

Abstract
The coordination-driven self-assembly of four different trigonal prisms from three equivalents of one of four different tetrapyridyl star connectors and six equivalents of a platinum linker dication in nitromethane is presented. This face-directed approach affords high yield without template assistance. The prisms have been characterized by multinuclear and DOSY NMR and dual ESI-FT-ICR mass spectrometry. The use of a conformationally chiral star connector leads to a conformationally chiral prism when connector arm ends attached to a vertex have a strongly correlated twist sense and chirality is communicated across polyhedral faces, edges and vertices. Molecular mechanics results suggest that in the smallest prism 3d collective effects dominate and the all-P and all-M conformers are strongly favored. NMR data prove that the two edges of the pyridine rings in the triflate salts of 3a - 3d are distinct. An Eyring plot of rates obtained from line-shape analysis and 1-D EXCHSY NMR yields an activation enthalpy ΔH‡ of ∼12 kcal/mol and activation entropy ΔS‡ of ∼-15 cal/mol.K for the edge interconversion process, compatible with pyridine rotation around the Pt-N bond. For 3c, this behavior is observed only up to ∼318 K. At higher temperatures, the Eyring plot is again linear but follows a very different straight line, with ΔH‡ of ∼35 kcal/mol and ΔS‡ of ∼60 cal/mol.K. This highly unusual result is further investigated and discussed in the following companion paper.
Introduction
Coordination-driven self-assembly via the directional bonding approach is a useful strategy for the preparation of 2- and 3-D supramolecules.1,2,3,4,5,6,7,8,9,10,11,12,13,14 Some of the 3-D examples include cubes,15,16 adamantanoids,17 double squares,18 cuboctahedra,19 and dodecahedra.20 These are highly symmetrical species composed of building blocks that define some or all of their edges. Lower symmetry assemblies, for example trigonal21,22,23,24 and tetragonal22,23 prisms, are less common and are more easily approached using face-directed techniques in which component linkers span the faces of the desired aggregate. Varying the component dimensions then alters the size and shape of the final assembly. This methodology allows the creation of 3-D supramolecular hosts of relatively low symmetry which ultimately should show enhanced guest selectivity.
Planar tetratopic ligands 1 have been of interest to us in other contexts,25 and several are readily available. Their reaction with an approximate26 90° linker 2 in a 1:2 ratio may lead to trigonal (3), tetragonal (4), pentagonal (5), hexagonal (6), and possibly even larger coordination prisms, in which the tetratopic ligands span the side panels and the linkers occupy the vertices (Figure 1). The use of face-defining star-shaped connectors in general, and of tetratopic tetrapyridyl ligands in particular, offers some interesting opportunities for the production of chiral prisms. Although prisms whose edges are mere lines are achiral, prisms whose edges are constructed from molecules and whose vertices are metal atoms are potentially chiral, and this has already been exploited by two groups of authors. Chiral ligands at square planar vertex metal atoms,27 later also using edges of C2h symmetry,28,29 were utilized by Stang and co-workers, first with molecular squares, and then with polyhedra.30 Optically active three-dimensional cages with vertex metal atoms carrying achiral ligands in a chiral octahedral arrangement have been explored by Raymond and co-workers.7,31,32
Figure 1.

Reaction of tetratopic ligand 1 with linker 2 in a 1:2 ratio may lead to coordination prisms 3 - 6.
We now introduce the concept of face-based cage chirality, which has a different origin, as it is defined by the cage faces and not vertices or edges. Star connectors used to define cage faces are frequently propeller-shaped, with all arms attached to the central moiety twisted in the same sense. Then, they impart conformational P or M helical chirality33 to each cage face. In an isolated star connector such as 1, the P-M interconversion by rotation around single bonds is ordinarily very facile, and the enantiomers cannot be separated and stored at room temperature. In a polyhedron, which consists of a collection of faces, two limiting cases are conceivable. In the “independent face limit”, the inversion of the chiral sense of a star connector in any face occurs independently of the chirality adopted by any other face. In the “coupled face limit”, star connectors located in different faces affect each other strongly through linkers located at shared edges or vertices. Collective effects result, and a P-M interconversion at one face is only possible with simultaneous interconversion at some or all other faces.
We find that the self-assembly of four trigonal prismatic cages 3 from four distinct star connectors 1 and the 90° platinum linker 2 proceeds well without template assistance (Scheme 1). We argue below that the tetrapyridyl ligands 1 vary in the expected degree of communication of chirality from face to face. Molecular modeling results strongly suggest that the smallest cage 3d is in the coupled face limit and will exhibit collective behavior, and thus sets the stage for a future experimental testing of the notion of face-based cage chirality.
Scheme 1.

Self-assembly of tetrapyridyl ligands 1 with cis-(Me3P)2(OTf)2 2 leads to trigonal prisms.
Each prism 3 is composed of three molecules of the star connector 1 and six units of the linker 2. The structures have been characterized by multinuclear and DOSY NMR and dual electrospray ionization Fourier transform ion cyclotron resonance (dualESI-FT-ICR) mass spectrometry.34 In the case of 3d, evidence for the formation of a larger dimeric aggregate under ESI conditions is also presented.
The rotational degree of freedom of the pyridine rings in the arms of the cross connectors is essential for the understanding of the conformational mobility and face-based chirality of cages such as 3. The activation parameters are unexceptional in all cases except 3c, whose automerization rate exhibits a bilinear Eyring plot with strikingly high ΔH‡ and ΔS‡ values in the high-temperature regime. This most unusual result is addressed in the following companion paper.35
Results
Stirring one equivalent of each of 1a - 1d with two equivalents of 2 in CD3NO2 for 18 h at room temperature results in quantitative (by NMR) formation of molecular cages 3a - 3d, respectively. Their structure was examined by (i) NMR spectroscopy, (ii) mass spectrometry, and (iii) diffusion constant measurement. In other solvents (CH2Cl2, CHCl3), the products are stable, but they do not form as readily and cleanly.
NMR Spectroscopy
In all cases the 31P and 1H NMR spectra are consistent with the formation of a single discrete product. The 31P NMR spectra displayed a sharp peak near -28 ppm with concomitant 195Pt satellites. This upfield shift of approximately 8 ppm relative to 2 is consistent with back donation from the platinum atoms upon coordination. The 1H NMR spectra showed two sets of signals for the pyridyl rings. Although these protons would have magnetically different environments even if the pyridine rings were located in the planes of the prism faces, the preferences of the platinum linkers discussed below make it very likely that these rings do not lie in the prism faces but are partially or orthogonally twisted. The two magnetic environments then correspond to protons located on the inside and the outside of the prism. There is ample precedent for this behavior, and for restricted ring rotational equilibration.21,36,37
The pyridyl proton resonances of 3 are shifted slightly to lower field as expected due to the loss of electron density that occurs upon coordination. The only exception was the resonance of one Hα nucleus of 3d at 8.20 ppm, which shifts to higher field relative to the starting ligand 1d (8.51 ppm). Platinum back donation causes the resonance of the phosphine methyl groups to move approximately 0.1 ppm to higher field. Interestingly, the resonances of the vinylic protons of 3b experience quite large low field shifts (0.2-0.4 ppm).
The three CpCo moieties in both 3c and 3d are all equivalent and appear to be either all located inside or all outside of the square faces of the self-assembled cage. The proton chemical shifts are entirely normal and show no indications of ring current effects ascribable to the proximity of other rings. This strongly suggests that the Cp rings are located quite far apart, hence outside the prisms.
Molecular Mechanics
Molecular mechanics modeling (UFF,38,39 using the computer program TINK40) was used for two purposes. The first was to aid with the consideration of the issue discussed just above, the location of the CpCo moieties in 3c and 3d. We found that in the larger prism 3c there is enough internal space for both locations to be possible (Figure 2). The CpCo-inside isomer is calculated to be the less stable of the two by ∼1 kcal/mol, but this small difference could be changed by differential solvation effects and in any event is within the error of the method. While we cannot rule out the CpCo-inside isomer with absolute certainty in the absence of X-ray data that we have been unable to obtain, we consider this structure rather unlikely for the reasons discussed above. The only way these residues can be located in the smaller prism 3d is on the outside (Figure 3).
Figure 2.
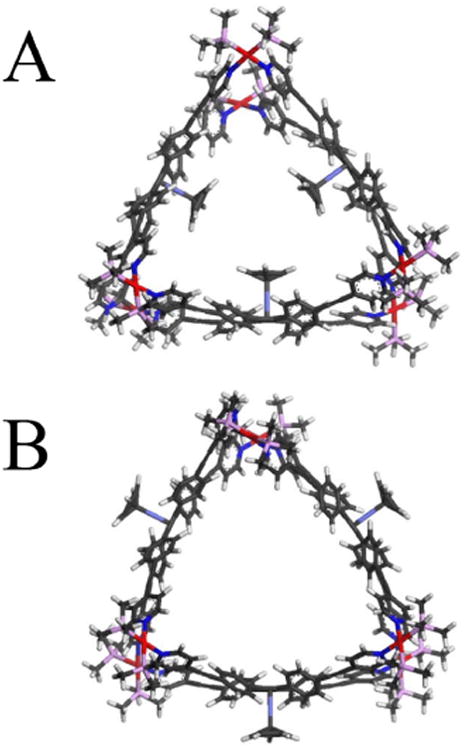
Calculated structures of (A) 3c with CpCo residues inside and (B) 3c with CpCo residues outside the cage.
Figure 3.
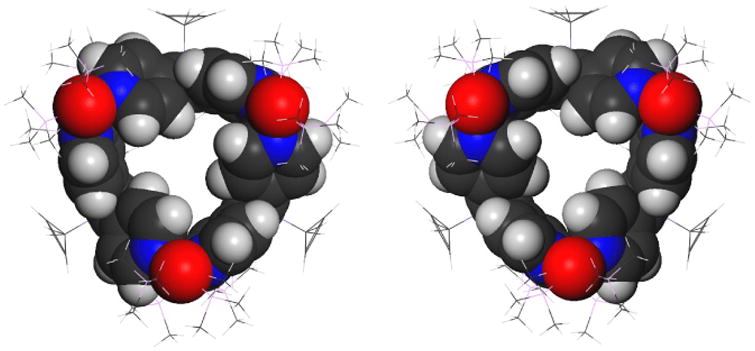
PPP and MMM Enantiomers of 3d.
The second issue to which molecular mechanics was applied is the face-based chirality of the prism 3d. Geometry optimization yielded a single pair of enantiomeric conformers, PPP and MMM, where the labels P and M refer to the three tetrapyridylcyclobutadiene propellers located in the individual prism faces (Figure 3). The four pyridine rings in a face are not equally twisted out of the plane of the central cyclobutadiene ring. Two located on opposed vertices of the ring are twisted by 45 ° (Type I) and the other two are only by 15 ° (Type II), such that the average twist is still similar to that in the free connector 1d. Each platinum atom is in a nearly perfectly square planar environment and is coordinated to one pyridine ring of each type. The Type I pyridine makes a dihedral angle of 82 ° with the NPtN plane whereas the Type II pyridine makes an angle of 65 ° and both thus deviate from the optimal angle of 90 °. If the cyclopentadiene rings were circular, the prism 3d would have D3 symmetry.
All attempts to find PPM and MMP conformers failed, and we strongly suspect that structures in which the absolute chirality of one of the three faces differs from the other two are not local minima on the potential energy surface. All local minima other than PPP/MMM that were found were higher by at least 20 kcal/mol and contained one or two faces in which the four pyridine rings were not twisted in the same sense and no longer resembled a propeller.
Mass Spectrometry
Evidence for the trimeric nature of the products was obtained with dualESI-FT-ICR mass spectrometry. This gentle ionization technique allowed the intact prisms (minus several triflate counterions) to be observed. The high resolving power afforded by FT-ICR mass spectrometry allowed unequivocal charge state determination. Supramolecules of this size (approximately 5000 Da) display larger isotopic shifts (defined as the difference between average and monoisotopic masses) relative to proteins of similar molecular mass, due to the presence of numerous platinum atoms. Thus, in this situation the most abundant isotope is considered for the determination of molecular mass, rather than the monoisotopic mass. This could have an error of ±1 amu or 200 ppm for molecules with molecular masses on the order of 5000 amu.
The internally calibrated dualESI-FT-ICR mass spectrum of 3a is shown in Figure 4a. The inset shows an expansion of an isotopically resolved species (top) carrying a 4+ charge centered at m/z = 1180.8486, assigned to [3a-4OTf-]4+. It is in excellent agreement with the theoretical isotope distribution (bottom) predicted for this species. A mass measurement accuracy after internal calibration (MMAint) of -0.3 ppm for the most abundant isotope is also consistent with the proposed structure. Extensive fragmentation at lower m/z values was evident, suggesting that 3a may still dissociate even under the softer ionization conditions employed.
Figure 4.

DualESI-FT-ICR mass spectra of cages 3 with experimental and theoretical isotopic distributions (inset). Internal calibrant abbreviations are designated AngI (Angiotensin I), G (Glucagon) and Ins (Insulin). MMAint is the mass measurement accuracy using internal calibration, MMAext is the mass measurement accuracy using external calibration, * is the most abundant isotope, and • is electronic noise.
In contrast, the assembly 3b displayed a simpler mass spectrum (Figure 4b). Due to the poor signal-to-noise ratio for this structure, the hexapole accumulation was significantly increased, precluding internal calibration. Two charge states assignable to 3b were detected, namely [3b-3OTf-]3+ (m/z = 1632.2074) and the more abundant [3b-4OTf-]4+ (m/z = 1186.8201). Both correlate well with their theoretical distributions and have good MMA values for the most abundant isotope despite the absence of internal calibrants.
The mass spectrum of the largest cage 3c is displayed in Figure 4c. A small peak with a 4+ charge, centered near m/z 1482.4063, was assigned to [3c-4OTf-]4+ (MMAext -3 ppm). We were unable to detect any of the larger prisms 4-6 in the mass spectra of 3a-3c under a variety of electrospray and instrumental conditions.
Finally, the mass spectrum of 3d is shown in Figure 4d. Similar to 3b, minimal fragmentation of the intact structure was observed. An isotopically resolved cluster at m/z = 1626.0660 displaying a 3+ charge is assigned to [3d-3OTf-]3+. The unavoidable ion abundance mismatch between the experimental and theoretical distributions is due to ion counting statistics and/or artifacts involving coulombic interactions in the mass spectrometer. Consequently, the experimentally second most abundant isotope (m/z = 1625.7377) has the best MMAint (-0.7 ppm) with the theoretically most abundant isotope. Interestingly, another peak centered at m/z = 1981.0706 with a 5+ charge was observed. This is uncharacteristic of the discrete prism 3d. The isotopic pattern is consistent with an ionic formula derived from six molecules of 1d and twelve molecules of 2 minus five triflate ions (the internally calibrated MMA of the most abundant isotope was -3 ppm).
There are at least three structures that fit this stoichiometry. The first is a cube-like structure 7a, feasible due to the square panel nature of 1d. Indeed, Grieco and collaborators15 have reported that the reaction of 1d with Pd(en)(NO3)2 in a 1:2 ratio affords the cubic supramolecule 7b which they characterized with NMR and nested ion mobility / time-of-flight mass spectrometry (Figure 5). The second possibility is a hexagonal prism of type 6. This is more compatible with our NMR signals than 7b, which shows only one set of signals for the pyridyl rings, apparently due to a low rotation barrier around the Pd-N bond. However, as we shall describe below, prism 6 is hard to reconcile with the DOSY NMR results. Moreover, based on entropy considerations, there is no apparent reason why a prism of this size would be formed instead of or in conjunction with the smaller tetragonal (4) and pentagonal (5) prisms, for which no mass spectral evidence was found. Finally, the formation of 6 or 7 from 3d would require complex rearrangements not likely to occur under electrospray conditions.
Figure 5.

Self-assembled cubes 7a and 7b.
A third and most likely explanation for the unexpected peak is the dimeric aggregate [3d2-5OTf-]5+ held together by weak electrostatic forces between the prisms and counterions. These forces become more important as the solution evaporates in the electrospray process due to the lack of competition with the solvent. Such dimerization, observed previously in 2-D systems,41 is a consequence of mild ionization conditions.
Diffusion Constant Determination
In an attempt to obtain additional confirmation of their trimeric stoichiometry, ensembles 3 were subjected to DOSY NMR measurements.42,43 Numerous chemical and biological systems23,24,44,45,46,47,48 have been studied with this method, including our nanoscopic dodecahedra.20 The present results are summarized in Table 1. They were obtained with the DOSY Bipolar Pulse Pair Stimulated Echo (Dbppste) pulse sequence with carefully calibrated gradients and temperature control (298 K). Viscosity variation among the various solutions measured had a maximum effect of ±5% on the observed D values, as judged by the values obtained for the residual CHD2NO2 solvent proton resonance. The accuracy of the NMR diffusion experiments and the sources of error are described in more detail in the Experimental Part.
Table 1.
Radii of Prisms 3 and 6 as Deduced from Diffusion Coefficients of Triflate Salts and as Predicted by Molecular Mechanics.
| Compd. | Da,bCHD2NO2 | Da,c | R from Dd | predicted R from UFF e | |
|---|---|---|---|---|---|
| trimer 3 | hexamer 6 | ||||
| 3a | 20.9 ±0.8 | 4.0 ±0.6 | 8.8 | 10.8 | 20.4 |
| 3b | 19.7 ±0.8 | 2.6 ±0.6 | 13.5 | 10.6 | 20.2 |
| 3c | 19.5 ±0.8 | 1.9 ±0.6 | 18.3 | 14.8 | 26.1 |
| 3d | 21.4 ±0.8 | 3.8 ±0.6 | 9.2 | 9.2 | 15.1 |
Diffusion coefficient in units of 10-10 m2s-1, obtained from DOSY NMR measurements in CD3NO2 at 298 ± 0.25 K.
The diffusion coefficient of the solvent molecules in the solution measured. The value in pure solvent is 22.2 ±0.8 10-10 m2s-1 and the slight variation is attributed to differences in solution viscosity.
The diffusion coefficient of the solutes 3.
Radius in Å, calculated from the Stokes-Einstein equation, assuming viscosity of the pure solvent. These values are only approximate. The error is believed to be dominated by the unknown uncertainties associated with the use of this equation (see text).
Each predicted radius R was estimated from a UFF38,39,40 optimized geometry of the prisms 3 and 6. Each experimental radius R was calculated from the diffusion coefficient D using the Stokes-Einstein equation approximation, and therefore contains possible contributions from firmly associated counterions. The values of R are only approximate, because the Stokes-Einstein equation was derived for a neutral spherical macroscopic object, whereas our prisms are charged, only very approximately spherical, and of molecular dimensions. In view of this intrinsic inaccuracy in the conversion of D to R, the inaccuracy of the value of D obtained in the DOSY experiments and the uncertainties associated with the imperfect knowledge of temperature and viscosity (see Experimental Part) are considered negligible.
In general the prisms are quite symmetrical and possess large cavities. The distance from each prismatic center to the platinum nuclei is approximately equal to the radius of the smallest sphere required to enclose each supramolecule. For 3a this is estimated to be 10.8 Å while for 6a it is 20.4 Å. The experimental radius of 8.8 Å indicates that 3a is the most likely structure. In fact, the predicted radii of all trimeric prisms 3 are in much better agreement with the experimental values than hexamers 6. This reinforces the conclusions based on our mass spectral data.
Intracage Dynamics by Variable Temperature NMR
In addition to providing suitable substrates for future studies of the concept of face-based chirality, facile access to prisms 3 also offered an opportunity to examine the internal mobility of species self-assembled through metal coordination chemistry. Only a few such studies have been reported, despite continued interest in the area.49,50,51,52,53,54
Since the 1H NMR AB multiplets due to the α and β proton pairs of the pyridyl moiety are inequivalent for prisms 3a - 3d, we were able to obtain the enthalpy (ΔH‡) and entropy (ΔS‡) of activation for pyridyl edge interchange in each assembly in CD3NO2 with a DNMR study. NMR peak assignments of the exchanging protons were verified by 2D ROESY experiments, relying on the opposite phase of the NOE and the chemical exchange correlations. Since the exchanging proton pairs appeared as non-first order AB multiplets, computer simulation was used to determine their chemical shifts and J coupling constant before DNMR line shape simulation. The rates were deduced from spectra obtained at a series of temperatures spanning the range from the low-temperature to the high-temperature limit. They were crosschecked at selected temperatures by repeating the measurements at three different magnetic field strengths. These yielded different line shapes, but identical rate constants.
The rates were independently verified and extended to lower temperatures by transient exchange (1-D EXCHSY) measurements, using an array of mixing times for chemical exchange. In these experiments, the exchange rate constant is measured directly by fitting an appropriate expontential function55 to the rise of the ratio of the resonance peak due to exchange to a selectively inverted resonance peak. They permit the measurement of exchange rates that are well below the limit accessible by lineshape simulation.
The Eyring plots are shown in Figure 6 and the activation parameters are collected in Table 2. Linear fits (R2 ≥ 0.97) were obtained for the triflate salts of all prisms in CD3NO2, except 3c (R2 = 0.91) for which a bilinear fit with a crossing point at ∼318 K is clearly superior (R2 ≥ 0.99 in both regions).
Figure 6.
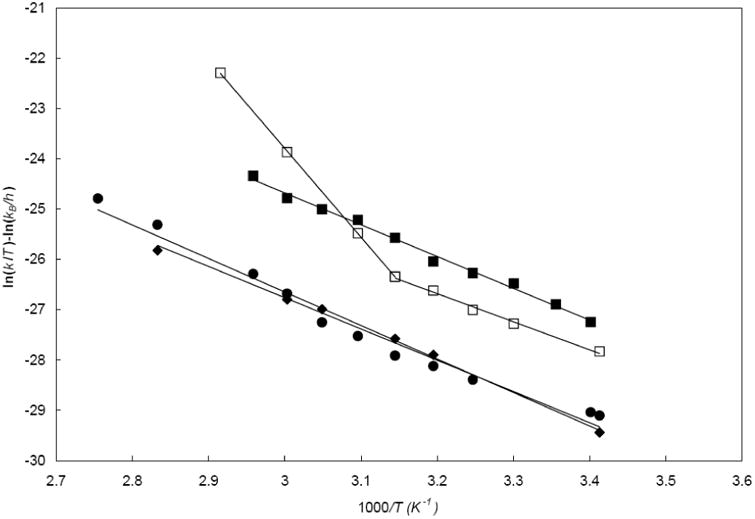
Eyring plots for pyridine edge interconversion rate constants in CD3NO2 for 3a/12 TfO− (diamond), 3b/12 TfO− (square), 3c/12 TfO− (empty square), and 3d/12 TfO− (circle).
Table 2.
Eyring Activation Parameters for Interconversion of Pyridine Edges in 3a-3d Triflates in CD3NO2.
| ΔH‡ kcal/mol | ΔS‡ cal/mol.K | R2 | ||
|---|---|---|---|---|
| 3a 12 TfO- | 12.4 (± 2.1) | -16.1 (± 5.5) | 0.995 | |
| 3b 12 TfO- | 11.6 (± 2.0) | -14.4 (± 4.9) | 0.994 | |
| 3c 12 TfO- | < ∼318 K | 11.1 (± 1.9) | -17.5 (± 6.0) | 0.993 |
| > ∼318 K | 35.1 (± 2.5) | 58.1 (± 8.1) | 1.000 | |
| 3d 12 TfO- | 13.2 (± 2.2) | -13.2 (± 4.5) | 0.973 |
In order to investigate the possibility that the nonlinear Eyring behavior arises from partial decomposition into some platinum colloid which then promotes a heterogeneous process, a drop of mercury, known56 to suppress such processes through physisorption or amalgamation, was added to a solution of 3c in CD3NO2 and pyridyl proton exchange rates were remeasured. They were unaffected and lay within 5% of previously measures rates.
Discussion
Cage Structure
The combined evidence provided by NMR, MS, and diffusion coefficient values, as well as analogy to prior work, leaves no doubt in our minds that the self-assembled products have the trigonal prismatic structure 3. The efficiency with which they form in the absence of a template is remarkable.
The results can be compared with those of a related study by Fujita and co-workers,22 in which the tetrapyridyl unit, 3,3′,5,5′-tetrakis(3-pyridyl)biphenyl (8), and their well developed Pd(en)(NO3)2 linker were self-assembled into coordination boxes. Simple combination of these two components in a 1:2 ratio yielded a tetragonal prism as the major product. When biphenyl was included in the reaction, a trigonal prism was formed quantitatively instead and two molecules of biphenyl were encapsulated in the cavity. These assemblies, together with uncharacterized minor products, were all in dynamic equilibrium.22 In contrast, our trigonal prisms are formed without template assistance and are not in detectable equilibrium with other structures. The stronger coordination bonding of the Pt-N versus the Pd-N systems is a likely explanation for the differences.15,22 The work presented herein illustrates the subtle deviations that can occur in abiological self-assembly between seemingly similar systems and shows why it is imperative to examine a variety of reaction substrates and conditions.
Pyridine Ligand Rotation
Moving from considerations of favored prism structure to a discussion of its internal mobility, we note that rotation of the pyridine ligands leads to the exchange of their distinct edges and is relevant for the racemization process discussed below. Because each prism contains 12 pyridine rings whose rotations need not be independent, a full kinetic analysis requires far more experimental information than is available presently and represents a formidable task. At present, we are forced to assume that each pyridine rotates independently and shall thus deal only with their average behavior. As will be argued below, this assumption is undoubtedly poor in the case of 3d, and we shall deemphasize this prism in what follows.
The activation parameters for the pyridine edge exchange reaction in the triflate salts of 3a, 3b, and 3d, whose Eyring plots can be fitted with a single straight line, are all identical within our experimental accuracy (Table 2) and lie within the error bar of those found for the low temperature process in the triflate of 3c, 11.1 kcal/mol and -17.5 cal/mol.K, respectively. To our knowledge, there is only one previous report of ΔH‡ and ΔS‡ values for a platinum-bipyridyl based assembly, where Tárkányi et al.54 found ΔH‡ values of 12.5 kcal/mol and 14.1 kcal/mol and ΔS‡ values of -13.9 cal/mol.K and -17.2 cal/mol.K by selective inversion recovery (SIR) NMR experiments for hexafluorophosphate salts of a self-assembled rectangle and triangle, respectively. These values are essentially the same as those found in this study. They have been assumed to correspond to a simple reaction path in which the pyridine edge exchange by rotation around an intact Pt-N bond, and this interpretation appears perfectly natural to us. This simplicity is however only apparent, and we have found that the rates of pyridine rotation are significantly affected by the counterions present, implying a more complex process. This will be considered in the following companion paper,35 along with the unusual bilinear nature of the Eyring plot for 3c, which is of particular interest. There is a hint of bilinearity also in the Eyring plot for 3d, but it is much less pronounced.
An additional aspect of our results appears worthy of discussion and future pursuit, namely the chirality of the prisms, which is of a type different from those recognized up to now.31 We consider first the chirality of the isolated star connectors 1a - 1d, and then, the chirality of the assembled prisms.
Chirality of the Star Connectors 1
The chirality-inducing twist of the four pyridyl substituents out of the plane of the central ring of these star connectors is ultimately due to steric crowding. This is absent in 1a, in which one can expect the four pyridine rings to be coplanar with the benzene ring at the achiral equilibrium geometry, and the barrier to twisting the pyridine rings to be low, and affected only by the weak pyridine - benzene conjugation through the triple bond. In cages built with 1a, the pyridine rings will be nearly free to adopt the twist angles favored by the linker 2, and there should be little if any communication of the sense of twist from any of the four arms to the others through the central benzene ring. A similar situation can be expected with 1b, although the four double bonds will most likely not be entirely coplanar and there may be a small degree of correlation between the twists of the four pyridine rings.
In contrast, based on the crystal structures of tetraphenylcyclobutadienecyclopentadienylcobalt and similar sandwich complexes,57 there is no doubt that the four phenyl rings attached to the central ring in 1c will be twisted by close to 30 – 40°, all in the same sense of twist, making the favored conformations of this star connector distinctly chiral. However, judging by variable-temperature NMR spectra of this class of compounds,58 the interconversion of enantiomers is extremely facile. To whatever degree conjugation through the triple bond favors coplanarity, its terminal pyridine rings will be twisted similarly. However, it will take little energy to twist a pyridine ring out of planarity with its phenyl partner, and it should still be quite easy to adapt the orientation of the pyridine moieties to the requirements of the linker 2.
Only the connector 1d is expected to act differently. Here again, judging by known X-ray structures,57 the rings attached to the cyclobutadiene square are twisted by about 30 – 40°, all in the same sense, P or M, and according to NMR evidence conformational racemization must be very facile. However, since their four nitrogen atoms will be directly attached to the Pt atoms of the four linkers 2 at the vertices, the sense of twist that defines their chirality can be expected to be transferred through the four vertices to adjacent faces quite forcefully.
Chirality in the Prisms 3
If the edge or vertex linkers that tie the faces together into a cage discern their chirality, there will be an energy difference between a PP (or MM) and a PM face combination at a shared edge, and also among the various face combinations at a shared vertex. A collective behavior will result, and definite patterns of P and M faces on the cage will be energetically favored to different degrees. Cage racemization, which calls for a P-M interconversion at all the faces, could then require significantly higher activation energy than the facile P-M interconversion of an isolated star connector. If the P-M interconversion is attempted at a single face at a time, energy penalties will have to be paid at each mismatched edge or vertex, and if it is attempted at two or more faces at the same time to avoid these mismatches, the activation energies of the interconversions will add up.
The linker 2 has no chirality on its own. Even after it is attached to two pyridine ligands, it keeps a plane of symmetry, as the pyridine rings prefer to orient perpendicular to the P-Pt-P plane.26,52,59 However, according to molecular models, if one of these rings is twisted about its N-Pt bond, steric constraints cause the other ring to twist in the same sense. In a related complex 9, it costs about 14 kcal/mol to rotate one of the rings relative to its N-Pt axis, keeping the other intact.52 In our trigonal prismatic cages 3 we would expect the pyridine rings to be oriented at angles that reflect a compromise between the preferences of the star connectors 1 and the linkers 2.
In 3a - 3c this compromise should be easy to reach without serious energetic consequences. The pyridine rings should be close to orthogonal to the prism face since the preferences of the connectors 1a -1c are not strong, and they should not introduce any particular ground-state strain. The barriers for rotation about the Pt-N bonds should be perhaps even a little lower than the 14 kcal/mol found for 9, due to the additional π-electron conjugation gained when the pyridine is turned parallel to the face of the prism. In these three prisms the rotations at the individual vertices should not be strongly correlated and should occur nearly independently (independent face limit).
In 3d, in the earlier reported cubic cage 7, and in other polyhedra built from 1d, molecular modeling suggests that the situation is different. The preference of the star connector 1d for a 30 – 40° twist out of the prism face is quite distinct and in prisms whose faces are represented by this connector a P or M chirality on one face should definitely favor a like chirality at all adjacent faces. This is indeed the result obtained in our molecular mechanics calculations for 3d (Figure 3). It is interesting to see how the molecule of 3d copes with the conflicting stereochemical preferences of the square connectors (propeller shape) and the platinum atoms (pyridine plane perpendicular to the NPtN plane). The connector compromises by twisting two of the pyridines more (Type I) and two very little (Type II), and the platinum atom compromises by not insisting on having both pyridines perpendicular to the NPtN plane, twisting one a little and the other one significantly. The mechanism of chirality transfer from face to face is clearly apparent.
We expect the favored stereochemistry of all cages built from 1d to be all-P or all-M. Their ground states should be destabilized since preferences of the star connectors 1 and the metal linkers 2 clash and a serious compromise is necessary. However, since pyridine rotation is now expected to require a destruction of the propeller-like arrangement of the four pyridine rings at the center of one or more of the square connectors, or collective motion of the pyridine ligands, the transition state for racemization by rotation should be destabilized as well, and in the end the activation parameters for the interchange of the inner and outer pyridine protons in 3d by pyridine rotation could be similar to those observed for the other prisms. This indeed appears to be the case in Table 2.
At the moment, the anticipated favored all-P and all-M chirality of 3d and similar polyhedra is merely a result of modeling and calculations, and it would be interesting to stabilize it and detect it experimentally, perhaps after inclusion of a small chiral molecule inside the prism cavity.
Conclusions
Trigonal prisms 3 are easily accessible in high yield from tetrapyridyl star connectors 1 and the platinum acceptor 2 via template-free self-assembly, and have been characterized by multinuclear and DOSY NMR spectroscopy and dualESI-FT-ICR mass spectrometry. The products 3 are expected to be entropically and enthalpically favored over larger coordination prisms 4 - 6 and open-chain oligomeric systems, respectively. The presence of a positive cooperative effect influencing their formation is unlikely.60
An examination of the prisms 3 revealed two points of particular interest. They permit the study of endo-exo pyridine edge interchange, and they introduce the previously unappreciated face-based conformational cage chirality. Such conformational chirality should be a general phenomenon in polyhedra built from star-shaped connectors and edge or vertex based linkers. For 1d and similar star connectors with strongly correlated twist sense on the individual arms, all-P and all-M faced polyhedra 3d are expected to be favored.
The availability of prism 3c, whose pyridine edge interchange displays a highly unusual kinetic behavior, offers unique opportunities for additional insight into the properties of self-assembled polyhedra, characterized and discussed in the following companion paper.35 We propose that they pertain to the first steps of cage unraveling and thus the last steps in the mechanism of its self-assembly, and to the structure of the solvent around and/or inside the cage.
Experimental Part
General
Ligands 1b,61 1d,57 and the linker cis-(Me3P)2Pt(OTf)2 262 were prepared according to known procedures. IR spectra were recorded on a Mattson GL-3020 FTIR spectrometer. The dualESI-FT-ICR mass spectrometry instrumental setup has been described previously.34,63 NMR spectra were recorded on a Varian Unity 300 spectrometer operating at 300 MHz for 1H and 121.4 MHz for 31P and Inova 400 spectrometer operating at 161.9 MHz for 31P. The 1H chemical shifts are reported relative to the residual protons of CD3NO2 (δ 4.33 ppm). An external, unlocked sample of H3PO4 was used to reference 31P spectra (δ 0.0 ppm). DOSY NMR experiments were performed on an Inova 500 MHz spectrometer.
Temperature-dependent 1H NMR spectra for line-shape analysis were recorded with 300, 400, and 600 MHz instruments, over a range spanning the low to the high temperature limit. Temperatures were calibrated by standard methods using methanol below the ambient and glycerol above and were known to better than 0.25 K. Dynamic NMR simulations were performed using the DNMR3 utilities in the Spinworks 2.4 software.64 Complete assignment of the exchanging proton spin systems was accomplished by simulation of the low temperature ROESY spectrum in CD3NO2, relying on the opposite phase of the NOE correlations and the chemical exchange correlations. The chemical shifts and J coupling constants were accurately determined with the NUMARIT spin-simulation package in Spinworks 2.4, and this was followed by DNMR lineshape simulation of the AB spin systems (see Supporting Information). Exchange rate constants k were determined at each temperature using the DNMR3 simulation algorithm, and the results were independent of magnetic field strength.
Transient exchange (1D-EXCHSY) experiments were performed using the DPFGSE sequence,65 which uses shaped, selective excitation pulses to selectively invert the magnetization corresponding to ½ of a pair of exchanging protons, and monitoring the buildup of inverted magnetization of the exchanging partner. Experiments were performed using a minimum of six mixing times over a range of 0.05 to 0.5 s. Selective inversion pulses were calculated using Varian's “PBOX” software, implementing sech180 shaped pulses centered on the desired resonance of duration adequate to effect selective inversion of the desired resonance, while not affecting proton magnetization associated with nearby peaks in the spectrum.
DOSY NMR measurements of the diffusion coefficient D were performed using the DOSY Bipolar Pulse Pair Stimulated Echo pulse sequence (Dbppste) sequence and a Nalorac® Triple-Inverse probe with a Z-axis PFG coil. No gradient shaping was applied, as it had led to no improvement in previous calibrations with this probe and to no detectable problems due to gradient eddy-current effects. PFG bipolar pulse pairs of 2.0 msec each were applied using gradient strengths ranging from 9.03 to 44.25 gauss/cm, where careful calibration showed linearity better than 99.8%; this probe and gradient-pulse-amplifier are capable of gradient strengths up to 68 gauss/cm. Gradient stability tests with this probe and amplifier yielded reproducibility better than > 99.9% over 20 repetitions. A constant diffusion delay Δ of 0.050 s was used in all experiments. The sample temperature was set to 298 K using ethylene glycol and standard calibration methods. The use of bipolar gradient-pulse-pairs eliminates many sources of error, and gradient strengths were calibrated using the width (in Hz) of a sample of known length along the NMR-tube (Z) axis. Calibrations were further checked by multiple, redundant measurements using the residual 1H NMR signal from 99.9% D2O (D = 18.72 × 10-10 m2/s,66 also reported as 19×10-10 m2/s67). Repeated measurements at temperatures of 297, 298, & 299 K yielded temperature-corrected values of D = 18.5 ± 0.6 m2/s (ranging from 17.9 to 19.1 ×10-10). The values of D determined by the DOSY experiment were used to calculate the hydrodynamic radii (R) from the Stokes-Einstein equation: D = kBT/6πRη, where kB is the Boltzmann constant, T is the absolute temperature, and η is the published68 viscosity of nitromethane (0.0062 gs-1cm-1). The effect of variable viscosity from sample to sample was examined using the D value observed for the residual protons in the solvent resonance, which ranged from 19.5 to 21.4 × 10-10 m2/s, with an average value of 20.4 m2/s. This results in a limiting error due to variable viscosity of ±5%.
Calculations
Geometries were minimized with the TINK 40 program using the UFF force field38 with Powell and truncated Newton optimization methods. Some structures were subjected to short dynamics runs in an effort to relax them and find additional minima. Charges for each species were calculated by charge equilibration39 and were kept throughout.
Synthesis of 1,2,4,5-Tetrakis(4-pyridylethynyl)benzene 1a
The reported conditions61 were difficult to reproduce. Instead, 1,2,4,5-tetraiodobenzene69 (0.40 g, 0.69 mmol), 4-ethynylpyridine (0.36 g, 3.49 mmol), Pd(PPh3)4 (0.17 g, 0.15 mmol) and CuI (0.03 g, 0.16 mmol) were placed in an oven-dried Schlenk flask under N2. THF (8 mL) and triethylamine (2.4 mL) were added. The reaction mixture was heated in an oil bath at 60 °C for 24 h. The precipitated product was filtered, taken up in chloroform, treated with activated carbon and filtered again. Concentration of the filtrate and addition of hexane yielded 1a (0.08 g, 24 %) of sufficient purity (1H NMR) for further reaction.
Synthesis of Tetrakis[4-(4-pyridylethynyl)phenyl]cyclobutadienecyclopentadienylcobalt(I) 1c
To a flame-dried 50 mL three-neck flask with a condenser was added tetrakis(4-iodophenyl)cyclobutadienecyclopentadienylcobalt(I) (100 mg, 0.10 mmol),57 4-ethynylpyridine hydrochloride (63 mg, 0.44 mmol), (PPh3)2PdCl2 (7 mg, 0.01 mmol), and CuI (4 mg, 0.02 mmol). The flask was evacuated for 10 min and purged with argon. The solids were dissolved in 10 mL of THF and then 10 mL of triethylamine was added. The solution was refluxed for 8 h and the reaction progress was monitored by NMR. Upon completion, the flask was cooled and all solvents were removed under reduced pressure. The resulting dark solid was chromatographed on silica gel with 9/1 chloroform/methanol. The first band off the column contained triphenylphosphine salts, followed by the product as an orange solid. The solid was crystallized by the slow evaporation of CH2Cl2 from an ∼3:1 MeOH:CH2Cl2 solution to yield 60 mg (68%) of 1d as violet crystals. Mp: 310°C (dec). 1H NMR (CDCl3, 500 MHz): δ 8.60 (dd, 3J = 5.2 Hz, 8H, Hα), 7.37 - 7.42 (m, 24H, Ar and Hβ), 4.64 (s, 5H, Cp). 13C{1H} NMR (CDCl3, 400 MHz): δ 150.02, 137.53, 131.96, 131.62, 128.89, 125.67, 120.28, 94.30, 87.62, 83.77, 74.80. IR (KBr, cm-1): 3034, 2215, 1712, 1590, 1513, 1405, 1213, 1108, 988, 846, 816, 750, 662, 630, 588, 546, 522. Anal. Calcd. for C61H21N4Co•2(CH3OH): C, 79.74; H, 4.78; N, 5.90. Found: C, 80.08; H, 5.01; N, 5.96.
General Procedure for the Preparation of Trigonal Prisms 3
The appropriate pyridyl linker 1 (1 equiv) and cis-(Me3P)2Pt(OTf)2 (2 equiv) were placed in a 1-dram vial. CD3NO2 (2 mL) was added and the mixture stirred for 18 h at room temperature. The solution was transferred to an NMR tube for analysis. For 3a and 3b the solvent was then evaporated, while 3c and3d were precipitated with diethyl ether. In all cases the product was washed with diethyl ether and then dried under reduced pressure.
3a. Yield 91%. 1H NMR (CD3NO2)5 9.28 (br d, 3J = 6.6 Hz, 12H, H), 8.94 (br d, 3J = 6.5 Hz, 12H, Hα,), 7.99 (s, 6H, Hphenyl), 7.77 (m, 24H, Hβ), 1.78 (d, 2JH–P = 11.3 Hz, 108H, PCH3); 31P{1H} NMR (CD3NO2) δ -27.7 (s, 195Pt satellites, 1JPt–P = 3128 Hz). IR (KBr, cm-1): 3101 w, 3005 w, 2923 w, 2216 w, 1612 s, 1510 w, 1429 m, 1262 s, 1226 s, 1162 s, 1063 w, 1032 s, 975 m, 953 s, 861 w, 838 w, 638 s, 574 w, 551 w, 518 m. Anal. Calcd. for C150H162F36N12O36P12Pt6S12•4H2O: C, 33.41; H, 3.18; N, 3.12. Found: C, 33.07; H, 3.26; N, 3.16.
3b. Yield 97%. 1H NMR (CD3NO2) 5 8.84 (m, 24H, Hα), 8.27 (d, 3J = 4.9 Hz, 12H, Hβ), 8.20 (s, 6H, Hphenyl), 8.12 (d, 3J = 15.8 Hz, 12H, C=CH), 7.61 (d, 3J = 4.9 Hz, 12H, Hβ,), 7.32 (d, 3J = 15.8 Hz, 12H, C=CH), 1.72 (d, 2JH-P = 11.2 Hz, 108H, PCH3). 31P{1H} NMR (CD3NO2) δ -28.0 (s, 195Pt satellites, 1JPt-P = 3125 Hz). IR (KBr, cm-1): 2981 w, 2926 w, 1611 s, 1434 w, 1384 w, 1260 s, 1226 m, 1165 m, 1067 w, 1031 s, 974 m, 954 m, 638 m, 520 w. Anal. Calcd. for C150H186F36N12O36P12Pt6S12•2CH3NO2•2(CH3CH2)2O: C, 34.23; H, 3.81; N, 3.49. Found: C, 34.00; H, 4.12; N, 3.46.
3c. Yield 93%. 1H NMR (CD3NO2): δ 8.87 (d, 3J = 3.7 Hz, 24H, Hα), 7.73 (d, 3J = 6.3 Hz, 12H, Hβ), 7.69 (d, 3J = 5.8 Hz, 12H, Hβ,), 7.50-7.37 (m, 48H, phenyleneH), 4.82 (s, 15H, CpH), 1.74 (d, 2JH-P = 11.2 Hz, 108H, PCH3). 31P{1H} NMR (CD3NO2)δ-27.8 (s, 195Pt satellites, 1JP-Pt = 3150 Hz). IR (KBr, cm-1): 2929 w, 2216 m, 2171 w, 1612 m, 1597 m, 1515 m, 1431 w, 1259 s, 1225 m, 1160 m, 1063 w, 1030 s, 974 m, 952 m, 844 w, 752 w, 681 w, 638 m, 553 w. Anal. Calcd. for C231H219Co3F36N12O36P12Pt6S12•5CH3NO2•1 1H2O: C, 40.32; H, 3.67; N, 3.39. Found: C, 40.20; H, 4.00; N, 2.99.
3d. Yield 97%. 1H NMR (CD3NO2) 5 8.80 (d, 3J = 5.3 Hz, 12H, H), 8.29 (d, 3J = 4.8 Hz, 12H, Hα, or Hβ), 8.20 (d, 3J = 4.8 Hz, 12H, Hβ, or Hα,), 7.57 (d, 3J = 5.3 Hz, 12H, Hβ), 4.50 (s, 15H, CpH), 1.73 (d, 2JH-P = 10.8 Hz, 108H, PCH3). 31P{1H} NMR (CD3NO2) δ −28.1 (s, 195Pt satellites, 1JPt-P = 3132 Hz). IR (KBr, cm-1): 3105 w, 2997 w, 2926 w, 1610 s, 1508 w, 1432 w, 1385 w, 1265 s, 1226 m, 1162 m, 1063 w, 1032 s, 974 m, 953 m, 861 w, 840 w, 638 s, 592 w, 577 w, 518 w. Anal. Calcd. for C135H171Co3F36N12O36P12Pt6S12•(CH3CH2)2O: C, 30.92; H, 3.38; N, 3.11. Found: C, 30.96; H, 3.73; N, 3.49.
Supplementary Material
Acknowledgments
We are grateful to Prof. R. G. Bergman for suggesting the mercury drop test. Financial support from the National Science Foundation (CHE-0446688, and OISE 0532040), National Institute of Health (5R01GM57052), the W. M. Keck Foundation, the USARO (DAAD19-01-1-0521), DoD High Performance Modernization Office (Challenge Project C1R and ARONC014), the European Commission (STRP NMP4-013880 and MCRTN-CT-2005-019481), the GAAV (IAA400550616) and Ministry of Education of the Czech Republic (KONTAKT ME-857) is gratefully acknowledged.
Footnotes
Supporting Information Available. NMR and infrared spectra of 3a-d. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Seidel SR, Stang PJ. Acc Chem Res. 2002;35:972. doi: 10.1021/ar010142d. [DOI] [PubMed] [Google Scholar]
- 2.Swiegers GF, Malefetse TJ. Coord Chem Rev. 2002;225:91. [Google Scholar]
- 3.Holliday BJ, Mirkin CA. Angew Chem Int Ed. 2001;40:2022. [PubMed] [Google Scholar]
- 4.Cotton FA, Lin C, Murillo CA. Acc Chem Res. 2001;34:759. doi: 10.1021/ar010062+. [DOI] [PubMed] [Google Scholar]
- 5.Swiegers GF, Malefetse TJ. Chem Rev. 2000;100:3483. doi: 10.1021/cr000023w. [DOI] [PubMed] [Google Scholar]
- 6.Leininger S, Olenyuk B, Stang PJ. Chem Rev. 2000;100:853. doi: 10.1021/cr9601324. [DOI] [PubMed] [Google Scholar]
- 7.Caulder DL, Raymond KN. Acc Chem Res. 1999;32:975. [Google Scholar]
- 8.Caulder DL, Raymond KN. J Chem Soc, Dalton Trans. 1999:1185. [Google Scholar]
- 9.Baxter PNW, Lehn JM, Baum G, Fenske D. Chem Eur J. 1999;5:102. [Google Scholar]
- 10.Fujita M. Chem Soc Rev. 1998;6:417. [Google Scholar]
- 11.Fujita M, Tominaga M, Hori A, Therien B. Acc Chem Res. 2005;38:369. doi: 10.1021/ar040153h. [DOI] [PubMed] [Google Scholar]
- 12.Maurizot V, Yoshizawa M, Kawano M, Fujita M. Dalton Trans. 2006;2750 doi: 10.1039/b516548m. [DOI] [PubMed] [Google Scholar]
- 13.Kawano M, Fujita M. Coord Chem Rev. 2007;251:2592. [Google Scholar]
- 14.Stang PJ, Olenyuk B. Acc Chem Res. 1997;30:502. [Google Scholar]
- 15.Johannessen SC, Brisbois RG, Fischer JP, Grieco PA, Counterman AE, Clemmer DE. J Chem Soc. 2001;123:3818. doi: 10.1021/ja004007s. [DOI] [PubMed] [Google Scholar]
- 16.Roche S, Haslam C, Adams H, Heath SL, Thomas J. A Chem Commun. 1998;1681 [Google Scholar]
- 17.Schweiger M, Seidel SR, Schmitz M, Stang PJ. Org Lett. 2000;2:1255. doi: 10.1021/ol005781n. [DOI] [PubMed] [Google Scholar]
- 18.Fujita M, Yu SY, Kusukawa T, Funaki H, Ogura K, Yamaguchi K. Angew Chem Int Ed. 1998;37:2082. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2082::AID-ANIE2082>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 19.Olenyuk B, Whiteford JA, Fechtenkotter A, Stang PJ. Nature. 1999;398:796. doi: 10.1038/19740. [DOI] [PubMed] [Google Scholar]
- 20.Olenyuk B, Levin MD, Whiteford JA, Shield JE, Stang PJ. J Chem Soc. 1999;121:10434. [Google Scholar]
- 21.Kuehl CJ, Kryschenko YK, Radhakrishnan U, Seidel SR, Huang SD, Stang PJ. Proc Natl Acad Sci U S A. 2002;99:4932. doi: 10.1073/pnas.012540799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamanoi Y, Sakamoto Y, Kusukawa T, Fujita M, Sakamoto S, Yamaguchi K. J Chem Soc. 2001;123:980. doi: 10.1021/ja003043o. [DOI] [PubMed] [Google Scholar]
- 23.Caskey DC, Michl J. J Org Chem. 2005;70:5442. doi: 10.1021/jo050409c. [DOI] [PubMed] [Google Scholar]
- 24.Caskey DC, Wang B, Zheng X, Michl J. Collect Czech Chem Commun. 2005;70:1970. [Google Scholar]
- 25.Michl J, Magnera TF. Proc Natl Acad Sci U S A. 2002;99:4788. doi: 10.1073/pnas.052016299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto T, Arif AM, Stang PJ. J Chem Soc. 2003;125:12309. doi: 10.1021/ja0302984. [DOI] [PubMed] [Google Scholar]
- 27.Stang PJ, Olenyuk B, Arif AM. Organometallics. 1995;14:5281. [Google Scholar]
- 28.Stang PJ, Olenyuk B. Angew Chem Int Ed Engl. 1996;35:732. [Google Scholar]
- 29.Olenyuk B, Whiteford JA, Stang PJ. J Chem Soc. 1996;118:8221. [Google Scholar]
- 30.Stang PJ, Olenyuk B, Muddiman DC, Smith RD. Organometallics. 1997;16:3094. [Google Scholar]
- 31.Seeber G, Tiedmann BEF, Raymond KN. Top Curr Chem. 2006;265:147. [Google Scholar]
- 32.Davis AV, Fiedler D, Ziegler M, Terpin A, Raymond KN. J Am Chem Soc. 2007;129:15354. doi: 10.1021/ja0764815. [DOI] [PubMed] [Google Scholar]
- 33.Eliel EL, Wilen SH. Stereochemistry of Organic Compounds. Wiley; New York: 1994. p. 1120. [Google Scholar]
- 34.Nepomuceno AI, Muddiman DC, Bergen HR, III, Craighead JR, Burke MJ, Caskey PE, Allan JA. Anal Chem. 2003;75:3411. doi: 10.1021/ac0342471. [DOI] [PubMed] [Google Scholar]
- 35.Vacek, J.; Caskey, D. C.; Horinek, D.; Shoemaker, R. K.; Stang, P. J.; Michl, J. submitted for publication.
- 36.Chi KW, Addicott C, Kryschenko YK, Stang PJ. J Org Chem. 2004;69:964. doi: 10.1021/jo035607n. [DOI] [PubMed] [Google Scholar]
- 37.Kuehl CJ, Huang SD, Stang PJ. J Chem Soc. 2001;123:9634. doi: 10.1021/ja0114355. [DOI] [PubMed] [Google Scholar]
- 38.Rappe AK, Casewit CJ, Colwell KS, Goddard WA, III, Skiff WM. J Am Chem Soc. 1992;114:10024. [Google Scholar]
- 39.Rappe AK, Goddard WA., III J Phys Chem. 1991;95:3358. [Google Scholar]
- 40.Vacek J, Michl J. New J Chem. 1997;21:1259. [Google Scholar]
- 41.Schalley CA, Müller T, Linnartz P, Witt M, Schäfer M, Lützen A. Chem Eur J. 2002;8:3538. doi: 10.1002/1521-3765(20020802)8:15<3538::AID-CHEM3538>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 42.Tanner JE. J Chem Phys. 1970;52:2523. [Google Scholar]
- 43.Mayzel O, Cohen Y. J Chem Soc, Chem Commun. 1994:1901. [Google Scholar]
- 44.Caskey DC, Shoemaker RK, Michl J. Org Lett. 2004;6:2093. doi: 10.1021/ol049539i. [DOI] [PubMed] [Google Scholar]
- 45.Buschhaus B, Bauer W, Hirsch A. Tetrahedron. 2003;59:3899. [Google Scholar]
- 46.Menger FM, Azov VA. J Am Chem Soc. 2002;124:11159. doi: 10.1021/ja0206238. [DOI] [PubMed] [Google Scholar]
- 47.Cohen Y, Ayalon A. Angew Chem Int Ed. 1995;34:816. [Google Scholar]
- 48.Rymdén R, Stilbs P. Biophys Chem. 1985;21:145. doi: 10.1016/0301-4622(85)85016-x. [DOI] [PubMed] [Google Scholar]
- 49.Park SJ, Shin DM, Sakamoto S, Yamaguchi K, Chung YK, Lah MS, Hong JI. Chem Eur J. 2005;11:235. doi: 10.1002/chem.200400801. [DOI] [PubMed] [Google Scholar]
- 50.Megyes T, Jude H, Grósz T, Bakó I, Radnai T, Tárkányi G, Pálinkás G, Stang PJ. J Am Chem Soc. 2005;127:10731. doi: 10.1021/ja0523690. [DOI] [PubMed] [Google Scholar]
- 51.Tiede DM, Zhang R, Chen LX, Yu L, Lindsey JS. J Chem Soc. 2004;126:14054. doi: 10.1021/ja048209q. [DOI] [PubMed] [Google Scholar]
- 52.Fuss M, Siehl HU, Olenyuk B, Stang PJ. Organometallics. 1999;18:758. [Google Scholar]
- 53.Fan J, Whiteford JA, Olenyuk B, Levin MD, Stang PJ, Fleischer EB. J Chem Soc. 1999;121:2741. [Google Scholar]
- 54.Tárkányi G, Jude H, Pálinkás G, Stang PJ. Org Lett. 2005;7:4971. doi: 10.1021/ol051910u. [DOI] [PubMed] [Google Scholar]
- 55.Clare B, Sarker N, Shoemaker R, Hagadorn JR. Inorg Chem. 2004;43:1159. doi: 10.1021/ic035035p. [DOI] [PubMed] [Google Scholar]
- 56.Widegrin JA, Finke RG. J Mol Cat A: Chem. 2003;198:317. [Google Scholar]
- 57.Harrison RM, Brotin T, Noll B, Michl J. Organometallics. 1997;16:3401. [Google Scholar]
- 58.Zheng X, Mulcahy ME, Horinek D, Galeotti F, Magnera TF, Michl J. J Chem Soc. 2004;126:4540. doi: 10.1021/ja039482f. [DOI] [PubMed] [Google Scholar]
- 59.Chi KW, Addicott C, Arif AM, Das N, Stang PJ. J Org Chem. 2003;68:9798. doi: 10.1021/jo0353785. [DOI] [PubMed] [Google Scholar]
- 60.Ercolani G. J Chem Soc. 2003;125:16097. doi: 10.1021/ja038396c. [DOI] [PubMed] [Google Scholar]
- 61.Amoroso AJ, Cargill Thompson AMW, Maher JP, McCleverty JA, Ward MD. Inorg Chem. 1995;34:4828. [Google Scholar]
- 62.Stang PJ, Cao DH, Saito S, Arif AM. J Chem Soc. 1995;117:6273. [Google Scholar]
- 63.Kryschenko YK, Seidel SR, Muddiman DC, Nepomuceno AI, Stang PJ. J Chem Soc. 2003;125:9647. doi: 10.1021/ja030209n. [DOI] [PubMed] [Google Scholar]
- 64.Kirk Marat, Spinworks 2.4 Software, University of Manitoba. Available from: < http//www.umanitoba.ca/chemistry/nmr/spinworks>.
- 65.Stott K, Keeler J, Van QN, Shaka AJ. J Mag Res. 1997;125:302. [Google Scholar]
- 66.Bruker Instruments, personal communication.
- 67.Longsworth LG. J Phys Chem. 1960;64:1914. [Google Scholar]
- 68.West RC, editor. CRC Handbook of Chemistry and Physics. 67th. CRC Press; Boca Raton, Florida: 1986. p. F-40. [Google Scholar]
- 69.Mattern DL. J Org Chem. 1983;48:4772. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.