

Metal elements represent some of the most fundamental chemical building blocks of life and are required to sustain the growth, development, and sustenance of all living organisms and ecosystems across the planet.1 Like other major chemical units in biology, metals are distributed in a heterogeneous fashion across biological systems in both a spatial and temporal manner. Depending on their function, select metals are enriched in specific locations within organisms, tissues, and cells, while being depleted in other locations. However, unlike organic biomolecules, metals cannot be synthesized at the location where they are used. Instead, they must be acquired from an organism’s environment, carefully trafficked to the correct tissue and cell, and loaded into the correct protein or other biological target.2–7 Moreover, too much or too little of a given metal nutrient in a given location can be detrimental to the living system.8–11
As metals are acquired, they may be part of the labile or static metal pool or both (Figure 1). The labile pool is the collection of metal ions in a cell that are weakly bound and can undergo kinetically appreciable ligand exchange, thereby moving between proteins and small molecules with relative ease. In contrast, the static metal pool is made up of ions that are tightly bound to ligands, usually proteins, and do not dissociate. Typically, the static metal pool is viewed as a thermodynamic sink and the final destination for metals traveling through the labile metal pool.12 The total metal pool is thus made up of the sum of the labile and static metal pools, and metal homeostasis is maintained by a balance of the acquisition and trafficking pathways with the excretion pathways for metalloproteins or other metal–ligand complexes. Disruption of any of these fundamental processes can lead to complex, multifaceted, and often widespread effects that are detrimental to health and development.8,13,14 As such, elucidating mechanisms of metal acquisition, mobilization, and/or sequestration is vitally important to understanding the contribution of metals to healthy and disease states within living systems.
Figure 1.

The total metal pool comprises the labile and static metal pools. Physical techniques that map the distribution of the total metal pool in cells, tissues, and organisms include technologies that measure atomic mass (LA-ICPMS and SIMS) and technologies that probe electronic structure (e.g., XFM, XAS, and EFTEM, among others). Fluorescent sensors map the labile metal pool. Representative data were reproduced from the following publications: CF3/Ctl-CF3 ± BCS: from Dodani, S. C.; Firl, A.; Chan, J.; Nam, C. I.; Aron, A. T.; Onak, C. S.; Ramos-Torres, K. M.; Paek, J.; Webster, C. M.; Feller, M. B.; Chang, C. J. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 16280–16285 (ref 164). Copyright 2016 National Academy of Sciences. CS790AM: from Hirayama, T.; Van de Bittner, G. C.; Gray, L. W.; Lutsenko, S.; Chang, C. J. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 2228–2233 (ref 170). Copyright 2012 National Academy of Sciences. CSR1: from Krishnamoorthy, L.; Cotruvo, J. A.; Chan, J.; Kaluarachchi, H.; Muchenditsi, A.; Pendyala, V. S.; Jia, S.; Aron, A. T.; Ackerman, C. M.; Vander Wal, M. N.; Guan, T.; Smaga, L. P.; Farhi, S. L.; New, E. J.; Lutsenko, S.; Chang, C. J. Nat. Chem. Biol. 2016, 12, 586–592 (ref 167). Copyright 2016 Nature Publishing Group. CS3: from Dodani, S. C.; Domaille, D. W.; Nam, C. I.; Miller, E. W.; Finney, L. A.; Vogt, S.; Chang, C. J. Proc. Natl. Acad. Sci. U.S.A. 2011, 29, 686–700 (ref 160). Copyright 2016 National Academy of Sciences. Trx-Puro: from Spangler, B.; Morgan, C. W.; Fontaine, S. D.; Vander Wal, M. N.; Chang, C. J.; Wells, J. A.; Renslo, A. R. Nat. Chem. Biol. 2016, 12, 680–685 (ref 138). Copyright 2016 Nature Publishing Group. FIP-1: from Aron, A. T.; Loehr, M. O.; Bogena, J.; Chang, C. J. J. Am. Chem. Soc. 2016, 138, 14338–14346 (ref 136). Copyright 2016 The American Chemical Society. LA-ICPMS: from Portbury, S. D.; Hare, D. J.; Sgambelloni, C.; Finkelstein, D. I.; Adlard, P. A. Metallomics 2016, 8, 193–200 (ref 61) with the permission of The Royal Chemical Society. SIMS: from Biesemeier, A.; Eibl, O.; Eswara, S.; Audinot, J.-N.; Wirtz, T.; Pezzoli, G.; Zucca, F. A.; Zecca, L.; Schraermeyer, U. J. Neurochem. 2016, 138, 339–353 (ref 71). Copyright 2016 Wiley. XFM and XAS: from James, S. A.; Roberts, B. R.; Hare, D. J.; de Jonge, M. D.; Birchall, I. E.; Jenkins, N. L.; Cherny, R. A.; Bush, A. I.; McColl, G. Chem. Sci. 2015, 6, 2952–2962 (ref 195) with the permission of The Royal Chemical Society. EFTEM: from Treiber, C. D.; Salzer, M. C.; Riegler, J.; Edelman, N.; Sugar, C.; Breuss, M.; Pichler, P.; Cadiou, H.; Saunders, M.; Lythgoe, M.; Shaw, J.; Keays, D. A. Nature 2012, 484, 367–370 (ref 110). Copyright 2016 Nature Publishing Group.
In this Review, we present an overview of analytical methods for imaging the distributions of metals in biological systems. Specifically, we highlight the power of using multiple complementary analytical techniques, in concert, to map total metal pools and distinguish which subsets of those total pools are static and tightly bound versus dynamic and weakly bound. To illustrate this general approach, we focus our attention on the two most abundant redox-active transition metals in living systems, iron and copper. Both iron and copper have been traditionally studied in bioinorganic chemistry as static cofactors that are tightly bound by metallochaperones and buried in protein active sites to protect cells against oxidative stress,15,16 but they now are emerging as dynamic transition metal signals that can reversibly affect the function of proteins in allosteric regions outside active sites. Applying a suite of analytical techniques to probe the quantity, location, and oxidation state of total and labile transition metal pools can reveal new principles that define the metallobiochemistry of metabolism and signaling.
In this context, available technologies for metal imaging can be divided into two categories. The first category comprises techniques for measuring total metal pools, including laser ablation inductively coupled plasma mass spectrometry (LA-ICPMS), secondary ion mass spectrometry (SIMS), X-ray fluorescence microscopy (XFM), X-ray absorbance spectroscopy (XAS), particle-induced X-ray emission (PIXE), and various electron microscopy (EM) methods (Figure 1). The second category comprises techniques for measuring labile metal pools using small-molecule and genetically encodable sensors and indicators (Figure 1). We survey the state-of-the-art for metal imaging technologies and conclude by highlighting select case studies to show how multiple complementary methods can be applied to study iron and copper pools over the various time scales that span transition metal metabolism to transition metal signaling.
TECHNOLOGIES FOR IMAGING TOTAL METAL POOLS
Multiple technologies have been applied to map the relative distribution of elements in biological samples.17,18 In each of these techniques, the sample is placed in the path of a high-energy beam (e.g., laser, ion, X-ray, or electron) and then moved relative to this source, so that the beam scans across the sample. As with all scanning techniques, trade-offs are made between analysis time, spatial resolution, extent of sample coverage, and sampling statistics at each location, which govern sensitivities and limits of detection. We point to an elegant recent tutorial review of the trade-offs of sensitivity, selectivity, and spatial resolution for metal imaging in biology.19
In this context, we provide a survey of the state-of-the-art for technologies that can be used to image total metal content in biological samples. These technologies can be divided into two categories: (1) methods that measure the mass of the atom using mass spectrometry and (2) methods that probe the electronic structure of that atom. These categories organize our discussion below, and we highlight considerations for each technique that are specific to copper and iron.
We begin with a few brief comments on sample preparation, a topic that is relevant to all total metal imaging methods. Since metals are not covalently anchored inside of cells, leaching and relocalization of these elements during sample preparation must be approached with caution.20–27 Best practices for minimal perturbation of metal localization include flash-freezing or freeze-drying samples.28,29 Additionally, for X-ray-based techniques that provide information about metal oxidation state and binding environments, radiation-induced reduction, as well as oxygen exposure leading to metal oxidation, which is of particular concern for iron and copper, could potentially introduce artifacts and confound interpretation.30–32
Technologies That Probe Atomic Mass
Mass spectrometry-based methods for metal mapping provide direct information about the metal isotope. However, these techniques cannot provide information about the oxidation state or coordination geometry of the metal in the tissue, since the metal is removed from the tissue and ionized during detection.
Laser Ablation Inductively-Coupled Plasma Mass Spectrometry (LA-ICPMS)
LA-ICPMS maps the concentration of individual isotopes in samples, most commonly tissue slices, making it a workhorse for imaging metals in biology (Figure 2A).33–37 Quadrupole, sector field,38 and time-of-flight (TOF)39 analyzers have all been used for LA-ICPMS to give single-isotope mass resolution (e.g., 56Fe vs 57Fe and 63Cu vs 65Cu). This capability has been particularly useful for isotope uptake studies.40–42 TOF or multicollector sector field mass spectrometers analyze multiple isotopes from exactly the same location and provide the highest precision isotope ratio mapping;43,44 however, the use of quadrupole mass spectrometers (Q-MS) for isotope ratio mapping is being explored,42,45,46 since Q-MS is the most common mass analyzer used for ICPMS.
Figure 2.

Schematic illustration and representative data for technologies that map metals by measuring atomic mass. (A) Schematic illustration of LA-ICPMS. (B) Schematic illustration of NanoSIMS. (C) LA-ICPMS was used in a longitudinal study of mouse brain slices. Using the healthy contralateral hemisphere as a control, changes in the metal content of the injured ipsilateral hemisphere were observed at multiple time points during the wound-healing process. Reproduced from Portbury, S. D.; Hare, D. J.; Sgambelloni, C.; Finkelstein, D. I.; Adlard, P. A. Metallomics 2016, 8, 193–200 (ref 61) with the permission of The Royal Chemical Society. (C) Representative data highlighting the use of NanoSIMS for colocalizing metal and nonmetal signals at subcellular resolution. PO2− marks lipid bodies, while S− marks neuromelanin. Copper localizes in a pattern that is most similar to S–, indicating that Cu associates with neuromelanin rather than lipid bodies within neurons. Reproduced from Biesemeier, A.; Eibl, O.; Eswara, S.; Audinot, J.-N.; Wirtz, T.; Pezzoli, G.; Zucca, F. A.; Zecca, L.; Schraermeyer, U. J. Neurochem. 2016, 138, 339–353 (ref 71). Copyright 2016 Wiley.
To achieve the highest signal for LA-ICPMS metal mapping, the most abundant isotope is typically monitored. For iron and copper, these are 56Fe, with 92% natural abundance, and 63Cu, with 69% natural abundance, respectively. However, polyatomic interferences present a formidable analytical challenge since Q-MS has relatively low mass-resolving power (typically <300 M/ΔM) and cannot discriminate elemental ions from polyatomic ions.47 For example, [40Ar16O]+ flies at the same mass-to-charge ratio as [56Fe]+ and Q-MS detectors cannot distinguish them. Because the plasma for ICPMS is based on argon, and biological samples contain significant amounts of oxygen, the background signal from [40Ar16O]+ is particularly detrimental to 56Fe imaging. Similar concerns exist for [40Ar23Na]+ interference with [63Cu]+ imaging. Recently, kinetic energy discrimination (KED) using H2 gas flow was developed to allow 56Fe imaging with Q-MS analyzers.48 Although the method dramatically decreases the overall signal, the ratio of signal/background is improved, and useful images have been obtained.
The raw output of LA-ICPMS is in units of counts per second, which may be converted to metal concentrations using standards. For biological samples, matrix-matched standards made from soft materials are preferred over glass standards, which are hard and exhibit different ablation properties compared to soft tissue. Standards are typically prepared by adding known amounts of metal to homogenized tissue49,50 or gelatin51 and subsequently slicing the standards to the same thickness as the tissue to be analyzed. Additionally, normalization to an internal standard isotope that is evenly distributed in biological tissue (such as 13C or a deposited thin layer of Au) can correct for variations in ablation efficiency across the sample.52,53 Approaches for quantification of LA-ICPMS data have been recently reviewed.54 Although detection limits as low as 0.01 μg g−1 have been reported for rare and precious elements (e.g., Th, U, and Ag),55,56 practical detection limits for earth-abundant metals such as copper and iron are in the range of 0.1–2 μg g−1 depending on experimental setup;38,48,50,56–58 this is primarily due to inherent differences in ionizability in the plasma, interferences from the gas background, and environmental contamination of the surfaces used for sample mounting (e.g., glass slides, double-sided tape).
An attractive feature of LA-ICPMS is the wide variety of samples that can be analyzed. Sample requirements are minimal and include being able to fit inside the sample chamber (standard commercial chambers are approximately 100 cm2 × 1 cm high) on a flat surface so that the sample surface remains in the focal plane of the beam during imaging. For tissue slices, a thickness of 20 μm or greater is sufficient to provide signal above the background, and a laser power that fully ablates the sample thickness without perturbing the rest of the sample provides the most consistent results.54,59 Thicker samples can also be used, and depth profiling by ablating through thick samples can provide useful information about changes in elemental concentration in the z-dimension.60 LA-ICPMS is a destructive technique, so any other imaging must be done before LA-ICPMS analysis.
Generally, the dimensions of the laser beam define the lateral spatial resolution of the image, although scan rates that under-or oversample the ablated area also contribute to spatial resolution.59 Typical beam diameters used for biological imaging are 5 μm–150 μm. This range of beam diameters makes LA-ICPMS quite versatile; large spot sizes provide a rapid and economical way of scanning large tissue sections or a high volume of samples (Figure 2C),61 while small spot sizes allow detailed analysis of a region of interest. Methods have been published for optimizing trade-offs between spatial resolution and acquisition time.62 Subcellular imaging of gold and silver nanoparticles and antibodies labeled with precious metals has been achieved,59,63–65 but subcellular resolution is not yet available for imaging of endogenous metals. Improvements in the limits of detection of LA-ICPMS may yield instruments capable of quantifying endogenous metals at subcellular resolution.58,64,66,67
Secondary Ion Mass Spectrometry (SIMS)
SIMS maps individual isotopes with higher mass resolution and higher spatial resolution than LA-ICPMS. Although matrix effects make absolute quantification challenging, the NanoSIMS instrument readily achieves subcellular imaging of endogenous metals and nonmetals.68–71 As such, NanoSIMS is exceptional for its ability to track isotope uptake, sequestration, and mobilization with subcellular resolution.70
In SIMS, the sample is affixed to a sample carrier which is pumped into a high-vacuum chamber at ambient temperature (custom-built cryocooled chambers exist but are not routinely used72). A primary ion pulse or reactive primary ion beam delivers charge to the sample, causing secondary ions to be emitted (Figure 2B). These secondary ions are focused into a mass spectrometer, and maps of ion counts are generated. Two basic types of SIMS include static SIMS and dynamic SIMS.69 Static SIMS uses an ion pulse and TOF detector; a full ion spectrum is collected with each pulse, allowing the investigator to view the relative levels of all species at each location.73 This technique is especially useful for exploratory work and profiling work, but the yield of secondary ions is inherently low in static SIMS, limiting its sensitivity. Dynamic SIMS uses a reactive ion beam with a sector field detector. The reactive ion beam sputters into the sample, embedding ions in the sample surface and dramatically increasing the yield of secondary ions, which improves the sensitivity of the instrument. However, the sector field detector collects only five or seven masses at a time (depending on the model), so the investigator must decide ahead of time which masses to monitor. Although these two techniques can be delineated, instruments designed for static SIMS can be run in dynamic mode, so the distinction is not absolute. In the area of metal imaging, dynamic SIMS provides the sensitivity necessary for high spatial resolution imaging. In particular, the CAMECA NanoSIMS 50 and 50L dynamic SIMS instruments have set the standard in the field for sensitivity and spatial resolution of SIMS imaging,68,69 and we focus our discussion on applications with this platform.
Like ICPMS, SIMS counts the number of ions of a specific mass. The mass resolving power of SIMS is much higher than typical ICPMS (~3500 M/ΔM at 100% transmission)74 and can thus discriminate elemental ions from polyatomic interferences, providing a clean readout of each isotope of interest. The sensitivity of SIMS depends on the type of ionizing beam used, and it varies from element to element. Secondary cations are generated by an anionic beam, so elements that readily adopt a positive charge, such as copper and iron, are easiest to map with an anionic primary ion beam (O− on the NanoSIMS). The converse is true of secondary anions, which are analyzed by a Cs+ beam on the NanoSIMS instrument. Although carbon, phosphorus, and sulfur are much less ionizable in an anionic primary beam, they are orders of magnitude more abundant in a biological sample than iron and copper, so images of these elements can be acquired in anionic mode. The carbon signal is used to normalize for matrix effects at different locations in the sample, and maps of other nonmetals can provide useful information about the elemental composition of areas of high metal concentration (Figure 2D).71 Absolute quantification of SIMS data is challenging because the generation of secondary ions is highly dependent on the matrix in which they are embedded. Although glass and metal standards are most frequently used, in an ideal case, matrix-matched standards for biological samples would provide a more accurate calibration curve. As such, recent studies mapping the distribution of metals in algae70 and neurons71 report data in counts-per-second rather than metal concentrations.
SIMS analysis is performed on relatively small biological samples (from single cells to a few cm2) due to the relatively small sample chamber. Since the lateral spatial resolution of NanoSIMS can routinely approach 100 nm, this technique excels in the analysis of single cells and subcellular metal localization. The samples must be plated on a conductive surface or coated with a thin conductive layer (e.g., Au) to diffuse potential buildup of charge from the ion beam. Additionally, the sample must be robust to high vacuum, which means biological samples must be dry. Because SIMS is a surface technique (accessing only the top 100–500 nm of the sample), samples as thin as 200 nm can be used.70 Thicker samples can be scanned at the surface, or depth profiling can be used to probe metal concentration within the sample.
Technologies That Probe Electronic Structure
A complementary set of technologies is based on measuring energy absorption and emission by the metal of interest. The amount of energy that is absorbed or emitted is characteristic of each element and reflects the energies of the element’s orbitals. These techniques cannot provide isotopic information, as they do not interact with the atomic nucleus. However, they do interact with the atom’s electronic structure and can provide information about the oxidation state and coordination geometry of the metal in its native environment, since the metal is not removed from the sample during analysis.
X-ray Fluorescence Microscopy (XFM)
XFM is the most commonly used technique for imaging iron and copper in biological systems. Although it requires the use of a synchrotron facility, many synchrotron facilities around the world have stations dedicated to biological investigation, and improvements in XFM hardware continue to enable new experimental systems to be pursued.75–78 In XFM, the sample is placed on a sample holder in the path of a high-intensity X-ray beam at ambient pressure and, traditionally, ambient temperature (Figure 3A). When an X-ray from the beam collides with an atom in the sample, energy is transferred from the X-ray to the atom. This energy transfer causes an electron from the inner shell of the atom to be ejected, leaving a hole. An outer-shell electron from the atom relaxes to fill the hole. The atom emits an X-ray, which is detected by an energy dispersive detector, allowing for simultaneous, multielement analysis.79 The energy of the emitted X-ray is the energy gap between the outer shell and inner shell orbitals involved, and it is characteristic of the atom that emits the X-ray. Because metals have multiple outer shells, multiple X-ray emission energies are possible. However, the most intense X-ray emission from a metal occurs at its Kα line, which is typically used for analysis. Kα denotes emission of an X-ray due to an electron moving into the 1s orbital (K shell) from the 2p orbital. The Kα lines for iron and copper are 6.404 and 8.048 keV, respectively.80,81 Fortunately, the fluorescence emission lines for the transition metals fall well outside the crowded emission region from lighter, more abundant elements (up to ~4 keV), allowing excellent quantification of iron and copper by XFM in biological samples (Figure 3B). With the use of spectral fitting, these elements can be quantified with limits of detection reaching 0.1–1 μg g−1.80
Figure 3.

Schematic illustration and representative data for technologies that map metals by probing electronic structure. (A) Schematic illustration of the basic setup for technologies that probe the electronic structure of metals. The high-energy beam is composed of X-rays for XFM and XAS; a particle beam is used for μ-PIXE; and an electron beam is used for EDX, STEM-EELS, and EFTEM. (B) An X-ray emission spectrum illustrating the location of the Fe and Cu Kα-lines, which are well-defined, lying outside the crowded low-energy region. Reproduced from Biesemeier, A.; Eibl, O.; Eswara, S.; Audinot, J.-N.; Wirtz, T.; Pezzoli, G.; Zucca, F. A.; Zecca, L.; Schraermeyer, U. J. Neurochem. 2016, 138, 339–353 (ref 71). Copyright 2016 Wiley. (C) φ-XANES maps of wildtype worms and worms lacking the iron storage protein ferritin; computationally defined ROIs are color coded according to their Fe2+/Fe3+ ratio. Cool colors indicate a low Fe2+/Fe3+ ratio, similar to ferritin (as shown in the spectral comparisons on the right), while warm colors indicate high Fe2+/Fe3+ ratios. Warmer pixels are more prevalent in worms lacking ferritin (percentages are illustrated in the ROI bar graphs on the right). Reproduced from James, S. A.; Hare, D. J.; Jenkins, N. L.; de Jonge, M. D.; Bush, A. I.; McColl, G. Sci. Rep. 2016, 6, 20350 (ref 76). Copyright 2016 Nature Publishing Group. (D) XFM images of iron-treated PC12 rat neural tumor cells overexpressing alpha-synuclein. While sulfur and zinc are relatively homogeneously distributed throughout the cell, iron is confined to subcellular puncta. DIC, differential interference contrast. Reproduced from Ortega, R.; Carmona, A.; Rodeau, S.; Perrin, L.; Dučić, T.; Carboni, E.; Bohic, S.; Cloetens, P.; Lingor, P. Mol. Neurobiol. 2016, 53, 1925–1934 (ref 89). Copyright 2016 Springer. (E) TEM (left) and EFTEM (right) images of ferritin molecules in macrophages. The dark spots in the TEM image correlate well with the high signal in the EFTEM Fe image, confirming a high iron concentration in these electron-dense puncta. Reproduced from Treiber, C. D.; Salzer, M. C.; Riegler, J.; Edelman, N.; Sugar, C.; Breuss, M.; Pichler, P.; Cadiou, H.; Saunders, M.; Lythgoe, M.; Shaw, J.; Keays, D. A. Nature 2012, 484, 367–370 (ref 110). Copyright 2016 Nature Publishing Group.
Quantification of XFM data can be achieved by calibration with known standards, such as metal thin films80 or organic standard reference materials, such as bovine liver.82 The resulting values have units of μg cm−2, which cannot be directly converted to μg g−1 unless the thickness and density of the sample is known. By assuming that the sample thickness reflects the slicing thickness during tissue preparation, and by using known tissue densities of similar tissues, these unit conversions have been extrapolated.83 However, in one comparison between XFM and LA-ICPMS in brain tissue, a systematic discrepancy in absolute metal quantification was observed, even though both techniques revealed similar metal maps. Values measured for copper using XFM were 60% of the values measured by LA-ICPMS, whereas XFM values for iron were 150% of the LA-ICPMS values.82 Nevertheless, both of these technologies provide excellent metal maps at their respective length scales and offer powerful tools for probing metal biology.
Sample preparation for XFM analysis is quite straightforward, since the only requirement is that the sample be mounted on a substrate that does not absorb or emit X-rays in the energy ranges to be analyzed. Typically, silicon nitride or Ultralene windows are used for cells and small tissue slices (typical tissue thickness is 10–30 μm).80 However, very large samples can also be analyzed, for example, slices of human heart (5 mm thick) sealed inside a polypropylene bag (30 μm thick plastic).84 Samples are commonly dried or fixed, although the development of cryocooled analysis chambers has permitted the analysis of frozen, unfixed, hydrated samples.85–87 Some beamlines have an optical setup to image larger samples, while others are optimized for smaller samples, such as single cells, and provide high spatial resolution images, down to 50 nm (Figure 3D).85,88,89 The Maia detector at the Australian Synchrotron is a notable exception, being designed to accommodate samples as large as 600 × 300 mm2 (analyzed, for example, at 100 μm resolution), while still achieving <1 μm resolution in other samples.75,76
Recent advances in XFM methodology have enabled the analysis of more structurally complex samples and provided insight into the cellular substructure in which metals are stored. For example, tomographic methods measure metal concentrations in intact 3-dimensional structures.77,90–93 Additionally, the combination of XFM with ptychography has allowed the unambiguous overlay of subcellular structure with subcellular element distribution, alleviating the longstanding problem of aligning transmitted light microscopy images with XFM images to assign subcellular metal localization.28
X-ray Absorbance Spectroscopy (XAS)
Like XFM, XAS requires the high-energy X-ray beam of a synchrotron X-ray facility. In XAS, however, the energy that is absorbed by the analyte is measured, rather than the energy that is emitted. Measuring the absorbed energy provides information about the oxidation state of the analyte and, potentially, its coordination geometry and ligands. XAS data can be collected in transmission mode or fluorescence mode. Because copper and iron are quite dilute in a biological sample, they cannot be detected in transmission mode. Thus, we discuss fluorescence-mode XAS here, which achieves detection limits in the low or sub μg g−1.80,94 The X-ray beam used for XAS has a defined, but variable, energy. During spectrum collection, the energy of the beam starts at a low energy and is increased, by small intervals, to higher and higher energies. When the energy of the beam matches the electron binding energy of a particular atom in the sample, an electron is ejected from the inner shell of the atom, leaving a hole. An outer-shell electron relaxes to fill the hole, emitting an X-ray. Because fluorescence emission is proportional to energy absorption, the number of X-rays that are emitted from the sample can be used as a proxy for the number of photons that were absorbed from the incident beam. Each beam energy stimulates X-ray emission from a specific element (or set of elements); the more X-rays that are emitted when the sample is exposed to a certain beam energy, the more atoms of the corresponding element there are in the sample. An element’s largest absorbance occurs at its K-edge.95 “K” denotes ejection of an electron from the 1s orbital (K shell), and the word “edge” is used because a large change in absorbance occurs at this energy, causing an “edge” in the spectrum. The Kedges for iron and copper occur at 7.112 and 8.993 keV, respectively.81 Two types of XAS have been delineated: X-ray absorption near edge structure (XANES), which is also called near-edge X-ray absorption fine structure (NEXAFS) and extended X-ray absorption fine structure (EXAFS). A XANES spectrum starts a few eV before the absorption edge of the element of interest and ends ~150 eV above the absorption edge; XANES provides information about the oxidation state of the atoms of that element. An EXAFS spectrum continues from 150 to 800 eV above the absorption edge and provides information about the coordination environment of the atoms of that element.96
The largest limitation to XAS imaging of biological samples has been sample damage due to extensive irradiation during image acquisition. A XANES spectrum for one element typically requires reading the absorption at 100 different beam energies, exposing each location in a sample to 100-times the radiation that it would normally receive during an XFM image.19 At these radiation doses, sample morphology is altered, and metals may be photoreduced, fundamentally interfering with the purpose of XANES.30,31 Two experimental approaches have been pursued to overcome this limitation. First, holding the sample at cryogenic temperatures minimizes sample damage.28,97 Second, exceptionally sensitive detectors have reduced the time necessary to acquire a XANES spectrum,75 demonstrating a 100-fold reduction in the total radiation used for image acquisition.76 This method, named φ-XANES, has allowed investigators to map the oxidation state of iron in living, anaesthetized C. elegans.
Typically, XAS data are analyzed by comparing the experimental spectrum to a spectrum of a known compound. However, because of the vast number of iron coordination environments present in a biological sample, this method of analysis can be difficult to apply effectively. With φ-XANES data, a new mode of analysis has become available. Principle component analysis was used to identify aggregate modes of iron coordination present in each φ-XANES image.98 These modes were computationally defined spectra, rather than being derived from the spectrum of a control compound. Each pixel of the image was assigned a color based on its aggregate mode of iron coordination (ie. its spectrum), and a map of coordination modes was generated. By color-coding the image according to the ratio of Fe2+/Fe3+ in each computationally defined coordination mode, an intuitive map of iron oxidation states in C. elegans was created (Figure 3C).76 Direct comparison of the maps from wildtype and ftn-2 worms lacking ferritin reveals a shift to a higher Fe2+/Fe3+ ratio throughout the worm, without altering the spatial arrangement of more-oxidizing and less-oxidizing environments within the worm (see section Metal Dynamics Over Long Time Scales: Transition Metal Nutrition and Aging for further discussion).
Micro Particle-Induced X-ray Emission (μ-PIXE)
Micro-PIXE uses a particle beam to stimulate X-ray emission. Thus, μ-PIXE requires the use of a particle accelerator, making it perhaps the least accessible technique for most metallobiology studies. The main advantage of PIXE is that it is quantitative without the need for standards. The backscattered and transmitted particles from the beam can be analyzed using Rutherford backscattering spectroscopy (RBS) and scanning transmission ion microscopy (STIM) to provide a measurement of the local sample mass. Analyte mass is normalized to sample mass, providing concentrations in units of μg g−1.99 PIXE operates in a vacuum, so samples must be dried. A sample thickness of 20–30 μm is sufficiently thin to allow STIM measurements of tissue density,100 and this thickness has been used for imaging fixed101 and freeze-dried100,102 biological samples mounted on thin plastic films. Using a beam size of 1–5 μm2, μ-PIXE achieves limits of detection for iron and copper that are 1–10 μg g−1.80,102
Electron Microscopy (EM)
EM methods for metal analysis use the same principles as XFM and XAS, but the primary beam is an electron beam. These methods can be performed on traditional electron microscopes, but these microscopes must be outfitted with correctly located energy dispersion detectors or energy filters, which may not be available in some EM facilities. Similar to XFM, energy dispersive X-ray spectroscopy (abbreviated EDX, EDS, EDXS, or XEDS) measures the X-rays emitted from the sample due to its interaction with the electron beam.103 Compared to XFM, the background signal for EDX is quite high. Inelastic electron interactions within the SEM generate a background signal (called Bremsstrahlung, which is German for “braking radiation”) that greatly diminishes sensitivity for heavy elements. Because of this background radiation, EDX is significantly less sensitive than XFM for iron and copper, providing limits of detection of 0.01 wt % (100 μg g−1) for these elements.104,105 However, EDX can be used to determine elemental composition of very small subcellular regions that are enriched for iron or copper, such as neuromelanin melanosomes71 and cataracts.106 Thus, EDX may provide by far the highest spatial resolution of all metal mapping techniques (~10 nm),107 but only for subcellular structures in which the local metal concentration exceeds 100 μg g−1.
Unlike EDX, electron energy loss spectroscopy (EELS) can be very sensitive for elements like P, Ca, and Fe,107 detecting as little as a single iron atom within purified hemoglobin.108 EELS is a transmission technique that measures the energy lost by electrons as they interact with electrons in the sample. Core losses (50–600 eV) are characteristic of specific elements and can be used to determine elemental composition at a given location in the sample.18 EELS images can be acquired by two techniques: scanning transmission electron microscopy EELS (STEM-EELS) or energy filtered transmission electron microscopy (EFTEM). STEM-EELS uses a scanning electron beam to record a full EELS spectrum at each location in the sample. Data acquisition is unbiased because a full EEL spectrum is acquired at each location, but a large dose of radiation is also administered to the sample, potentially causing significant sample damage.109 EFTEM uses a fixed beam to irradiate a large area of the sample and an energy filter to analyze only the electrons that correspond to an energy loss from the element of interest (after background subtraction). EFTEM greatly reduces the radiation dose received by the sample, but only a few energies are analyzed, biasing the analysis toward the elements that are expected to be in the sample. For an excellent comparison of these techniques, see da Cunha et al.18
Sample preparation for EELS is nontrivial, following the same process as sample preparation for any transmission electron microscopy (TEM) technique. Electron microscopy is performed in a vacuum, and electrons do not penetrate thick samples. Thus, samples must be fixed (by aldehyde fixation or high-pressure freezing), embedded in resin, and sectioned to very thin slices (50–100 nm). Typically, tissue sections are stained with heavy elements to increase contrast during imaging. However, contrast elements such as osmium and uranium must be avoided for iron analysis because of spectral interferences.18 EELS imaging has proven particularly powerful for the imaging of ferritin,110 the principle iron storage protein in mammalian cells (Figure 3E).111 Individual molecules of ferritin can be resolved within a tissue section, and the amount of iron loaded into each ferritin complex can be estimated based on the size of the iron particle that is visible by EM.112 Subramaniam and co-workers demonstrated that degenerating neurons of mice have lower ferritin inside their axons, while ferritin instead builds up outside the axon.113 Previous studies using light microscopy had suggested that neurodegeneration correlated with ferritin accumulation in neuronal axons; only electron microscopy provided the resolution to demonstrate that the increase in ferritin occurred outside of neuronal axons. Additionally, EFTEM tomography has enabled the visualization of 3-dimensional subcellular structure at unprecedented resolution, which may allow ferritin to be visualized within the subcellular context.114,115 As our understanding of the labile pool grows, EELS is sure to provide valuable information about the role of ferritin in iron sequestration and mobilization.116,117
TECHNOLOGIES FOR IMAGING LABILE IRON AND COPPER POOLS
Analytical methods for assessing labile metal pools118,119 can complement the suite of techniques for direct imaging of total metal pools. The labile metal pool consists of metal ions that are weakly bound to intracellular ligands, such that these ions can be rapidly removed or sequestered by competing metal chelators in the biological environment. Such metal pools may also undergo ligand exchange with fluorescent probes that respond to metal binding and/or reactivity with a change in fluorescence, enabling metal detection with spatial and temporal resolution. These probes can be reversible sensors or irreversible dosimeters. Desirable properties of an effective fluorescent metal probe include (1) high selectivity for the metal of interest, even in the presence of competing metals, other analytes in the cellular milieu, or local changes in pH, redox, and hydrophilicity/hydrophobicity, (2) a large turn-on increase or ratiometric wavelength shift in fluorescence to provide spatial information, in contrast to probes that turn off (i.e., disappear) in the presence of analyte, (3) compatibility with common microscopy laser lines and/or filter sets in terms of excitation/emission wavelengths, (4) visible, red-shifted spectral profiles to minimize sample photodamage and interference from native cellular autofluorescence in blue wavelengths, and (5) predictable localization in a given biological specimen. For redox-active metals such as iron and copper, an additional challenge is to avoid electron- and energy-transfer quenching pathways from transient odd-electron species that can arise from ground or excited states. We will restrict our discussion to probes for labile iron and copper pools that target the Fe(II) and Cu(I) oxidation states, which are dominant within the cell owing to the reducing intracellular environment.
We focus on synthetic small-molecule reagents, which have potential for broad application to many cell, tissue, and organism models as they do not require transfection or other manipulations to be introduced into a specimen. At the same time, however, the complexity of biological systems means that there is no one-size-fits-all probe for all systems, and each chemical reagent has to be tested and validated with proper controls in each biological setting and application. Indeed, potential confounding factors and artifacts can include increases and shifts in fluorescence signals due to accumulation, relocalization, or aggregation of dyes. As such, studies that employ imaging of labile metal pools benefit from biological controls with genetic and/or pharmacological manipulation, as well as complementary direct metal imaging methods. Additionally, the pursuit of ratiometric indicators with an internal standard and/or control probes that enable disentangling of dye- versus receptor-dependent signal changes is highly encouraged.
Fluorescent indicators fall into one of two basic categories: (1) recognition-based and (2) reaction-based (see Figure 4). Recognition-based probes respond to the reversible coordination of a metal to a receptor, whereas reaction-based probes bind a metal to trigger a chemical event that leads to a fluorescence change. Recognition-based detectors are valued for their reversibility but require careful matching of appropriate Kd values in order to avoid stripping and redistributing tightly bound metal pools. Reaction-based indicators can be valuable for detecting small changes in metal levels when the reaction is catalytic with respect to the metal, allowing the amplification of signal and integration of signal over time. Such indicators do not permanently bind the metal and thereby avoid perturbing the labile metal pool or undergoing metal-induced fluorescence quenching; however, after the reaction, diffusion of the probe away from the metal leads to a loss of spatial information. Although a wide variety of fluorescent iron and copper probes have been reported in the literature, only a limited subset of these diverse candidates has been satisfactorily characterized in cells, tissues, or animals with comparative images where pharmacological or biological treatments induce metal excess or deficiency, and our discussion focuses on these reagents.
Figure 4.
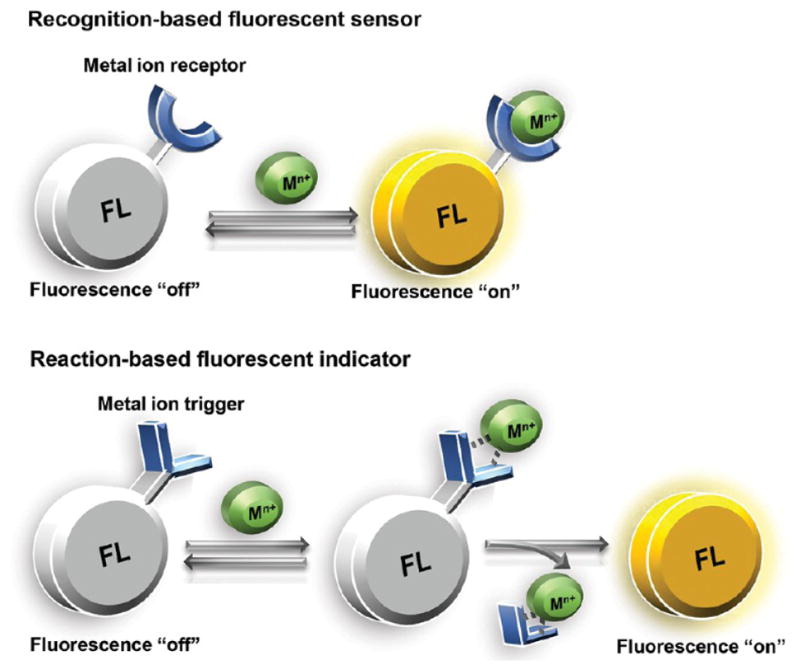
Illustration of recognition-based and reaction-based fluorescent sensors for metal ions. (A) Recognition-based turn-on sensors fluoresce when the metal is bound; they are reversible, turning off when the metal is released. (B) Reaction-based indicators fluoresce after a metal-catalyzed, irreversible chemical event. The fluorophore does not remain associated with the metal after the reaction takes place.
Fluorescent Probes for Labile Iron
Iron, the body’s most abundant transition metal element, presents unique challenges as an analyte, beyond the common challenges of imaging biological metals. This metal readily cycles between Fe2+ and Fe3+ under biological conditions, and although mounting evidence has suggested that the intracellular labile iron pool120 consists mainly of Fe2+,121 the redox activity of iron and its ability to adopt high- or low-spin configurations makes iron a potent potential fluorescence quencher by electron and energy transfer. Fe2+ is also a weakly coordinating metal on the Irving-Williams series, so developing effective receptors that can selectively bind this ion over competing Cu2+, Ni2+, and Zn2+ in particular, is difficult. During probe characterization, potential complications involving iron solubility122 or uncontrolled Fenton oxidations must be considered. As such, interest in visualizing Fe2+ in living systems is high, but developing selective and sensitive Fe2+-responsive probes remains a significant challenge.
Recognition-Based Iron Sensors
Recognition-based fluorescent sensors for turn-on detection of Fe2+ remain elusive, but several turn-off sensors have been employed to observe changes in labile Fe2+ levels in cell lysates, live cells, and even model organisms. The two most commonly used sensors, based on a fluorescein scaffold, are the commercially available Calcein and Phen Green SK dyes (Figure 5). Neither sensor shows high iron selectivity, as Calcein responds strongly to Cu2+, Co2+, and Ni2+ (>95% fluorescence decrease for each),123 and Phen Greek SK responds strongly to Cu+ and Cu2+ (97% and 70% decrease, respectively).124 Oxidation state specificity for Fe2+ over Fe3+ is also modest, as Calcein shows nearly quantitative fluorescence quenching in the presence of Fe2+ but 40–50% quenching in the presence of an equivalent amount of Fe3+. Despite the limited selectivity of Calcein and Phen Greek SK for Fe2+, iron-specific chelators can be used with these probes to identify changes in labile iron pools. Additional Fe2+ sensors include the pyridinone-based indicator CP655 (Figure 5) that exhibits improved selectivity for Fe2+ over other cations, with only Cu2+ presenting mild cross-recognition (42% decrease).125 However, CP655 is not selective for Fe2+ over Fe3+ and also shows pH sensitivity. Nevertheless, this reagent has been employed to probe labile iron with uniform cellular staining.126 Finally, RPA, RDA, and PIRO (Figure 5) are rhodamine-based fluorescent iron sensors that localize to the mitochondria owing to the positive charge delocalized over the fluorophore structure.127,128 This set of probes displays decreasing affinity for Fe2+ (RPA > RDA > PIRO), enabling visualization of endogenous iron (with RPA and RDA) or exogenous iron (with PIRO) depending on the biological situation. Each of the probes recognizes Cu2+ to some extent (RPA, 73% decrease; RDA, 87%; PIRO, 27%) but has good selectivity over other metals. The rhodamine-based iron sensors have identified a rise in mitochondrial labile iron when heme synthesis is blocked, with a control rhodamine 123 dye showing that mitochondrial membrane potential is not disrupted under these conditions.
Figure 5.

Structures of recognition-based (upper section) and reaction-based (lower section) Fe2+ probes.
Reaction-Based Iron Indicators
The growing palette of chemodosimeters for Fe2+ detection exploits the potent redox activity of this metal ion. A variety of mechanisms, including N-oxide deoxygenation, TEMPO radical reduction, oxygen-dependent oxidative C–O bond cleavage, and endoperoxide cleavage, have been reported. RhoNox-1 (Figure 5) was the first reaction-based Fe2+ probe to be used in a cellular system.129 This rhodamine-based probe relies on the ability of Fe2+ to selectively deoxygenate an N-oxide, converting it to a tertiary amine with a concomitant 30-fold fluorescence turn-on response. RhoNox-1 derivatives such as HMRhoNox-M (Figure 5), which displays a more stable pH profile and a 60-fold turn-on in fluorescence, has been used to visualize iron uptake via transferrin endocytosis130 and iron accumulation in ovarian endometriosis.131 A related rhodamine probe, Rh-T (Figure 5), contains a pendant paramagnetic TEMPO group that quenches fluorescence.132,133 Fe2+ reduces the TEMPO radical to a diamagnetic hydroxylamine, resulting in a 2.5-fold fluorescence turn-on with good selectivity for Fe2+ over other metal cations, as well as cellular reductants such as ascorbate and NADH. In human fibroblasts, Rh-T responds to the addition of exogenous iron, but its signal does not decrease in response to iron chelation.
Our laboratory published Iron Probe-1 (IP-1, Figure 5), a first-generation Fe2+ fluorescent indicator inspired by the oxidative reactivity of mononuclear nonheme iron enzymes.134 In the presence of Fe2+ and O2, IP-1 undergoes a C–O bond-cleavage reaction to release a reduced fluorescein alcohol derivative, resulting in a 6-fold fluorescence turn-on with high selectivity over competing metal ions in the cell. Only free Co2+ elicits a response from IP-1 in vitro, but more importantly, the probe does not respond to cyanocobalamin (vitamin B12), the biologically relevant form of cobalt in cells. In HepG2/C3A liver cells, IP-1 localizes to the lysosome, where it is able to sense both increases and decreases in iron levels from iron supplementation and chelation, respectively. Additionally, IP-1 was capable of detecting increases in labile iron following treatment with hepcidin or vitamin C, two natural compounds known to increase labile iron stores.
To improve upon the 3-component reaction of IP-1 and provide a direct reaction-based detector for Fe2+, we recently presented FIP-1 (Figure 5), a unique ratiometric fluorescent indicator for this metal ion. FIP-1 is a FRET-based probe that uses an endoperoxide trigger135 to cleave a linker between 5-aminomethyl coumarin and fluorescein in the presence of Fe2+.136 This direct and oxygen-independent 2-component reaction proceeds rapidly in the presence of Fe2+ and results in a 2-fold FRET change. Moreover, FIP-1 distributes evenly within cells and can clearly distinguish both increases and decreases in endogenous labile iron pools. Using FIP-1, we demonstrated that cancer cells possess higher levels of labile iron stores compared to noncancerous cells. Finally, we provided the first evidence of elevations in labile iron during the induction of ferroptosis,136 a form of iron-dependent cell death.137 A related puromycin-based endoperoxide probe (Trx-Puro, Figure 5) reveals that overexpression of ferritin or ferroportin, iron storage and iron export proteins, respectively, decreases labile iron stores in a variety of cancer cell models.138
Fluorescent Probes for Labile Copper
Like iron, copper can cycle between two oxidation states under biological conditions, Cu+ and Cu2+, presenting a selectivity challenge for probe design and characterization. Previous reports provide evidence that Cu+ is the dominant intracellular copper oxidation state for labile pools,139,140 although Cu+ is prone to disproportionation in water, which requires stabilization by appropriate ligands.141 Additionally, both oxidation states of copper are capable of quenching fluorescence, making the design of recognition-based probes particularly challenging as charge-transfer processes can generate transient Cu+ and Cu2+ species. As the field of fluorescence-based Cu+ probes has been thoroughly reviewed,119,142–144 we focus our discussion on probes that have been employed for biological application.
Recognition-Based Copper Sensors
The first sensor for Cu+ was introduced by Fahrni and co-workers in 2005.139 Using a triarylpyrazoline dye and macrocyclic thioether copper-binding motif, CTAP-1 gave a 4.6-fold fluorescent turn-on in the presence of excess Cu+, with no response or interference from other biologically relevant cations (Figure 6). In NIH 3T3 fibroblasts, CTAP-1 showed a significant fluorescence increase in cells cultured in high-copper media compared to basal media. Additionally, its signal showed good agreement with the subcellular distribution of total copper observed by XFM. Subsequent careful studies elucidated photophysical properties to improve signal-to-noise responses145–147 and provide updated CTAP-2 and CTAP-3 versions with improved hydrophilicity (Figure 6).148,149 In particular, CTAP-2, bearing four pendant hydroxyl groups on the thioether macrocycle, was capable of detecting the metalated Atox1 copper metallochaperone on a gel, and CTAP-3, bearing both hydroxyl and sulfonate groups, dissolves directly in water with no nanoparticle formation.
Figure 6.

Structures of recognition-based Cu+ sensors.
In parallel studies, our laboratory developed the first fluorescent copper sensors with visible excitation and emission profiles, which have enabled the study of copper homeostasis in a broad range of biological models used in our work as well as independent investigations by others. The first-generation BODIPY-based copper sensor, Coppersensor-1 (CS1, Figure 6), features a bis(2-((2-(ethylthio)ethyl)thio)ethyl)amine (BETA) receptor150,151 and shows high selectivity for Cu+ over all biologically relevant cations, with a 10-fold turn-on in the presence of Cu+ in vitro. Initial studies showed that CS1 can identify copper-loaded HEK 293 cells compared to control cells, and this work was validated by another study152 that also showed that this first-generation probe has different localization patterns in two other cell lines, M17 and U87MG. It is not surprising, with the complexity of biological systems, that there is not a one-size-fits-all chemical tool for all applications; as such, it is critical to implement both chemical and biological controls when using a given chemical probe for a given biological model. Indeed, with proper controls in place, CS1 has been employed as a pilot screening tool for assessing fluctuations in labile copper pools in bacteria,153 yeast,154–156 plant,157 and mammalian systems.158 Inspired by work by Nagano on treating fluorescent sensors as electron-transfer cassettes,159 we developed a next-generation Coppersensor-3 (CS3, Figure 6) probe by replacing the fluoro substituents on the BODIPY core with methoxy substituents to improve its brightness (Φ = 0.40 for CS3 vs Φ = 0.13 for CS1) and signal-to-noise response to Cu+ (75-fold turn-on for CS3 vs 10-fold for CS1).160 Notably, the more electron-rich BODIPY core also manifests itself in a tighter Cu+ Kd for CS3 (9 × 10−14 M vs 3 × 10−12 M for CS1). The combination of higher optical brightness, greater turn-on response to Cu+, and tighter Kd for CS3 has enabled its use for assessing basal pools of labile copper in a variety of cell types, including neurons,160 tumor cells,161 mouse fibroblasts,162 liver cells,163 and algae.70 These studies are bolstered by genetic and pharmacological controls as well as independent measures of the total copper pool.
Biological systems are complex mixtures of proteins, nucleic acids, glycans, lipids, and other organic species. Therefore, a balance between the hydrophobicity and hydrophilicity of any probe must be met for the use of a given probe for a given application. Indeed, in the case of copper sensors, similar to CTAP-1, BODIPY-based CS1 and CS3 do not homogeneously stain cells and thus are best suited for use in some biological models but not in others. In an effort to discriminate between dye-dependent fluorescent changes and metal-dependent fluorescent changes, we have introduced the concept of “synthetic mutagenesis” to create matched control probes. A first example is the development of Control Coppersensor-3 (Ctrl-CS3, Figure 6), which utilizes the same methoxy BODIPY core as CS3, but where the thioether sulfurs are replaced by isosteric carbons, which is akin to a methionine-to-alanine switch.70 Thus, Ctl-CS3 cannot bind to copper and does not turn on in the presence of Cu+. In Chlamydomonas reinhardtii, CS3 and Ctl-CS3 were used in pilot screening studies, in conjunction with direct metal imaging techniques, to identify the accumulation of copper in subcellular vesicular compartments, termed cuprosomes; the fluorescence of CS3 increased in the vesicles of experimentally treated cells compared to control cells, but the fluorescence of Ctl-CS3 did not.70 Neither probe responded to mutant algae with lipid-trafficking deficiencies, showing that fluorescent changes were not due to changes in the hydrophobicity of subcellular environments. Our hope is that continued development of control probes to use in conjunction with analyte-responsive probes will help with the interpretation of imaging data using such reagents.
To expand the palette of fluorescent copper probes to more hydrophilic cores with the goal of improving their use in more hydrophobic environments, such as thicker tissue and animal specimens, our laboratory reported a first-generation Copper Rhodol series: CR1–CR5.164 The most responsive of these sensors, Copper Rhodol 3 (CR3, Figure 6), gave a 13-fold turn-on response to Cu+. Replacement of the methyl substituent on the receptor-bearing aryl ring with a bulkier, more electron-withdrawing fluoromethyl substituent, gave Copper Fluor 3 (CF3, Figure 6) with a 40-fold turn-on response to Cu+. Partition coefficient measurements demonstrated that CR3 and CF3 were significantly more hydrophilic (log D = 0.96 and 1.15, respectively) than the BODIPY-based CS3 (log D = 3.46). Additionally, both CR3 and CF3 responded to copper selectively in the presence of model liposomes, proteins, glutathione, and cell lysates, whereas control probes based on these scaffolds (Ctl-CR3 and Ctl-CF3, Figure 6) did not respond to copper under similar conditions. These new reagents helped to identify an exchangeable pool of copper in developing hippocampal neurons and retinal slices, which regulates normal spontaneous activity in neural circuits.164
Replacement of the oxygen atom on the rhodol core with a silicon isostere165,166 led to the development of Copper Silicon Rhodol-1 (CSR1, Figure 6), a highly photostable fluorescent copper sensor that enables imaging of changes in copper pools in the same sample over long periods of time (Figure 6).167 CSR1 retains a selective and sensitive response to Cu+ (12-fold turn-on) on a hydrophilic probe (log D = 1.15) and was successfully used to monitor changes in labile copper pools in adipocytes, where it stained the cytosol but not lipid droplets. CSR1 discriminated cells pretreated with copper, chelator or vehicle, and it responded to on-stage addition of the membrane-permeable copper chelator, tris((ethylthio)ethyl)-amine (TEMEA). Finally, CSR1 revealed a decrease in labile copper in adipocytes upon stimulation of the beta-adrenergic receptor, concomitant with an increase in lipolysis. Fluorescence from the control probe Ctl-CSR1 (Figure 6) remained stable during parallel experiments, demonstrating the copper-specificity of CSR1 fluorescence in adipocytes. With these pilot imaging studies in hand, we went on to demonstrate that copper is an endogenous modulator of lipolysis through a cAMP signaling cascade where copper acts at the level of the cysteine 768 residue to reversibly inhibit the activity of phosphodiesterase PDE3B.167
The toolbox of fluorescent copper sensors continues to expand, including sensors with near-IR optical profiles for use in thicker tissue and whole-animal settings, as well as ratiometric and organelle-targeted sensors. For copper sensing in thicker tissue, ACu1 is a 2-photon probe by Cho and coworkers that excites at 750 nm in 2-photon mode (1-photon mode, 365 nm) (Figure 6).168 Localizing to both mitochondria and Golgi, ACu1 has been used to visualize copper in live hippocampal slices from rats. Additionally, Wan and co-workers published a Cy7 Cu+ sensor using the BETA receptor (structure 3 in Figure 6), which was used to visualize copper addition and ascorbate-triggered copper mobilization in MG63 osteosarcoma cells.169 Our laboratory developed a Cy7 Cu+ sensor, Coppersensor 790 acetoxymethyl ester (CS790AM, Figure 6), which enabled the first fluorescence imaging of labile copper pools in living mice.170 CS790AM displays a 17-fold turn-on to copper with a highly red-shifted optical profile (λabs = 760 nm, λem = 790 nm). When injected into mice, CS790AM revealed both increased copper from copper injection and decreased copper from injection of the copper-specific chelator ATN-224, the FDA-approved choline salt of tetrathiomolyb-date. Additionally, CS790AM could discriminate between wildtype mice and Wilson’s disease model mice, which lack the copper exporter ATP7B. Compared to wildtype, both the abdomens and isolated livers of ATP7B−/− mice exhibited higher fluorescence signal from CS790AM, indicating an accumulation of copper, which was confirmed by bulk ICPMS and online LC–MS/AA measurements on digested liver tissue.170
To image labile copper pools at the subcellular level, our laboratory reported Mitochondrial Coppersensor-1 (Mito-CS1, Figure 6),171 the first organelle-targetable copper sensor, by appending a triphenylphosphonium tag172 onto an asymmetric BODIPY scaffold173 as a cationic, lipophilic tag to localize the probe to the mitochondria based on mitochondrial membrane potential. Mito-CS1, in conjunction with other biochemical assays, revealed that cells prioritize mitochondria for copper homeostasis over other cellular compartments. This reagent enabled observation of expansion and depletion of the mitochondrial copper pool by copper supplementation and chelation. Interestingly, comparative studies in fibroblasts lacking the mitochondrial copper metallochaperones SCO1 and SCO2 and the copper export pump ATP7A showing that total and labile mitochondrial copper pools remain constant even in situations where whole-cell copper pools are altered. Important control experiments with the cationic dye rhodamine 123 demonstrated that mitochondrial membrane potential was not altered due to copper treatment or deletion of SCO1 or SCO2. A related water-soluble derivative of CS1, OBEP-CS1 (Figure 6), bears an alkylpyridinium group to drive it to the mitochondria in live cells but turns off in response to Cu+.174
Ratiometric probes are highly valued for their intrinsic internal standard that can correct for variations in dye localization and other experimental imaging conditions. Attachment of the BETA receptor to a naphthyl fluorophore yielded Naphthyl-CS1 (Figure 6), which localizes to both mitochondria and the Golgi apparatus in SH-SY5Y cells and was able to sense changes in copper status with copper supplementation.175 InCCu1, a ratiometric mitochondrial copper sensor developed by New and co-workers (Figure 6),176 can specifically monitor increases in mitochondrial copper upon supplementation. Moreover, InCCu1 was used to suggest that cisplatin interferes with copper transport to the mitochondria upon copper supplementation. Finally, Dns-LLC, a Golgi-targeted peptide-based sensor (Figure 6),177 shows a 12-fold turn-on in the presence of copper with an exceptionally tight binding affinity (12 fM); it responds to increases and decreases in Golgi copper levels following copper supplementation or chelation, respectively.
Reaction-Based Copper Indicators
Reaction-based approaches have proved useful for the development of fluorescent probes that go beyond traditional designs based on lock-and-key recognition.143,178–181 An elegant bioinspired approach to reaction-based Cu+ detection based on oxidative cleavage of the tetradentate ligand, tris[(2-pyridyl)methyl]amine (TPA), was reported by Taki and Yamamoto in 2010 (Figure 7).182 Upon Cu+ binding to the probe FluTPA1, oxidative C–O bond cleavage separates the TPA fragment from the fluorophore and releases the fluorescent dye with a 100-fold turn-on. FluTPA1 and its membrane-permeable FluTPA2 analogue, based on Tokyo Green,183 show good selectivity over other metal cations, as well as biological oxidants, including hydrogen peroxide, hypochlorite, and hydroxyl radical. FluTPA2 exhibits a notable fluorescence turn-on in HeLa cells after treatment with copper.182 The TPA ligand has subsequently been used to cage 2-(2′-hydroxyphenyl) benzothiazole,184 coumarin,185 xanthone,186 resorufin,187 cyanine-quinone,188 and imino-coumarin189 (Figure 7), although only the latter three have been applied to cells. Additionally, a mitochondrially targeted reaction-based Cu+ probe, RdlTPA-TPP (Figure 7), has been developed using the TPA ligand, circumventing the localization problem associated with this probe.190 We have expanded the scope of oxidative cleavage reactions to detect cobalt191 and iron.134 In addition, we have recently utilized the TPA trigger to develop the first bioluminescent probe for Cu+, Copper Caged Luciferin-1 (CCL-1), which enables the imaging of labile copper levels in cells and living animals.192 CCL-1 responds selectively to Cu+ in vivo with high signal-to-noise, and the combination of a small-molecule caged substrate and genetically encoded enzymatic reporter affords a platform for longitudinal imaging of the same living animal over time with cell- and tissue-specific resolution. In conjunction with biochemical and physiological assays, CCL-1 revealed a liver-specific copper deficiency that accompanies the onset of metabolic symptoms of glucose intolerance and weight gain in a diet-induced mouse model of nonalcoholic fatty liver disease (NAFLD).
Figure 7.

Structures and representative data from reaction-based indicators for Cu+. (A) All reaction-based triggers for Cu+ are based on the TPA trigger (designated “T”, in blue). This trigger has been appended to many small molecule reporters, including fluorescein (FluTPA1), Tokyo Green (FluTPA2), cyanine-quinone (TPACy), an imino-coumarin precursor (CP1), benzothiazole (HBTCu), coumarin (Probe 1), xanthone (XanCu), resorufin (ResCu), rhodol with a mitochondrial tag (RdlTPA-TPP), and, most recently, luciferin (CCL-1). (B) The use and mechanism-of-action of CCL-1 for imaging Cu+ in live animals is illustrated. (C) In mice expressing liver-specific luciferase, CCL-1 signal is observed only in the liver and is dependent on copper levels (top panel); its signal increases in response to copper supplementation with copper chloride and decreases in response to copper chelation with ATN-224, a derivative of tetrathiomolybdate. (C, Bottom panel) After 8 weeks of a high-fat diet, mice have lower CCL-1 liver signal than mice fed a control diet for 8 weeks, even though both groups of mice began the study with the same CCL-1 liver signal. Reproduced from Heffern, M. C.; Park, H. M.; Au-Yeung, H. Y.; Van de Bittner, G. C.; Ackerman, C. M.; Stahl, A.; Chang, C. J. Proc. Natl. Acad. Sci. U.S.A. 2016, in press, DOI: 10.1073/pnas.1613628113 (ref 192). Copyright 2016 Academy of Sciences.
APPLYING MULTIPLE ANALYTICAL IMAGING METHODS TO STUDY TRANSITION METALS IN BIOLOGY
The levels, localizations, and redox- and ligand-based speciations of copper and iron in biological systems change over time, as cells, tissues, and organisms grow, age, and respond to their environments. Techniques for visualizing total and/or labile metal pools can enable these changes to be mapped with spatial and temporal resolution and, thus, can aid in gaining insights into the roles of these redox-active metals over the wide range of time scales spanning signaling to metabolism to nutrition and aging. We emphasize the use of a combination of multiple techniques to address these exciting and open questions, as together they can provide a more holistic picture of transition metal dynamics, ranging from localization to oxidation state to bioavailability. Here, we highlight some recent representative examples of studies that combine multiple metal imaging methods to study and understand how copper and iron are sequestered and mobilized in biological systems in physiological and pathological situations.
Metal Dynamics Over Long Time Scales: Transition Metal Nutrition and Aging
Accumulation of iron in tissues as a function of age has been known since the early 1900s,193,194 but mechanisms by which iron acquisition contributes to senescence and death remain an active area of research. A combination of fluorescent probes and histochemical stains, along with advanced X-ray fluorescence and absorbance imaging techniques, has produced substantial progress toward addressing these questions. Recently, McColl, Bush, and co-workers examined iron accumulation and aging in C. elegans, specifically focusing on the role of ferritin in iron storage.195 Using XFM, the investigators observed an accumulation of iron in old worms (12 days) compared to young adult worms (4 days), particularly in the intestinal cells (Figure 8A). Control measurements show no change in calcium observed in the same time frame. Perl’s stain showed an age-dependent increase in nonheme iron, revealing iron accumulation specifically in intestinal cell nuclei, dense inclusions in the head, and the germ nuclei of postreproductive adults. The punctate iron distribution was confirmed by high-resolution XFM in a subsequent study.26 To determine whether the labile iron pool expanded with increased total iron, the investigators stained live worms with Calcein, which fluoresces less in the presence of labile iron. Indeed, Calcein fluorescence was lower in old worms than young worms, confirming an increase in labile iron with age (Figure 8B).195
Figure 8.

Iron accumulates, and the Fe2+/Fe3+ ratio increases, in aging worms. (A) XFM images reveal total iron accumulation, but no change in calcium levels, in old worms compared to young worms. Compton scattering provides anatomical structure. (B) Calcein staining is lower in older worms, indicating an increase in labile iron with aging. (C) XANES imaging shows a shift to higher Fe2+/Fe3+ ratios in older worms, as observed by a shift in the Fe K-edge to lower energies. Reproduced from James, S. A.; Roberts, B. R.; Hare, D. J.; de Jonge, M. D.; Birchall, I. E.; Jenkins, N. L.; Cherny, R. A.; Bush, A. I.; McColl, G. Chem. Sci. 2015, 6, 2952–2962 (ref 195) with the permission of The Royal Chemical Society.
Having demonstrated the expansion of both total and labile iron pools with age, the same researchers examined the role of ferritin, the main iron storage protein,111 during aging. Size exclusion chromatography experiments revealed that the fraction of iron bound to ferritin decreases as age increases, despite an overall increase in iron levels. While the labile iron pool consists mainly of Fe2+, iron is stored in ferritin as Fe3+; therefore, a decrease in the fraction of ferritin-bound iron suggests that the Fe2+/Fe3+ ratio may increase with age.195 Relative levels of Fe2+ and Fe3+ oxidation states in live, hydrated, anaesthetized C. elegans were mapped using fluorescence XANES (φ-XANES) microscopy,195 at a radiation dose demonstrated to preserve sample structure (Figure 8C).76 The iron K-edge position shifts to higher energies with oxidation, so its energy can be used to assess the relative fraction of Fe2+ and Fe3+ in a sample.196 Interestingly, young wildtype worms had K-edge positions that corresponded to a mixture of Fe2+ and Fe3+, whereas old wildtype worms had Kedge positions that were lower energy and thus shifted toward Fe2+. Taken together, these data suggest that the role of iron in aging is not simply defined by an overall increase in iron levels but includes a shift from Fe3+ to Fe2+ in vivo, linked to changes in ferritin loading.
Metal Dynamics Over Medium Time Scales: Transition Metal Metabolism
At shorter time scales within the span of minutes to hours, cells actively accumulate, store, mobilize, and efflux copper and iron under specific environmental conditions. Metal imaging experiments with high spatial resolution, using a variety of X-ray, mass spectrometry, and chemical probe methods, have played significant roles in identifying and characterizing intracellular structures for metal storage and mobilization.26,162,197,198 To provide one representative example, the single-cell eukaryotic model organism Chlamydomonas reinhardtii accumulates copper under conditions of environmental zinc deprivation, as demonstrated by bulk ICPMS measurements.199 Interestingly, this organism responds to zinc deprivation in a manner associated with intracellular copper deficiency by upregulating copper import machinery and downregulating the synthesis of proteins requiring copper cofactors such as plastocyanin.70 The mismatch between (1) a measurable accumulation of bulk total copper and (2) a functional response characteristic of low intracellular copper suggested that the copper pools accumulating inside these cells might be sequestered into compartments where they are not accessible to the cell’s copper-sensing machinery. To address this question, pilot imaging studies using the small-molecule probe CS3 revealed an increase in fluorescent puncta in zinc-deprived cells, suggesting that copper accumulates in distinct subcellular structures under conditions of zinc deprivation (Figure 9A).70 This increase in fluorescence was not observed using the control probe Ctrl-CS3. Further control experiments with copper supplementation or chelation, along with genetic manipulations of copper homeostasis machinery or lipid transport, all confirm that CS3 responds in this model in a copper-dependent fashion. These experiments led to the direct observation of copper and calcium accumulation in electron-dense structures, termed cuprosomes, using NanoSIMS (Figure 9B).
Figure 9.

The localization of copper changes on the time scale of hours, in order to respond to changes in nutrient availability. (A) In the model organism C. reinhardtii, the copper sensor CS3 reveals the accumulation of copper in subcellular structures, termed cuprosomes, under conditions of zinc starvation (– Zn) compared to control (+ Zn). Cuprosomes were not observed by the control sensor Ctl-CS3. Chl, chlorophyll autofluorescence; DIC, differential interference contrast. (B) By NanoSIMS, calcium and copper are observed within electron-dense structures along the cell periphery. NanoSIMS scale bars are metal counts normalized to carbon counts. Reproduced from Hong-Hermesdorf, A. M.; Miethke, M.; Gallaher, S. D.; Kropat, J.; Dodani, S. C.; Chan, J.; Barupala, D.; Domaille, D. W.; Shirasaki, D. I.; Loo, J. A.; Weber, P. K.; Pett-Ridge, J.; Stemmler, T. L.; Chang, C. J.; Merchant, S. S. Nat. Chem. Biol. 2014, 10, 1034–1042 (ref 70). Copyright 2016 Nature Publishing Group.
Moreover, sequestration of copper in these intracellular cuprosomes is reversible. Upon zinc resupply, fluorescent puncta from CS3 staining started to decrease after 3 h, and the fluorescence signal became more evenly distributed throughout the cell by 24 h. NanoSIMS imaging also showed a decrease in copper at sites of copper accumulation following zinc resupply. Further NanoSIMS analysis with isotope labeling showed that, upon restoration of zinc, copper from these compartments is incorporated into newly synthesized plastocyanin preferentially over copper from extracellular media. Additionally, cells moved from low-zinc media to low-copper media had a growth advantage over cells moved from zinc-replete media to low-copper media, suggesting that the copper accumulated during zinc deprivation becomes bioavailable and may provide an evolutionary advantage under conditions of fluctuating nutrient availability. This example adds to the growing literature concerning the identification of new protein machineries that influence the storage and mobilization of copper in intracellular compartments, including CTR1,200 CTR2,162,201 ATP7B,163 metallothionein, and Atox1.197
Metal Dynamics Over Short Time Scales: Transition Metal Signaling
In contrast to the wide acceptance of nonredox alkali and alkaline earth metals as mobile metal signals (e.g., Na+, K+, Ca2+), the localization of redox-active transition metals like copper and iron has been thought to be highly restricted to buried protein active sites and other inaccessible stores in order to protect cells, tissues, and organisms against oxidative stress and damage. However, more recent findings have expanded this traditionally narrow view of metals in biology to a new paradigm of transition metal signaling. As signaling elements, copper and iron can be rapidly mobilized in response to external stimuli in order to convey information. The fast and reversible binding of these elements to proteins and other biological targets outside active sites influences the function of these targets in downstream signaling cascades.
Our laboratory reported a foundational discovery in redox transition metal signaling by identifying fast copper relocalization within a cell, following an external stimulus, as shown using a primary neuronal cell model.160 A combination of XFM and imaging using the small-molecule fluorescent probe CS3 revealed dynamic mobilization of copper within seconds from neuronal cell bodies to dendritic spines following depolarization of dissociated hippocampal neurons with potassium chloride (Figure 10A). XFM control experiments showed that zinc did not relocalize under the same conditions. Further experiments using both imaging methods established that copper mobilization following membrane depolarization was calcium-dependent, suggesting crosstalk between copper and canonical cell signaling pathways. This work provided direct imaging evidence that complemented older studies on bulk copper release from synaptosomes202,203 and explants,204 as well as the movement of copper-trafficking proteins in neurons.205–207 Additionally, membrane-impermeable copper chelators disrupt neural function,164,208 which provides evidence for a functional role for copper in neuronal signaling.209 Indeed, a more recent study from our laboratory has characterized the functional significance of labile copper in neural circuits within intact tissue using fluorescent sensors for dual two-photon copper and calcium imaging (Figure 10B). The data show that copper is an endogenous regulator of spontaneous activity, a fundamental property of all neural circuits, by acting as a type of brake to avoid hyperactivity, via the Ctr1 copper ion channel and NMDA receptor.164
Figure 10.

Metals undergo dramatic relocalization on short time scales. The copper sensor CS3 was used to visualize the movement of copper in primary hippocampal neurons from (A, upper panel) cell bodies in resting neurons to (B, upper panel) dendritic spines upon neuronal depolarization with potassium chloride. (A, lower panel; B, lower panel) These results were confirmed by XFM. Reproduced from Dodani, S. C.; Domaille, D. W.; Nam, C. I.; Miller, E. W.; Finney, L. A.; Vogt, S.; Chang, C. J. Proc. Natl. Acad. Sci. U.S.A. 2011, 29, 686–700 (ref 160). Copyright 2016 National Academy of Sciences. (C, upper panel) The copper sensor CF3 revealed a loosely bound copper pool in retinal neurons, which could be rapidly depleted (C, lower panel) upon incubation with an extracellular copper chelator. (D, upper panel) The signal from the matched control probe, Ctl-CF3, was not altered (D, lower panel) by chelator treatment. Reproduced from Dodani, S. C.; Firl, A.; Chan, J.; Nam, C. I.; Aron, A. T.; Onak, C. S.; Ramos-Torres, K. M.; Paek, J.; Webster, C. M.; Feller, M. B.; Chang, C. J. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 16280–16285 (ref 164). Copyright 2016 National Academy of Sciences. All color schemes scale from cool colors (low signal) to warm colors (high signal) with arbitrary units of fluorescence counts.
The concept of copper as a representative example of a transition metal signal has been expanded beyond the brain by the identification of copper as an essential regulator of lipolysis, the breakdown of fat to control body weight and energy metabolism.167 Dynamic fluxes of copper in stimulated adipocytes, as imaged by the copper-responsive fluorescent probe CSR1, reversibly inhibit the enzyme PDE3B, a phosphodiesterase responsible for shutting down the lipolysis pathway by breakdown of the second messenger cAMP.167 Further biochemical studies characterized a key cysteine residue at an allosteric site outside the active site of PDE3B that modulates its copper-dependent function, linking transition metal signaling to a molecular target. These studies directly tie copper to cAMP/PDE, lipolysis, and metabolic processes related to obesity, and parallel studies have linked copper to other disease pathways such as cancer proliferation210–212 and heart disease.213–215 Interestingly, many of these disease pathways are interconnected. Indeed, obesity is a risk factor for diseases including diabetes, heart disease, cancer, and liver disease. Thus, these fields are ripe for further imaging studies to directly monitor fluctuations in copper in response to biological stimuli and to elucidate how copper may be used to transmit information at the cellular, tissue, and whole-organism levels.
CONCLUDING REMARKS
Metals pervade all aspects of the central dogma of biology, as they are required for the synthesis of all DNA and RNA molecules, as well as the proper function of approximately half of all proteins. Because metals can neither be created nor destroyed in biological systems, metal localization is governed by complex acquisition and excretion systems. The ubiquity, necessity, and unique homeostasis of metals drive a desire to study these biomolecules, and the electronic structure and coordination chemistry of metals provide unique chemical signatures that can be harnessed by physical techniques and small molecule probes to map the contributions of metals to the dynamic blueprint of life.
We have reviewed the growing collection of analytical methods to monitor both total and labile metal pools in their native biological contexts. These techniques, when used in concert, can provide complementary information on quantity, location, and oxidation state of a given metal over a wide range of length scales and time regimes, particularly spanning the shorter time scales of signaling to medium time scales of metabolism to longer time scales of nutrition and aging. A key principle is the open collaboration between chemistry and biology, where disparate communities will continue to work together to evaluate the best available set of chemical imaging tools for a given biological system and question. Multiple independent measurements and controls from both chemical and biological perspectives will push the field forward. Finally, an emerging theme of these investigations, applied to redox-active transition metals such as copper and iron, is the concept of transition metal signaling. In this burgeoning paradigm, these potent and reactive elements are not only viewed as static cofactors buried within protein active sites and other biomolecules to perform structural and catalytic chemistry, but they are also recognized as dynamic signals that can interact with targets outside active sites on faster time scales to affect function. The coming years are sure to illuminate more aspects of this bioinorganic chemistry and metallobiochemistry beyond the active site.
Acknowledgments
We thank the National Institutes of Health (Grant GM 79465) for supporting our work on metal imaging probes. C.M.A. has been supported by a Fannie and John Hertz Foundation Graduate Fellowship as well as by a Chemical Biology Training Grant from the NIH (Grant T32 GM066698). C.J.C. is an Investigator of the Howard Hughes Medical Institute.
Biographies
Cheri M. Ackerman is a Fannie and John Hertz Foundation Graduate Fellow in the Chemical Biology program at the University of California, Berkeley. She studies under the mentorship of Prof. Christopher Chang, focusing on methods for mapping and manipulating copper distribution in cells and animals. Originally from Holland, Michigan, Cheri received her B.S. in Biochemistry and Spanish from Calvin College in Grand Rapids, Michigan, where she researched the nuclear-cytoplasmic shuttling of galectin-3 as a Beckman Scholar in the laboratory of Prof. Eric Arnoys. She is currently the Webmaster for the Chemistry Graduate Life Committee at UCB, where she serves on subcommittees that promote healthy faculty-graduate student mentoring and increased diversity and inclusion in the chemistry department.
Sumin Lee was raised in Seoul, Korea. She received her B.S. and M.S. degrees from Ewha Womans University where she carried out research on transition metal sensors and photoelectrocatalysis with Wonwoo Nam. Currently, she is a graduate student in the group of Prof. Christopher Chang at the University of California, Berkeley. Her research includes the development of small molecule fluorescent sensors and porous polymers for transition metal detection.
Christopher J. Chang is the Class of 1942 Chair Professor in the Departments of Chemistry and Molecular and Cell Biology at UC Berkeley, Howard Hughes Medical Institute Investigator, and Faculty Scientist in the Chemical Sciences Division of Lawrence Berkeley National Laboratory. He is a Senior Editor of ACS Central Science. Chris received his B.S. and M.S. from Caltech in 1997, working with Harry Gray, studied as a Fulbright scholar in Strasbourg, France, with Nobel Laureate Jean-Pierre Sauvage, and received his Ph.D. from MIT in 2002 with Dan Nocera. After postdoctoral studies with Steve Lippard, Chris joined the UC Berkeley faculty in 2004. His laboratory focuses on chemical biology and inorganic chemistry, with particular interests in molecular imaging and catalysis applied to neuroscience and sustainable energy. His group’s work has been honored by many awards such as Dreyfus, Beckman, Sloan, and Packard Foundations, Amgen, Astra Zeneca, and Novartis, Technology Review, ACS (Cope Scholar, Eli Lilly, Nobel Laureate Signature, Baekeland), RSC (Transition Metal Chemistry), and SBIC. Most recently Chris received the 2015 Blavatnik Award in Chemistry.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. University Science Books; Mill Valley, CA: 1994. [Google Scholar]
- 2.van den Berghe PVE, Klomp LWJ. Nutr Rev. 2009;67:658–672. doi: 10.1111/j.1753-4887.2009.00250.x. [DOI] [PubMed] [Google Scholar]
- 3.Lutsenko S. Metallomics. 2016;8:840–852. doi: 10.1039/c6mt00176a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Printz B, Lutts S, Hausman JF, Sergeant K. Front Plant Sci. 2016;7:601. doi: 10.3389/fpls.2016.00601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milto IV, Suhodolo IV, Prokopieva VD, Klimenteva TK. Biochemistry (Moscow) 2016;81:549–564. doi: 10.1134/S0006297916060018. [DOI] [PubMed] [Google Scholar]
- 6.Kaplan JH, Maryon EB. Biophys J. 2016;110:7–13. doi: 10.1016/j.bpj.2015.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Festa RA, Thiele DJ. Curr Biol. 2011;21:R877–R883. doi: 10.1016/j.cub.2011.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gozzelino R, Arosio P. Int J Mol Sci. 2016;17:130. doi: 10.3390/ijms17010130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poujois A, Devedjian JC, Moreau C, Devos D, Chaine P, Woimant F, Duce JA. Curr Treat Options Neurol. 2016;18:46. doi: 10.1007/s11940-016-0426-1. [DOI] [PubMed] [Google Scholar]
- 10.Wang P, Wang Z-Y. Ageing Res Rev. 2016 doi: 10.1016/j.arr.2016.10.003. [DOI] [Google Scholar]
- 11.Bashir K, Rasheed S, Kobayashi T, Seki M, Nishizawa NK. Front Plant Sci. 2016;7:1192. doi: 10.3389/fpls.2016.01192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banci L, Bertini I, Ciofi-Baffoni S, Kozyreva T, Zovo K, Palumaa P. Nature. 2010;465:645–648. doi: 10.1038/nature09018. [DOI] [PubMed] [Google Scholar]
- 13.Lenartowicz M, Krzeptowski W, Lipiński P, Grzmil P, Starzyński R, Pierzchała O, M⊘ller LB. Front Mol Neurosci. 2015;8:72. doi: 10.3389/fnmol.2015.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu X, Leegwater P, Fieten H. Int J Mol Sci. 2016;17:196. doi: 10.3390/ijms17020196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubino JT, Franz KJ. J Inorg Biochem. 2012;107:129–143. doi: 10.1016/j.jinorgbio.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 16.Baker HM, Anderson BF, Baker EN. Proc Natl Acad Sci U S A. 2003;100:3579–3583. doi: 10.1073/pnas.0637295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin Z, Caruso JA, Lai B, Matusch A, Becker JS. Metallomics. 2011;3:28–37. doi: 10.1039/c0mt00048e. [DOI] [PubMed] [Google Scholar]
- 18.da Cunha MML, Trepout S, Messaoudi C, Wu TD, Ortega R, Guerquin-Kern JL, Marco S. Micron. 2016;84:23–36. doi: 10.1016/j.micron.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Hare DJ, New EJ, de Jonge MD, McColl G. Chem Soc Rev. 2015;44:5941–5958. doi: 10.1039/c5cs00055f. [DOI] [PubMed] [Google Scholar]
- 20.Al-Ebraheem A, Dao E, Desouza E, Li C, Wainman BC, McNeill FE, Farquharson MJ. Physiol Meas. 2015;36:N51–N60. doi: 10.1088/0967-3334/36/3/N51. [DOI] [PubMed] [Google Scholar]
- 21.Colvin RA, Jin Q, Lai B, Kiedrowski L. PLoS One. 2016;11:e0159582. doi: 10.1371/journal.pone.0159582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hare DJ, Gerlach M, Riederer P. J Neural Transm. 2012;119:1515–1521. doi: 10.1007/s00702-012-0898-4. [DOI] [PubMed] [Google Scholar]
- 23.Matsuyama S, Shimura M, Fujii M, Maeshima K, Yumoto H, Mimura H, Sano Y, Yabashi M, Nishino Y, Tamasaku K, Ishizaka Y, Ishikawa T, Yamauchi K. X-Ray Spectrom. 2010;39:260–266. [Google Scholar]
- 24.James SA, Myers DE, de Jonge MD, Vogt S, Ryan CG, Sexton BA, Hoobin P, Paterson D, Howard DL, Mayo SC, Altissimo M, Moorhead GF, Wilkins SW. Anal Bioanal Chem. 2011;401:853–864. doi: 10.1007/s00216-011-4978-3. [DOI] [PubMed] [Google Scholar]
- 25.Hackett MJ, McQuillan JA, El-Assaad F, Aitken JB, Levina A, Cohen DD, Siegele R, Carter EA, Grau GE, Hunt NH, Lay PA. Analyst. 2011;136:2941–2952. doi: 10.1039/c0an00269k. [DOI] [PubMed] [Google Scholar]
- 26.Hare DJ, Jones MWM, Wimmer VC, Jenkins NL, de Jonge MD, Bush AI, McColl G. Metallomics. 2016;8:156–160. doi: 10.1039/c5mt00288e. [DOI] [PubMed] [Google Scholar]
- 27.Mizuhira V, Hasegawa H, Notoya M. Prog Histochem Cytochem. 2000;35:68–183. [PubMed] [Google Scholar]
- 28.Deng J, Vine DJ, Chen S, Nashed YSG, Jin Q, Phillips NW, Peterka T, Ross R, Vogt S, Jacobsen CJ. Proc Natl Acad Sci U S A. 2015;112:2314–2319. doi: 10.1073/pnas.1413003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feldmann, Kindness A, Ek P. J Anal At Spectrom. 2002;17:813–818. [Google Scholar]
- 30.George SJ, Fu J, Guo Y, Drury OB, Friedrich S, Rauchfuss T, Volkers PI, Peters JC, Scott V, Brown SD, Thomas CM, Cramer SP. Inorg Chim Acta. 2008;361:1157–1165. [Google Scholar]
- 31.Yano J, Kern J, Irrgang KD, Latimer MJ, Bergmann U, Glatzel P, Pushkar Y, Biesiadka J, Loll B, Sauer K, Messinger J, Zouni A, Yachandra VK. Proc Natl Acad Sci U S A. 2005;102:12047–12052. doi: 10.1073/pnas.0505207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowman SEJ, Bridwell-Rabb J, Drennan CL. Acc Chem Res. 2016;49:695–702. doi: 10.1021/acs.accounts.5b00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Becker JS. J Mass Spectrom. 2013;48:255–268. doi: 10.1002/jms.3168. [DOI] [PubMed] [Google Scholar]
- 34.Becker JS, Matusch A, Wu B. Anal Chim Acta. 2014;835:1–18. doi: 10.1016/j.aca.2014.04.048. [DOI] [PubMed] [Google Scholar]
- 35.Sussulini A, Becker JS, Becker JS. Mass Spectrom Rev. 2015 doi: 10.1002/mas.21481. [DOI] [Google Scholar]
- 36.Konz I, Fernández B, Fernández ML, Pereiro R, Sanz-Medel A. Anal Bioanal Chem. 2012;403:2113–2125. doi: 10.1007/s00216-012-6023-6. [DOI] [PubMed] [Google Scholar]
- 37.Pozebon D, Scheffler GL, Dressler VL, Nunes MAG. J Anal At Spectrom. 2014;29:2204–2228. [Google Scholar]
- 38.Becker JS, Zoriy M, Matusch A, Wu B, Salber D, Palm C, Becker JS. Mass Spectrom Rev. 2009;29:156–175. doi: 10.1002/mas.20239. [DOI] [PubMed] [Google Scholar]
- 39.Kindness A. Clin Chem. 2003;49:1916–1923. doi: 10.1373/clinchem.2003.022046. [DOI] [PubMed] [Google Scholar]
- 40.Fosset C, McGaw BA, Reid MD, McArdle HJ. J Inorg Biochem. 2005;99:1018–1022. doi: 10.1016/j.jinorgbio.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 41.Bondanese VP, Lamboux A, Simon M, Lafont JE, Albalat E, Pichat S, Vanacker JM, Telouk P, Balter V, Oger P, Albarède F. Metallomics. 2016;8:1177–1184. doi: 10.1039/c6mt00102e. [DOI] [PubMed] [Google Scholar]
- 42.Flórez MR, Aramendía M, Resano M, Lapeña AC, Balcaen L, Vanhaecke F. J Anal At Spectrom. 2013;28:1005–1015. [Google Scholar]
- 43.Barbaste M, Halicz L, Galy A, Medina B, Emteborg H, Adams CF, Lobinski R. Talanta. 2001;54:307–317. doi: 10.1016/s0039-9140(00)00651-2. [DOI] [PubMed] [Google Scholar]
- 44.Vanhaecke F, Balcaen L, Malinovsky D. J Anal At Spectrom. 2009;24:863–886. [Google Scholar]
- 45.Urgast DS, Ou O, Gordon MJ, Raab A, Nixon GF, Kwun IS, Beattie JH, Feldmann J. Anal Bioanal Chem. 2012;402:287–297. doi: 10.1007/s00216-011-5461-x. [DOI] [PubMed] [Google Scholar]
- 46.Wehe CA, Thyssen GM, Herdering C, Raj I, Ciarimboli G, Sperling M, Karst U. J Am Soc Mass Spectrom. 2015;26:1274–1282. doi: 10.1007/s13361-015-1141-y. [DOI] [PubMed] [Google Scholar]
- 47.U.S. Geological Survey. [accessed November 18, 2016];High Resolution ICP-MS Laboratory. http://crustal.usgs.gov/laboratories/icpms/high_resolution.html.
- 48.Lear J, Hare DJ, Fryer F, Adlard PA, Finkelstein DI, Doble PA. Anal Chem. 2012;84:6707–6714. doi: 10.1021/ac301156f. [DOI] [PubMed] [Google Scholar]
- 49.Becker JS, Zoriy MV, Pickhardt C, Palomero-Gallagher N, Zilles K. Anal Chem. 2005;77:3208–3216. doi: 10.1021/ac040184q. [DOI] [PubMed] [Google Scholar]
- 50.Hare DJ, Lear J, Bishop D, Beavis A, Doble PA. Anal Methods. 2013;5:1915–1921. [Google Scholar]
- 51.Van Malderen SJM, Vergucht E, De Rijcke M, Janssen C, Vincze L, Vanhaecke F. Anal Chem. 2016;88:5783–5789. doi: 10.1021/acs.analchem.6b00334. [DOI] [PubMed] [Google Scholar]
- 52.Austin C, Fryer F, Lear J, Bishop D, Hare D, Rawling T, Kirkup L, McDonagh A, Doble P. J Anal At Spectrom. 2011;26:1494–1494. [Google Scholar]
- 53.Frick DA, Günther D. J Anal At Spectrom. 2012;27:1294–1303. [Google Scholar]
- 54.Limbeck A, Galler P, Bonta M, Bauer G, Nischkauer W, Vanhaecke F. Anal Bioanal Chem. 2015;407:6593–6617. doi: 10.1007/s00216-015-8858-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Becker JS, Zoriy MV, Dehnhardt M, Pickhardt C, Zilles K. J Anal At Spectrom. 2005;20:912–917. [Google Scholar]
- 56.Dressler VL, Pozebon D, Mesko MF, Matusch A, Kumtabtim U, Wu B, Sabine Becker J. Talanta. 2010;82:1770–1777. doi: 10.1016/j.talanta.2010.07.065. [DOI] [PubMed] [Google Scholar]
- 57.Phung AT, Baeyens W, Leermakers M, Goderis S, Vanhaecke F, Gao Y. Talanta. 2013;115:6–14. doi: 10.1016/j.talanta.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 58.Bishop DP, Clases D, Fryer F, Williams E, Wilkins S, Hare DJ, Cole N, Karst U, Doble PA. J Anal At Spectrom. 2016;31:197–202. [Google Scholar]
- 59.Mueller L, Traub H, Jakubowski N, Drescher D, Baranov VI, Kneipp J. Anal Bioanal Chem. 2014;406:6963–6977. doi: 10.1007/s00216-014-8143-7. [DOI] [PubMed] [Google Scholar]
- 60.Konz I, Fernandez B, Fernández ML, Pereiro R, González-Iglesias H, Coca-Prados M, Sanz-Medel A. Anal Bioanal Chem. 2014;406:2343–2348. doi: 10.1007/s00216-014-7617-y. [DOI] [PubMed] [Google Scholar]
- 61.Portbury SD, Hare DJ, Sgambelloni C, Finkelstein DI, Adlard PA. Metallomics. 2016;8:193–200. doi: 10.1039/c5mt00234f. [DOI] [PubMed] [Google Scholar]
- 62.Lear J, Hare D, Adlard P, Finkelstein D, Doble P. J Anal At Spectrom. 2012;27:159–164. [Google Scholar]
- 63.Drescher D, Giesen C, Traub H, Panne U, Kneipp J, Jakubowski N. Anal Chem. 2012;84:9684–9688. doi: 10.1021/ac302639c. [DOI] [PubMed] [Google Scholar]
- 64.Wang HAO, Grolimund D, Giesen C, Borca CN, Shaw-Stewart JRH, Bodenmiller B, Günther D. Anal Chem. 2013;85:10107–10116. doi: 10.1021/ac400996x. [DOI] [PubMed] [Google Scholar]
- 65.Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schüffler PJ, Grolimund D, Buhmann JM, Brandt S, Varga Z, Wild PJ, Günther D, Bodenmiller B. Nat Methods. 2014;11:417–422. doi: 10.1038/nmeth.2869. [DOI] [PubMed] [Google Scholar]
- 66.Douglas DN, Managh AJ, Reid HJ, Sharp BL. Anal Chem. 2015;87:11285–11294. doi: 10.1021/acs.analchem.5b02466. [DOI] [PubMed] [Google Scholar]
- 67.Van Malderen SJM, Managh AJ, Sharp BL, Vanhaecke F. J Anal At Spectrom. 2016;31:423–439. [Google Scholar]
- 68.Herrmann AM, Ritz K, Nunan N, Clode PL, Pett-Ridge J, Kilburn MR, Murphy DV, O’Donnell AG, Stockdale EA. Soil Biol Biochem. 2007;39:1835–1850. [Google Scholar]
- 69.Boxer SG, Kraft ML, Weber PK. Annu Rev Biophys. 2009;38:53–74. doi: 10.1146/annurev.biophys.050708.133634. [DOI] [PubMed] [Google Scholar]
- 70.Hong-Hermesdorf A, Miethke M, Gallaher SD, Kropat J, Dodani SC, Chan J, Barupala D, Domaille DW, Shirasaki DI, Loo JA, Weber PK, Pett-Ridge J, Stemmler TL, Chang CJ, Merchant SS. Nat Chem Biol. 2014;10:1034–1042. doi: 10.1038/nchembio.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Biesemeier A, Eibl O, Eswara S, Audinot J-N, Wirtz T, Pezzoli G, Zucca FA, Zecca L, Schraermeyer U. J Neurochem. 2016;138:339–353. doi: 10.1111/jnc.13648. [DOI] [PubMed] [Google Scholar]
- 72.Dickinson M, Heard PJ, Barker JHA, Lewis AC, Mallard D, Allen GC. Appl Surf Sci. 2006;252:6793–6796. [Google Scholar]
- 73.Vickerman J, Winograd N. Cluster TOF-SIMS Imaging and the Characterization of Biological Materials. In: Mahoney CM, editor. Cluster Secondary Ion Mass Spectrometry: Principles and Applications. John Wiley & Sons, Inc; Hoboken, NJ: 2013. pp. 269–312. [Google Scholar]
- 74.CAMECA. [accessed November 18, 2016];NanoSIMS: Introduction to the instrumentation. 2016 http://www.cameca.com/literature/product-brochures.aspx.
- 75.Ryan CG, Siddons DP, Kirkham R, Li ZY, de Jonge MD, Paterson DJ, Kuczewski A, Howard DL, Dunn PA, Falkenberg G, Boesenberg U, De Geronimo G, Fisher LA, Halfpenny A, Lintern MJ, Lombi E, Dyl KA, Jensen M, Moorhead GF, Cleverley JS, Hough RM, Godel B, Barnes SJ, James SA, Spiers KM, Alfeld M, Wellenreuther G, Vukmanovic Z, Borg S. J Phys Conf Ser. 2014;499:012002. [Google Scholar]
- 76.James SA, Hare DJ, Jenkins NL, de Jonge MD, Bush AI, McColl G. Sci Rep. 2016;6:20350. doi: 10.1038/srep20350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bourassa D, Gleber S-C, Vogt S, Shin CH, Fahrni CJ. Metallomics. 2016;8:1122–1130. doi: 10.1039/c6mt00073h. [DOI] [PubMed] [Google Scholar]
- 78.Chen S, Paunesku T, Yuan Y, Jin Q, Finney L, Hornberger B, Flachenecker C, Lai B, Brister K, Jacobsen C, Woloschak G, Vogt S. Microsc Today. 2015;23:26–29. doi: 10.1017/S1551929515000401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fahrni CJ. Curr Opin Chem Biol. 2007;11:121–127. doi: 10.1016/j.cbpa.2007.02.039. [DOI] [PubMed] [Google Scholar]
- 80.Ralle M, Lutsenko S. BioMetals. 2009;22:197–205. doi: 10.1007/s10534-008-9200-5. [DOI] [PubMed] [Google Scholar]
- 81.Kaye GWC, Laby TH. X-ray absorption edges, characteristic X-ray lines and fluorescence yields. [accessed October 25, 2016];Tables of Physical & Chemical Constants. (16). 1995; 2005 Online Version 1.0, http://www.kayelaby.npl.co.uk/atomic_and_nuclear_physics/4_2/4_2_1.html.
- 82.Davies KM, Hare DJ, Bohic S, James SA, Billings JL, Finkelstein DI, Doble PA, Double KL. Anal Chem. 2015;87:6639–6645. doi: 10.1021/acs.analchem.5b01454. [DOI] [PubMed] [Google Scholar]
- 83.Vogt S, Ralle M. Anal Bioanal Chem. 2013;405:1809–1820. doi: 10.1007/s00216-012-6437-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.House MJ, Fleming AJ, de Jonge MD, Paterson D, Howard DL, Carpenter J-P, Pennell DJ, StPierre TG. J Cardiov Magn Reson. 2014;16:80. doi: 10.1186/s12968-014-0080-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen S, Deng J, Yuan Y, Flachenecker C, Mak R, Hornberger B, Jin Q, Shu D, Lai B, Maser J, Roehrig C, Paunesku T, Gleber SC, Vine DJ, Finney L, VonOsinski J, Bolbat M, Spink I, Chen Z, Steele J, Trapp D, Irwin J, Feser M, Snyder E, Brister K, Jacobsen C, Woloschak G, Vogt S. J Synchrotron Radiat. 2014;21:66–75. doi: 10.1107/S1600577513029676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paunesku T, Vogt S, Maser J, Lai B, Woloschak G. J Cell Biochem. 2006;99:1489–1502. doi: 10.1002/jcb.21047. [DOI] [PubMed] [Google Scholar]
- 87.Dučić T, Barski E, Salome M, Koch JC, Bähr M, Lingor P. J Neurochem. 2013;124:250–261. doi: 10.1111/jnc.12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martínez-Criado G, Villanova J, Tucoulou R, Salomon D, Suuronen J-P, Labouré S, Guilloud C, Valls V, Barrett R, Gagliardini E, Dabin Y, Baker R, Bohic S, Cohen C, Morse J. J Synchrotron Radiat. 2016;23:344–352. doi: 10.1107/S1600577515019839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ortega R, Carmona A, Roudeau S, Perrin L, Dučić T, Carboni E, Bohic S, Cloetens P, Lingor P. Mol Neurobiol. 2016;53:1925–1934. doi: 10.1007/s12035-015-9146-x. [DOI] [PubMed] [Google Scholar]
- 90.Kim SA, Punshon T, Lanzirotti A, Li L, Alonso JM, Ecker JR, Kaplan J, Guerinot ML. Science. 2006;314:1295–1298. doi: 10.1126/science.1132563. [DOI] [PubMed] [Google Scholar]
- 91.de Jonge MD, Holzner C, Baines SB, Twining BS, Ignatyev K, Diaz J, Howard DL, Legnini D, Miceli A, McNulty I, Jacobsen CJ, Vogt S. Proc Natl Acad Sci U S A. 2010;107:15676–15680. doi: 10.1073/pnas.1001469107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.de Jonge MD, Vogt S. Curr Opin Struct Biol. 2010;20:606–614. doi: 10.1016/j.sbi.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 93.Bourassa D, Gleber S-C, Vogt S, Yi H, Will F, Richter H, Shin CH, Fahrni CJ. Metallomics. 2014;6:1648–1655. doi: 10.1039/c4mt00121d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McRae R, Bagchi P, Sumalekshmy S, Fahrni CJ. Chem Rev. 2009;109:4780–4827. doi: 10.1021/cr900223a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. [accessed October 25, 2016];X-ray Absorption Spectroscopy: Introduction to Experimental Procedures. http://www-ssrl.slac.stanford.edu/mes/xafs/xas_intro.html.
- 96.Bourassa MW, Miller LM. Metallomics. 2012;4:721–738. doi: 10.1039/c2mt20052j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bacquart T, Devès G, Carmona A, Tucoulou R, Bohic S, Ortega R. Anal Chem. 2007;79:7353–7359. doi: 10.1021/ac0711135. [DOI] [PubMed] [Google Scholar]
- 98.Lerotic M, Jacobsen C, Schäfer T, Vogt S. Ultramicroscopy. 2004;100:35–57. doi: 10.1016/j.ultramic.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 99.Ortega R, Deves G, Carmona A. J R Soc, Interface. 2009;6:S649–S658. doi: 10.1098/rsif.2009.0166.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rajendran R, Minqin R, Ynsa MD, Casadesus G, Smith MA, Perry G, Halliwell B, Watt F. Biochem Biophys Res Commun. 2009;382:91–95. doi: 10.1016/j.bbrc.2009.02.136. [DOI] [PubMed] [Google Scholar]
- 101.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 102.Schnell Ramos M, Khodja H, Mary V, Thomine S. Front Plant Sci. 2013;4:168. doi: 10.3389/fpls.2013.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Susnea I, Weiskirchen R. Mass Spectrom Rev. 2016;35:666–686. doi: 10.1002/mas.21454. [DOI] [PubMed] [Google Scholar]
- 104.Eibl O, Schultheiss S, Blitgen-Heinecke P, Schraermeyer U. Micron. 2006;37:262–276. doi: 10.1016/j.micron.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 105.Biesemeier A, Schraermeyer U, Eibl O. Exp Eye Res. 2011;93:29–39. doi: 10.1016/j.exer.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 106.Jang HJ, Kim JM, Choi CY. Exp Eye Res. 2014;121:58–65. doi: 10.1016/j.exer.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 107.Sousa AA, Leapman RD. Ultramicroscopy. 2012;123:38–49. doi: 10.1016/j.ultramic.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leapman RD. J Microsc. 2003;210:5–15. doi: 10.1046/j.1365-2818.2003.01173.x. [DOI] [PubMed] [Google Scholar]
- 109.Aronova MA, Sousa AA, Zhang G, Leapman RD. J Microsc. 2010;239:223–232. doi: 10.1111/j.1365-2818.2010.03376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Treiber CD, Salzer MC, Riegler J, Edelman N, Sugar C, Breuss M, Pichler P, Cadiou H, Saunders M, Lythgoe M, Shaw J, Keays DA. Nature. 2012;484:367–370. doi: 10.1038/nature11046. [DOI] [PubMed] [Google Scholar]
- 111.Theil EC. Annu Rev Biochem. 1987;56:289–315. doi: 10.1146/annurev.bi.56.070187.001445. [DOI] [PubMed] [Google Scholar]
- 112.Iancu TC. Electron Microsc Rev. 1992;5:209–229. doi: 10.1016/0892-0354(92)90011-e. [DOI] [PubMed] [Google Scholar]
- 113.Zhang P, Land W, Lee S, Juliani J, Lefman J, Smith SR, Germain D, Kessel M, Leapman R, Rouault TA, Subramaniam S. J Struct Biol. 2005;150:144–153. doi: 10.1016/j.jsb.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Aronova MA, Leapman RD. MRS Bull. 2012;37:53–62. doi: 10.1557/mrs.2011.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Messaoudi C, Aschman N, Cunha M, Oikawa T, Sorzano COS, Marco S. Microsc Microanal. 2013;19:1669–1677. doi: 10.1017/S1431927613013317. [DOI] [PubMed] [Google Scholar]
- 116.Salgado JC, Olivera-Nappa A, Gerdtzen ZP, Tapia V, Theil EC, Conca C, Nuñez MT. BMC Syst Biol. 2010;4:147. doi: 10.1186/1752-0509-4-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kakhlon O, Cabantchik ZI. Free Radical Biol Med. 2002;33:1037–1046. doi: 10.1016/s0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- 118.Qian X, Xu Z. Chem Soc Rev. 2015;44:4487–4493. doi: 10.1039/c4cs00292j. [DOI] [PubMed] [Google Scholar]
- 119.Carter KP, Young AM, Palmer AE. Chem Rev. 2014;114:4564–4601. doi: 10.1021/cr400546e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jacobs A. Ciba Found Symp. 1976:91–106. doi: 10.1002/9780470720325.ch5. [DOI] [PubMed] [Google Scholar]
- 121.Ma Y, Abbate V, Hider RC. Metallomics. 2015;7:212–222. doi: 10.1039/c4mt00214h. [DOI] [PubMed] [Google Scholar]
- 122.Burdette SC. Eur J Inorg Chem. 2015;2015:5728–5729. [Google Scholar]
- 123.Breuer W, Epsztejn S, Millgram P, Cabantchik IZ. Am J Physiol. 1995;268:C1354–C1361. doi: 10.1152/ajpcell.1995.268.6.C1354. [DOI] [PubMed] [Google Scholar]
- 124.Petrat F, Rauen U, de Groot H. Hepatology. 1999;29:1171–1179. doi: 10.1002/hep.510290435. [DOI] [PubMed] [Google Scholar]
- 125.Ma Y, de Groot H, Liu Z, Hider Robert C, Petrat F. Biochem J. 2006;395:49–55. doi: 10.1042/BJ20051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ma Y, Liu Z, Hider RC, Petrat F. Anal Chem Insights. 2007;2:61–67. [PMC free article] [PubMed] [Google Scholar]
- 127.Petrat F, Weisheit D, Lensen M, de Groot H, Sustmann R, Rauen U. Biochem J. 2002;362:137–147. doi: 10.1042/0264-6021:3620137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rauen U, Springer A, Weisheit D, Petrat F, Korth H-G, de Groot H, Sustmann R. ChemBioChem. 2007;8:341–352. doi: 10.1002/cbic.200600311. [DOI] [PubMed] [Google Scholar]
- 129.Hirayama T, Okuda K, Nagasawa H. Chem Sci. 2013;4:1250–1256. [Google Scholar]
- 130.Niwa M, Hirayama T, Okuda K, Nagasawa H. Org Biomol Chem. 2014;12:6590–6597. doi: 10.1039/c4ob00935e. [DOI] [PubMed] [Google Scholar]
- 131.Mori M, Ito F, Shi L, Wang Y, Ishida C, Hattori Y, Niwa M, Hirayama T, Nagasawa H, Iwase A, Kikkawa F, Toyokuni S. Redox Biol. 2015;6:578–586. doi: 10.1016/j.redox.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen J-L, Zhuo S-J, Wu Y-Q, Fang F, Li L, Zhu C-Q. Spectrochim Acta, Part A. 2006;63:438–443. doi: 10.1016/j.saa.2005.04.057. [DOI] [PubMed] [Google Scholar]
- 133.Maiti S, Aydin Z, Zhang Y, Guo M. Dalton Trans. 2015;44:8942–8949. doi: 10.1039/c4dt03792h. [DOI] [PubMed] [Google Scholar]
- 134.Au-Yeung HY, Chan J, Chantarojsiri T, Chang CJ. J Am Chem Soc. 2013;135:15165–15173. doi: 10.1021/ja4072964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Borstnik K, Paik IH, Shapiro TA, Posner GH. Int J Parasitol. 2002;32:1661–1667. doi: 10.1016/s0020-7519(02)00195-9. [DOI] [PubMed] [Google Scholar]
- 136.Aron AT, Loehr MO, Bogena J, Chang CJ. J Am Chem Soc. 2016;138:14338–14346. doi: 10.1021/jacs.6b08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Trends Biochem Sci. 2016;41:274–286. doi: 10.1016/j.tibs.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Spangler B, Morgan CW, Fontaine SD, Vander Wal MN, Chang CJ, Wells JA, Renslo AR. Nat Chem Biol. 2016;12:680–685. doi: 10.1038/nchembio.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yang L, McRae R, Henary MM, Patel R, Lai B, Vogt S, Fahrni CJ. Proc Natl Acad Sci U S A. 2005;102:11179–11184. doi: 10.1073/pnas.0406547102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lutsenko S. Curr Opin Chem Biol. 2010;14:211–217. doi: 10.1016/j.cbpa.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Paredes E, Das SR. Bioorg Med Chem Lett. 2012;22:5313–5316. doi: 10.1016/j.bmcl.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 142.Cotruvo JA, Jr, Aron AT, Ramos-Torres KM, Chang CJ. Chem Soc Rev. 2015;44:4400–4414. doi: 10.1039/c4cs00346b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Aron AT, Ramos-Torres KM, Cotruvo JA, Chang CJ. Acc Chem Res. 2015;48:2434–2442. doi: 10.1021/acs.accounts.5b00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fahrni CJ. Curr Opin Chem Biol. 2013;17:656–662. doi: 10.1016/j.cbpa.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chaudhry AF, Verma M, Morgan MT, Henary MM, Siegel N, Hales JM, Perry JW, Fahrni CJ. J Am Chem Soc. 2010;132:737–747. doi: 10.1021/ja908326z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Verma M, Chaudhry AF, Morgan MT, Fahrni CJ. Org Biomol Chem. 2010;8:363–370. doi: 10.1039/b918311f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chaudhry AF, Mandal S, Hardcastle KI, Fahrni CJ. Chem Sci. 2011;2:1016–1024. doi: 10.1039/C1SC00024A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Morgan MT, Bagchi P, Fahrni CJ. J Am Chem Soc. 2011;133:15906–15909. doi: 10.1021/ja207004v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Morgan MT, McCallum AM, Fahrni CJ. Chem Sci. 2016;7:1468–1473. doi: 10.1039/c5sc03643g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zeng L, Miller EW, Pralle A, Isacoff EY, Chang CJ. J Am Chem Soc. 2006;128:10–11. doi: 10.1021/ja055064u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Miller EW, Zeng L, Domaille DW, Chang CJ. Nat Protoc. 2006;1:824–827. doi: 10.1038/nprot.2006.140. [DOI] [PubMed] [Google Scholar]
- 152.Price KA, Hickey JL, Xiao Z, Wedd AG, James SA, Liddell JR, Crouch PJ, White AR, Donnelly PS. Chem Sci. 2012;3:2748–2759. [Google Scholar]
- 153.Santo CE, Lam EW, Elowsky CG, Quaranta D, Domaille DW, Chang CJ, Grass G. Appl Environ Microbiol. 2011;77:794–802. doi: 10.1128/AEM.01599-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Quaranta D, Krans T, Santo CE, Elowsky CG, Domaille DW, Chang CJ, Grass G. Appl Environ Microbiol. 2011;77:416–426. doi: 10.1128/AEM.01704-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Beaudoin J, Ioannoni R, Lopez-Maury L, Bahler J, Ait-Mohand S, Guerin B, Dodani SC, Chang CJ, Labbe S. J Biol Chem. 2011;286:34356–34372. doi: 10.1074/jbc.M111.280396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cusick KD, Minkin SC, Dodani SC, Chang CJ, Wilhelm SW, Sayler GS. Environ Sci Technol. 2012;46:2959–2966. doi: 10.1021/es204027m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bernal M, Casero D, Singh V, Wilson GT, Grande A, Yang H, Dodani SC, Pellegrini M, Huijser P, Connolly EL, Merchant SS, Kramer U. Plant Cell. 2012;24:738–761. doi: 10.1105/tpc.111.090431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Achard MES, Stafford SL, Bokil NJ, Chartres J, Bernhardt PV, Schembri MA, Sweet MJ, McEwan AG. Biochem J. 2012;444:51–57. doi: 10.1042/BJ20112180. [DOI] [PubMed] [Google Scholar]
- 159.Gabe Y, Ueno T, Urano Y, Kojima H, Nagano T. Anal Bioanal Chem. 2006;386:621–626. doi: 10.1007/s00216-006-0587-y. [DOI] [PubMed] [Google Scholar]
- 160.Dodani SC, Domaille DW, Nam CI, Miller EW, Finney LA, Vogt S, Chang CJ. Proc Natl Acad Sci U S A. 2011;108:5980–5985. doi: 10.1073/pnas.1009932108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Huang CP, Fofana M, Chan J, Chang CJ, Howell SB. Metallomics. 2014;6:654–661. doi: 10.1039/c3mt00331k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ohrvik H, Nose Y, Wood LK, Kim BE, Gleber SC, Ralle M, Thiele DJ. Proc Natl Acad Sci U S A. 2013;110:E4279– E4288. doi: 10.1073/pnas.1311749110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Polishchuk EV, Concilli M, Iacobacci S, Chesi G, Pastore N, Piccolo P, Paladino S, Baldantoni D, van Ijzendoorn SCD, Chan J, Chang CJ, Amoresano A, Pane F, Pucci P, Tarallo A, Parenti G, Brunetti-Pierri N, Settembre C, Ballabio A, Polishchuk RS. Dev Cell. 2014;29:686–700. doi: 10.1016/j.devcel.2014.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dodani SC, Firl A, Chan J, Nam CI, Aron AT, Onak CS, Ramos-Torres KM, Paek J, Webster CM, Feller MB, Chang CJ. Proc Natl Acad Sci U S A. 2014;111:16280–16285. doi: 10.1073/pnas.1409796111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Koide Y, Urano Y, Hanaoka K, Terai T, Nagano T. ACS Chem Biol. 2011;6:600–608. doi: 10.1021/cb1002416. [DOI] [PubMed] [Google Scholar]
- 166.Koide Y, Urano Y, Hanaoka K, Terai T, Nagano T. J Am Chem Soc. 2011;133:5680–5682. doi: 10.1021/ja111470n. [DOI] [PubMed] [Google Scholar]
- 167.Krishnamoorthy L, Cotruvo JA, Chan J, Kaluarachchi H, Muchenditsi A, Pendyala VS, Jia S, Aron AT, Ackerman CM, Vander Wal MN, Guan T, Smaga LP, Farhi SL, New EJ, Lutsenko S, Chang CJ. Nat Chem Biol. 2016;12:586–592. doi: 10.1038/nchembio.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lim CS, Han JH, Kim CW, Kang MY, Kang DW, Cho BR. Chem Commun. 2011;47:7146–7148. doi: 10.1039/c1cc11568e. [DOI] [PubMed] [Google Scholar]
- 169.Cao X, Lin W, Wan W. Chem Commun. 2012;48:6247–6249. doi: 10.1039/c2cc32114a. [DOI] [PubMed] [Google Scholar]
- 170.Hirayama T, Van de Bittner GC, Gray LW, Lutsenko S, Chang CJ. Proc Natl Acad Sci U S A. 2012;109:2228–2233. doi: 10.1073/pnas.1113729109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Dodani SC, Leary SC, Cobine PA, Winge DR, Chang CJ. J Am Chem Soc. 2011;133:8606–8616. doi: 10.1021/ja2004158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Smith RAJ, Porteous CM, Gane AM, Murphy MP. Proc Natl Acad Sci U S A. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Domaille DW, Zeng L, Chang CJ. J Am Chem Soc. 2010;132:1194–1195. doi: 10.1021/ja907778b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Giuffrida ML, Rizzarelli E, Tomaselli GA, Satriano C, Trusso Sfrazzetto G. Chem Commun. 2014;50:9835–9838. doi: 10.1039/c4cc02147a. [DOI] [PubMed] [Google Scholar]
- 175.Satriano C, Sfrazzetto GT, Amato ME, Ballistreri FP, Copani A, Giuffrida ML, Grasso G, Pappalardo A, Rizzarelli E, Tomaselli GA, Toscano RM. Chem Commun. 2013;49:5565–5567. doi: 10.1039/c3cc42069h. [DOI] [PubMed] [Google Scholar]
- 176.Shen C, Kolanowski JL, Tran CMN, Kaur A, Akerfeldt MC, Rahme MS, Hambley TW, New EJ. Metallomics. 2016;8:915–919. doi: 10.1039/c6mt00083e. [DOI] [PubMed] [Google Scholar]
- 177.Jung KH, Oh E-T, Park HJ, Lee K-H. Biosens Bioelectron. 2016;85:437–444. doi: 10.1016/j.bios.2016.04.101. [DOI] [PubMed] [Google Scholar]
- 178.Chan J, Dodani SC, Chang CJ. Nat Chem. 2012;4:973–984. doi: 10.1038/nchem.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yang Y, Zhao Q, Feng W, Li F. Chem Rev. 2013;113:192–270. doi: 10.1021/cr2004103. [DOI] [PubMed] [Google Scholar]
- 180.Kim SK, Sessler JL. Chem Soc Rev. 2010;39:3784–3809. doi: 10.1039/c002694h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lee S, Yuen KKY, Jolliffe KA, Yoon J. Chem Soc Rev. 2015;44:1749–1762. doi: 10.1039/c4cs00353e. [DOI] [PubMed] [Google Scholar]
- 182.Taki M, Iyoshi S, Ojida A, Hamachi I, Yamamoto Y. J Am Chem Soc. 2010;132:5938–5939. doi: 10.1021/ja100714p. [DOI] [PubMed] [Google Scholar]
- 183.Urano Y, Kamiya M, Kanda K, Ueno T, Hirose K, Nagano T. J Am Chem Soc. 2005;127:4888–4894. doi: 10.1021/ja043919h. [DOI] [PubMed] [Google Scholar]
- 184.Maity D, Kumar V, Govindaraju T. Org Lett. 2012;14:6008–6011. doi: 10.1021/ol302904c. [DOI] [PubMed] [Google Scholar]
- 185.Yu K-K, Li K, Hou J-T, Yu X-Q. Tetrahedron Lett. 2013;54:5771–5774. [Google Scholar]
- 186.Maity D, Sarkar B, Maiti S, Govindaraju T. ChemPlusChem. 2013;78:785–788. doi: 10.1002/cplu.201300089. [DOI] [PubMed] [Google Scholar]
- 187.Maity D, Raj A, Karthigeyan D, Kundu TK, Govindaraju T. RSC Adv. 2013;3:16788–16794. [Google Scholar]
- 188.Maity D, Raj A, Karthigeyan D, Kundu TK, Govindaraju T. Supramol Chem. 2015;27:589–594. [Google Scholar]
- 189.Hu Z, Hu J, Wang H, Zhang Q, Zhao M, Brommesson C, Tian Y, Gao H, Zhang X, Uvdal K. Anal Chim Acta. 2016;933:189–195. doi: 10.1016/j.aca.2016.05.031. [DOI] [PubMed] [Google Scholar]
- 190.Taki M, Akaoka K, Mitsui K, Yamamoto Y. Org Biomol Chem. 2014;12:4999–5005. doi: 10.1039/c4ob00527a. [DOI] [PubMed] [Google Scholar]
- 191.Au-Yeung HY, New EJ, Chang CJ. Chem Commun. 2012;48:5268–5270. doi: 10.1039/c2cc31681a. [DOI] [PubMed] [Google Scholar]
- 192.Heffern MC, Park HM, Au-Yeung H-Y, Van de Bittner GC, Ackerman CM, Stahl A, Chang CJ. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1613628113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Spatz H. Z Gesamte Neurol Psychiatr. 1922;77:261–390. [Google Scholar]
- 194.Hallgren B, Sourander P. J Neurochem. 1958;3:41–51. doi: 10.1111/j.1471-4159.1958.tb12607.x. [DOI] [PubMed] [Google Scholar]
- 195.James SA, Roberts BR, Hare DJ, de Jonge MD, Birchall IE, Jenkins NL, Cherny RA, Bush AI, McColl G. Chem Sci. 2015;6:2952–2962. doi: 10.1039/c5sc00233h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Berry AJ, O’Neill HSC, Jayasuriya KD, Campbell SJ, Foran GJ. Am Mineral. 2003;88:967–977. [Google Scholar]
- 197.Miyayama T, Suzuki KT, Ogra Y. Toxicol Appl Pharmacol. 2009;237:205–213. doi: 10.1016/j.taap.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 198.Ralle M, Huster D, Vogt S, Schirrmeister W, Burkhead JL, Capps TR, Gray L, Lai B, Maryon E, Lutsenko S. J Biol Chem. 2010;285:30875–30883. doi: 10.1074/jbc.M110.114447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Malasarn D, Kropat J, Hsieh SI, Finazzi G, Casero D, Loo JA, Pellegrini M, Wollman FA, Merchant SS. J Biol Chem. 2013;288:10672–10683. doi: 10.1074/jbc.M113.455105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Nose Y, Kim B-E, Thiele DJ. Cell Metab. 2006;4:235–244. doi: 10.1016/j.cmet.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 201.Rees EM, Lee J, Thiele DJ. J Biol Chem. 2004;279:54221–54229. doi: 10.1074/jbc.M411669200. [DOI] [PubMed] [Google Scholar]
- 202.Kardos J, Kovács I, Hajós F, Kálmán M, Simonyi M. Neurosci Lett. 1989;103:139–144. doi: 10.1016/0304-3940(89)90565-x. [DOI] [PubMed] [Google Scholar]
- 203.Hopt A, Korte S, Fink H, Panne U, Niessner R, Jahn R, Kretzschmar H, Herms J. J Neurosci Methods. 2003;128:159–172. doi: 10.1016/s0165-0270(03)00173-0. [DOI] [PubMed] [Google Scholar]
- 204.Hartter DE, Barnea A. Synapse. 1988;2:412–415. doi: 10.1002/syn.890020408. [DOI] [PubMed] [Google Scholar]
- 205.Schlief ML, Craig AM, Gitlin JD. J Neurosci. 2005;25:239–246. doi: 10.1523/JNEUROSCI.3699-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Schlief ML, West T, Craig AM, Holtzman DM, Gitlin JD. Proc Natl Acad Sci U S A. 2006;103:14919–14924. doi: 10.1073/pnas.0605390103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Acevedo KM, Hung YH, Dalziel AH, Li QX, Laughton K, Wikhe K, Rembach A, Roberts B, Masters CL, Bush AI, Camakaris J. J Biol Chem. 2011;286:8252–8262. doi: 10.1074/jbc.M110.128512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Gaier ED, Rodriguiz RM, Zhou J, Ralle M, Wetsel WC, Eipper BA, Mains RE. J Neurophysiol. 2014;111:1927–1939. doi: 10.1152/jn.00631.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.D’Ambrosi N, Rossi L. Neurochem Int. 2015;90:36–45. doi: 10.1016/j.neuint.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 210.Turski ML, Brady DC, Kim HJ, Kim BE, Nose Y, Counter CM, Winge DR, Thiele DJ. Mol Cell Biol. 2012;32:1284–1295. doi: 10.1128/MCB.05722-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, Knapp S, Xiao K, Campbell SL, Thiele DJ, Counter CM. Nature. 2014;509:492–496. doi: 10.1038/nature13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ishida S, Andreux P, Poitry-Yamate C, Auwerx J, Hanahan D. Proc Natl Acad Sci U S A. 2013;110:19507–19512. doi: 10.1073/pnas.1318431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kim B-E, Turski ML, Nose Y, Casad M, Rockman HA, Thiele DJ. Cell Metab. 2010;11:353–363. doi: 10.1016/j.cmet.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zheng L, Han P, Liu J, Li R, Yin W, Wang T, Zhang W, James Kang Y. Pharmacol Ther. 2015;148:66–84. doi: 10.1016/j.pharmthera.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 215.Zhou Z, Johnson WT, Kang YJ. J Nutr Biochem. 2009;20:621–628. doi: 10.1016/j.jnutbio.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]