl, 150 mm NaCl, 1% Nonidet P-40, 3 mm EDTA, 5% glycerol, and protease inhibitor mixture). Lysate was cleared at 100,000 × g for 1.5 h. Immunoprecipitation was carried out using 1.8 ml of lysate at 2.8 mg/ml with 75 μl of PierceTM anti-HA magnetic beads. The magnetic beads were washed five times with end-over-end rotation: wash 1 (20 min) in lysis buffer, washes 2–4 (15 min) in TBS with 0.5% Tween 20, and wash 5 with PBS. The magnetic beads were then washed three more times in 300 μl of 50 mm ammonium bicarbonate with end-over-end rotation for 20 min. The magnetic beads were then brought up in 300 μl of 50 mm ammonium bicarbonate for on-bead digestion, and 2.5 μl of 250 ng/μl PierceTM chymotrypsin protease (catalog no. 90056) was added. Digest was left at room temperature overnight with end-over-end rotation. The next day, the supernatant was separated from the magnetic beads and collected using a magnetic stand. The magnetic beads were washed with 100 μl of 50 mm ammonium bicarbonate for 20 min, and the supernatant was pooled. Samples were immediately sent to the University of California Davis proteomics core for tandem mass spectrometry analysis (LC-MS/MS). Digested peptides were analyzed by LC-MS/MS on a Thermo Scientific Q Exactive Plus Orbitrap mass spectrometer in conjunction with Proxeon Easy-nLC II HPLC (Thermo Scientific) and Proxeon nanospray source. The digested peptides were loaded into a 100-μm × 25 mm Magic C18 100-Å 5U reverse phase trap, where they were desalted online before being separated using a 75 μm × 150 mm Magic C18 200-Å 3U reverse phase column. Peptides were eluted using a 60-min gradient with a flow rate of 300 nl/min. An MS survey scan was obtained for the m/z range 350–1600, and MS/MS spectra were acquired using a top 15 method, where the top 15 ions in the MS spectra were subjected to high-energy collisional dissociation. An isolation mass window of 1.6 m/z was used for the precursor ion selection, and a normalized collision energy of 27% was used for fragmentation. A 15-s duration was used for the dynamic exclusion. The total number of identified Nma1 and Nma1-specific peptide sequences (peptide spectrum matches) were determined using Byonic software (75) with 1% maximum spectra decoy false-discovery rate. The Skyline software (41) was used to calculate the peak area intensity of the acetylated N-terminal peptides identified by Byonic. The percentages of N-terminal acetylated Nma1 and Nma2 peptides shown in Fig. 4G were calculated from two independent experiments using the Scaffold software with peptide threshold set at 1% false-discovery rate.
Author contributions
T.C. designed and performed the experiments. C.J.T.R. performed experiments shown in Figs. 2 (A and B) and 4 (D and E). M.S. and B.S.P. performed the mass spectrometry studies and data analysis shown in Fig. 4G. S.J.L. helped conceive the experiments, analyzed the data, and co-wrote the manuscript with T.C. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. B. Polevoda for sharing the TPM1-5 plasmid, Dr. P. Venkatakrishnan for critical reading of this manuscript, and Dr. J. Roth and Dr. S. Collins for suggestions and discussions.
This study was supported by NIGMS, National Institutes of Health, Grant GM102297. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1.
- NMN
- nicotinamide mononucleotide
- NA
- nicotinic acid
- NAM
- nicotinamide
- QA
- quinolinic acid
- NR
- nicotinamide riboside
- NAT
- N-terminal acetyltransferase
- NMNAT
- nicotinamide mononucleotide adenylyltransferase
- rAPase
- repressible acid phosphatase
- oe
- overexpression
- EGFP
- enhanced green fluorescent protein.
References
- 1. Kato M., and Lin S. J. (2014) Regulation of NAD+ metabolism, signaling and compartmentalization in the yeast Saccharomyces cerevisiae. DNA Repair 23, 49–58 10.1016/j.dnarep.2014.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nikiforov A., Kulikova V., and Ziegler M. (2015) The human NAD metabolome: functions, metabolism and compartmentalization. Crit. Rev. Biochem. Mol. Biol. 50, 284–297 10.3109/10409238.2015.1028612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chini C. C., Tarragó M. G., and Chini E. N. (2017) NAD and the aging process: role in life, death and everything in between. Mol. Cell. Endocrinol. 455, 62–74 10.1016/j.mce.2016.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Imai S., and Guarente L. (2014) NAD+ and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471 10.1016/j.tcb.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Garten A., Schuster S., Penke M., Gorski T., de Giorgis T., and Kiess W. (2015) Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 11, 535–546 10.1038/nrendo.2015.117 [DOI] [PubMed] [Google Scholar]
- 6. Verdin E. (2015) NAD+ in aging, metabolism, and neurodegeneration. Science 350, 1208–1213 10.1126/science.aac4854 [DOI] [PubMed] [Google Scholar]
- 7. Cantó C., Menzies K. J., and Auwerx J. (2015) NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 22, 31–53 10.1016/j.cmet.2015.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang Y., and Sauve A. A. (2016) NAD+ metabolism: bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 1864, 1787–1800 10.1016/j.bbapap.2016.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown K. D., Maqsood S., Huang J. Y., Pan Y., Harkcom W., Li W., Sauve A., Verdin E., and Jaffrey S. R. (2014) Activation of SIRT3 by the NAD+ precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 20, 1059–1068 10.1016/j.cmet.2014.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Williams P. A., Harder J. M., Foxworth N. E., Cochran K. E., Philip V. M., Porciatti V., Smithies O., and John S. W. (2017) Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 355, 756–760 10.1126/science.aal0092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin J. B., Kubota S., Ban N., Yoshida M., Santeford A., Sene A., Nakamura R., Zapata N., Kubota M., Tsubota K., Yoshino J., Imai S. I., and Apte R. S. (2016) NAMPT-mediated NAD+ biosynthesis is essential for vision in mice. Cell Rep. 17, 69–85 10.1016/j.celrep.2016.08.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Belenky P., Racette F. G., Bogan K. L., McClure J. M., Smith J. S., and Brenner C. (2007) Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 129, 473–484 10.1016/j.cell.2007.03.024 [DOI] [PubMed] [Google Scholar]
- 13. Bedalov A., Hirao M., Posakony J., Nelson M., and Simon J. A. (2003) NAD+-dependent deacetylase Hst1p controls biosynthesis and cellular NAD+ levels in Saccharomyces cerevisiae. Mol. Cell Biol. 23, 7044–7054 10.1128/MCB.23.19.7044-7054.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Medvedik O., Lamming D. W., Kim K. D., and Sinclair D. A. (2007) MSN2 and MSN4 link calorie restriction and TOR to sirtuin-mediated lifespan extension in Saccharomyces cerevisiae. PLoS Biol. 5, e261 10.1371/journal.pbio.0050261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anderson R. M., Bitterman K. J., Wood J. G., Medvedik O., and Sinclair D. A. (2003) Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423, 181–185 10.1038/nature01578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gallo C. M., Smith D. L. Jr., and Smith J. S. (2004) Nicotinamide clearance by Pnc1 directly regulates Sir2-mediated silencing and longevity. Mol. Cell Biol. 24, 1301–1312 10.1128/MCB.24.3.1301-1312.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lu S. P., and Lin S. J. (2011) Phosphate-responsive signaling pathway is a novel component of NAD+ metabolism in Saccharomyces cerevisiae. J. Biol. Chem. 286, 14271–14281 10.1074/jbc.M110.217885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bieganowski P., Seidle H. F., Wojcik M., and Brenner C. (2006) Synthetic lethal and biochemical analyses of NAD and NADH kinases in Saccharomyces cerevisiae establish separation of cellular functions. J. Biol. Chem. 281, 22439–22445 10.1074/jbc.M513919200 [DOI] [PubMed] [Google Scholar]
- 19. Lu S. P., Kato M., and Lin S. J. (2009) Assimilation of endogenous nicotinamide riboside is essential for calorie restriction-mediated life span extension in Saccharomyces cerevisiae. J. Biol. Chem. 284, 17110–17119 10.1074/jbc.M109.004010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kulikova V., Shabalin K., Nerinovski K., Dölle C., Niere M., Yakimov A., Redpath P., Khodorkovskiy M., Migaud M. E., Ziegler M., and Nikiforov A. (2015) Generation, release, and uptake of the NAD precursor nicotinic acid riboside by human cells. J. Biol. Chem. 290, 27124–27137 10.1074/jbc.M115.664458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsang F., James C., Kato M., Myers V., Ilyas I., Tsang M., and Lin S. J. (2015) Reduced Ssy1-Ptr3-Ssy5 (SPS) signaling extends replicative life span by enhancing NAD+ homeostasis in Saccharomyces cerevisiae. J. Biol. Chem. 290, 12753–12764 10.1074/jbc.M115.644534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kato M., and Lin S. J. (2014) YCL047C/POF1 is a novel nicotinamide mononucleotide adenylyltransferase (NMNAT) in Saccharomyces cerevisiae. J. Biol. Chem. 289, 15577–15587 10.1074/jbc.M114.558643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Polevoda B., Cardillo T. S., Doyle T. C., Bedi G. S., and Sherman F. (2003) Nat3p and Mdm20p are required for function of yeast NatB Nα-terminal acetyltransferase and of actin and tropomyosin. J. Biol. Chem. 278, 30686–30697 10.1074/jbc.M304690200 [DOI] [PubMed] [Google Scholar]
- 24. Singer J. M., and Shaw J. M. (2003) Mdm20 protein functions with Nat3 protein to acetylate Tpm1 protein and regulate tropomyosin-actin interactions in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 100, 7644–7649 10.1073/pnas.1232343100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Polevoda B., Norbeck J., Takakura H., Blomberg A., and Sherman F. (1999) Identification and specificities of N-terminal acetyltransferases from Saccharomyces cerevisiae. EMBO J. 18, 6155–6168 10.1093/emboj/18.21.6155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Van Damme P., Lasa M., Polevoda B., Gazquez C., Elosegui-Artola A., Kim D. S., De Juan-Pardo E., Demeyer K., Hole K., Larrea E., Timmerman E., Prieto J., Arnesen T., Sherman F., Gevaert K., and Aldabe R. (2012) N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc. Natl. Acad. Sci. U.S.A. 109, 12449–12454 10.1073/pnas.1210303109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aksnes H., Hole K., and Arnesen T. (2015) Molecular, cellular, and physiological significance of N-terminal acetylation. Int. Rev. Cell Mol. Biol. 316, 267–305 10.1016/bs.ircmb.2015.01.001 [DOI] [PubMed] [Google Scholar]
- 28. Hermann G. J., King E. J., and Shaw J. M. (1997) The yeast gene, MDM20, is necessary for mitochondrial inheritance and organization of the actin cytoskeleton. J. Cell Biol. 137, 141–153 10.1083/jcb.137.1.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Singer J. M., Hermann G. J., and Shaw J. M. (2000) Suppressors of mdm20 in yeast identify new alleles of ACT1 and TPM1 predicted to enhance actin-tropomyosin interactions. Genetics 156, 523–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tempel W., Rabeh W. M., Bogan K. L., Belenky P., Wojcik M., Seidle H. F., Nedyalkova L., Yang T., Sauve A. A., Park H. W., and Brenner C. (2007) Nicotinamide riboside kinase structures reveal new pathways to NAD+. PLos Biol. 5, e263 10.1371/journal.pbio.0050263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Delorme-Axford E., Guimaraes R. S., Reggiori F., and Klionsky D. J. (2015) The yeast Saccharomyces cerevisiae: an overview of methods to study autophagy progression. Methods 75, 3–12 10.1016/j.ymeth.2014.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reggiori F., and Klionsky D. J. (2013) Autophagic processes in yeast: mechanism, machinery and regulation. Genetics 194, 341–361 10.1534/genetics.112.149013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Suzuki S. W., Onodera J., and Ohsumi Y. (2011) Starvation induced cell death in autophagy-defective yeast mutants is caused by mitochondria dysfunction. PLoS One 6, e17412 10.1371/journal.pone.0017412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. An Z., Tassa A., Thomas C., Zhong R., Xiao G., Fotedar R., Tu B. P., Klionsky D. J., and Levine B. (2014) Autophagy is required for G1/G0 quiescence in response to nitrogen starvation in Saccharomyces cerevisiae. Autophagy 10, 1702–1711 10.4161/auto.32122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu H., and Bretscher A. (1992) Characterization of TPM1 disrupted yeast cells indicates an involvement of tropomyosin in directed vesicular transport. J. Cell Biol. 118, 285–299 10.1083/jcb.118.2.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Drees B., Brown C., Barrell B. G., and Bretscher A. (1995) Tropomyosin is essential in yeast, yet the TPM1 and TPM2 products perform distinct functions. J. Cell Biol. 128, 383–392 10.1083/jcb.128.3.383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu H. P., and Bretscher A. (1989) Disruption of the single tropomyosin gene in yeast results in the disappearance of actin cables from the cytoskeleton. Cell 57, 233–242 10.1016/0092-8674(89)90961-6 [DOI] [PubMed] [Google Scholar]
- 38. Pruyne D. W., Schott D. H., and Bretscher A. (1998) Tropomyosin-containing actin cables direct the Myo2p-dependent polarized delivery of secretory vesicles in budding yeast. J. Cell Biol. 143, 1931–1945 10.1083/jcb.143.7.1931 [DOI] [PubMed] [Google Scholar]
- 39. Polevoda B., and Sherman F. (2003) N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol. 325, 595–622 10.1016/S0022-2836(02)01269-X [DOI] [PubMed] [Google Scholar]
- 40. Hong H., Cai Y., Zhang S., Ding H., Wang H., and Han A. (2017) Molecular basis of substrate specific acetylation by N-terminal acetyltransferase NatB. Structure 25, 641–649.e3 10.1016/j.str.2017.03.003 [DOI] [PubMed] [Google Scholar]
- 41. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 10.1093/bioinformatics/btq054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee K. E., Heo J. E., Kim J. M., and Hwang C. S. (2016) N-terminal acetylation-targeted N-end rule proteolytic system: the Ac/N-end rule pathway. Mol. Cells 39, 169–178 10.14348/molcells.2016.2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen S. J., Wu X., Wadas B., Oh J. H., and Varshavsky A. (2017) An N-end rule pathway that recognizes proline and destroys gluconeogenic enzymes. Science 355, eaal3655 10.1126/science.aal3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim H.-K., Kim R.-R., Oh J.-H., Cho H., Varshavsky A., and Hwang C.-S. (2014) The N-terminal methionine of cellular proteins as a degradation signal. Cell 156, 158–169 10.1016/j.cell.2013.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hwang C.-S., Shemorry A., and Varshavsky A. (2010) N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973–977 10.1126/science.1183147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shemorry A., Hwang C.-S., and Varshavsky A. (2013) Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol. Cell 50, 540–551 10.1016/j.molcel.2013.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Monda J. K., Scott D. C., Miller D. J., Lydeard J., King D., Harper J. W., Bennett E. J., and Schulman B. A. (2013) Structural conservation of distinctive N-terminal acetylation-dependent interactions across a family of mammalian NEDD8 ligation enzymes. Structure 21, 42–53 10.1016/j.str.2012.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Z., Kulkarni K., Hanrahan S. J., Thompson A. J., and Barford D. (2010) The APC/C subunit Cdc16/Cut9 is a contiguous tetratricopeptide repeat superhelix with a homo-dimer interface similar to Cdc27. EMBO J. 29, 3733–3744 10.1038/emboj.2010.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Natalini P., Ruggieri S., Raffaelli N., and Magni G. (1986) Nicotinamide mononucleotide adenylyltransferase: molecular and enzymatic properties of the homogeneous enzyme from baker's yeast. Biochemistry 25, 3725–3729 10.1021/bi00360a037 [DOI] [PubMed] [Google Scholar]
- 50. Emanuelli M., Carnevali F., Lorenzi M., Raffaelli N., Amici A., Ruggieri S., and Magni G. (1999) Identification and characterization of YLR328W, the Saccharomyces cerevisiae structural gene encoding NMN adenylyltransferase: expression and characterization of the recombinant enzyme. FEBS Lett. 455, 13–17 10.1016/S0014-5793(99)00852-2 [DOI] [PubMed] [Google Scholar]
- 51. Emanuelli M., Amici A., Carnevali F., Pierella F., Raffaelli N., and Magni G. (2003) Identification and characterization of a second NMN adenylyltransferase gene in Saccharomyces cerevisiae. Protein Expr. Purif. 27, 357–364 10.1016/S1046-5928(02)00645-9 [DOI] [PubMed] [Google Scholar]
- 52. Huckaba T. M., Gay A. C., Pantalena L. F., Yang H.-C., and Pon L. A. (2004) Live cell imaging of the assembly, disassembly, and actin cable–dependent movement of endosomes and actin patches in the budding yeast, Saccharomyces cerevisiae. J. Cell Biol. 167, 519–530 10.1083/jcb.200404173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Prosser D. C., Drivas T. G., Maldonado-Báez L., and Wendland B. (2011) Existence of a novel clathrin-independent endocytic pathway in yeast that depends on Rho1 and formin. J. Cell Biol. 195, 657–671 10.1083/jcb.201104045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huckaba T. M., Lipkin T., and Pon L. A. (2006) Roles of type II myosin and a tropomyosin isoform in retrograde actin flow in budding yeast. J. Cell Biol. 175, 957–969 10.1083/jcb.200609155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rubinsztein D. C., Mariño G., and Kroemer G. (2011) Autophagy and aging. Cell 146, 682–695 10.1016/j.cell.2011.07.030 [DOI] [PubMed] [Google Scholar]
- 56. Fogel A. I., Dlouhy B. J., Wang C., Ryu S. W., Neutzner A., Hasson S. A., Sideris D. P., Abeliovich H., and Youle R. J. (2013) Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol. Cell Biol. 33, 3675–3688 10.1128/MCB.00079-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Helbig A. O., Rosati S., Pijnappel P. W., van Breukelen B., Timmers M. H., Mohammed S., Slijper M., and Heck A. J. (2010) Perturbation of the yeast N-acetyltransferase NatB induces elevation of protein phosphorylation levels. BMC Genomics 11, 685 10.1186/1471-2164-11-685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xu Y. F., Létisse F., Absalan F., Lu W., Kuznetsova E., Brown G., Caudy A. A., Yakunin A. F., Broach J. R., and Rabinowitz J. D. (2013) Nucleotide degradation and ribose salvage in yeast. Mol. Syst. Biol. 9, 665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Effelsberg D., Cruz-Zaragoza L. D., Tonillo J., Schliebs W., and Erdmann R. (2015) Role of Pex21p for piggyback import of Gpd1p and Pnc1p into peroxisomes of Saccharomyces cerevisiae. J. Biol. Chem. 290, 25333–25342 10.1074/jbc.M115.653451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Giardina B. J., Stanley B. A., and Chiang H.-L. (2014) Glucose induces rapid changes in the secretome of Saccharomyces cerevisiae. Proteome Sci. 12, 9–9 10.1186/1477-5956-12-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Belenky P., Stebbins R., Bogan K. L., Evans C. R., and Brenner C. (2011) Nrt1 and Tna1-independent export of NAD+ precursor vitamins promotes NAD+ homeostasis and allows engineering of vitamin production. PLoS One 6, e19710 10.1371/journal.pone.0019710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Imai S., Armstrong C. M., Kaeberlein M., and Guarente L. (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 10.1038/35001622 [DOI] [PubMed] [Google Scholar]
- 63. Smith J. S., Brachmann C. B., Celic I., Kenna M. A., Muhammad S., Starai V. J., Avalos J. L., Escalante-Semerena J. C., Grubmeyer C., Wolberger C., and Boeke J. D. (2000) A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. U.S.A. 97, 6658–6663 10.1073/pnas.97.12.6658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Steiger M. A., Jackman J. E., and Phizicky E. M. (2005) Analysis of 2′-phosphotransferase (Tpt1p) from Saccharomyces cerevisiae: evidence for a conserved two-step reaction mechanism. RNA 11, 99–106 10.1261/rna.7194605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yoon M. J., Yoshida M., Johnson S., Takikawa A., Usui I., Tobe K., Nakagawa T., Yoshino J., and Imai S. (2015) SIRT1-mediated eNAMPT secretion from adipose tissue regulates hypothalamic NAD+ and function in mice. Cell Metab. 21, 706–717 10.1016/j.cmet.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., and Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 10.1002/(SICI)1097-0061(19980130)14:2%3C115::AID-YEA204%3E3.0.CO%3B2-2 [DOI] [PubMed] [Google Scholar]
- 67. Burke D., Dawson D., and Sterns T. (2000) Methods in Yeast Genetics, pp. 171–174, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 68. Güldener U., Heck S., Fielder T., Beinhauer J., and Hegemann J. H. (1996) A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 24, 2519–2524 10.1093/nar/24.13.2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Longtine M. S., McKenzie A. 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., and Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 10.1002/(SICI)1097-0061(199807)14:10%3C953::AID-YEA293%3E3.0.CO%3B2-U [DOI] [PubMed] [Google Scholar]
- 70. Sheff M. A., and Thorn K. S. (2004) Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast 21, 661–670 10.1002/yea.1130 [DOI] [PubMed] [Google Scholar]
- 71. Easlon E., Tsang F., Skinner C., Wang C., and Lin S. J. (2008) The malate-aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev. 22, 931–944 10.1101/gad.1648308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Winzeler E. A., Shoemaker D. D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J. D., Bussey H., Chu A. M., Connelly C., Davis K., Dietrich F., Dow S. W., et al. (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906 10.1126/science.285.5429.901 [DOI] [PubMed] [Google Scholar]
- 73. Ghislain M., Talla E., and François J. M. (2002) Identification and functional analysis of the Saccharomyces cerevisiae nicotinamidase gene, PNC1. Yeast 19, 215–224 10.1002/yea.810 [DOI] [PubMed] [Google Scholar]
- 74. To-E A., Ueda Y., Kakimoto S. I., and Oshima Y. (1973) Isolation and characterization of acid phosphatase mutants in Saccharomyces cerevisiae. J. Bacteriol. 113, 727–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bern M., Kil Y. J., and Becker C. (2012) Byonic: advanced peptide and protein identification software. Curr. Protoc. Bioinformatics, Chapter 13, Unit 13.20 10.1002/0471250953.bi1320s40 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.