


Abstract
The dynamics and mechanism of how site-specific DNA-bending proteins initially interrogate potential binding sites prior to recognition have remained elusive for most systems. Here we present these dynamics for Integration Host factor (IHF), a nucleoid-associated architectural protein, using a μs-resolved T-jump approach. Our studies show two distinct DNA-bending steps during site recognition by IHF. While the faster (∼100 μs) step is unaffected by changes in DNA or protein sequence that alter affinity by >100-fold, the slower (1–10 ms) step is accelerated ∼5-fold when mismatches are introduced at DNA sites that are sharply kinked in the specific complex. The amplitudes of the fast phase increase when the specific complex is destabilized and decrease with increasing [salt], which increases specificity. Taken together, these results indicate that the fast phase is non-specific DNA bending while the slow phase, which responds only to changes in DNA flexibility at the kink sites, is specific DNA kinking during site recognition. Notably, the timescales for the fast phase overlap with one-dimensional diffusion times measured for several proteins on DNA, suggesting that these dynamics reflect partial DNA bending during interrogation of potential binding sites by IHF as it scans DNA.
INTRODUCTION
Numerous cellular processes are initiated by special proteins that bind with high specificity to their target sites on DNA. These proteins discriminate between specific and non-specific sites using a combination of ‘direct readout’, in which the protein contacts the chemically unique moieties of specific bases, and ‘indirect readout’, in which the protein recognizes sequence-dependent shape and variations in local DNA flexibility and ‘read’ DNA sequence by the ease with which they can deform the potential binding sites to match their binding interface (1). These DNA deformations are generally accompanied by conformational rearrangements in the bound protein (‘induced-fit’ mechanism) to facilitate favorable interactions and enhance the specificity (2,3). Proteins searching for their target sites within genomic DNA face a daunting task: they need to scan rapidly across a vast excess of non-specific sites, but not scan so fast as to ‘overlook’ their target sites. How proteins play this balancing act remains a puzzle.
Several lines of evidence suggest that proteins use some combination of 3-D (in solution) and 1-D (along the DNA) diffusion to rapidly hone in on their target site (4–9). Experimental estimates of 1-D diffusion constants range from 2 × 105 to 107 bp2/s for several different proteins ‘sliding’ or ‘hopping’ on DNA, indicating stepping times or ‘residence’ times per base-pair (bp) site in the range from 50 ns to 300 μs (10–16). Such rapid movement, which suggests a relatively smooth energy landscape on DNA, needs to be reconciled with a low free energy minimum once the protein is stably bound to its target site. The transition from a non-specific and mobile conformation to a specific conformation at the target site must be sufficiently fast to prevent the protein from diffusing away. The mechanism by which proteins slow down to more thoroughly examine potential binding sites, initiate recognition, and eventually form the myriad of specific contacts is not well understood.
Theoretical considerations of this ‘speed-stability’ paradox have implicated that a protein, while scanning DNA for its target site, intermittently switches conformations from a rapidly diffusing ‘search’ mode into a less mobile ‘interrogation’ or ‘recognition’ mode, to give it more time to closely examine a potential site (17,18). Consistent with this notion, nuclear magnetic resonance (NMR) studies have shown highly dynamic non-specific protein–DNA complexes, with conformational fluctuations on sub-millisecond timescales, commensurate with 1-D diffusion times (19–21). Evidence for highly dynamic protein–DNA complexes with implications for ‘facilitated diffusion’ also comes from DNA micromanipulation experiments that suggest rapid microscopic association/dissociation events, likely coupled to 1-D diffusion, and macroscopic association/dissociation events (22–24). Several single-molecule studies indicate a wide range of 1-D diffusion constants for a given protein on DNA, likely reflecting intermittently stalled binding modes, although the temporal and spatial resolution of these studies are insufficient to directly observe the transiently stalled proteins (16,25–28).
Direct experimental observations of protein–DNA conformational dynamics and trajectory during ‘interrogation’ en route to ‘recognition’ for site-specific DNA-bending proteins have been particularly challenging, owing to the lack of requisite time-resolution or sensitivity of many techniques. Although there is an extensive body of literature on measurements of the dynamics of protein–DNA interactions, using primarily stopped-flow (29–36) or single-molecule techniques (22–24,37–42), these studies are largely unable to separate bimolecular association/dissociation kinetics from unimolecular conformational dynamics occurring on timescales of a few milliseconds or faster. To our knowledge, only a handful of such studies could resolve DNA distortional dynamics that reflected the initial stages of site-specific recognition; these include kinetics of base-flipping during recognition of uracil by base excision repair pathway protein UDG (43); DNA bending upon specific binding by a restriction enzyme, EcoRV, to mechanically stretched DNA (37); DNA bending upon specific binding by Integration Host Factor (IHF) to one of its cognate sites on phage λ DNA (the H’ site) (33,44); DNA bending during mismatch (T-bulge) recognition by mismatch repair protein MutS (45); nucleotide-flipping during damage recognition by nucleotide excision repair protein XPC/Rad4 (46). Other studies have reported much slower DNA bending kinetics, occurring on timescales of >100 ms (30,39,41,47–49), which likely represent slow reorganization in the specific complex and not the initial recognition event.
A common and intriguing theme from the above-mentioned studies is that DNA deformations accompanying site-specific recognition are slow, on timescales longer than a few milliseconds and hence significantly slower than previously reported sub-millisecond 1-D diffusion times. Thus, the question remained: How does a rapidly diffusing protein slow down long enough at a potential binding site to enable ‘slow’ conformational rearrangements that lead to recognition?
A recent study that took advantage of the superior time resolution and sensitivity of the laser temperature-jump (T-jump) approach did in fact unveil fast (100–500 μs) DNA unwinding dynamics induced by DNA repair protein XPC/Rad4 (50). These rapid dynamics, which preceded a slower ∼10 ms nucleotide-flipping event that signaled DNA damage recognition (46,50), were observed on complexes with Rad4 bound non-specifically to DNA and were interpreted as evidence for conformational fluctuations between search and interrogation modes of the bound protein. Here, we present kinetic evidence for an analogous sequential two-step interrogation then recognition during site-specific binding and DNA bending by the Escherichia coli Integration Host Factor (IHF), described below.
IHF is an abundant nucleoid-associated architectural protein that also acts as a host factor for lysogeny by bacteriophage λ. It recognizes several sites on phage λ DNA, primarily by indirect readout, bending the DNA at its cognate site by nearly 180° over ∼35 bp, bringing distal regions of the DNA together to form higher-order nucleoprotein complexes (51). The crystal structure of IHF bound to one such cognate site (the H’ site) reveals that the DNA is sharply kinked at two sites ∼9-bp apart, and this severely bent DNA is stabilized by intercalation of conserved proline residues located on two β-ribbon arms that wrap around the DNA (Figure 1A). IHF is also known to bind non-specifically to DNA, although with 103–104-fold lower affinity than for specific binding sites (52–54). In its non-specific binding mode, IHF plays a role in chromatin compaction (55). Because of the severe DNA bending and the indirect nature of site-specific recognition in this complex, IHF serves as an excellent model system for analyzing the mechanism by which DNA-bending proteins probe sequence-dependent DNA deformability to recognize their target sites (56–58).
Figure 1.

IHF–DNA complexes and equilibrium FRET measurements. (A) Model of the IHF–H′ complex based on the cocrystal structure (PDB code: 1IHF) but without the nick in the DNA that was required for crystallization. The DNA is shown in gray, with locations of modifications in the DNA sequences used in this study highlighted in color: pink, mismatches introduced in the TT8AT, TTloop and ATloop substrates; red, T→A mutation in the H’44A substrate. Locations of three positively charged residues, shown in yellow, that were substituted for a neutral residue are also shown: (top) βK84A, (middle) αK5A and (bottom) αR21C. (B) Sequences of H’ substrates and its variants are shown. The 5′-end of the top strand is fluorescein-dT (yellow), and the 5′-end of the bottom (complementary) strand is TAMRA-dT (green). The consensus region is shown in gray. The blue arrows indicate location of the kinks in the DNA when in complex with IHF. Modifications in the DNA in pink/red are as described for (A). (C) Steady-state fluorescence emission spectra are shown for fluorescein-labeled H’ substrate (H'_D; 1 μM) in the absence (purple) and presence (orange) of 1.5 μM IHF, with excitation of fluorescein (donor) at 485 nm. Emission spectra are also shown for fluorescein/TAMRA-labeled H’ substrate (H'_DA; 1 μM) in the absence (blue) and presence (red) of 1.5 μM IHF, with excitation at 485 nm, and with direct excitation of TAMRA (acceptor) at 555 nm, in the absence (green) and presence (black) of IHF. All spectra are shown for measurements at 20°C. (D) The donor intensity of the H’ DNA-only sample (black) and IHF–H’ sample (red) are plotted versus temperature. The intensities are the area under the measured spectra, integrated from 510–535 nm, and are normalized such that the two data sets match at 20°C. The open symbols in each set represent reversibility checks and were measured after the sample was heated up to 60°C and then cooled back down to 20°C. The error bars are standard deviations from two independent sets of measurements. (Inset) FRET efficiency (E) of the IHF–H’ complex is plotted as a function of temperature. a.u., arbitrary unit.
Previous stopped-flow and laser T-jump measurements on IHF binding to its cognate H’ site revealed that DNA bending in the complex occurs on 1–10 ms (33,44), similar to the timescales for thermal disruption of a single A:T base pair in B-DNA, and probably corresponding to the IHF-induced disruption of stacking at the kinks (59–61). As discussed above, these rates for site-specific recognition were slow in comparison with the range of reported 1-D diffusion times for various proteins scanning DNA, leaving open the question whether these studies had missed a faster step. Preliminary evidence that an ∼100 μs step exists in the DNA bending dynamics of IHF–H’ was reported previously by our group (62,63), although the origin of this fast step was not thoroughly investigated until now.
Here, we present T-jump measurements on IHF–H’ and a series of variants that demonstrate that the rapid ∼100 μs phase is from DNA bending by IHF while bound non-specifically. We find that the fast phase rates are not changed by modifications in the consensus regions of H’ DNA or mutations in IHF that lower the positive charge of its wrapping surface, despite significant changes in binding affinity and specificity. Conditions that lower specificity are, however, reflected in corresponding increases in the relative amplitudes of the fast phase. Notably, this rapid phase overlaps with previously reported 1-D diffusion times of proteins scanning DNA and may represent rapid conformational switching between search and initial interrogation modes of IHF (and the bound DNA) as it ‘probes’ DNA while searching for its site. The results reported here provide us with key missing steps in the dynamical trajectory of site-specific recognition by IHF, with implications for the search mechanism by other site-specific DNA-bending proteins.
MATERIALS AND METHODS
Materials
The DNA sequences used in this study are shown in Figure 1. They were labeled with fluorescein (F) and TAMRA (R) attached to the thymidine overhangs at the 5′-end of the top and bottom strands, respectively, through six-carbon phosphoradimite linkers. DNA duplexes were prepared and annealed as described in Supplementary Methods 1.1. The IHF protein was prepared as described previously (64). Droplets of proteins were first flash-frozen in liquid nitrogen prior to storage in cryogenic tubes at −80°C. Individual frozen droplets were diluted into the binding buffer, as needed. All measurements were performed in binding buffer: 20 mM Tris–HCl (pH 8.0), 1 mM ethylenediaminetetraacetic acid (EDTA), 0.01% NP-40 (plus 2 mM dithiothreitol (DTT) in αR21C mutant protein samples), with [KCl] ranging from 50 to 200 mM.
Equilibrium measurements
The steady-state fluorescence emission spectra and anisotropies were measured on a FluoroMax4 spectrofluorimeter (Jobin Yvon, Inc., NJ, USA). The FRET efficiencies (FRET E) were obtained from the measured spectra under each condition, as described in Supplementary Methods 1.2. The circular dichroism (CD) measurements were carried out on JASCO J-810 spectropolarimeter, as described in Supplementary Methods 1.3. The dissociation constants (Kd) for all the IHF–DNA complexes at 100 mM KCl, in Table 1, are tabulated from previously reported equilibrium measurements on these complexes, in which the acceptor ratio (related to FRET E) for each complex were measured as a function of salt concentration; these ‘salt-titration’ profiles enabled Kd measurements in the sub-nM region, as described in ref. (64). Additionally, Kd of IHF–H’, obtained from the ratio of the on- and off-rates of this complex, measured in previous stopped-flow studies (33), are also included in Table 1.
Table 1. Dissociation constants (Kd) and relaxation rates (kr) for IHF–DNA complexes.
| Complex | K d (in M)a | K d (in M)a | K d (in M)a | k r (s−1) at 25°C | k r (s−1) at 37°C | ||
|---|---|---|---|---|---|---|---|
| (0.1 M KCl) | (0.2 M KCl) | (0.3 M KCl) | fast | slow | fast | slow | |
| IHF–H' | 2.7 (0.2)b × 10−11c 2.5 × 10−11 | 4.6 (0.4) × 10−8 | 3.0 (2.0) × 10−6 | 5.8 (1.6) × 103 | 39 (23) | 6.2 (2.3) × 103 | 132 (85) |
| IHF–TT8AT | 2.5 (1.2) × 10−12 | 2.0 (0.7) × 10−9 | 1.3 (0.2) × 10−7 | 6.2 (4.7) × 103 | 320 (110) | 8.6 (7.4) × 103 | 930 (249) |
| IHF–H'44A | 7.4 (2.8) × 10−9 | 5.8 (3.3) × 10−6 | 4.3 (3.3) × 10−4 | 5.3 (0.7) × 103 | 37 (7.4) | 4.4 (0.6) × 103 | 114 (10) |
| αK5A–H' | 3.5 (3.0) × 10−8 | 1.5 (0.5) × 10−6 | 1.3 (0.1) × 10−5 | 4.9 (1.9) × 103 | 43 (5.2) | 7.7 (1.6) × 103 | 79 (13) |
| βK84A–H' | 1.6 (0.3) × 10−8 | 1.1 (0.1) × 10−6 | 1.3 (0.2) × 10−5 | 3.0 (0.9) × 103 | 43 (1.3) | 3.5 (0.9) × 103 | 67 (12) |
| αR21C–H' | 1.0 (0.2) × 10−11 | 2.0 (0.1) × 10−8 | 1.8 (0.2) × 10−6 | 9.8 (5.7) × 103 | 70 (46) | 9.7 (5.6) × 103 | 224 (185) |
Temperature-jump measurements
Rapid T-jump of ∼5−10°C was achieved in sample cuvettes of path length 0.5 mm, and the fluorescence emission intensities of the donor (fluorescein) were measured as a function of time, with excitation at 488 nm, to acquire the relaxation traces (44,64). To cover the span of over three orders of magnitude in time, we recorded the T-jump kinetics traces over different timescales and then combined these traces (see Supplementary Figure S1). The combined relaxation traces were analyzed using maximum entropy method (MEM) to obtain a model-independent distribution of relaxation times that best described our relaxation traces. These traces were also analyzed using a sum of discrete exponential decay curves. The details of the laser T-jump spectrometer and the analysis of the relaxation traces are described in Supplementary Methods 1.4−1.12.
RESULTS
DNA bending by IHF monitored by FRET measurements
The ∼180° bending of the DNA in the IHF–H’ cocrystal structure (Figure 1A) shortens the end-to-end distance of the 35-bp H’ DNA from ∼100 Å to ∼50 Å (51). Therefore FRET measurements between fluorophore-labeled DNA ends provide a sensitive probe for direct measurement of DNA bending in complex with the protein (33,44,65). As in the previous kinetics studies of DNA bending by IHF, we labeled the 5′-termini of one DNA strand with fluorescein and the other with TAMRA (Figure 1B), and used FRET assay to measure changes in the end-to-end distance and hence the changes in the bent conformations of the IHF–H’ complex (66). All measurements reported here were performed in the binding buffer: 20 mM Tris–HCl (pH 8.0), 1 mM EDTA, 0.01% NP-40 and KCl concentrations 50, 100, 150 and 200 mM.
Important initial controls are shown in Figure 1C and D. A representative set of fluorescence emission spectra of donor only and donor–acceptor labeled H’ DNA (H’_D and H’_DA, respectively) in the presence and absence of IHF are shown at 20°C in Figure 1C. Excitation of the donor (fluorescein) in H’_D, at 485 nm, yields similar spectra with and without IHF (Figure 1C, orange and purple, respectively), with fluorescence emission intensities overlapping within 3%. Direct excitation of the acceptor (TAMRA) in H’_DA, at 555 nm, reveals fluorescence emission spectra with intensities that are ∼6% lower with IHF than without (Figure 1C, black and green, respectively). These results indicate that the quantum yields of the donor or the acceptor are only slightly affected by interactions with IHF. In the absence of IHF, a comparison of the spectra of H’_D and H’_DA, with excitation at 485 nm, reveals ∼4% decrease in the donor emission intensity (at 517 nm) with a shoulder appearing at ∼580 nm, corresponding to acceptor emission (Figure 1C, purple and blue, respectively). These results indicate some FRET between donor and acceptor even in unbent H’ DNA, with FRET efficiency (E), calculated as described in Supplementary Methods 1.3, of 0.038 ± 0.004. In the presence of IHF, the H’_DA spectrum exhibits a significant decrease in the donor emission intensity with a corresponding increase of the acceptor emission intensity, indicating FRET E of 0.57 ± 0.02 for the IHF–H’ complex at 20°C. As the temperature is raised from 20 to 60°C, the donor intensity in H’_DA in the absence of IHF decreases monotonically and reflects the temperature-dependent decrease in the quantum yield of the donor (Figure 1D). In the presence of IHF, the donor intensity in H'_DA first decreases and mirrors the behavior in DNA alone, up to ∼30°C; above that temperature, the donor intensity in the complex starts to increase relative to that of DNA alone as a result of DNA unbending in the IHF–H’ complex (44,64), and the FRET E of the complex decreases to 0.47 ± 0.04 at 60°C. We previously showed from measurements at different concentrations of IHF and DNA that this temperature dependent decrease in FRET at 100 mM KCl is from unbending of DNA while still bound to IHF, and not from dissociation of the IHF–H’ complex (44,67). Bimolecular dissociation of the complex starts to be significant at salt concentrations greater than about 250 mM KCl (67). Thus, for the range of [KCl] in this study (50–200 mM), we are primarily monitoring unimolecular DNA bending/unbending processes.
IHF bound to DNA is stabilized against thermal denaturation
To ensure that the IHF protein is stable under the conditions where T-jump measurements are done, we carried out thermal denaturation studies of IHF in the presence and absence of H’ DNA, using near-UV CD measurements (Supplementary Figure S2A). These studies augment our previous protein-only denaturation studies using near- and far-UV CD, as well as protein and complex denaturation studies using intrinsic Tyr fluorescence (Supplementary Figure S2B) (64). Altogether, these studies demonstrate that, while IHF has a melting temperature of ∼55–62°C in the absence of DNA, it is significantly stabilized when DNA is bound, with the melting temperature of IHF in the complex shifting to ∼70°C. Therefore, in the ∼30−60°C temperature range where the T-jump measurements are done, the IHF protein in the complex remains stable.
DNA bending/unbending kinetics in IHF–DNA complexes, measured with laser T-jump
Relaxation kinetics in IHF–DNA complexes in response to a laser T-jump perturbation are monitored using time-resolved FRET. Briefly, ∼10 ns IR laser pulses are used to rapidly increase the temperature of a small volume of the sample by 5–10°C within the duration of each pulse. The temporal response of the ensemble of molecules as they re-equilibrate from the conformational distribution characteristic of the initial temperature (Ti) to that of the new higher temperature (Tf) is monitored by recording the donor fluorescence emission, as described previously (44,64). Control measurements on donor-labeled DNA-only samples (or free fluorescein) show that the donor intensity drops immediately after the arrival of the IR pulse, on timescales faster than the first time point of ∼20 μs measured in our spectrometer, followed by a slow relaxation back to the pre-laser intensity levels (Supplementary Figure S3A). The initial rapid drop in intensity reflects the decrease in the quantum yield of the donor in response to the T-jump to a higher temperature, as also observed in equilibrium measurements on these control samples (Figure 1D, black), and is used as a measure of the size of the T-jump (see Supplementary Methods 1.8 and Figure S3B). The relaxation traces measured on the control samples reflect the heat dissipation from the heated volume of the sample and the decay of the temperature back to that of the surrounding bath. These decays are well described by the ‘T-jump recovery’ function (Supplementary Methods 1.9 and Equation S6), with a characteristic time constant of ∼206 ± 24 ms (Supplementary Figure S3C). Thus, the temperature of the heated volume stays approximately constant until ∼10 ms before dissipation; after ∼10 ms, we expect distortions in the measured relaxation traces from the decay of the T-jump itself. Nonetheless, despite the apparent T-jump time-window of ∼20 μs − 10 ms, we are able to characterize conformational relaxation kinetics in the IHF–DNA complexes with time constants of up to ∼20 ms, as detailed in Supplementary Methods 1.11.
In the case of IHF–DNA complex, the donor intensity exhibits a rapid drop immediately after the IR pulse as in the control samples as well as the slow decay back to the pre-laser intensity levels. Additionally, these samples also show conformational relaxation kinetics in the above-mentioned T-jump time window of ∼20 μs − 10 ms (Figure 2); these kinetics reflect the change in population of the complexes from the ensemble of conformations at the initial (low) temperature Ti to that at the final (high) temperature Tf (Supplementary Figure S4). In IHF–DNA complexes, the DNA unbends when the sample is heated up, and the FRET decreases (Figure 1D, inset); this decrease in FRET is reflected in an increase in the donor intensity (Figure 1D, red). Therefore, the donor intensity in our T-jump traces increases during the conformational relaxation process from a level characteristic of the equilibrium population at Ti to that of Tf (Supplementary Figure S4), and eventually decays to the pre-laser level with a time constant of ∼210 ms, similar to the T-jump decay observed in the control experiments.
Figure 2.

T-jump measurements on the IHF–H’ complex. (A and B) Donor fluorescence emission intensities of double-labeled H’ samples in the presence of IHF measured in response to a T-jump perturbation are plotted as a function of time, for T-jump from 34 to 38°C (A) and 48 to 52°C (B). Data are shown for measurements in 100 mM KCl. Red continuous lines are fits to the relaxation traces from the MEM analyses, as described in the text. (C and D) Control measurements are shown on donor-only strand of the H’ sequence, for T-jump from 25 to 29°C (C) and 57 to 62°C (D). Red continuous lines are fits to the T-jump recovery function (Supplementary Equation S6), with recovery time constant fixed at 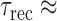 210 ms. (E and F) The distribution of the log of the relaxation times, log(τ), that best fit the relaxation traces in (A) and (B) are plotted in (E) and (F), respectively (red). The blue lines are fits to the distributions in terms of two Gaussians (shown in green), with independently varied peak positions and widths. The range of the distributions that fall outside the timescales of the measured relaxation traces is shown as dashed lines.
210 ms. (E and F) The distribution of the log of the relaxation times, log(τ), that best fit the relaxation traces in (A) and (B) are plotted in (E) and (F), respectively (red). The blue lines are fits to the distributions in terms of two Gaussians (shown in green), with independently varied peak positions and widths. The range of the distributions that fall outside the timescales of the measured relaxation traces is shown as dashed lines.
DNA bending kinetics in the IHF–H’ complex are biphasic
The relaxation kinetics of IHF–DNA complexes were first revealed as single-exponential decays in stopped-flow (33) and laser T-jump measurements (44), with relaxation times of ∼0.3−50 ms in the temperature range 10−60°C. With subsequent advances in instrumentation, we uncovered that these kinetics were in fact biphasic, with a previously unresolved fast phase appearing at around 100 μs in IHF–H’ (50,62). The fast phase appeared to be sequence-independent, suggesting that it reflected non-specific DNA bending by IHF. Those results were included in a preliminary form in a book chapter (63). Here, we augment those data with results on IHF mutants that also show biphasic kinetics, as well as data on how the amplitudes of the fast phase are modulated by varying salt concentrations. These additional studies, together with critical control experiments that rule out intrinsic dye dynamics as the source of the fast phase, validate and expand our preliminary interpretation.
Representative kinetics traces on IHF–H’ in 100 mM KCl, measured on timescales of 20 μs – 40 ms, are shown for two temperature conditions in Figure 2A and B, together with control measurements on a donor-only strand of H’ DNA on the same timescale (Figure 2C and D). As discussed above, the control samples show the initial drop in donor intensity in response to the rapid T-jump and slow re-equilibration of the sample temperature back to the initial (equilibrium) temperature. The characteristic time constant of these re-equilibration kinetics does not depend in any systematic way on the initial and final temperature conditions (Supplementary Figure S3C).
The conformational relaxation traces in the IHF–H’ complexes in our T-jump time-window exhibit an increase in donor intensity, consistent with a decrease in FRET between the fluorescent labels as the DNA unbends in the complex, as discussed above. Single-exponential fits to these relaxation traces show deviations at short times and require a minimum of two exponentials to fit the data (Supplementary Figure S5). All relaxation traces were therefore analyzed in terms of two-exponential fits convoluted with the T-jump recovery function (Supplementary Methods 1.6 and Equation S3). The rates and amplitudes corresponding to the two phases were obtained using a Monte Carlo search in parameter space, starting from 30 independent sets of randomly chosen values for each of the parameters (Supplementary Methods 1.7). The results from this analysis for IHF–H’ in 100 mM KCl are summarized in Supplementary Figure S6.
Independently, the relaxation traces were also analyzed using maximum entropy method (MEM), which provides a distribution of relaxation rates that best fit the kinetics traces without a priori assumptions as to the number of discrete exponentials required (68–70). The distribution of relaxation times obtained from MEM analysis reveal two dominant peaks, providing additional evidence for deviations from single-exponential decays (Figure 2E and F). The relaxation rates and amplitudes corresponding to the two phases, obtained from the MEM distributions (Supplementary Methods 1.10) are shown in Figure 3. The results from the MEM analysis are consistent with those from the discrete two-exponential analysis (Supplementary Figure S6), although the MEM approach provides a more robust set of rates and amplitudes, as anticipated. An Arrhenius plot of the relaxation rates for the two phases shows that the fast phase occurs at around 100 μs and is weakly dependent on the temperature, while the slow phase appears to be activated (Figure 3A). The amplitude of the slow phase is considerably larger at lower temperatures (>70% at 30°C), and decreases to ∼50% at 53°C (Figure 3B). The accuracy with which we can determine ∼100 μs relaxation times for the fast phase, given that the earliest time point in our relaxation traces is 20 μs, is discussed in Supplementary Methods 1.11.
Figure 3.

Temperature dependence of the relaxation rates and relative amplitudes for the IHF–H’ complex. (A) The relaxation rates for the fast (green) and slow (blue) phases, calculated from the MEM distributions of the corresponding relaxation traces, as described in the text, are plotted as a function of inverse (final) temperature. The continuous lines are an Arrhenius fit to the relaxation rates, with activation energies 2.8 ± 6.4 kcal/mol (fast phase) and 19.4 ± 1.6 kcal/mol (slow phase). The Arrhenius fits were done as described in Supplementary Methods 1.12; the errors in the activation energies are standard deviations of values obtained from fits to two independent sets of measurements. (B) The corresponding relative amplitudes for the fast (green) and slow (blue) phases are shown. The continuous lines connect the data points and are drawn to guide the eye.
The fast phase is not from dye dynamics
Previous anisotropy studies have shown that while fluorescein is reasonably well approximated as a freely rotating dye when attached to DNA at the 5′-end with a C-6 linker, that is not the case with TAMRA, which can stack against DNA duplex (65,71–73). We carried out steady-state anisotropy studies on donor–acceptor-labeled H’ in the presence and absence of IHF. Consistent with the earlier studies, in the absence of IHF we obtain low steady-state anisotropy of 0.053 ± 0.002 for fluorescein and significantly larger value of 0.156 ± 0.004 for TAMRA. In the presence of IHF, these values increase to 0.077 ± 0.003 and 0.209 ± 0.006, respectively. Therefore, it is conceivable that there is some contribution to the observed relaxation kinetics from T-jump induced perturbation of dye—macromolecule interactions, which could alter the quantum yield of the dyes and/or their orientations/mobility. To examine the possibility of misleading signals from such dye dynamics, we performed a series of control measurements (Supplementary Methods 1.13). First, we measured the temporal responses of the donor and acceptor dyes, separately, to T-jump perturbations: (i) using donor-only labeled IHF–H’, we excited the donor and monitored its fluorescence (Supplementary Figure S7A), and (ii) using donor–acceptor labeled IHF–H’, we directly excited the acceptor and monitored its fluorescence (Supplementary Figure S7B). No relaxation kinetics were observed in either case other than the slow T-jump recovery kinetics. Second, to detect any T-jump-induced changes in the relative orientations of the dyes that were unrelated to IHF-induced changes in DNA conformations, we measured T-jump kinetics on a shorter (14-bp) DNA, in the absence of IHF. The length of the 14-mer was chosen such that the FRET E between the donor and acceptor labels was 0.48 ± 0.05, close to the FRET E of 0.57 ± 0.02 in the IHF–H’ complex, and in the range where small changes in the separation or relative orientations of the dyes would result in a detectable FRET change. Once again, T-jump measurements on this DNA sample did not result in relaxation kinetics other than the T-jump recovery kinetics (Supplementary Figure S7C and D). Third, to rule out contributions to the fast phase from potential changes in the mobility/orientation of the dyes in the presence of the protein, we moved the dyes further away from the protein by using a longer variant of the H’ DNA (55 versus 35 bp in other experiments) (65). The nature of the biphasic kinetics in the IHF–DNA complex remained unchanged even with the longer DNA (Supplementary Figure S8), reaffirming that the interactions of the dyes with the protein are not a contributing factor to the observed relaxation kinetics. Taken together, these measurements suggest that either there are no dye unstacking/reorientational dynamics induced by T-jump in the DNA-only or IHF–DNA samples, or that these dynamics, if they occur, are faster than the first resolved time point of ∼20 μs in our spectrometer. We therefore conclude that the fast phase is indeed from DNA conformational dynamics in the complex.
Introducing mismatches at the site of the kinks affects the slow phase but not the fast phase
A plausible explanation for the two relaxation phases observed in the IHF–H’ complex is the sequential bending of first one flanking arm of the DNA and then the other. To examine this possibility we designed modified H’ constructs in which 2-bp mismatches were introduced at (i) both the kink sites (TT8AT), (ii) on one side (TTloop) or (iii) the other side (ATloop), as summarized in Figure 1B. Previous binding affinity studies showed that the Kd for the IHF–TT8AT complex is 11- to 15-fold tighter than that for the IHF–H’ complex (Table 1) (64,74). These results suggested that introducing mismatches at the site of the kinks likely lowers the energetic cost of bending/kinking DNA. Furthermore, equilibrium FRET measurements performed on the IHF–TT8AT complex showed that, as in the case of IHF–H’, the change in FRET with increasing temperature in the range of 20 to 50°C was independent of the IHF and DNA concentrations (64). Thus, at concentrations used in the T-jump measurements (5–20 μM), the FRET-based relaxation kinetics in the IHF–TT8AT complex are primarily from unimolecular bending/unbending of DNA while still bound to the protein.
T-jump measurements on the IHF–TT8AT complex also revealed deviations from single-exponential relaxation kinetics (Supplementary Figures S9 and 10). Both MEM and discrete exponential analyses on these relaxation traces showed consistent biphasic behavior (Supplementary Figure S11). The relaxation rates of the fast phase overlapped with corresponding rates measured for the IHF–H’ complex (Figure 4A). The slow phase, however, was ∼5-fold faster in the IHF–TT8AT complex than in the IHF–H’ complex. Thus, mismatches introduced at the kink sites accelerate DNA bending rates in the slow phase, although the enhanced DNA bending rates do not fully account for the >11- fold increase in binding affinity of IHF for TT8AT.
Figure 4.

Temperature dependence of the relaxation rates and relative amplitudes for variants of IHF–H’ complex. Data similar to those in Figure 3 are shown for IHF–TT8AT (A and B), IHF–TTloop (C and D) and IHF–ATloop (E and F). The activation energies are 17.9 ± 1.4 kcal/mol (fast phase) and 8.3 ± 2.7 kcal/mol (slow phase) for IHF–TT8AT, 11.9 ± 5.7 kcal/mol (fast phase) and 6.6 ± 3.5 kcal/mol (slow phase) for IHF–TTloop, and 11.9 ± 5.7 kcal/mol (fast phase) and 8.4 ± 5.4 kcal/mol (slow phase) for IHF–ATloop. The dashed lines in each panel represent the data measured for IHF–H’, reproduced from Figure 3.
T-jump measurement on IHF–ATloop and IHF–TTloop also revealed biphasic relaxation kinetics (Supplementary Figures S12 and 13), with behavior similar to that of IHF–TT8AT: the fast phase in IHF–ATloop and IHF–TTloop appeared unchanged compared with the fast phase of IHF–H’, while the slow phase was two to four times faster than the corresponding slow phase of IHF–H’ (Figure 4C and E). In all three variants of the H’ substrate, only the slower phase was affected by the insertion of the 2-bp mismatch, whether on one kink site or the other or both, while the fast phase remained unchanged. These results demonstrate that the two steps do not reflect sequential bending of the two flanking arms of DNA; rather, the fast step, which appears to be sequence independent, or at least insensitive to the sequence at the kink site, is attributed to non-specific DNA bending by IHF, while the slow step, which is responsive to the ease with which one or the other kink site can be distorted, is attributed to DNA bending/kinking at the kink sites to form the ‘recognition’ complex. These conclusions are reaffirmed by further experiments, as described below.
A DNA modification away from the kink sites has no effect on either of the two rates but increases the fast phase amplitudes
Next, we carried out equilibrium and T-jump studies with the H’44A sequence, which has a single T→A mutation in the TTR consensus region of the H’ sequence (where T = thymine and R = any purine) that can only be contacted by the protein after a strong bend has been introduced (Figure 1B). This mutation was previously shown to exhibit a 100- to 250-fold decrease in binding affinity for IHF (64,75). Furthermore, the FRET E in the IHF–H’44A complex was smaller than that measured for the IHF–H’ complex under identical solvent conditions (∼0.45 versus ∼0.57, at 25°C) (64). These results suggested that, despite the very similar crystal structures of IHF–H’ and IHF–H’44A complexes (75), the average conformation of H’44A in the complex is less bent under solution conditions, which is consistent with weaker interactions between IHF and one of the flanking ‘arms’ of the bent DNA.
In H’44A, the modification in the DNA sequence is separated by 6 bp from the location of the nearest kink (Figure 1B). Thus, the IHF–H’44A complex provides a useful control to examine the DNA bending kinetics in a complex that is considerably weaker in comparison with the IHF–H’ complex, but one in which the bendability of DNA at the kink sites is left undisturbed (64). T-jump measurements on the IHF–H′44A complex also revealed biphasic kinetics (Supplementary Figures S14 and 15), with rates similar to those for the IHF–H′ complex (Figure 5A). These results reaffirm that the slow phase kinetics remain unaltered unless the kink site is modified, despite the much weaker IHF–H’44A complex. However, on average the fast phase amplitude increased compared to the IHF–H’ complex, especially at higher temperatures (Figure 5B), consistent with the expectation that a decrease in the stability of the complex increases the fraction of non-specifically bound complexes, and that this fraction increases as the temperature is raised.
Figure 5.

Temperature dependence of the relaxation rates and relative amplitudes for the IHF–H’44A complex. (A) The relaxation rates for the fast and slow phases, and (B) the corresponding relative amplitudes, are shown for IHF–H’44A. Symbols and colors are as described for Figure 3. The dashed lines in each panel represent the data measured for IHF–H’, reproduced from Figure 3. The activation energies from the data in (A) are 4.1 ± 0.4 kcal/mol (fast phase) and 15.8 ± 1.9 kcal/mol (slow phase).
DNA bending rates in the slow phase reflect base-pair opening rates in matched/mismatched DNA
Next, we compared the fast and the slow relaxation rates measured on IHF–H’ and IHF–TT8AT with single base pair opening rates measured by NMR imino-proton exchange measurements (59–61,76). The relaxation rate for the slow phase in IHF–H’ at 30°C is remarkably similar to the A:T base pair opening rate measured at one of the kink sites of the H’ sequence, in the absence of IHF (Figure 6). Previously we had proposed that spontaneous DNA kinking from thermal fluctuations presented the primary bottleneck in forming the fully bent recognition complex in IHF (44,64). The results presented here suggest that this spontaneous kinking event likely corresponds to the slower step in the biphasic kinetics observed.
Figure 6.
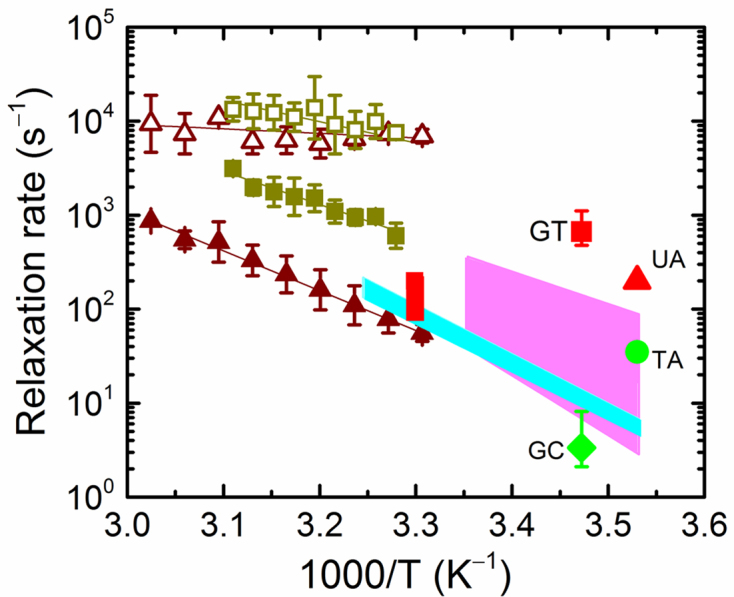
Comparison of T-jump relaxation rates for IHF–DNA and base-pair opening rates. The relaxation rates for the fast (open symbols) and slow (filled symbols) phases measured for IHF–H′ (brown triangles) and IHF-TT8AT (olive green squares) are reproduced from Figures 3A and 4A, respectively. The shaded areas represent the range of base-pair opening rates from imino proton exchange measurements, from Coman and Russu (59) (pink, A:T; cyan, C:G). The red vertical bar is the A:T base-pair opening rate for the H′ sequence, at the site of one of the kinks, from Dhavan et al. (60). The opening rates from NMR measurements are also shown for G:T mismatch (red square) from Moe and Russu (76); U:A mismatch (red triangle) from Parker et al. (61); T:A base-pair (green circle) from Parker et al. (61); and G:C base pair (green diamond) from Moe and Russu (76).
Opening rates for a mismatched pair in DNA are expected to be much faster than the corresponding rates in Watson–Crick matched base pairs (76–78), as a result of loss of hydrogen bonding and stacking interactions at mismatched sites. NMR measurements on DNA with mismatches indeed show enhanced breathing dynamics with ∼166-fold increase in the opening rate of a G:T mismatch compared to that of a G:C pair for the same sequence context (76), and a ∼6-fold increase in the opening rate for a U:A pair compared with that of a corresponding T:A pair (61). The slow rates of the biphasic kinetics observed in IHF–TT8AT, when extrapolated to low temperatures where the NMR measurements were done, agree remarkably well with the previously reported G:T and U:A opening rates (Figure 6). It is important to note here that the opening rates for matched or mismatched pairs strongly depend on the sequence context (76–78). Nonetheless, these data support the notion that the rate-limiting step in forming the specific complex is the spontaneous kinking of DNA at the kink sites, which in turn is rate-limited by the thermal disruption of the stacking interactions at that site (44,64).
What is the origin of the biphasic relaxation kinetics?
Two plausible scenarios could explain the biphasic kinetics observed for binding-site recognition by IHF. In the first scenario (Figure 7; scheme 1), the fast phase corresponds to non-specific bending (without significant unstacking) of DNA by IHF, followed by the slow recognition step that is rate-limited by intrinsic fluctuations in DNA at the kink sites. In the second scenario (Figure 7, scheme 2), a slow, rate-limiting DNA-kinking step occurs first, followed by a rapid reorganization of the protein and the DNA to form the stable recognition complex. In both cases, the fast step is predicted to remain unaffected by changes in the DNA sequence at the kink sites. To further explore the origin of the two phases, we carried out T-jump measurements as a function of the ionic strength and for different IHF mutants, as discussed in the sections below.
Figure 7.

Proposed pathways for DNA bending mechanism by IHF. Plausible alternative pathways for the transition from a non-specific complex with IHF bound to straight DNA to the fully bent and wrapped specific are illustrated. In the top scheme 1, the intermediate state is a partially bent conformation while still non-specifically bound; in the bottom scheme 2, the intermediate state is kinked at the specific kink sites.
An increase in [KCl] diminishes the fast phase amplitudes
The binding of proteins to DNA depends strongly on the ionic environment in solution, due to the polyelectrolyte nature of DNA, with changes in [salt] differentially affecting the non-specific and the specific binding affinities (79–83). Non-specific binding of proteins to DNA is primarily mediated by contacts to the DNA backbone via electrostatic interactions between basic residues in the protein and phosphate groups in the DNA; the strength of these interactions decreases with increasing [salt]. On the other hand, sequence-specific interactions are primarily mediated by contacts to the individual bases and typically involve hydrogen bonds and/or van der Waals interactions between amino acid side chains and specific DNA sequences. Hence, in general, these interactions are expected to be substantially less sensitive to [salt]. This trend is reflected in a generally larger slope of log(Kd) versus log[salt] observed for non-specific than for specific binding (84–87), indicating that DNA-binding specificity typically improves with increasing [salt].
Thermodynamics and kinetics measurements on the IHF–H’ complex are consistent with these notions. Previous isothermal titration calorimetry (ITC) and FRET-based binding studies showed that the overall Kd for the IHF–H’ complex increases with increasing [salt], and that ∼8–9 counterions are released when this complex is formed (67,83). Further analysis of the [salt]-dependent thermodynamics and kinetics of IHF–H’ in terms of a two-step sequential binding-then-bending kinetic scheme indicated that nearly all the counter-ions are released during the formation of the non-specific complex (88). These results are consistent with the ITC studies that showed that at low to moderate [K+] (60–100 mM), there is a strong competition between specific and non-specific binding of IHF due to low specificity ratio (Ks/Kns ∼102) and a very small non-specific site size (∼10 bp), and that increasing the [salt] had a larger effect on non-specific than on specific binding (83).
Against the backdrop of these previous results, we carried out equilibrium and T-jump experiments on IHF–H’ at three additional salt concentrations: 50, 150 and 200 mM KCl (Supplementary Figures S17–19). Our hypothesis is that if the fast phase in the observed kinetics is from conformational fluctuations between non-specific binding modes, then its amplitude should diminish with increasing [salt]. The T-jump relaxation kinetics measured in the 50–200 mM KCl are expected to be from unimolecular processes as we previously showed that, for salt concentrations up to at least 200 mM KCl, no bimolecular dissociation was detected in IHF–H’ with increasing temperature up to 60°C. However, increasing the [salt] does affect the magnitude of the DNA bend in the ensemble as the temperature is raised. This salt-dependent behavior is evident in the dramatic increase in donor intensity in the IHF–H’ complex at 200 mM KCl that already starts at ∼25°C compared with a less dramatic increase observed in 50 mM KCl and not until ∼35°C (Supplementary Figure S16). These data indicate that T-jump more readily perturbs the bent complex at high salt.
MEM analyses of the relaxation traces measured at 50 mM KCl reveal two distinct peaks in the distribution of relaxation times, clearly indicating two kinetics phases (Supplementary Figure S17), similar to the results in 100 mM KCl (Figure 2). In the case of 150 mM KCl, a predominantly single phase is observed at low temperature with barely detectable amplitude in the fast phase; at high temperature the amplitude in the fast phase increases slightly and appears as a shoulder in the distribution of the relaxation times (Supplementary Figure S18). At 200 mM salt, the amplitude of the fast phase remains small at all temperatures, and only a predominantly single distribution is observed in the MEM analysis, albeit with a broader distribution at higher temperatures (Supplementary Figure S19). The salient result here is that biphasic kinetics are observed at low [salt], with the relaxation rates and relative amplitudes of the fast and the slow phases observed at 50 mM KCl virtually indistinguishable from those at 100 mM KCl (Supplementary Figure S20); at higher [salt] (150–200 mM KCl), the amplitude of the fast phase diminishes significantly (Supplementary Figures S18 and 19). Thus, the fractional population that contributes to the fast phase decreases at high [salt]. These findings support scheme 1 in Figure 7, in which the fast phase corresponds to non-specific DNA bending by IHF, while the slow phase corresponds to the conversion from the partially bent non-specific complex to the specific complex through further bending and subsequent wrapping of cognate DNA.
Protein mutations that destabilize ionic interactions enhance the fast phase amplitudes
We next examined how IHF mutants designed to perturb the complex affected the relaxation kinetics. We studied three IHF mutants in which positively charged residues (Lys or Arg) at increasing distances from the kink sites were replaced with neutral residues: αK5A, βK84A and αR21C (Figure 1A). For the three mutant complexes: αK5A–H’, βK84A–H’ and αR21C–H’, the dissociation constants Kd (Table 1) were previously determined to be 35 ± 30 nM, 16 ± 3 nM and 10 ± 2 pM, respectively, compared with 27 ± 2 pM for the wild-type (wt) IHF–H’ complex (64). Thus, two of the mutants (αK5A and βK84A) exhibited significant loss of stabilizing interactions with bound H’ substrate, while no loss of stability was detected for the αR21C–H’ complex. Previous equilibrium measurements also showed that the decreases in FRET with increasing temperature for both αK5A–H’ and βK84A–H’ complexes were independent of protein and DNA concentrations in the range 20–70°C, indicating that, as for IHF–H’, this decrease was from unbending of DNA within the complex and not from any significant bimolecular disruption (64).
Previous T-jump studies reported on IHF mutants could only resolve a single relaxation phase (63,64). Here, we show that these mutants also yielded biphasic kinetics over the entire temperature range of the measurements (Figure 8 and Supplementary Figures S21–23). For the less stable mutant complexes, αK5A–H’ and βK84A–H’, the fast phase overlapped well with the fast phase of the wt IHF–H’ complex. The slow phase on the other hand, while exhibiting similar rates as the wt at the lower temperatures (near ∼30°C), was nearly 10-fold slower at the highest temperature of ∼50°C (Figure 8). The activation energies of the slow phase for αK5A–H’ and βK84A–H’ were 8.0 ± 2.1 and 7.6 ± 0.9 kcal/mol, respectively, significantly smaller than the 19.4 ± 1.6 kcal/mol for the slow step in the wt IHF–H’. The origin of this decrease in activation energy measured in the mutants is not immediately apparent. Notably, for both mutants, the relative amplitudes of the fast phase exceed that of the slow phase at high temperatures and are much larger than the fast phase amplitudes for wt IHF–H’ (Figure 8). This increase in the fast phase amplitudes in the mutants is consistent with scheme 1 (Figure 7) and indicates that, as the temperature is raised, a larger fraction of the bound mutant proteins is in the non-specific binding modes. In the case of αR21C-H’, with similar stability as the wt IHF–H’, both the relaxation rates and their relative amplitudes exhibit similar behavior as the wt complex (Figure 8E and F). This result is not surprising since the mutation site in this protein variant is at the edge of the DNA-wrapping surface and hence neither the thermodynamics nor the kinetics are affected by the loss of the positive charge at the location of this mutation.
Figure 8.

Temperature dependence of the relaxation rates and relative amplitudes for the mutant IHF–H’complexes. Data similar to those in Figure 3 are shown for αK5A-H’ (A and B), βK84A-H’ (C and D) and αR21C-H’ (E and F). The activation energies are 7.7 ± 4.5 kcal/mol (fast phase) and 7.9 ± 2.1 kcal/mol (slow phase) for αK5A-H’, 2.3 ± 3.4 kcal/mol (fast phase) and 7.6 ± 0.9 kcal/mol (slow phase) for βK84A-H’ and 3.9 ± 2.6 kcal/mol (fast phase) and 15.6 ± 0.2 kcal/mol (slow phase) for αR21C-H’.
DISCUSSION
Non-specific ‘interrogation’ and specific ‘recognition’ by IHF
The mechanism by which site-specific DNA-binding proteins scan DNA and at the same time probe potential binding sites for a match is not well understood. While several single-molecule studies have provided unique insights into how proteins scan DNA, they lack the temporal and spatial resolution to watch the protein ‘in action’ and to unveil the conformational dynamics during interrogation and recognition. Stopped-flow and single-molecule studies designed to examine the kinetics of protein–DNA interactions have yielded the rates of association/dissociation dynamics on several DNA-bending proteins (22–24,29–35,37–41); however, with a few notable exceptions, these studies were also unable to resolve conformational dynamics that confer recognition within the complex. Uncovering dynamics that reflect preliminary interrogation are even more challenging, since these initial interactions should be fast and commensurate with the timescales on which proteins diffuse on DNA. In our studies, we have taken advantage of the microsecond time-resolution and exquisite sensitivity of the T-jump perturbation approach to begin to examine such preliminary interrogation steps along the binding-site recognition trajectory (50,63).
This study builds on previous studies on the binding and bending mechanisms of the architectural protein IHF. Initial stopped-flow and T-jump studies on the IHF–H’ system resolved the first direct observation of DNA bending kinetics separable from DNA binding and occurring on timescales of a few milliseconds (33,44,58). Subsequent T-jump studies on IHF–H’ with improved sensitivity enabled us to reveal two distinct phases in the DNA-bending kinetics, with a previously undetected fast phase appearing at ∼100 μs (62,63,89). These biphasic kinetics were observed under conditions where T-jump perturbation does not dissociate the complex and therefore reflected unimolecular conformational changes in the IHF–H’ complex. Thus, the mechanism by which IHF recognizes and binds tightly to its cognate site appears to involve at least two steps. Here, we have explored the origin of the two-phase bending kinetics.
The fast phase rates appear sequence independent, while the slow phase rates reflect DNA kinkability at key kink sites
T-jump measurements with cognate DNA with mismatches inserted at the sites of the kinks provide clues to plausible origins of the two phases. The TT8AT substrate (with 2-bp mismatches at both kink sites), together with the TTloop (with the mismatches located on only one kink site) and the ATloop (with the mismatches located on the other kink site), also exhibit biphasic kinetics in T-jump measurements in the presence of IHF (Figure 4). However, in all three mismatched variants, only the slower phase is accelerated by the insertion of the mismatches, whether on one kink site or the other or both, while the fast phase remains unchanged. These results suggest that the two steps do not reflect sequential bending of the two flanks of DNA on either side of the kinks; instead, they point to rapid partial bending that appears to be independent of DNA flexibility, followed by slow bending/kinking that appears to be rate-limited by base-pair breathing (or unstacking) dynamics at one or the other kink sites (Figure 6). Mismatches at the kink sites accelerate this slower step. We assign it to the formation of the fully bent recognition complex.
Two plausible explanations for the biphasic kinetics are illustrated in Figure 7: scheme 1, in which the rapid phase corresponds to partial non-specific bending of DNA by IHF, which precedes full bending/kinking to form the specific recognition complex; scheme 2, in which the slow sequence-dependent kinking step occurs first followed by rapid reorganization of the still loosely bound protein–DNA complex to form the stable specific complex. A third scenario for the observed biphasic kinetics could be two competing mechanisms: a fast ‘lock-and-key’ recognition to already pre-bent DNA versus a slower induced fit with protein-induced DNA bending. We consider this scenario unlikely since we see no evidence of a substantial fraction of pre-bent DNA in the FRET studies of H’ DNA in the absence of IHF. We also do not observe an increase in the amplitude of the fast phase when we introduce mismatches at the kink sites, as in TT8AT, which might be expected to increase the population of pre-bent DNA in solution. Instead, our T-jump studies presented here, together with prior studies of non-specific DNA bending by IHF and structurally related proteins such as the histone-like protein HU (83,90–98), support scheme 1 of Figure 7 as the more likely explanation for the observed biphasic kinetics.
Destabilizing modifications in DNA or protein increase the non-specifically bound populations
Measurements with modified DNA sequences that do not alter the kink sites as well as IHF mutants that destabilize the specific complex also support scheme 1 over scheme 2. In H’44A, where the modification is in the TTR consensus region of the H’ sequence, which is 6 bp away from the nearest kink site, biphasic kinetics are observed with rates that overlap those measured for the IHF–H’ complex (Figure 5A). However, this modification, which destabilizes the complex by >100-fold, affects the relative amplitudes with an increase in the fast phase amplitudes observed at higher temperatures in IHF–H’44A (Figure 5B). This result is inconsistent with a lock-and-key mechanism but supports Figure 7, scheme 1: a decrease in binding affinity for the H’44A site (and likely a corresponding decrease in specificity) is expected to shift the equilibrium population of the IHF–H’44A bound complexes toward non-specifically bound conformations, resulting in a larger amplitude for re-equilibration within that population in response to the T-jump perturbation.
Further support for this conclusion comes from measurements on the IHF mutants. In scheme 2 of Figure 7, the fast phase is assigned to rapid reorganization or ‘induced-fit’ dynamics to form the specific IHF–DNA complex, and which is preceded by the slow, rate-limiting, DNA kinking of the cognate sequence. In this scheme, making mutations in the protein wrapping surface that stabilize or destabilize the complex are expected to alter the rates of the fast phase. In the αK5A and βK84A mutants, positively charged residues in the DNA-wrapping surface are mutated to neutral residues, thus disrupting ionic interactions that destabilize the specific complex by >500-fold. T-jump measurements on the mutant complexes reveal that the fast phase rates remain unchanged, albeit with a dramatic increase in the relative amplitudes of the fast phase in both mutants as the temperature is raised, in comparison with the wild-type protein. The observation that the fast phase rates are unaffected in the mutants but the relative amplitudes increase is consistent with scheme 1 and indicates that a reduction of positively charged residues in the DNA wrapping surface of IHF tilts the equilibrium toward the non-specific binding modes.
Non-specific DNA bending by IHF and its structurally homologous cousin HU
Previous in vitro studies have shown that IHF binds to DNA both specifically and non-specifically and that even in the non-specific binding mode IHF bends the DNA to some extent. There is ample experimental evidence that non-specific binding of IHF and other histone like proteins serve to compact nucleoid structure by a series of partial bends (83,97). The first evidence for non-specific DNA bending by IHF came from mechanical single molecule force-extension measurements on λ DNA in the presence of IHF (90). These studies demonstrated that addition of IHF shortened the contour length of λ DNA, and showed that the effect of IHF binding on the force response of λ DNA was similar to that predicted for DNA-bending proteins (92,96). Thus, this study provided evidence that non-specific IHF binding altered DNA structure, although to a smaller extent than that expected from specific IHF binding.
The non-specific binding modes of IHF may closely resemble conformations adopted by another DNA bending protein, the histone-like protein HU (90,91,93–95,98). Structurally, HU is very similar to IHF and is known to sharply bend DNA (57,99). However, unlike IHF, HU exhibits no sequence-specificity but binds preferentially to DNA with inserted distortions such as mismatched base pairs and extra helical bases, especially if these distortions are space about 9 bp apart, to accommodate the sharply kinked structure reminiscent of the IHF–H’ complex (97,99). In these highly-bent HU–DNA structures, as in the IHF–H’ complex, two invariant proline residues on the β arm of HU insert between base pairs, stabilizing the sharply bent DNA (51,99). Koh et al. (97) have extensively characterized the HU binding modes using ITC and equilibrium FRET measurements. Their ITC studies, carried out on HU from E. coli, showed that protein–DNA complex exhibited at least three different non-specific binding modes with site sizes of ∼34, 10 and 6 bp. The relative populations of these distinct non-specific binding modes were affected by salt concentration, salt type, pH, temperature and binding density ([HU]/DNA) (97). FRET measurements on these complexes showed that the 34-bp non-specific binding mode of HU appeared to be highly bent (by ∼143°), with nearly as much bending as seen in the specific IHF–H’ complex. The implication from these studies on HU is that similar multiple non-specific binding modes are also accessible to IHF. In fact, ITC studies with IHF, which showed that non-specific IHF binding is favored at low KCl concentration and high IHF–DNA stoichiometry, also showed that a smaller occluded size of DNA (∼10 bp) was observed in the non-specific binding mode compared to the specific complex (83,100). From these studies we conclude that there must be a distribution of populations between specific and non-specific binding modes of IHF, and that T-jump measurements detect the redistribution of these populations, with the fast phase corresponding to rearrangements within the non-specific binding modes (Figure 9).
Figure 9.
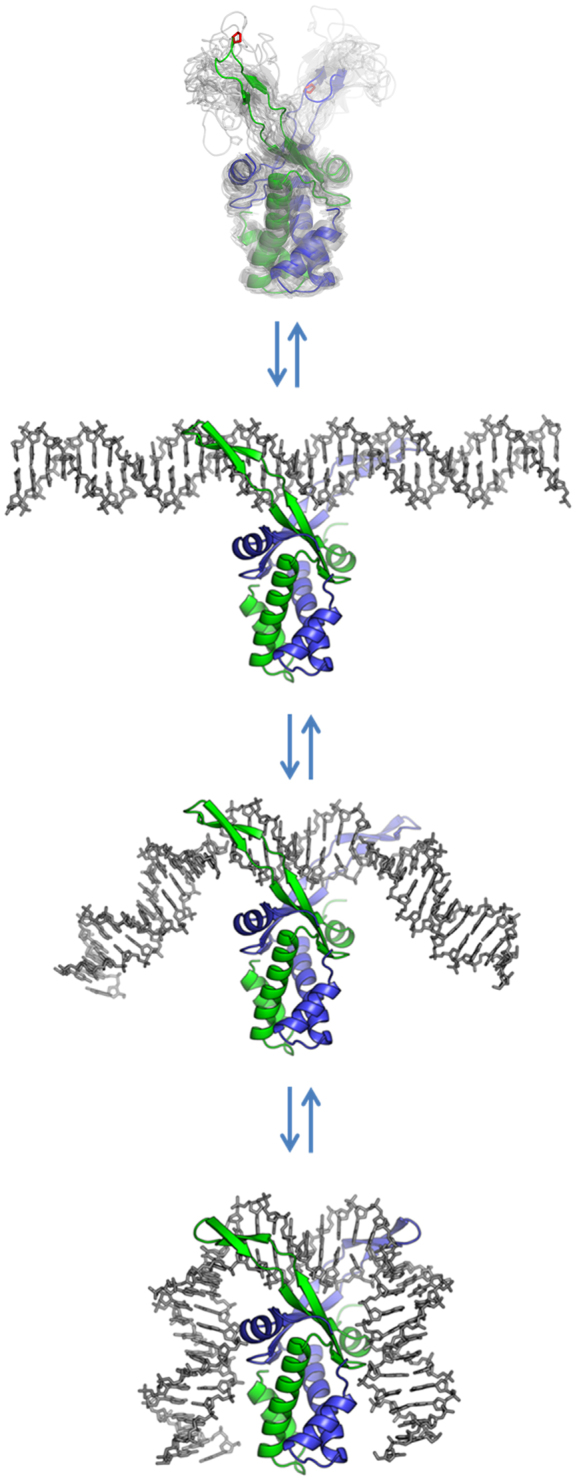
Illustration of two-step bending of DNA by IHF. The unbound IHF protein is illustrated by an ensemble of NMR structures of HU (PDB code: 1HUE); the structure of IHF in the absence of DNA has not been solved. The protein is shown to first bind to DNA in a non-specific manner, with the DNA still straight (the so-called encounter complex). The protein then bends the DNA while still bound non-specifically, giving rise to the fast phase in our T-jump measurements, followed by the slow specific kinking step, to form the fully bent, wrapped specific complex.
DNA bending in non-specific versus specific protein–DNA complexes: separating electrostatic from non-electrostatic contributions
DNA bending in protein–DNA complexes has contributions from electrostatics, for example, from asymmetric neutralization of the phosphate charges on one side of the DNA by an excess of positive charges on the protein binding interface (101–104), as well as from intercalation of specific amino acids to stabilize sharp kinks in DNA (55). A series of careful thermodynamics measurements on DNA binding and bending mechanisms of sequence-specific and non-specific proteins belonging to the high-mobility group box class of architectural proteins, although structurally unrelated to IHF, nonetheless provide valuable insights into the electrostatic and non-electrostatic contributions to both DNA bending and specificity (105). These studies reveal that DNA bending by non-specific-binding proteins in this class is mediated primarily by electrostatics, as illustrated by a strong dependence on the extent of DNA bending in complex on ionic strength; in contrast, DNA bending by specific-binding proteins is found to have significant contribution to stability and specificity from non-electrostatic interactions such as van der Waals interactions between apolar groups to form a more tightly packed interface (105). Consistent with these studies, previous thermodynamics and kinetics studies on IHF–H’ that examined the ionic-strength dependence of the association/dissociation and DNA bending/unbending rates demonstrated that the bulk of the salt-dependence on the binding affinity appeared in the formation of the non-specific complex, highlighting the electrostatic nature of the initial interactions, followed by slower rearrangements that were weakly dependent on [salt] (88). It is therefore conceivable that for many DNA-bending proteins, the initial non-specific binding, mediated primarily by electrostatics, results in a weakly bent DNA such as that observed here for IHF, while slower recognition steps that lead to full bending to form the specific complex are primarily non-electrostatic in nature, although these sequence of events along the recognition trajectory remain to be tested for most proteins.
CONCLUSION
Our finding of the fast phase in the IHF–H’ complex dynamics is an exciting discovery in the sequence of events along the target recognition trajectory of a site-specific DNA-bending protein, and represents the first example, to our knowledge, of the measurement of DNA bending rates in a non-specific complex. While non-specific binding of IHF and partial DNA bending is important for DNA compaction in the cells, it also likely plays a critical role in the mechanism by which IHF scans DNA in search of its target site, as suggested by the ‘facilitated diffusion’ models of the search process. Structural and thermodynamics studies of proteins bound non-specifically to DNA show that the protein–DNA interfaces are less intimately apposed than when they are in the specific complex, due to lack of specific close-fitting interactions with the DNA, leaving room for retention of substantial interfacial water between them. This relatively loose structure allows the protein–DNA to remain mobile and enables rapid scanning of the DNA while bound non-specifically. In the case of IHF, we envision the scan/search mode of the protein as one in which the relatively unstructured β-arms of IHF wrap and unwrap around the DNA, testing each site for conformability. The measurements reported here are blind to the dynamics of the protein, and only detect the bending/unbending of the bound DNA via the FRET labels attached to the DNA ends. Direct measurements of the wrapping/unwrapping dynamics of the β-arms of the protein, for example, by introducing Trp residues at suitable positions on the arms, could be a key experiment to further examine the origin of the fast phase, and to add additional insight into the search/interrogation and recognition mechanism of its cognate site by IHF.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Ying Z. Pigli (University of Chicago) for purification of IHF proteins and its mutants.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [MCB-1158217 to A.A., MCB-1715649 to A.A., P.A.R.]. Funding for open access charge: NSF [MCB-1158217].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lawson C.L., Berman H.M.. Rice PA, Correll CC. Indirect readout of DNA sequence by proteins. Protein-Nucleic Acid Interactions. 2008; Cambridge: Royal Society of Chemistry; 66–90. [Google Scholar]
- 2. Spolar R.S., Record M.T. Jr. Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994; 263:777–784. [DOI] [PubMed] [Google Scholar]
- 3. Garvie C.W., Wolberger C.. Recognition of specific DNA sequences. Mol. Cell. 2001; 8:937–946. [DOI] [PubMed] [Google Scholar]
- 4. Berg O.G., Winter R.B., von Hippel P.H.. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981; 20:6929–6948. [DOI] [PubMed] [Google Scholar]
- 5. Winter R.B., Berg O.G., von Hippel P.H.. Diffusion-driven mechanisms of protein translocation on nucleic acids. 3. The Escherichia coli lac repressor–operator interaction: kinetic measurements and conclusions. Biochemistry. 1981; 20:6961–6977. [DOI] [PubMed] [Google Scholar]
- 6. von Hippel P.H., Berg O.G.. Facilitated target location in biological systems. J. Biol. Chem. 1989; 264:675–678. [PubMed] [Google Scholar]
- 7. Stanford N.P., Szczelkun M.D., Marko J.F., Halford S.E.. One- and three-dimensional pathways for proteins to reach specific DNA sites. EMBO J. 2000; 19:6546–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Halford S.E., Marko J.F.. How do site-specific DNA-binding proteins find their targets. Nucleic Acids Res. 2004; 32:3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Halford S.E. An end to 40 years of mistakes in DNA-protein association kinetics. Biochem. Soc. Trans. 2009; 37:343–348. [DOI] [PubMed] [Google Scholar]
- 10. Segal E., Fondufe-Mittendorf Y., Chen L., Thastrom A., Field Y., Moore I.K., Wang J.P., Widom J.. A genomic code for nucleosome positioning. Nature. 2006; 442:772–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blainey P.C., van Oijen A.M., Banerjee A., Verdine G.L., Xie X.S.. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:5752–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gorman J., Chowdhury A., Surtees J.A., Shimada J., Reichman D.R., Alani E., Greene E.C.. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol. Cell. 2007; 28:359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tafvizi A., Huang F., Leith J.S., Fersht A.R., Mirny L.A., van Oijen A.M.. Tumor suppressor p53 slides on DNA with low friction and high stability. Biophys. J. 2008; 95:L01–L03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bonnet I., Biebricher A., Porte P.L., Loverdo C., Benichou O., Voituriez R., Escude C., Wende W., Pingoud A., Desbiolles P.. Sliding and jumping of single EcoRV restriction enzymes on non-cognate DNA. Nucleic Acids Res. 2008; 36:4118–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schonhoft J.D., Stivers J.T.. Timing facilitated site transfer of an enzyme on DNA. Nat. Chem. Biol. 2012; 8:205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nelson S.R., Dunn A.R., Kathe S.D., Warshaw D.M., Wallace S.S.. Two glycosylase families diffusively scan DNA using a wedge residue to probe for and identify oxidatively damaged bases. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E2091–E2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Slutsky M., Mirny L.A.. Kinetics of protein-DNA interaction: facilitated target location in sequence-dependent potential. Biophys. J. 2004; 87:4021–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou H.X. Rapid search for specific sites on DNA through conformational switch of nonspecifically bound proteins. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:8651–8656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kalodimos C.G., Biris N., Bonvin A.M., Levandoski M.M., Guennuegues M., Boelens R., Kaptein R.. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science. 2004; 305:386–389. [DOI] [PubMed] [Google Scholar]
- 20. Friedman J.I., Majumdar A., Stivers J.T.. Nontarget DNA binding shapes the dynamic landscape for enzymatic recognition of DNA damage. Nucleic Acids Res. 2009; 37:3493–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iwahara J., Zweckstetter M., Clore G.M.. NMR structural and kinetic characterization of a homeodomain diffusing and hopping on nonspecific DNA. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:15062–15067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Graham J.S., Johnson R.C., Marko J.F.. Concentration-dependent exchange accelerates turnover of proteins bound to double-stranded DNA. Nucleic Acids Res. 2011; 39:2249–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCauley M.J., Rueter E.M., Rouzina I., Maher L.J. 3rd, Williams M.C.. Single-molecule kinetics reveal microscopic mechanism by which High-Mobility Group B proteins alter DNA flexibility. Nucleic Acids Res. 2013; 41:167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murugesapillai D., McCauley M.J., Maher L.J. 3rd, Williams M.C.. Single-molecule studies of high-mobility group B architectural DNA bending proteins. Biophys. Rev. 2017; 9:17–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leith J.S., Tafvizi A., Huang F., Uspal W.E., Doyle P.S., Fersht A.R., Mirny L.A., van Oijen A.M.. Sequence-dependent sliding kinetics of p53. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16552–16557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ghodke H., Wang H., Hsieh C.L., Woldemeskel S., Watkins S.C., Rapic-Otrin V., Van Houten B.. Single-molecule analysis reveals human UV-damaged DNA-binding protein (UV-DDB) dimerizes on DNA via multiple kinetic intermediates. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E1862–E1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cuculis L., Abil Z., Zhao H., Schroeder C.M.. Direct observation of TALE protein dynamics reveals a two-state search mechanism. Nat. Commun. 2015; 6:7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kong M., Liu L., Chen X., Driscoll K.I., Mao P., Bohm S., Kad N.M., Watkins S.C., Bernstein K.A., Wyrick J.J. et al. Single-molecule imaging reveals that Rad4 employs a dynamic DNA damage recognition process. Mol. Cell. 2016; 64:376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perez-Howard G.M., Weil P.A., Beechem J.M.. Yeast TATA binding protein interaction with DNA: fluorescence determination of oligomeric state, equilibrium binding, on-rate, and dissociation kinetics. Biochemistry. 1995; 34:8005–8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parkhurst K.M., Brenowitz M., Parkhurst L.J.. Simultaneous binding and bending of promoter DNA by the TATA binding protein: real time kinetic measurements. Biochemistry. 1996; 35:7459–7465. [DOI] [PubMed] [Google Scholar]
- 31. Dhavan G.M., Crothers D.M., Chance M.R., Brenowitz M.. Concerted binding and bending of DNA by Escherichia coli integration host factor. J. Mol. Biol. 2002; 315:1027–1037. [DOI] [PubMed] [Google Scholar]
- 32. Hiller D.A., Fogg J.M., Martin A.M., Beechem J.M., Reich N.O., Perona J.J.. Simultaneous DNA binding and bending by EcoRV endonuclease observed by real-time fluorescence. Biochemistry. 2003; 42:14375–14385. [DOI] [PubMed] [Google Scholar]
- 33. Sugimura S., Crothers D.M.. Stepwise binding and bending of DNA by Escherichia coli integration host factor. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:18510–18514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang S.N., Crothers D.M.. The role of nucleotide cofactor binding in cooperativity and specificity of MutS recognition. J. Mol. Biol. 2008; 384:31–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hancock S.P., Hiller D.A., Perona J.J., Jen-Jacobson L.. The energetic contribution of induced electrostatic asymmetry to DNA bending by a site-specific protein. J. Mol. Biol. 2011; 406:285–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tang G.Q., Deshpande A.P., Patel S.S.. Transcription factor-dependent DNA bending governs promoter recognition by the mitochondrial RNA polymerase. J. Biol. Chem. 2011; 286:38805–38813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van den Broek B., Noom M.C., Wuite G.J.. DNA-tension dependence of restriction enzyme activity reveals mechanochemical properties of the reaction pathway. Nucleic Acids Res. 2005; 33:2676–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dixit S., Singh-Zocchi M., Hanne J., Zocchi G.. Mechanics of binding of a single integration-host-factor protein to DNA. Phys. Rev. Lett. 2005; 94:118101. [DOI] [PubMed] [Google Scholar]
- 39. Tolic-Norrelykke S.F., Rasmussen M.B., Pavone F.S., Berg-Sorensen K., Oddershede L.B.. Stepwise bending of DNA by a single TATA-box binding protein. Biophys. J. 2006; 90:3694–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reinhard B.M., Sheikholeslami S., Mastroianni A., Alivisatos A.P., Liphardt J.. Use of plasmon coupling to reveal the dynamics of DNA bending and cleavage by single EcoRV restriction enzymes. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:2667–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sass L.E., Lanyi C., Weninger K., Erie D.A.. Single-molecule FRET TACKLE reveals highly dynamic mismatched DNA-MutS complexes. Biochemistry. 2010; 49:3174–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blair R.H., Goodrich J.A., Kugel J.F.. Single-molecule fluorescence resonance energy transfer shows uniformity in TATA binding protein-induced DNA bending and heterogeneity in bending kinetics. Biochemistry. 2012; 51:7444–7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stivers J.T., Pankiewicz K.W., Watanabe K.A.. Kinetic mechanism of damage site recognition and uracil flipping by Escherichia coli uracil DNA glycosylase. Biochemistry. 1999; 38:952–963. [DOI] [PubMed] [Google Scholar]
- 44. Kuznetsov S.V., Sugimura S., Vivas P., Crothers D.M., Ansari A.. Direct observation of DNA bending/unbending kinetics in complex with DNA-bending protein IHF. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:18515–18520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sharma A., Doucette C., Biro F.N., Hingorani M.M.. Slow conformational changes in MutS and DNA direct ordered transitions between mismatch search, recognition and signaling of DNA repair. J. Mol. Biol. 2013; 425:4192–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen X., Velmurugu Y., Zheng G., Park B., Shim Y., Kim Y., Liu L., Van Houten B., He C., Ansari A. et al. Kinetic gating mechanism of DNA damage recognition by Rad4/XPC. Nat. Commun. 2015; 6:5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Parkhurst K.M., Richards R.M., Brenowitz M., Parkhurst L.J.. Intermediate species possessing bent DNA are present along the pathway to formation of a final TBP-TATA complex. J. Mol. Biol. 1999; 289:1327–1341. [DOI] [PubMed] [Google Scholar]
- 48. Delgadillo R.F., Whittington J.E., Parkhurst L.K., Parkhurst L.J.. The TATA-binding protein core domain in solution variably bends TATA sequences via a three-step binding mechanism. Biochemistry. 2009; 48:1801–1809. [DOI] [PubMed] [Google Scholar]
- 49. Porschke D. Structures during binding of cAMP receptor to promoter DNA: promoter search slowed by non-specific sites. Eur. Biophys. J. 2012; 41:415–424. [DOI] [PubMed] [Google Scholar]
- 50. Velmurugu Y., Chen X., Slogoff Sevilla P., Min J.H., Ansari A.. Twist-open mechanism of DNA damage recognition by the Rad4/XPC nucleotide excision repair complex. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E2296–E2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rice P.A., Yang S., Mizuuchi K., Nash H.A.. Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell. 1996; 87:1295–1306. [DOI] [PubMed] [Google Scholar]
- 52. Yang S.W., Nash H.A.. Comparison of protein binding to DNA in vivo and in vitro: defining an effective intracellular target. EMBO J. 1995; 14:6292–6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang S., Cosstick R., Gardner J.F., Gumport R.I.. The specific binding of Escherichia coli integration host factor involves both major and minor grooves of DNA. Biochemistry. 1995; 34:13082–13090. [DOI] [PubMed] [Google Scholar]
- 54. Murtin C., Engelhorn M., Geiselmann J., Boccard F.. A quantitative UV laser footprinting analysis of the interaction of IHF with specific binding sites: re-evaluation of the effective concentration of IHF in the cell. J. Mol. Biol. 1998; 284:949–961. [DOI] [PubMed] [Google Scholar]
- 55. Johnson R.C., Stella S., Heiss J.K.. Rice PA, Correll CC. Bending and compaction of DNA by proteins. Protein-Nucleic Acid Interactions. 2008; Cambridge: Royal Society of Chemistry; 176–220. [Google Scholar]
- 56. Yang C.C., Nash H.A.. The interaction of E. coli IHF protein with its specific binding sites. Cell. 1989; 57:869–880. [DOI] [PubMed] [Google Scholar]
- 57. Swinger K.K., Rice P.A.. IHF and HU: flexible architects of bent DNA. Curr. Opin. Struct. Biol. 2004; 14:28–35. [DOI] [PubMed] [Google Scholar]
- 58. Khrapunov S., Brenowitz M., Rice P.A., Catalano C.E.. Binding then bending: a mechanism for wrapping DNA. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:19217–19218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Coman D., Russu I.M.. A nuclear magnetic resonance investigation of the energetics of basepair opening pathways in DNA. Biophys. J. 2005; 89:3285–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dhavan G.M., Lapham J., Yang S., Crothers D.M.. Decreased imino proton exchange and base-pair opening in the IHF-DNA complex measured by NMR. J. Mol. Biol. 1999; 288:659–671. [DOI] [PubMed] [Google Scholar]
- 61. Parker J.B., Bianchet M.A., Krosky D.J., Friedman J.I., Amzel L.M., Stivers J.T.. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature. 2007; 449:433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vivas P. Mechanism of integration host factor, a DNA-bending protein, probed with laser temperature-jump. 2009; Chicago: University of Illinois at Chicago; Ph.D. thesis. [Google Scholar]
- 63. Ansari A., Kuznetsov S.V.. Williams MC, Maher LJ. Dynamics and mechanism of DNA-bending proteins in binding site recognition. Biophysics of DNA-Protein Interactions. 2010; NY: Springer; 107–142. [Google Scholar]
- 64. Vivas P., Velmurugu Y., Kuznetsov S.V., Rice P.A., Ansari A.. Mapping the transition state for DNA bending by IHF. J. Mol. Biol. 2012; 418:300–315. [DOI] [PubMed] [Google Scholar]
- 65. Lorenz M., Hillisch A., Goodman S.D., Diekmann S.. Global structure similarities of intact and nicked DNA complexed with IHF measured in solution by fluorescence resonance energy transfer. Nucleic Acids Res. 1999; 27:4619–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hillisch A., Lorenz M., Diekmann S.. Recent advances in FRET: distance determination in protein-DNA complexes. Curr. Opin. Struct. Biol. 2001; 11:201–207. [DOI] [PubMed] [Google Scholar]
- 67. Vivas P., Kuznetsov S.V., Ansari A.. New insights into the transition pathway from nonspecific to specific complex of DNA with Escherichia coli integration host factor. J. Phys. Chem. B. 2008; 112:5997–6007. [DOI] [PubMed] [Google Scholar]
- 68. Livesey A.K., Brochon J.C.. Analyzing the distribution of decay constants in pulse-fluorimetry using the maximum entropy method. Biophys. J. 1987; 52:693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Steinbach P.J., Ionescu R., Matthews C.R.. Analysis of kinetics using a hybrid maximum-entropy/nonlinear-least-squares method: application to protein folding. Biophys. J. 2002; 82:2244–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Steinbach P.J. Inferring lifetime distributions from kinetics by maximizing entropy using a bootstrapped model. J. Chem. Inf. Comput. Sci. 2002; 42:1476–1478. [DOI] [PubMed] [Google Scholar]
- 71. Vamosi G., Gohlke C., Clegg R.M.. Fluorescence characteristics of 5-carboxytetramethylrhodamine linked covalently to the 5′ end of oligonucleotides: multiple conformers of single-stranded and double-stranded dye-DNA complexes. Biophys. J. 1996; 71:972–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Unruh J.R., Gokulrangan G., Lushington G.H., Johnson C.K., Wilson G.S.. Orientational dynamics and dye-DNA interactions in a dye-labeled DNA aptamer. Biophys. J. 2005; 88:3455–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Preus S., Wilhelmsson L.M.. Advances in quantitative FRET-based methods for studying nucleic acids. Chembiochem. 2012; 13:1990–2001. [DOI] [PubMed] [Google Scholar]
- 74. Grove A., Galeone A., Mayol L., Geiduschek E.P.. Localized DNA flexibility contributes to target site selection by DNA-bending proteins. J. Mol. Biol. 1996; 260:120–125. [DOI] [PubMed] [Google Scholar]
- 75. Lynch T.W., Read E.K., Mattis A.N., Gardner J.F., Rice P.A.. Integration host factor: putting a twist on protein-DNA recognition. J. Mol. Biol. 2003; 330:493–502. [DOI] [PubMed] [Google Scholar]
- 76. Moe J.G., Russu I.M.. Kinetics and energetics of base-pair opening in 5′-d(CGCGAATTCGCG)-3′ and a substituted dodecamer containing G.T mismatches. Biochemistry. 1992; 31:8421–8428. [DOI] [PubMed] [Google Scholar]
- 77. Bhattacharya P.K., Cha J., Barton J.K.. 1H NMR determination of base-pair lifetimes in oligonucleotides containing single base mismatches. Nucleic Acids Res. 2002; 30:4740–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cao C., Jiang Y.L., Stivers J.T., Song F.. Dynamic opening of DNA during the enzymatic search for a damaged base. Nat. Struct. Mol. Biol. 2004; 11:1230–1236. [DOI] [PubMed] [Google Scholar]
- 79. Lohman T.M., Mascotti D.P.. Thermodynamics of ligand-nucleic acid interactions. Methods Enzymol. 1992; 212:400–424. [DOI] [PubMed] [Google Scholar]
- 80. Record M.T. Jr, Anderson C.F., Lohman T.M.. Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: the roles of ion association or release, screening, and ion effects on water activity. Q. Rev. Biophys. 1978; 11:103–178. [DOI] [PubMed] [Google Scholar]
- 81. Manning G.S. The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Q. Rev. Biophys. 1978; 11:179–246. [DOI] [PubMed] [Google Scholar]
- 82. Jen-Jacobson L., Jacobson L.A.. Rice PA, Cornell CC. Role of water and effects of small ions in site-specific protein-DNA interactions. Protein-Nucleic Acid Interactions. 2008; Cambridge: Royal Society of Chemistry; 13–46. [Google Scholar]
- 83. Holbrook J.A., Tsodikov O.V., Saecker R.M., Record M.T. Jr. Specific and non-specific interactions of integration host factor with DNA: thermodynamic evidence for disruption of multiple IHF surface salt-bridges coupled to DNA binding. J. Mol. Biol. 2001; 310:379–401. [DOI] [PubMed] [Google Scholar]
- 84. Engler L.E., Sapienza P., Dorner L.F., Kucera R., Schildkraut I., Jen-Jacobson L.. The energetics of the interaction of BamHI endonuclease with its recognition site GGATCC. J. Mol. Biol. 2001; 307:619–636. [DOI] [PubMed] [Google Scholar]
- 85. Engler L.E., Welch K.K., Jen-Jacobson L.. Specific binding by EcoRV endonuclease to its DNA recognition site GATATC. J. Mol. Biol. 1997; 269:82–101. [DOI] [PubMed] [Google Scholar]
- 86. Lesser D.R., Kurpiewski M.R., Jen-Jacobson L.. The energetic basis of specificity in the Eco RI endonuclease–DNA interaction. Science. 1990; 250:776–786. [DOI] [PubMed] [Google Scholar]
- 87. Record M.T. Jr, Anderson C.F., Mills P., Mossing M., Roe J.H.. Ions as regulators of protein-nucleic acid interactions in vitro and in vivo. Adv. Biophys. 1985; 20:109–135. [DOI] [PubMed] [Google Scholar]
- 88. Vivas P., Velmurugu Y., Kuznetsov S.V., Rice P.A., Ansari A.. Global analysis of ion dependence unveils hidden steps in DNA binding and bending by integration host factor. J. Chem. Phys. 2013; 139:121927. [DOI] [PubMed] [Google Scholar]
- 89. Velmurugu Y. Dynamics and mechanism of DNA bending-proteins in binding-site recognition. 2016; Chicago: University of Illinois at Chicago; Ph.D. thesis. [Google Scholar]
- 90. Ali B.M., Amit R., Braslavsky I., Oppenheim A.B., Gileadi O., Stavans J.. Compaction of single DNA molecules induced by binding of integration host factor (IHF). Proc. Natl. Acad. Sci. U.S.A. 2001; 98:10658–10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wojtuszewski K., Hawkins M.E., Cole J.L., Mukerji I.. HU binding to DNA: evidence for multiple complex formation and DNA bending. Biochemistry. 2001; 40:2588–2598. [DOI] [PubMed] [Google Scholar]
- 92. Yan J., Marko J.F.. Effects of DNA-distorting proteins on DNA elastic response. Phys. Rev. E Stat. Nonlin. Soft. Matter Phys. 2003; 68:011905. [DOI] [PubMed] [Google Scholar]
- 93. Skoko D., Wong B., Johnson R.C., Marko J.F.. Micromechanical analysis of the binding of DNA-bending proteins HMGB1, NHP6A, and HU reveals their ability to form highly stable DNA-protein complexes. Biochemistry. 2004; 43:13867–13874. [DOI] [PubMed] [Google Scholar]
- 94. Sagi D., Friedman N., Vorgias C., Oppenheim A.B., Stavans J.. Modulation of DNA conformations through the formation of alternative high-order HU-DNA complexes. J. Mol. Biol. 2004; 341:419–428. [DOI] [PubMed] [Google Scholar]
- 95. Arthanari H., Wojtuszewski K., Mukerji I., Bolton P.H.. Effects of HU binding on the equilibrium cyclization of mismatched, curved, and normal DNA. Biophys. J. 2004; 86:1625–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yan J., Kawamura R., Marko J.F.. Statistics of loop formation along double helix DNAs. Phys. Rev. E. 2005; 71:061905. [DOI] [PubMed] [Google Scholar]
- 97. Koh J., Saecker R.M., Record M.T. Jr. DNA binding mode transitions of Escherichia coli HU(alphabeta): evidence for formation of a bent DNA–protein complex on intact, linear duplex DNA. J. Mol. Biol. 2008; 383:324–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Xiao B., Johnson R.C., Marko J.F.. Modulation of HU-DNA interactions by salt concentration and applied force. Nucleic Acids Res. 2010; 38:6176–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Swinger K.K., Lemberg K.M., Zhang Y., Rice P.A.. Flexible DNA bending in HU-DNA cocrystal structures. EMBO J. 2003; 22:3749–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Vander Meulen K.A., Saecker R.M., Record M.T. Jr. Formation of a wrapped DNA-protein interface: experimental characterization and analysis of the large contributions of ions and water to the thermodynamics of binding IHF to H' DNA. J. Mol. Biol. 2008; 377:9–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Mirzabekov A.D., Rich A.. Asymmetric lateral distribution of unshielded phosphate groups in nucleosomal DNA and its role in DNA bending. Proc. Natl. Acad. Sci. U.S.A. 1979; 76:1118–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Manning G.S., Ebralidse K.K., Mirzabekov A.D., Rich A.. An estimate of the extent of folding of nucleosomal DNA by laterally asymmetric neutralization of phosphate groups. J. Biomol. Struct. Dyn. 1989; 6:877–889. [DOI] [PubMed] [Google Scholar]
- 103. Strauss J.K., Maher L.J. 3rd. DNA bending by asymmetric phosphate neutralization. Science. 1994; 266:1829–1834. [DOI] [PubMed] [Google Scholar]
- 104. Kosikov K.M., Gorin A.A., Lu X.J., Olson W.K., Manning G.S.. Bending of DNA by asymmetric charge neutralization: all-atom energy simulations. J. Am. Chem. Soc. 2002; 124:4838–4847. [DOI] [PubMed] [Google Scholar]
- 105. Dragan A.I., Read C.M., Makeyeva E.N., Milgotina E.I., Churchill M.E., Crane-Robinson C., Privalov P.L.. DNA binding and bending by HMG boxes: energetic determinants of specificity. J. Mol. Biol. 2004; 343:371–393. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.