

ABSTRACT
Lipopolysaccharide (LPS) of Porphyromonas gingivalis exists in at least two known forms, O-LPS and A-LPS. A-LPS shows heterogeneity in which two isoforms designated LPS1,435/1,449 and LPS1,690 appear responsible for tissue-specific immune signalling pathways activation and increased virulence. The modification of lipid A to tetra-acylated1,435/1,449 and/or penta-acylated1,690 fatty acids indicates poor growth conditions and bioavailability of hemin. Hemin protects P. gingivalis from serum resistance and the lipid A serves as a site for its binding. The LPS1,435/1,449 and LPS1,690 isoforms can produce opposite effects on the human Toll-like receptors (TLR) TLR2 and TLR4 activation. This enables P. gingivalis to select the conditions for its entry, survival, and that of its co-habiting species in the host, orchestrating its virulence to control innate immune pathway activation and biofilm dysbiosis. This review describes a number of effects that LPS1,435/1,449 and LPS1,690 can exert on the host tissues such as deregulation of the innate immune system, subversion of host cell autophagy, regulation of outer membrane vesicle production, and adverse effects on pregnancy outcome. The ability to change its LPS1,435/1,449 and/or LPS1,690 composition may enable P. gingivalis to paralyze local pro-inflammatory cytokine production, thereby gaining access to its primary location in periodontal tissue.
KEYWORDS: P. gingivalis, lipid A, heterogeneity, penta-acylated form, tetra-acylated form, innate immunity subversion
Introduction
Porphyromonas gingivalis is considered to be a keystone Gram-negative, anaerobic intracellular pathogen in adult periodontitis [1]. It has a plethora of virulence factors [2] of which lipopolysaccharide (LPS), gingipains, fimbriae, hemagglutinins, and outer membrane vesicles (OMVs) are of major importance. LPS is located in the outer membrane of Gram-negative bacteria and is a potent stimulator of host’s innate immune signal transduction pathways in a tissue/cell-specific manner [3]. This is seen in bone, epithelial cell barrier breakdown, and keratinocytes [4,5]. Historically, LPS has been thought to consist of three variable and conserved regions [6,7]. These include lipid A (common to all forms of LPS from Gram-negative bacteria), which can consist of fatty acid esters; these fatty acid esters in turn are either attached to phosphorylated glucosamine disaccharides or 3-OH isobranched C17:0 and other fatty acids with amide-links to both saccharide units in lipid A: a conserved core oligosaccharide that links lipid A to the O-antigen; and a highly variable O-polysaccharide or O-antigen [8] (Figure 1).
Figure 1.
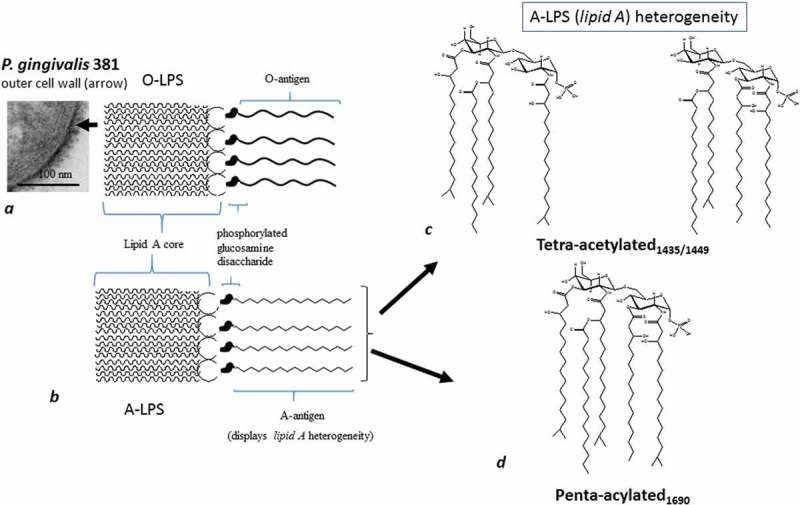
Schematically shows two main forms of LPS (O-LPS and A-LPS) and their position on the outer cell wall (arrow). (a) Section of P. gingivalis (FDC 381) demonstrated by transmission electron microscopy. Thick arrow points to lipid A with fatty acid esters attached to phosphorylated glucosamine disaccharides and the O-antigen. The latter is O-LPS (with O-antigen tetrasaccharide repeating units); (b) A-LPS (with surface anionic polysaccharide [APS] repeating units). There are two lipid As in A-LPS as shown in (c) and (d). (c) A tetra-acylated form with two different molecular weights, hence two structures. (d) the penta-acylated form.
Of the LPS macromolecule, lipid A is responsible for the endotoxic activities, and its recognition by host cells leads to differential immuno-inflammatory responses [4,7–9]. From LPS, it is the heterogeneity within the A-LPS which represents the major virulence factors that promote inflammation and bone loss. Hence in immunological terms, the A-LPS heterogeneous forms represent ‘pathogen associated molecular patterns’ (PAMPs) and have been extensively studied for their role in the pathogenesis of periodontitis (for a review, see Nichols et al. [10]), however not without controversy. Whereas some studies reported that P. gingivalis LPS stimulates secretion of pro-inflammatory cytokines [11], others found contradictory results with regard to cytokine release [5,12]. Besides, LPS acting as an agonist for Toll-like receptor (TLR) TLR2 or as an antagonist and/or agonist for TLR4 activation [13–16] added to further contradiction. Although TLR2 activation by P. gingivalis LPS is possible, the difference lies in the form of LPS presented to the host. For example, the protein-free LPS is unable to activate TLR2, whereas the bound LPS on the live bacterium can mediate TLR2 activation via a novel class of lipoprotein lipase-sensitive molecules highlighting the importance of active infection [17]. In essence, and according to Lasica et al. [18], the tightly associated or covalently attached protein to LPS on live P. gingivalis accounts for the TLR2 activation. In terms of bone loss, in vitro and in vivo experiments have provided conflicting data. For example, when P. gingivalis is co-cultured with bone cells in vitro, the effect on bone resorbing cells appears to be mediated through engagement of both TLR2 and TLR4 [15,19]. However, similar experiments conducted in experimental animals orally infected with P. gingivalis predominantly show bone loss to be mediated by TLR2 [20–22]. If the in vivo data are confirmed in human alveolar bone resorption mechanisms, this is another example of P. gingivalis LPS lipid A heterogeneity showing differential inflammatory signalling according to tissue specificity.
P. gingivalis has two major forms of LPS, O-LPS (Figure 1(a)) [23] and A-LPS (Figure 1(b)) [24–26]. O-LPS is a conventional O-antigen polysaccharide found in most bacteria with Gram-negative characteristics, and A-LPS is an anionic polysaccharide (APS) (Figure 1(b)). Both O and A-LPS forms are linked to lipid A. While the O-antigen repeating polysaccharide unit of O-LPS contains →3)-α-d-Galp-(1→6)-α-d-Glcp-(1→4)-α-l-Rha-(1→3)-β-d-GalNAcp-(1→, the polysaccharide repeating unit of A-LPS consists of a phosphorylated branched d-Man-containing oligomer made up of an α1→6-linked d-mannose backbone. To the latter branch of the backbone, α1→2-linked d-Man side chains of varying lengths with one or two residues are attached at position 2 [27].
In addition to the structural differences in P. gingivalis A-LPS mentioned above, a variety of different lipid A structures have been reported [28,29]. These lipid A structures, in the literature, are referred to as phosphorylated tetra-acylated and phosphorylated penta-acylated proteins. The tetra-acylated form of A-LPS has a molecular weight of 1,435 and 1,449 Da, and the penta-acylated form of A-LPS has a molecular weight of 1,690 Da [30]. For clarity, the tetra-acylated form of A-LPS has been designated LPS1,435/1,449 (Figure 1(c)) and the penta-acylated form LPS1,690 (Figure 1(d)) [30]. The role of these variable regions appears to involve signalling pathway activation in various effector cells, organs, and diseases (Table 1). It has to be stressed that the heterogeneity is related to the bioavailability of essential growth nutrients such as hemin [25], phosphate availability [31], and to an extent on temperature [32]. The aim of this review is to assess the importance of the heterogeneity in P. gingivalis LPS lipid A.
Table 1.
Heterogenous tetra- and penta acylated A-LPS isoforms involved in immune signal transduction pathways in various cell types.
| A-LPS | Signalling pathway activated | Genes (up regulated) | Genes (down regulated) | Effector cells/organ/disease | Reference |
|---|---|---|---|---|---|
| LPS1,435/1,449 and LPS1,690 | NF-κB | ELK1, HRAS, IL-1β, TLR-4, TLR-5, TLR-9, TNF, TRAF6, and UBE2N | BTK, IL-2, IRAK1, LTA, CD180, MAPK8IP, NFKBIL1, SIGRR, TIRAP, TLR-1, and TLR-7 | Human gingival fibroblasts/gingivae/ periodontitis |
[4] |
| LPS1,435/1,449 | p38 MAPK and ERK1/2 P’ways | Unaffected genes: SAPK/JNK and AKT | [4] | ||
| LPS1,690 | NF-κB and p38 MAPK and ERK1/2 P’ways NF-κB and p38 MAPK |
GM-CSF, CXCL10, G-CSF, IL-6, IL-8, CCL2 and TLR4 NFKBIA, NFKB1, and IKBKB. MAP2K4 and MAPK8, E-selectin |
Human gingival fibroblasts/gingivae/periodontitis Human oral keratinocytes |
[4, 5, 50] |
Lipid A phosphatase is required for colonization and commensal overgrowth of bacteria in periodontitis
P. gingivalis has the ability to change its A-LPS lipid A phosphate composition (LPS1,435/1,449 or LPS1,690) in response to differing environmental conditions. It is this property, which makes the bacterium highly adaptable to different inflammatory niches. P. gingivalis can produce A-LPS lipid A structures that are agonists (LPS1,690) or antagonists (LPS1,435/1,449) to activation of TLR4 [25,31,32] because under the influence of P. gingivalis, it helps to create an inflammophilic environment for selection of its co-species and control any competition. Despite the fact that phosphatases are important for successful colonization of P. gingivalis, Zenobia et al. [33] demonstrated in a rabbit ligature model of experimental periodontitis that exposure of the oral cavity to the mutant strains (with locked lipid A structures) did not increase the microbial load. This suggests that the mutant strains were unable to remodel their lipid A structure, which would otherwise be capable of inducing a favourable co-inhabiting environment. This begs the question: why would P. gingivalis change its lipid A phosphate composition? The answer lies in the Zenobia et al. [33] study that detected significant qualitative changes in the microbial composition from the mutant strains, in that all P. gingivalis strains and their LPS preparations resulted in the development of periodontitis. Furthermore, this keystone bacterium even in its mutant form changed the lipid A composition by utilizing the phosphatases from its microenvironment. Hence, successful colonization took place whereby the microbial load increased in the rabbit ligature model of periodontitis [31]. These results suggest that not only disruption of host homeostasis is highly plausible but also that an evolving commensal microbial community can become dysbiotic even under the influence of a mutant form of P. gingivalis resulting in disease.
The lipid A structure is modulated by hemin and temperature
Heme [Fe(II)-protoporphyrin IX] and hemin [Fe(III)-protoporhyrin IX-Cl] are important for the growth, survival, and virulence of P. gingivalis [34,35]. P. gingivalis cannot synthesize the protoporhyrin IX ring and does not have siderophores for alternative bioavailability of hemin [36–39]. Therefore, P. gingivalis depends on the host for a supply of heme [40]. The lysis of multiple erythrocytes, in order to feed P. gingivalis heme, would lead to decrease in oxygen for the host, which would in turn contribute to ischemia. Therefore, the bacterium gains advantage for ideal growth conditions. However, if the supply of hemin is less than adequate, as tested in vitro, for the pathogen, at least, it is proposed that the A-LPS lipid A structure of P. gingivalis becomes modified according to the hemin concentration in the growth medium [25]. Different isoforms of A-LPS have been demonstrated in experiments where P. gingivalis cultures contained either low or high hemin concentrations (Figure 1(c,d). At high hemin concentration, LPS1,435/1,449 (high hemin) was synthesized, and it elicited a weak immune cell activation via TLR2. Biochemical analysis of the lipid A region demonstrated a mono-phosphorylated, tetra-acylated structure (Figure 1(c)) [25]. Alternatively, P. gingivalis culture from low hemin concentration, LPS1,690 (low hemin), was found to be more immunogenic [25]. Biochemical analysis showed that the LPS activity, surprisingly, was of antagonist nature with a mono-phosphorylated penta-acylated lipid A (Figure 1(d)) [25]. It is worth noting that P. gingivalis LPS1,435/1,439 and LPS1,690 mixtures obtained from culture conditions with high hemin content also demonstrated a potent immunogenic response. These observations, taken together, suggest that P. gingivalis LPS1,435/1,439/1,690, may have tools to paralyze the host’s innate defences and expose tissues to an inflammophilic milieu [41,42]. One advantage of the heterogeneity displayed in P. gingivalis A-LPS would be that at least it would provide resistance from septicaemia/endotoxin shock to the host.
Coats et al. [31] found that P. gingivalis uses endogenous lipid A 1 activity to produce a unique non-phosphorylated lipid in A-LPS. This immunologically silent lipid A has the capacity to provide a highly effective mechanism, which is used by this bacterium to evade TLR4 sensing and to resist killing by cationic anti-microbial peptides released by the host. As the lipid A 1-phosphatase activity was suppressed by hemin, it has been suggested that hemin-dependent regulation of lipid A 1-dephosphorylation could change the A-LPS lipid A activity from TLR4 evasive to TLR4 suppressive. This may alter the critical interaction between P. gingivalis, the local microbial community, and the innate immune system of the host. Hemin acquisition proteins such as gingipain K (Kgp) and/or the heme receptor (HmuR) molecules [25,43] appear to be the sensors of its concentration in the environment, although other proteins for hemin binding or its transport may also exist.
A-LPS of P. gingivalis serves as a matrix for the deposition of µ-oxo-bisheme [[Fe(III)PPIX]2O] and is accountable for the black pigment seen within these cells. µ-Oxo-bisheme pigmentation is a form of a bacteriocin and a virulence factor of P. gingivalis [40]. Both Arg and Lys gingipains are required for the deposition of this characteristic black pigment. Absence of A-LPS in the extracellular surface of P. gingivalis diminishes the scaffold/anchoring mechanism that otherwise retains Arg- and Lys-gingipains [40]. This implies that A-LPS serves as a site for deposition/binding of hemin. Since pigmentation depends on the presence of A-LPS and Arg- and Lys-gingipains, these observations suggest a significant role of A-LPS in the eventual virulence of P. gingivalis.
Curtis et al. [32] discovered that P. gingivalis at normal body temperature mainly produced non-phosphorylated and mono-phosphorylated tetra-acylated lipid A structures. This phenomenon was tested by culturing P. gingivalis at higher temperatures (39 or 41°C), so mimicking the conditions at the sites of periodontal inflammation. This resulted in generating increased amounts of both mono-phosphorylated and penta-acylated lipid A. The final nail in the coffin for P. gingivalis was the activation of the TLR4 at higher temperatures, which could potentially kill it via β-defensins 2 and 3 activities. If modification of lipid A by the host’s body temperature variation affects the virulence and susceptibility to killing, then this inflammophilic bacterium may have evolved hidden mechanisms to thrive under precisely those conditions in vivo.
LPS heterogeneity clarifies earlier contradictory results related to host innate immune mediators
It has become apparent that the A-LPS of P. gingivalis plays an important role in deregulating the innate immune system of the host. This is further achieved by changing the host’s defence signalling mechanisms through its heterogeneous A-LPS lipid A structures [31,44]. For example, Herath et al. [4] observed that the NF-ĸB signalling pathway was noticeably activated in human gingival fibroblasts (HGFs) by LPS1,690 but not by LPS1,435/1,449. The heterogeneity displayed in the A-LPS also modulated the secretion of a different profile of the pro-inflammatory cytokine expression such as IL-6 and IL-8 in HGFs [45]. Furthermore, P. gingivalis LPS1,690 significantly up regulated the expression of IL-6 and IL-8 mRNA at the gene level in HGFs, whereas LPS1,435/1,449 failed to do so [45]. In human monocytes, the A-LPS induced production of IL-1α, IL-1β, IL-6, and IL-8, although at a lower rate than that induced by lipid A obtained from total A-LPS [26]. In addition, pro-inflammatory genes significantly up regulated by LPS1,690 (GM-CSF, CXCL10, G-CSF, IL-6, IL-8, and CCL2) were down regulated by LPS1,435/1,449 [4] (see Table 1). HGFs matrix metalloproteinase (MMP)-3 and its protein were strikingly up regulated by penta-acylated P. gingivalis LPS1,690 and hexa-acylated Escherichia coli LPS but not by tetra-acylated P. gingivalis LPS1,435/1,449 [30], suggesting plausible crosstalk between LPS signalling from Gram-negative pathogens during mixed infections. Furthermore, human beta-defensins, hBD-1, hBD-2, and hBD-3 mRNAs, were significantly up regulated in human epithelia by P. gingivalis LPS1,690 but down regulated by LPS1,435/1,449 [46]. Differential signalling pathway activation in various cell types [4] helps explain (at least in part) the contradictory results reported by earlier studies [28,29] and demonstrates tissue-specific modes of inflammatory signals initiated essentially by the same immunogen. The advantage to the keystone bacterium would likely be in suppressing, some signalling pathways for adapting to different tissue environments. This may explain the role P. gingivalis plays in establishing disparate organ-specific inflammatory pathologies.
In human oral keratinocytes (HOKs), P. gingivalis penta-acylated mono-phosphorylated LPS1,690 has been shown to act as an agonist for E-selectin expression, while tetra-acylated mono-phosphoryl structures LPS1,435/1,449 act as antagonists [47]. Interestingly, structurally similar, penta-acylated, mono-phosphorylated LPS forms from P. gingivalis and the genus Bacteroides (from the gastrointestinal tract) caused vastly different types of innate immune reactions that were ascribed to subtle differences in the molecular weight fatty acid content of A-LPS [48]. This supports further the view that P. gingivalis A-LPS is unlikely to cause sepsis and formation of intra-abdominal abscesses in the host.
The different isoforms of LPS can also affect periodontal pathogenesis by disrupting pattern recognition receptors (PRRs) such as LPS-binding protein (LBP) [49,50]. LPS1,4 35/1,449 down regulated recombinant human (rh) LBP-induced IL-6 and IL-8 mRNAs significantly more than P. gingivalis LPS1,690 [50]. P. gingivalis LPS1,690-rh LBP interaction also caused a dramatic up regulation of the transcript of the cell surface molecule CD180 and a significant down regulation of the myeloid differentiation factor 1 (MD-1) transcript. This probably occurred through fine-tuning of the CD180–MD1 complex and appropriate TLRs [50]. LPS1,690 was shown to stimulate LBP in HOKs by affecting the signalling pathways of NF-ĸB and p38 MAPK whereas LPS1,435/1,449 was unable to perform this task [51]. Changing the lipid A structure of P. gingivalis may, therefore, be a crucial strategy for this keystone periodontal pathogen to escape from the hostile innate host defences.
Interestingly, an oral infection in apoliporotein E knockout (ApoE−/−) mice with a P. gingivalis strain expressing antagonistic lipid A caused vascular inflammation, macrophage accumulation, and progression of atherosclerosis [52], whereas a strain producing agonistic lipid A increased the levels of pro-inflammatory mediators and activated the inflammasome in a caspase-11-dependent manner. This led to host cell lysis and reduced bacterial survival. Thus, P. gingivalis avoided immune detection and promoted chronic inflammation in the vasculature. Chronic diseases may, therefore, be ascribed to pathogen-related strategies for immune evasion leading to low-grade inflammation.
Differences in lipid A have different effects on TLR signalling
The innate immune system senses the invasion of pathogenic microorganisms via TLRs that recognize specific PAMPs. Heterogeneity in the P. gingivalis A-LPS can determine the virulence of this bacterium. This is seen especially via the elegant experiments related to hemin bioavailability [25]. Thus, P. gingivalis LPS1,435/1,469 and LPS1,690 can modulate TLR2 and/or TLR4 depending on the immediate environmental conditions and the cell type [25,30,47,50] to facilitate (epithelial/matrix) barrier breakdown for its access to its eventual periodontal niche. Tetra- and penta-acylated lipid A structures of P. gingivalis differentially activated TLR4-mediated NF-kB signal transduction and modulated the expression of IL-6 and IL-8 in HGFs [4]. TLR2 and cluster of differentiation (CD) 14 mRNA were regulated differentially in human gingival epithelia while the modulation of hBD-2 expression was suggested to occur through co-operation of both TLR2 and TLR4 [46]. In another study, multiple lipid A species interacted functionally with both TLR2 and TLR4 [15]. However, as already mentioned, the TLR2 agonist activity in P. gingivalis is most likely due to a lipoprotein, and treatment of LPS with lipoproteinase as free LPS substantially attenuated the TLR2 engaging activity [17]. These findings explain previous observations that P. gingivalis LPS can act both as a TLR4 agonist and antagonist, with an end result of varying cytokine secretion profiles [5,11,12]. Also in HOKs, P. gingivalis LPS1,690-induced LBP expression occurred through both TLR2 and TLR4 [51], while recombinant rh LBP significantly up regulated the expression of IL-6 and IL-8 in HOKs through the TLR2 signalling pathway [50]. These observations repeatedly highlight the adaptability of P. gingivalis not only to disrupt cell-specific barriers but also to select the level of potency conducive to the required inflammophillic milieu and disease initiation/progression. The shape of the lipid A component of LPS has been proposed to determine the physiological functioning of LPS. Conical-shaped LPS (e.g. from E. coli) may stimulate cells through TLR4 while cylindrically shaped LPS e.g. from P. gingivalis may stimulate cytokine production through TLR2 [53]. Strictly speaking, cylindrical LPS molecules, albeit from other bacterial sources (Rhodobacter sphaerodides), demonstrate antagonistic properties at TLR levels. Accordingly, the LPS from P. gingivalis can act as a TLR2 or TLR4 agonist or antagonist depending upon the TLR cell type being activated in the host, or if multiple bacterial lipid A species are present. The joint signalling pathways elicited by TLR2 and TLR4 agonists may diverge to enable distinct patterns of expression, a property elicited by P. gingivalis A-LPS heterogeneity.
LPS influences host cell autophagy processes
P. gingivalis often subverts autophagy processes in host cells in order to survive. In a study by Blasi et al. [54], variants of P. gingivalis LPS altered lipidation of autophagy protein, i.e. microtubule-associated protein 1 light chain 3 (LC3). Whereas LPS1,690 stimulated production of very large autophagic protein LC3-positive vacuoles and cargo sorting protein MREG puncta in macrophages, the LPS1,690-mediated LC3 lipidation was reduced when LPS1,435/1,449 was present. This indicated that the prevalence of a particular LPS moiety could affect the degradative capacity of host cells, thereby influencing bacterial survival. This is noteworthy since peripheral blood mononuclear cells from periodontitis patients show increased levels of autophagy-related gene expression and high levels of mitochondrial reactive oxygen species, with further increase in LC3 upon stimulation of gingival fibroblasts with P. gingivalis LPS [55]. The ability to dysregulate autophagy in phagocytic cells has implications for development of distant organ inflammatory diseases in which either P. gingivalis or its LPS have been detected or been tested experimentally [56,57].
A-LPS provides serum resistance to P. gingivalis
P. gingivalis is unique in that it displays commonality for the biosynthesis of A-LPS and the glycosylation of Arg gingipains [58]. Given that these two virulence factors, independently of each other, can be potent immune effectors in the host, why has P. gingivalis acquired this co-operation with its LPS and Arg gingipains as well? Experiments conducted with deletion mutants in the loci porR (PG1138) and wbpB (PG2119), which are devoid of A-LPS but possess O-LPS [59,60], exerted less gingipain enzymatic activity. These loci are, therefore, probably involved in the biosynthesis of A-LPS [60,61]. Furthermore, the mutants were susceptible to killing by the complement system, suggesting that A-LPS has a role in serum resistance to P. gingivalis [61,62]. Also, in the non-pigmented P. gingivalis strain HG66, which lacks A-LPS but possesses O-LPS, a nonsense mutation in the wbpB gene was detected, and Wbp pathway gene mutants were found to be A-LPS deficient [60]. Both penta-acylated lipid A and non-phosphorylated lipid A failed to activate TLR4 and provided the pronounced ability of these bacteria to resist polymorphonuclear neutrophil-mediated killing. The above experimental outcome suggests that A-LPS and Arg gingipains may be acting as switches that exert influence on the endotoxin/exotoxin activity for maintaining bacterial survival and retaining pathogenicity. The pigment of this bacterium plays a vital role in maintaining A-LPS heterogeneity and therefore its potent virulence.
A-LPS affects production of OMVs
The main component of the outer leaflet of outer membrane vesicles (OMVs) is LPS, while the inner leaflet consists of a layer of phospholipids. Rangarajan et al. [27] suggested a novel mechanism for the production of OMVs in P. gingivalis implying dephosphorylation of the lipid A region of A-LPS that is controlled or regulated by the OM protein PG0027. This may be responsible for destabilization of the outer membrane followed by blebbing and generation of OMVs. Such an explanation is based on PG0027, which seems to control the amount of phosphorylated and non-phosphorylated species of lipid A in A-LPS, and thereby the production of OMVs in P. gingivalis. It is also plausible that these vesicles are produced in regions of the outer membrane enriched in A-LPS that are devoid of non-phosphorylated lipid A. Conversely, dephosphorylation of lipid A through a PG0027-dependent process may be required for optimal processing of OMVs. The relative proportions of non-phosphorylated and phosphorylated lipid A are obviously crucial for outer membrane blebbing and generation in P. gingivalis.
Biosynthesis of A-LPS and glycosylation of Arg gingipains share common steps
Gingipains are important virulence factors of P. gingivalis in addition to LPS. Genesis of these factors may share common pathways as suggested previously, in respect to µ-oxo-bisheme, responsible for the black pigment. Since a monoclonal antibody (MAb1B5) raised against the Arg-gingipain RgpA cross-reacted with a cell-surface polysaccharide of P. gingivalis strain W50 (A-LPS) [24,63], it was suggested that the maturation pathway of the Arg-gingipains could be linked to the biosynthesis of a surface carbohydrate [24]. The cross-reacting APS, different from the LPS and serotype capsule polysaccharide, was found to be a phosphorylated branch of mannan backbone. The backbone consisted of α-1,6-linked mannose residues, and the side chains of α-1,2-linked mannose oligosaccharides of various lengths. One of the side chains of the repeating unit carried Manα1–2Manα1-phosphate linked through phosphorous to a mannose backbone at position 2. This lead to the proposal that the Manα1–2Manα1-phosphate fragment constitutes part of the epitope recognized by MAb1B5. This phosphorylated branched mannan represented a new polysaccharide that appears immunologically connected to the post-translational additions of Arg-gingipains [24,63]. Accordingly, there seem to be common steps in the biosynthesis of A-LPS and the glycosylation of Arg gingipains in P. gingivalis.
Dysregulation of the TLR function may cause adverse pregnancy outcomes
P. gingivalis has been associated with adverse pregnancy outcomes (APOs) [64,65]. A hemin-rich medium promotes the growth of TLR4 antagonist forms of P. gingivalis [25]. This may have consequences for the placenta, which is a blood-rich tissue with abundance of red blood cells. The placental environment may, therefore, be rich in tetra- and penta-acylated residues in the A-LPS of P. gingivalis [64]. The ability of P. gingivalis to generate two different isoforms of A-LPS that either activate or antagonize TLR4 could be particularly important to the maternal–foetal interface [64]. P. gingivalis dysregulation of the TLR function in uterus, placenta, and foetal membranes may promote APO [64–67]. In rats, LPS from P. gingivalis increased maternal blood pressure, induced placental and foetal growth restriction, and increased foetal resorptions, without inducing proteinuria and inflammation [68]. It is likely, therefore, that P. gingivalis LPS and/or A-LPS may play a role in pregnancy complications related to periodontal pathogens.
Concluding remarks
A-LPS of P. gingivalis is remarkably heterogeneous. This was originally discovered following various biochemical approaches used to extract LPS. The different isoforms of LPS varied biochemically because of the methodology utilized, and the environmental factors experienced during their culture, for example presence of hemin and temperature conditions. Analytical techniques were instrumental in deducing that P. gingivalis lipid A consists of multiple structures including a phosphorylated penta-acylated form and a phosphorylated tetra-acylated form. These species were absent in O-LPS. The fact that different strains of P. gingivalis displayed varying capacity to elicit host inflammatory responses also suggests that the structure of the major virulence factors O-LPS and A-LPS could differ. This property is reflected in their interactions with the PRRs. The variation could actually be one of multiple strategies that P. gingivalis uses to evade innate host defence in periodontal tissues, thereby contributing to periodontal pathogenesis.
The recognition that there are at least two different forms of P. gingivalis LPS, O-LPS and A-LPS, is significant. This is particularly important because A-LPS has been shown to have important effects on virulence through its effect on OMV production, serum resistance, and by serving as a matrix for deposition of µ-oxo-bisheme on the cell surface. The heterogeneous lipid A structures may also have distinct and opposing effects on TLR receptors playing a critical role in the early innate immune response to invading pathogens. Only subtle changes in lipid A structures are required to influence the host immune response to a large extent, and it has been speculated that P. gingivalis uses the ability of changing its lipid A composition to paralyze local pro-inflammatory cytokine production, thereby gaining access to periodontal tissue. Although the mechanisms that control the natural lipid A heterogeneity in P. gingivalis are not clear, they may exert a form of immunomodulation that helps P. gingivalis adapt and survive in different host environments and induce chronic inflammation.
Unfortunately, there is no evidence yet to demonstrate that a shift in lipid A classes occurs in subgingival plaque samples when comparing healthy/gingivitis or chronic periodontitis sites. This is a critical biological correlate required to be demonstrated before arguments about the importance of different lipid A classes can be accepted. Lipid A preparation from wild-type P. gingivalis contains unphosphorylated and phosphorylated lipid A constituents as well as tetra- and penta-acylated classes. Considering the documented opposing effects of these preparations, and the likelihood of the presence of multiple species within each isolate, it is not clear how the combinations of lipid A classes affect the engagement of innate immune receptors in its primary niche and/or at disparate sites. One possibility is certain, P. gingivalis is a ‘microbial wizard’ at adapting to tissue-specific cell receptor signal transduction pathways to colonise niches where growth conditions may be less than adequate albeit in vitro, in vivo animals, or the human host. This wizard’s wand may well be the heretogenity displayed in P. gingivalis lipid A structures.
Biographies
Ingar Olsen is professor emeritus and guest researcher at Department of Oral Biology, Faculty of Dentistry, University of Oslo. He is senior research investigator, Department of Microbiology, Forsyth Institute, Cambridge, MA; DDS from the Faculty of Dentistry, University of Oslo in 1966; Dr. Odont in 1976; professor in oral microbiology 1988; Dean for research 2002–2009, previously main supervisor of more than 20 Ph.D. students.
Sim K. Singhrao is a senior research fellow in the School of Dentistry, University of Central Lancashire. Singhrao has successfully supervised two Ph.D. studentships and has three current studentships.
Funding Statement
There is no external funding nevertheless; SKS wishes to thank the University of Central Clancashire for their continued financial support.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1]. Hajishengallis G, Darveau RP, Curtis MA.. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10(10):717–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]. How KY, Song KP, Chan KG. Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front Microbiol. 2016;7:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3]. Beutler B. Endotoxin, toll-like receptor 4, and the afferent limb of innate immunity. Curr Opin Microbiol. 2000;3(1):23–28. [DOI] [PubMed] [Google Scholar]
- [4]. Herath TD, Darveau RP, Seneviratne CJ, et al. Tetra- and penta-acylated lipid A structures of Porphyromonas gingivalis LPS differentially activate TLR4-mediated NF-κB signal transduction cascade and immuno-inflammatory response in human gingival fibroblasts. PLoS One. 2013;8(3):e58496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]. Reife RA, Shapiro RA, Bamber BA, et al. Porphyromonas gingivalis lipopolysaccharide is poorly recognized by molecular components of innate host defense in a mouse model of early inflammation. Infect Immun. 1995;63(12):4686–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6]. Rietschel ET, Brade H. Bacterial endotoxins. Sci Am. 1992;267(2):54–61. [DOI] [PubMed] [Google Scholar]
- [7]. Rietschel ET, Kirikae T, Schade FU, et al. Bacterial endotoxin: molecular relationships of structure to activity and function. Faseb J. 1994;8(2):217–225. [DOI] [PubMed] [Google Scholar]
- [8]. Dixon DR, Darveau RP. Lipopolysaccharide heterogeneity: innate host responses to bacterial modification of lipid a structure. J Dent Res. 2005;84(7):584–595. [DOI] [PubMed] [Google Scholar]
- [9]. Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol. 2003;3(2):169–176. [DOI] [PubMed] [Google Scholar]
- [10]. Nichols FC, Bajrami B, Clark RB, et al. Free lipid A isolated from Porphyromonas gingivalis lipopolysaccharide is contaminated with phosphorylated dihydroceramide lipids: recovery in diseased dental samples. Infect Immun. 2012;80(2):860–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11]. Kocgozlu L, Elkaim R, Tenenbaum H, et al. Variable cell responses to P. gingivalis lipopolysaccharide. J Dent Res. 2009;88(8):741–745. [DOI] [PubMed] [Google Scholar]
- [12]. Darveau RP, Belton CM, Reife RA, et al. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis . Infect Immun. 1998;66(4):1660–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Hirschfeld M, Ma Y, Weis JH, et al. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000. 15;165(2):618–622. [DOI] [PubMed] [Google Scholar]
- [14]. Hirschfeld M, Weis JJ, Toshchakov V, et al. Signaling by toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect Immun. 2001;69(3):1477–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]. Darveau RP, Pham TT, Lemley K, et al. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immun. 2004;72(9):5041–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]. Triantafilou M, Gamper FG, Lepper PM, et al. Lipopolysaccharides from atherosclerosis-associated bacteria antagonize TLR4, induce formation of TLR2/1/CD36 complexes in lipid rafts and trigger TLR2-induced inflammatory responses in human vascular endothelial cells. Cell Microbiol. 2007;90(8):2030–2039. [DOI] [PubMed] [Google Scholar]
- [17]. Jain S, Coats SR, Chang AM, et al. A novel class of lipoprotein lipase-sensitive molecules mediates Toll-like receptor 2 activation by Porphyromonas gingivalis . Infect Immun. 2013;81(4):1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. Lasica AM, Goulas T, Mizgalska D, et al. Structural and functional probing of PorZ, an essential bacterial surface component of the type-IX secretion system of human oral-microbiomic Porphyromonas gingivalis . Sci Rep. 2016;6:37708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19]. Lin J, Bi L, Yu X, et al. Porphyromonas gingivalis exacerbates ligature-induced, RANKL-dependent alveolar bone resorption via differential regulation of Toll-like receptor 2 (TLR2) and TLR4. Infect Immun. 2014;82(10):4127–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20]. Gibson FC 3rd, Ukai T, Genco CA. Engagement of specific innate immune signaling pathways during Porphyromonas gingivalis induced chronic inflammation and atherosclerosis. Front Biosci. 2008;13:2041–2059. [DOI] [PubMed] [Google Scholar]
- [21]. Ukai T, Yumoto H, Gibson FC 3rd, et al. Macrophage-elicited osteoclastogenesis in response to bacterial stimulation requires Toll-like receptor 2-dependent tumor necrosis factor-alpha production. Infect Immun. 2008;76(2):812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22]. Papadopoulos G, Weinberg EO, Massari P, et al. Macrophage-specific TLR2 signaling mediates pathogen-induced TNF-dependent inflammatory oral bone loss. J Immunol. 2013;190(3):1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23]. Paramonov N, Bailey D, Rangarajan M, et al. Structural analysis of the polysaccharide from the lipopolysaccharide of Porphyromonas gingivalis strain W50. Eur J Biochem. 2001;268(17):4698–4707. [DOI] [PubMed] [Google Scholar]
- [24]. Paramonov N, Rangarajan M, Hashim A, et al. Structural analysis of a novel anionic polysaccharide from Porphyromonas gingivalis strain W50 related to Arg-gingipain glycans. Mol Microbiol. 2005;58(3):847–863. [DOI] [PubMed] [Google Scholar]
- [25]. Al-Qutub MN, Braham PH, Karimi-Naser LM, et al. Hemin-dependent modulation of the lipid A structure of Porphyromonas gingivalis lipopolysaccharide. Infect Immun. 2006;74(8):4474–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26]. Rangarajan M, Aduse-Opoku J, Paramonov N, et al. Identification of a second lipopolysaccharide in Porphyromonas gingivalis W50. J Bacteriol. 2008;190(8):2920–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27]. Rangarajan M, Aduse-Opoku J, Hashim A, et al. LptO (PG0027) is required for lipid A 1-phosphatase activity in Porphyromonas gingivalis W50. J Bacteriol. 2017;199(11):pii: e00751–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28]. Ogawa T. Chemical structure of lipid A from Porphyromonas (Bacteroides) gingivalis lipopolysaccharide. FEBS Lett. 1993;332(1–2):197–201. [DOI] [PubMed] [Google Scholar]
- [29]. Kumada H, Haishima Y, Umemoto T, et al. Structural study on the free lipid A isolated from lipopolysaccharide of Porphyromonas gingivalis . J Bacteriol. 1995;177(8):22098–22106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]. Herath TD, Wang Y, Seneviratne CJ, et al. The expression and regulation of matrix metalloproteinase-3 is critically modulated by Porphyromonas gingivalis lipopolysaccharide with heterogeneous lipid A structures in human gingival fibroblasts. BMC Microbiol. 2013;13:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31]. Coats SR, Jones JW, Do CT, et al. Human Toll-like receptor 4 responses to P. gingivalis are regulated by lipid A 1- and 4ʹ-phosphatase activities. Cell Microbiol. 2009;11(11):1587–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32]. Curtis MA, Percival RS, Devine D, et al. Temperature-dependent modulation of Porphyromonas gingivalis lipid A structure and interaction with the innate host defenses. Infect Immun. 2011;79(3):1187–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33]. Zenobia C, Hasturk H, Nguyen D, et al. Porphyromonas gingivalis lipid A phosphatase activity is critical for colonization and increasing the commensal load in the rabbit ligature model. Infect Immun. 2014;82(2):650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Gibbons RJ, Macdonald JB. Hemin and vitamin K compounds as required factors for the cultivation of certain strains of Bacteroides melaninogenicus . J Bacteriol. 1960;85:164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35]. Shah HN, Bonnett R, Mateen B, et al. The porphyrin pigmentation of subspecies of Bacteroides melaninogenicus . Biochem J. 1979;180(1):45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. Bramanti TE, Holt SC. Roles of porphyrins and host iron transport proteins in regulation of growth of Porphyromonas gingivalis W50. J Bacteriol. 1991;173(22):7330–7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37]. Genco CA. Regulation of hemin and iron transport in Porphyromonas gingivalis . Adv Dent Res. 1995;9(1):41–47. [DOI] [PubMed] [Google Scholar]
- [38]. Schifferle RE, Shostad SA, Bayers-Thering MT, et al. Effect of protoporphyrin IX limitation on porphyromonas gingivalis . J Endod. 1996;22(7):352–355. [DOI] [PubMed] [Google Scholar]
- [39]. Olczak T, Simpson W, Liu X, et al. Iron and heme utilization in Porphyromonas gingivalis . FEMS Microbiol Rev. 2005;29(1):119–144. [DOI] [PubMed] [Google Scholar]
- [40]. Rangarajan M, Aduse-Opoku J, Paramonov NA, et al. Hemin binding by Porphyromonas gingivalis strains is dependent on the presence of A-LPS. Mol Oral Microbiol. 2017;32(5):365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41]. Lamont RJ, Chan A, Belton CM, et al. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. 1995;63(10):3878–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42]. Saito A, Inagaki S, Ishihara K. Differential ability of periodontopathic bacteria to modulate invasion of human gingival epithelial cells by Porphyromonas gingivalis . Microb Pathog. 2009;47(6):329–333. [DOI] [PubMed] [Google Scholar]
- [43]. Simpson W, Olczak T, Genco CA. Lysine-specific gingipain K and heme/hemoglobin receptor HmuR are involved in heme utilization in Porphyromonas gingivalis . Acta Biochim Pol. 2004;51(1):253–262. [PubMed] [Google Scholar]
- [44]. Trent MS, Stead CM, Tran AX, et al. Diversity of endotoxin and its impact on pathogenesis. J Endotoxin Res. 2006;12(4):205–223. [DOI] [PubMed] [Google Scholar]
- [45]. Herath TD, Wang Y, Seneviratne CJ, et al. Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity differentially modulates the expression of IL-6 and IL-8 in human gingival fibroblasts. J Clin Periodontol. 2011;38(8):694–701. [DOI] [PubMed] [Google Scholar]
- [46]. Lu Q, Darveau RP, Samaranayake LP, et al. Differential modulation of human β-defensins expression in human gingival epithelia by Porphyromonas gingivalis lipopolysaccharide with tetra- and penta-acylated lipid A structures. Innate Immun. 2009;15(6):325–335. [DOI] [PubMed] [Google Scholar]
- [47]. Reife RA, Coats SR, Al-Qutub M, et al. Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity: differential activities of tetra- and penta-acylated lipid A structures on E-selectin expression and TLR4 recognition. Cell Microbiol. 2006;8(5):857–868. [DOI] [PubMed] [Google Scholar]
- [48]. Berezow AB, Ernst RK, Coats SR, et al. The structurally similar, penta-acylated lipopolysaccharides of Porphyromonas gingivalis and Bacteroides elicit strikingly different innate immune responses. Microb Pathog. 2009;47(2):68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49]. Ding PH, Jin LJ. The role of lipopolysaccharide-binding protein in innate immunity: a revisit and its relevance to oral/periodontal health. J Periodontal Res. 2014;49(1):1–9. [DOI] [PubMed] [Google Scholar]
- [50]. Ding PH, Darveau RP, Wang CY, et al. 3LPS-binding protein and its interactions with P. gingivalis LPS modulate pro-inflammatory response and Toll-like receptor signaling in human oral keratinocytes. PLoS One. 2017;12(4):e0173223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51]. Ding PH, Wang CY, Darveau RP, et al. Nuclear factor-κB and p38 mitogen-activated protein kinase signaling pathways are critically involved in Porphyromonas gingivalis lipopolysaccharide induction of lipopolysaccharide-binding protein expression in human oral keratinocytes. Mol Oral Microbiol. 2013;28(2):129–141. [DOI] [PubMed] [Google Scholar]
- [52]. Slocum C, Coats SR, Hua N, et al. Distinct lipid A moieties contribute to pathogen-induced site-specific vascular inflammation. PLoS Pathog. 2014;10(7):e1004215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53]. Netea MG, van Deuren M, Kullberg BJ, et al. Does the shape of lipid A determine the interaction of LPS with Toll-like receptors? Trends Immunol. 2002;23(3):135–139. [DOI] [PubMed] [Google Scholar]
- [54]. Blasi I, Korostoff J, Dhingra A, et al. Variants of Porphyromonas gingivalis lipopolysaccharide alter lipidation of autophagic protein, microtubule-associated protein 1 light chain 3, LC3. Mol Oral Microbiol. 2016;31(6):486–500. [DOI] [PubMed] [Google Scholar]
- [55]. Bullon P, Cordero MD, Quiles JL, et al. Autophagy in periodontitis patients and gingival fibroblasts: unraveling the link between chronic diseases and inflammation. BMC Med. 2012;10:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56]. Poole S, Singhrao SK, Kesavalu L, et al. Determining the presence of periodontopathic virulence factors in short-term post-mortem Alzheimer’s disease brain tissue. J Alzheimers Dis. 2013;36:665–677. [DOI] [PubMed] [Google Scholar]
- [57]. Wu Z, Ni J, Liu Y, et al. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun. 2017;pii: S0889 1591 (17)30189–7 DOI: 10.1016/j.bbi.2017.06.002. [DOI] [PubMed] [Google Scholar]
- [58]. Paramonov N, Aduse-Opoku J, Hashim A, et al. Identification of the linkage between A-polysaccharide and the core in the A-lipopolysaccharide of Porphyromonas gingivalis W50. J Bacteriol. 2015;197(10):1735–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59]. Shoji M, Ratnayake DB, Shi Y, et al. Construction and characterization of a nonpigmented mutant of Porphyromonas gingivalis: cell surface polysaccharide as an anchorage for gingipains. Microbiology. 2002;148(Pt 4):1183–1191. [DOI] [PubMed] [Google Scholar]
- [60]. Shoji M, Sato K, Yukitake H, et al. Involvement of the Wbp pathway in the biosynthesis of Porphyromonas gingivalis lipopolysaccharide with anionic polysaccharide. Sci Rep. 2014;4:5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61]. Slaney JM, Curtis MA. Mechanisms of evasion of complement by Porphyromonas gingivalis . Front Biosci. 2008;13:188–196. [DOI] [PubMed] [Google Scholar]
- [62]. Slaney JM, Gallagher A, Aduse-Opoku J, et al. Mechanisms of resistance of Porphyromonas gingivalis to killing by serum complement. Infect Immun. 2006;74(9):5352–5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63]. Curtis MA, Thickett A, Slaney JM, et al. Variable carbohydrate modifications to the catalytic chains of the RgpA and RgpB proteases of Porphyromonas gingivalis W50. Infect Immun. 1999;67(8):3816–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64]. Reyes L, Phillips P, Wolfe B, et al. Porphyromonas gingivalis and adverse pregnancy outcome. J Oral Microbiol. 2017;10(1):1374153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65]. Daalderop LA, Wieland BV, Tomsin K, et al. Periodontal disease and pregnancy outcomes: overview of systematic reviews. JDR Clin Translational Res. 2017;XX(X):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66]. Gonzalez JM, Xu H, Ofori E, et al. Toll-like receptors in the uterus, cervix, and placenta: is pregnancy an immunosuppressed state? Am J Obstet Gynecol. 2007;197(3):296.e1–6. [DOI] [PubMed] [Google Scholar]
- [67]. Duriez M, Quillay H, Madec Y, et al. Human decidual macrophages and NK cells differentially express Toll-like receptors and display distinct cytokine profiles upon TLR stimulation. Front Microbiol. 2014;5:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68]. Kunnen A, van Pampus MG, Aarnoudse JG, et al. The effect of Porphyromonas gingivalis lipopolysaccharide on pregnancy in the rat. Oral Dis. 2014;20(6):591–601. [DOI] [PubMed] [Google Scholar]