


Abstract
RNA molecules play important and diverse regulatory roles in the cell. Inspired by this natural versatility, RNA devices are increasingly important for many synthetic biology applications, e.g. optimizing engineered metabolic pathways, gene therapeutics or building up complex logical units. A major advantage of RNA is the possibility of de novo design of RNA-based sensing domains via an in vitro selection process (SELEX). Here, we describe development of a novel ciprofloxacin-responsive riboswitch by in vitro selection and next-generation sequencing-guided cellular screening. The riboswitch recognizes the small molecule drug ciprofloxacin with a KD in the low nanomolar range and adopts a pseudoknot fold stabilized by ligand binding. It efficiently interferes with gene expression both in lower and higher eukaryotes. By controlling an auxotrophy marker and a resistance gene, respectively, we demonstrate efficient, scalable and programmable control of cellular survival in yeast. The applied strategy for the development of the ciprofloxacin riboswitch is easily transferrable to any small molecule target of choice and will thus broaden the spectrum of RNA regulators considerably.
INTRODUCTION
RNA devices became a key focus of synthetic biology in recent years. They have been used to implement genetic circuits and networks based on small regulatory RNAs, e.g. toehold-switches (1) or STARs (2), synthetic riboswitches (3) and allosterically controlled ribozymes (4–7). The fast progression of this development resulted in the transition from simple proof of concept to sophisticated and useful applications targeting complex problems (8). RNA devices are unique due to their modular nature that allows the simple and straightforward linkage of different domains, e.g. between a sensor and an actuator. Thus, a whole range of different functions may be united in one RNA molecule. Although it is very common to select natural regulatory domains and adapt them for different purposes, there is also the option of de novo generation of RNA sensor domains. In principle, the powerful in vitro selection method (SELEX) (9,10) allows the selection of a suitable sensor RNA for any desired target molecule. Sensor RNAs thus designed recognize their target with great affinity and specificity. However, despite the fact that several dozen small molecule-binding aptamers have been generated to date (11), only a handful of those are suitable for the design of RNA devices (12). Theophylline- and tetracycline-binding aptamers have been the most successful here (13). They allow the construction of synthetic riboswitches that may be used for control of transcription termination (14), or translation initiation (15), mRNA splicing (16) or control of mRNA stability (17). In contrast to natural riboswitches that are mainly found in bacteria (18), synthetic riboswitches could be developed for all three domains of life (3).
A wealth of adroit proof-of-concept studies demonstrating the application of synthetic riboswitches are available nowadays. However, all studies to date are exclusively limited to the theophylline or tetracycline aptamer systems, which effectively prevents a wider application of synthetic riboswitches. To remedy this shortage of applications and to stimulate and invigorate the field of synthetic riboswitch development, the repertoire of aptamers suitable and available for riboswitch design needs to be extended. First and foremost, methodology for the identification of such aptamer domains is required.
The main reason for the limited suitability of most aptamers is that both excellent binding properties and conformational switching are essential, yet the latter is a feature not addressed during the process of in vitro selection (3). To find aptamers that combine superior binding properties and the ability to undergo conformational switching, cellular screening after in vitro selection is required. Such screening systems have been established (19–21) and their functionality has been successfully demonstrated, e.g. for the neomycin aptamer (22). Now, we have extended the method to include next-generation sequencing (NGS). NGS has revolutionized not only aptamer selection, but proved the proverbial game changer for research across most disciplines of the life sciences (23). The application of NGS allows the collection of detailed information for the individual selection rounds. Thus, it was possible to choose selection rounds that showed a certain degree of enrichment, yet maintained maximum diversity. We assume that this approach will allow a substantial acceleration of the transition between in vitro and in vivo, while simultaneously reducing screening efforts.
In the present study, we demonstrate the approach described above for ciprofloxacin (CFX) as it is a well-known fluoroquinolone antibiotic, FDA-approved (24) and with a favorable toxicity profile for many organisms (25,26). Thus, it guarantees portability and broad applicability. Furthermore, cellular uptake is granted in both lower and higher eukaryotes (27). We were able to generate a CFX-binding aptamer with riboswitching properties. Structural probing revealed a pseudoknot structure that enfolded and essentially sealed the binding pocket. It showed functionality both in yeast and a human cell line and is sufficiently efficient to control cell fate by blocking pyrimidine metabolism or a resistance gene.
In sum, we demonstrate here the de novo development of a novel small molecule-dependent synthetic riboswitch. We characterized a robust procedure for development that may be used as a template for application with any other ligand. Thus, our findings present an ideal basis and a springboard to jumpstart the wide application of aptamer-based riboregulators.
MATERIALS AND METHODS
Pool preparation
For in vitro selection experiments, we used a 1:1 mixture of a completely randomized and a partially-structured library (28). In short, the completely randomized library consists of 64 nucleotides (nt) whereas the partially structured library contained a 12-nt long fixed sequence (5′-CTG CTT CGG CAG-3′) flanked by 26 random nt on each side. Both libraries are flanked by constant regions (5′ constant: 5′-GGG AGA CGC AAC TGA ATG AA-3′/3′ constant: 5′-TCC GTA ACT AGT CGC GTC AC-3′) for amplification using the oligonucleotides Pool_fwd 5′-GTA TAA TAC GAC TCA CTA TAG GGA GAC GCA ACT GAA TGA A-3′ and Pool_rev 5′-GTG ACG CGA CTA GTT ACG GA-3′). Both pools were amplified using the following PCR conditions: 10 mM Tris–Cl (pH 9.0), 50 mM KCl, 1.5 mM MgCl2, 0.1% Triton X-100, 0.2 mM dNTPs (each), 30 nM pool template, 2 μM Pool_fwd, 2 μM Pool_rev, 50 U/ml Taq DNA Polymerase (NEB). 1015 pool template molecules were amplified in a 60 mL PCR reaction for only 7 cycles to reduce PCR-induced bias. PCR efficiency was calculated according to Hall et al. (29).
After large-scale amplification, DNA pool template was ethanol-precipitated, dissolved in MQ-H2O [de-ionized water purified with ion exchange resin and filtered through a Biofilter (ELGA) to remove possible RNase contamination] and subsequently phenol:chloroform extracted (30). The purified DNA template was transcribed using T7 RNA polymerase as described previously (31). Afterwards, the transcribed RNA was ethanol-precipitated, dissolved in formamide containing 25 mM EDTA and loaded on a 6% denaturing polyacrylamide gel (8 M urea). The RNA was visualized by UV shadowing, sliced out and eluted overnight in 300 mM Na-acetate (pH 6.5). Hereafter, eluted RNA was ethanol-precipitated, the pellet was redissolved in a suitable amount of water and molarity was calculated.
CFX immobilization
CFX was immobilized on Profinity™ Epoxide Resin (Bio-Rad). For this, 2 g dry resin was swollen in MQ-H2O, twice washed with MQ-H2O and vacuum-filtered. After a second wash with coupling buffer (50 mM KCl, 132 mM NaOH, pH 13.0), the resin was mixed 1:2 with 5 mM CFX solution in coupling buffer. The reaction was protected from light and incubated over night at room temperature (RT) on an H5600 rotator (Labnet). Afterwards, the resin was washed with MQ-H2O, vacuum-filtered and remaining active groups were blocked by incubation with 1 M ethanolamine (MEA) for 4 h. Finally, the CFX-coupled resin was washed according to the supplier's instructions with alternating buffer change from pH 4.0 (100 mM acetate, 500 mM NaCl) to pH 8.0 (100 mM phosphate, 500 mM NaCl). Lastly, the resin was washed with MQ-H2O and stored in 0.02% (w/v) NaN3 at 4°C in the dark for up to 3 months.
In vitro selection
For the first round of in vitro selection, 1.2 × 1015 RNA molecules from the initial pool (1:1 mixture of completely randomized and pre-structured pool, see above) were spiked with ∼250 kCPM of 5′ 32P-labeled RNA pool in MQ-H2O. RNA folding was performed by heating the mixture to 95°C for 5 min and subsequently placed on ice water for additional 5 min. After the folding step, yeast tRNA was added to a final concentration of 1 mg/ml and the volume was adjusted to 1 column volume (CV, 500 μl) with 1× binding buffer (40 mM HEPES pH 7.4, 125 mM KCl, 5 mM MgCl2, 5% DMSO), respectively. For depletion of RNAs able to bind the affinity matrix, the RNA library was first incubated for 30 min with 1 CV of a non-derivatized column (mock). The mock column consisted of Profinity™ Epoxide Resin that had been treated only with MEA instead of CFX. After negative selection, unbound RNAs were added to 1 CV CFX-coupled resin and incubated for 30 min at RT. Next, the column was washed with 10 CV binding buffer and bound RNAs were eluted with either 4 CV 20 mM EDTA (round 1–5) or 4 CV 1 mM CFX (round 6–10) in 1× binding buffer.
Eluted RNA was ethanol-precipitated with Na-acetate in the presence of 15 μg GlycoBlue™ Coprecipitant (Ambion) and washed twice with 70% (v/v) ethanol. The air-dried pellets were dissolved in a total volume of 50 μl MQ-H2O and reverse-transcribed and amplified (RT-PCR). For RT-PCR, 50 μl eluted RNA was mixed with 1× PCR buffer (10 mM Tris–Cl pH 9.0, 50 mM KCl, 0.1% Triton X-100), 1× first strand buffer (Invitrogen), 2 mM DTT (Roche), 1 μM Pool_fwd, 1 μM Pool_rev, 1.5 mM MgCl2 and 0.3 mM dNTPs (each). The reaction was heated to 65°C for 5 min and then quickly placed on ice. After that, 5 U Taq DNA Polymerase (NEB) and 200 U SuperScript™ II (Thermo Fisher Scientific) were added to the reaction and RNA was reverse-transcribed and amplified (54°C for 10 min followed by 6–10 cycles of 95°C for 1 min, 58°C for 1 min and 72°C for 1 min). Product formation was monitored on a 3% agarose gel.
For the following rounds, RNA was transcribed following (31). In short, 10 μl of RT-PCR product was mixed with 40 mM Tris–Cl (pH 8.0), 5 mM DTT, 2.5 mM NTPs (each), 15 mM MgCl2, 100 U T7 RNA Polymerase (NEB), 40 U ribonuclease inhibitor (moloX) and 33 nM 32P-α-UTP (Hartmann analytics) in a total volume of 100 μl. Transcription was carried out at 37°C for 1 h. Afterwards, transcription was precipitated with NH4-acetate/ethanol, washed twice with 70% EtOH and the pellet was dissolved in a suitable amount of water. Five hundred kCPM RNA was folded, diluted in 1× binding buffer and subsequently loaded onto the column for the next round of SELEX.
Plasmid cloning and doped pool generation
All plasmids and oligonucleotides used in this study are listed in Supplementary Table S1 and Supplementary Table S2, respectively. For cloning, two 30 bp overlapping oligonucleotides were designed and amplified using Q5® High-Fidelity DNA polymerase (NEB) according to the supplier's instructions. The resulting PCR product was purified (QIAquick PCR Purification Kit, Qiagen), digested with AgeI-HF and NheI-HF (NEB) and ligated into equally digested pWHE601* with T4 DNA Ligase (NEB).
Doped pools were generated using the oligonucleotides AgeI_doped_fwd and NheI_[3.0/4.5/9.0/30.0]_doped_rev (Supplementary Table S3, Microsynth AG), respectively, with the construct ΔATG as template and amplified using Q5® High-Fidelity DNA polymerase (NEB) according to the supplier's instructions. Again, digestion and ligation into pWHE601* followed. Transformation of the ligation mixture was done after butanol precipitation into NEB® 10-beta Competent Escherichia coli (High Efficiency) according to the supplier's protocol. This ensured that the number of different plasmids yielded by this process were >50,000.
Cultivation of yeast and GFP measurements
The Saccharomyces cerevisiae strain RS453α (MATα ade2-1 trp1-1 can1-100 leu2-3 his3-1 ura3-52) (32) was transformed using Frozen-EZ Yeast Transformation II Kit (Zymo Research). Transformed cells were plated on SCD-ura plates [0.2% YNB w/o AA (Difco), 0.55% ammonium sulfate (Roth), 2% glucose (Roth), 12 μg/ml adenine (SIGMA), 1× MEM amino acids (SIGMA), 2% Agar (Oxoid)] and incubated at 30°C for 3 days in a humidified incubator. Single colonies were picked and cultured in 1.5 ml SCD-ura for 24 h (450 rpm, 30°C, 24-well plates) before they were diluted 1:1000 in fresh media with and without 1 mM CFX. Again, after 24 h incubation cells were washed twice with 1× PBS and diluted to an OD600 of ∼0.4 for fluorescence measurements.
Fluorescence measurements were performed on a Fluorolog FL3-22 (Horiba Jobin Yvon) with an excitation wavelength set to 474 nm (slit 2 nm) and an emission wavelength of 509 nm (slit 2 nm). The integration time was set to 0.5 sec and temperature was adjusted to 28°C. Afterward, OD600 for each culture was determined and fluorescence intensity was normalized to it. As negative control, pWHE601* (21) was analyzed in parallel as a blank and its value was subtracted from all data. Yeast cells containing pWHE601* are referred to as GFP– cells, whereas cells expressing GFP (transformed with pWHE601 (33)) are referred to as GFP+ cells. Both controls are treated equally as the riboswitch-controlled constructs. Each experiment was done in duplicates and reproduced at least three times.
In vivo screening
Library preparation and in vivo screening was performed according to the established protocol by Suess et al. (34) with modifications described in Schneider et al. (21). In short, libraries for in vivo screening were cloned by homologues recombination in yeast. For that, RT-PCR product of a defined round was amplified with CFX_HR_fwd (5′-CAA GCT ATA CCA AGC ATA CAA TCA ACT CCA AGC TAG ATC TAC CGG TGG GAG ACG CAA CTG AAT GAA-3′) and CFX_HR_rev (5′-CAA GAA TTG GGA CAA CTC CAG TGA AAA GTT CTT CTC CTT TGC TAG CGT GAC GCG ACT AGT TAC GGA-3′) to attach 46 bp overhang for recombination into pWHE601*. Target vector pWHE601* was digested using AgeI-HF and NheI-HF (both NEB) and transformed into RS463α with a 10-molar excess of insert using Frozen-EZ Yeast Transformation II Kit (Zymo Research). Transformed cells were spread on several SCD-ura plates in a way that assures a moderate colony density which simplifies picking clones. For the first screening round, cells were selected under the fluorescence binocular and checked for GFP expression. Clones with low and moderate fluorescence were picked and transferred to a 96-well plate with 200 μL SCD-ura. After sealing the plates with BREATHseal™ (Greiner Bio-One), cells were incubated for 24 h at 30°C on the plate shaker Titramax 1000 (Heidolph instruments) at 450 rpm. Next, cells were split 1:10 in fresh SCD-ura containing no ligand (control) or 1 mM CFX [preliminary experiments showed no influence on cell growth or GFP signal intensity with CFX concentrations up to 10 mM (Supplementary Figure S1)]. Again, after 24 h incubation cells were diluted 1:10 with 1x PBS and GFP fluorescence and OD600 was measured. As positive control pWHE601 (GFP+) and as negative control pWHE601* (GFP-) were measured in parallel and used for normalization (pWHE601) and for subtracting autofluorescence of yeast and media (pWHE601*). Positive hits were streaked out on SCD-ura plates and incubated. From this, four independent colonies were picked and screened again with the protocol above. Verified hits were taken for plasmid preparation using QIAprep Spin Miniprep Kit (Qiagen) and the user-developed protocol from Michael Jones (protocol PR04 ‘Isolation of plasmid DNA from yeast’). Plasmids were passaged trough E. coli DH5α and sequenced with GFP_rev (5′-CCA CTG ACA GAA AAT TTG TGC-3′). Unique candidates were then transformed back into yeast and GFP fluorescence was measured again.
NGS library preparation and data analysis
Barcodes were attached to all selection rounds by PCR using the oligonucleotides Seq_IL_fwd and Seq_IL_rev[0-10] (Supplementary Table S4). Forward and reverse oligonucleotides hybridizes at the 5′ and 3′ constant regions, respectively, thus the sequence of the T7 polymerase promoter was removed. The oligonucleotides Seq_IL_rev[0-10] introduced a 4-mer barcode to assign each sequence to the specific round after sequencing. After amplification, the samples were Gel-purified (Zymoclean Gel DNA Recovery Kit, Zymo Research) and mixed in equimolar amounts for Illumina sequencing reaction (GenXPro GmbH).
To monitor the enrichment process of single sequences and to characterize the SELEX process, we computed for each round of selection a Levenshtein distance distribution from sequence and structural data. The Levenshtein distance measures the difference between two sequences by calculating the smallest number of insertions, deletions, or substitutions necessary to transform one character string—such as a biomolecular sequence, or a RNA secondary structure—into another (35). We computed the Levenshtein distance between every sequence within each selection round. To compare RNA structures, all sequences were folded with RNAfold 2.3.4 (36) at 300 K with the thermodynamic parameter set from Andronescu et al. (37). Here, the Levenshtein distance was computed between the respective dot-bracket annotations of the RNA molecules. Afterward, the Levenshtein distance was normalized by their respective reads per million (RPM) value for both sequences. For every round, a histogram was generated, followed by calculating its cumulative frequency distribution (CFD) followed by normalization by the number of data to cumulative probabilities [P(x)]. To assess the distances between these Levenshtein distance distribution, we computed the Kolmogorov–Smirnoff statistic (38), CDF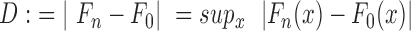 , where Fn is a cumulative distribution function (CDF) derived from cumulative probabilities of the respective Levenshtein distance distributions obtained from the nth SELEX round. Accordingly, F0 is the CDF obtained from the first round and sup is defined as the supremum of the set of distances.
, where Fn is a cumulative distribution function (CDF) derived from cumulative probabilities of the respective Levenshtein distance distributions obtained from the nth SELEX round. Accordingly, F0 is the CDF obtained from the first round and sup is defined as the supremum of the set of distances.
RNA synthesis for in vitro analysis
For in vitro analysis (in-line probing, fluorescence titration experiments and ITC measurements), RNA was transcribed from PCR-generated templates, all containing at least one 5′-terminal guanosyl residue to facilitate transcription in vitro using T7 RNA polymerase. For this, two oligonucleotides were designed with an overlap of 30 bp (Supplementary Table S5) and amplified using Q5® High-Fidelity DNA polymerase (NEB) according to the supplier's instructions. After ethanol precipitation, the DNA template was used for in vitro transcription with T7 RNA polymerase (NEB) as reported previously (31). The RNA was gel purified (39) and molarity was determined by spectrophotometric measurement using NanoDrop 1000 Spectrophotometer (Thermo Scientific).
In-line probing experiments
For in-line probing, RNA was dephosphorylated and 5′ 32P-labeled as previously described (40). After PAGE purification, 35 kcpm of each 5′ 32P-labeled RNA were incubated for 68 h at 22°C in in-line reaction buffer (10 mM Tris–Cl pH 8.3 @ 20°C, 10 mM MgCl2, 100 mM KCl). To generate a size marker, the 5′ 32P-labeled RNAs were subjected to alkaline hydroxylation by incubation for 3 min at 96°C in 50 mM Na2CO3 (pH 9.0), or incubated for 3 min at 55°C with 20 U RNase T1 at denaturing conditions to identify guanines (41). After in-line reaction, alkaline hydroxylation or RNase T1 treatment, reactions were ethanol precipitated and the pellet was dissolved in 5 M urea. All reactions were separated by denaturing polyacrylamide gel electrophoresis. Afterward, gels were dried and analyzed using phosphoimaging (GE Healthcare).
Fluorescence titration experiments
Dissociation constants (KD) for RNA-CFX complexes were determined by measuring the fluorescence quenching as a function of RNA concentration in the presence of a fixed CFX concentration. Fluorescence intensities were measured on a Fluorolog FL3-22 (Horiba Jobin Yvon) with an excitation wavelength set to 335 nm (slit 5 nm) and an emission wavelength of 420 nm (slit 5 nm). The integration time was set to 0.5 s and temperature was adjusted to 25°C. In between, the addition of RNA, the reaction was stirred for 1 min and equilibrated for an extra minute. For the titration experiments, 50 nM CFX in 1× binding puffer (F0) was mixed with increasing amounts of gel-purified RNA and fluorescence intensity was measured (F). Prior to the titration experiment, RNA solutions were heated to 95°C for 5 min and snap-cooled on ice for 5 min (RNA folding step). After that, binding buffer was added to a final concentration of 40 mM HEPES, 125 mM KCl, 5 mM MgCl2, pH 7.4.
Curve fitting was done using Prism (GraphPad Software) and nonlinear regression analysis with following equation by least squares fitting: Y = Bmax*Xh/(KDh + Xh), with Bmax = maximum binding, h = hill slope, X = concentration of RNA.
Isothermal titration calorimetry
RNA folding and buffer compositions were chosen according to the fluorescence titration experiments. 100 × 10−6 M CFX solution were prepared in the same buffer. ITC experiments were carried out with an MicroCal PEAQ-ITC (Malvern Instruments) with the sample cell (200 μl) containing 10 × 10−6 M RNA and 100 × 10−6 M CFX solution in the injector syringe (40 μl). After thermal equilibration at 25°C, an initial 150 s delay and one initial 0.4 μl injection, 12 serial injections of 3.0 μl at intervals of 150 s and at a stirring speed of 750 rpm were performed. Raw data were recorded as power (μcal s−1) over time (min). The heat associated with each titration peak was integrated and plotted against the corresponding molar ratio of CFX and RNA. The dissociation constant (KD) was extracted from a curve fit of the corrected data by use of the one-site binding model provided by MicroCal PEAQ-ITC Analysis Software 1.1.0.1262. Measurements were repeated at least twice.
Serial dilution growth assay
Overnight cultures were either grown in YPD [1% yeast extract (Oxoid), 2% peptone (BD), 2% glucose (Roth), 2% Agar] supplemented with 0.5 mg/ml G418 or in SCD-ura. Both YPD and SCD-ura were supplemented with CFX to a final concentration of 1 mM to condition the cells to the OFF-state. After overnight incubation, cultures were 1- to 5-fold diluted in fresh media and grown to an OD600 of 1–2. Cells were washed with 1× PBS and diluted to an OD600 of 10.0 followed by 6-fold 1:10 serial dilution in 1× PBS (denoted as 0.6 respectively). From the diluted cultures, 5 μl were spotted onto SCD-ura plates supplemented with 0.5 mg/ml G418 in the absence (control) or presence of 1 mM CFX. Growth differences were recorded following incubation of the plates for 2–3 days at 30°C.
Dual luciferase assay
One day before transfection, HeLa cells were transferred to a 24-well plate (40 000 cells/well in 1 ml DMEM). According to the manufacturer's protocol, 1 μl Lipofectamine 2000 (Invitrogen) and 250 ng pDNA was used for transfection. After 2 h, transfection medium (Opti-MEM) was replaced by fresh medium supplemented with or without 100 μM CFX (Sigma-Aldrich). Luminescence was measured 24 h post transfection using the Dual-Glo Luciferase Assay System according to the manufacturer's instructions (Promega). Luminescence was detected using an Infinite M200 Microplate Reader (Tecan). The ratio between firefly and Renilla luciferase activity was calculated for each well to normalize for transfection efficiency. Mean values and standard deviations were calculated from triplicates and normalized to the values of the corresponding vector without riboswitch. Each experiment was repeated at least three times.
RESULTS AND DISCUSSION
Identification of CFX-binding aptamers by in vitro selection (SELEX)
To select aptamers that recognize CFX, we immobilized CFX directly to an epoxy-activated, solid polyacrylamide support (Figure 1A). The reaction conditions were adjusted to a slight molar excess of CFX compared to accessible reactive epoxy groups on the column. In consequence, we assume that under these alkaline conditions, the epoxy group mainly reacts with the secondary amino group of the piperazinyl residue, exposing CFX to the solvent (42).
Figure 1.

Progress of in vitro selection. (A) Chemical structure of CFX. The arrow indicates the most likely attachment site to the epoxy-activated PAA-matrix. (B) Shown is the fraction of loaded RNA that could be eluted from CFX-derivatized columns after each selection round. RNA was eluted by either 20 mM EDTA (round 1–5) or 1 mM CFX (round 6–10). In the first three rounds, a negative selection was performed (*). In round 5 and 10, stringency was increased by doubling the number of column washes or a decrease in the concentration of immobilized CFX to one-tenth, respectively (‡). Pre-elution steps were performed in round seven and eight (#) (for further details see also Supplementary Table S6).
The RNA library with a starting diversity of 1.2 × 1015 RNA molecules included a 64 nt-long random region with half of it containing a small stem loop in the middle, a library composition already established (28). It was discussed that preformed stem-loops provide favourable conditions for aptamer selection by acting as nucleation sites for RNA structure formation (43–45). We had no a priori knowledge of the nature of the aptamer we were exploring, including both a completely randomized region and a preformed stem loop gave us the full scope to unrestrainedly select for the best fit.
In the first five rounds, we eluted unspecifically with EDTA to ensure elution of every RNA molecule, neglecting their binding properties. This approach should guarantee that the first enrichment of the pool introduces no bias toward low affinity aptamers because of the mild selection conditions. Furthermore, in the first three rounds, a negative selection step was carried out to remove RNA molecules that recognize the solid support. The amount of eluted RNA in these early rounds was as expected to be very low (details in Supplementary Table S6) since most of the RNA molecules of the randomized pools do not recognize the ligand. After a first enrichment in round 4 (Figure 1B), we increased stringency by increasing the number of washing steps (round 5) or switching to specific elution with CFX in round 6. Despite increased stringency, more RNA was eluted from the column. In consequence, we decided to implement a pre-elution step in rounds 7 and 8 to eliminate RNA species with fast Koff rates (11). Additionally, we reduced the amount of immobilized ligands to one-tenth in the last round of selection. A detailed summary of the selection process can be found in Supplementary Table S6.
For a first glimpse of the selection progress, we sequenced 23 candidates of round 10. We found 13 different sequences, eight of them were unique and about half of them contained the predefined stem loop, but no shared motifs could be found (Supplementary Table S7). Binding capacities of all individual candidates were analyzed by their interaction with the CFX-derivatized column (Supplementary Figure S2A). Most of the aptamers showed a strong interaction with the column and could be specifically eluted with CFX. Some candidates included in the analysis did not perform as expected, e.g. R10K3 showed a weak interaction and R10K9 an interaction similar to the entire pool binding capacity in round 10. However, R10K6, R10K7 and R10K4 showed an elution profile up to 4-fold improved. Four candidates were selected for quantification of CFX binding by fluorescence titration experiments. Here, the intrinsic fluorescence of CFX and the respective quenching upon RNA-binding were used to determine the dissociation constant (KD) of the respective aptamers. For all tested RNAs, the KD was below 100 nM (Supplementary Figure S2B). Our analysis suggested that the enrichment process yielded aptamers of the desired high binding affinity. However, we simultaneously managed to maintain a sufficient diversity of candidates with adequate structural flexibility, i.e. ideal conditions for a detailed examination of the sequence composition and subsequent riboswitch screening.
Deep sequencing of aptamer selection populations
Selection experiments aim to enrich aptamers with high binding affinity and specificity for their respective ligands. However, our practical experience in recent years clearly demonstrates that superior binding affinity of aptamers alone is insufficient for successful development into riboswitches. On the contrary, a subsequent screening step has proved indispensable (12,22,46). Implementing NGS into our workflow was instrumental in the identification of selection rounds that were best-suited for the laborious screening for switching aptamers, which considered both sequence and structure diversity in conjunction with library enrichment.
By Illumina sequencing, we obtained a total number of 4.2 million reads for all investigated rounds, of which 92% could be sorted according to their corresponding barcoding (Supplementary Table S4). Next, identical sequences were summed up and the total read count was normalized for each round to reads per million (RPM).
A statistical analysis was performed for the NGS data. We determined the enrichment of the 100 most abundant sequences (Top100, Figure 2A) and the proportion of the background or so-called orphans (Figure 2B). For the most abundant sequences, we see a clear exponential enrichment throughout the SELEX process, which is in line with our expectations. On the other hand, the number of orphans drops constantly up to round 6 and remains at a low level to the end of selection. In addition to the sequence-based analysis, we also performed a structural evaluation. For this, we predicted MFE structures of all sequences. We compared similarities of the dot-bracket annotation by calculating the Levenshtein distance LvDist(X,Y) (35) of all predictions within each round with each other. In our experience, sequence-based motif search will only yield useful results if the pool is enriched to a certain degree. Since we evaluated all rounds of selection, structural similarity rather than motifs will allow a better estimation of pools diversity (47). The histogram of the LvDist(X,Y) distribution was converted to a cumulative distribution function (CDF), so that the diversity of the pool of each round can be easily assessed (Figure 2C). As a distance measure between the obtained CDFs, we used Kolmogorv–Smirnoff's D (ks-test) which computes the supremum 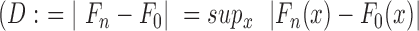 ) between two CDFs (Figure 2D).
) between two CDFs (Figure 2D).
Figure 2.

NGS analysis of CFX in vitro selection. (A) Over the course of the SELEX experiment, the most abundant sequences (Top100 of each round) were enriched in an exponential fashion. (B) Increased stringency over time reduced the amount of background (‘Orphans’) continuously till a plateau was reached (round 6–10). (C) The cumulative distribution function (CDF) for each round based on calculated Levenshtein distances on MFE structures is plotted for each round, resulting in an increased P(x) over the selection experiment. Shown are the results for the Top1000. ( D) Based on CDF, D was derived and its logarithm is plotted against the selection rounds for the Top1000. Here, D is computed between the first round and all remaining.
In contrast to the cumulative RPM of the Top100 enriched sequences, we observed neither a gradual nor an exponential increase in enrichment (compare Figure 2A with D). Rather, we found a more uneven distribution of enrichment. These findings can be correlated to the SELEX procedure (Figure 1B). The differences between the rounds can be assessed by looking at the distance measure D (Figure 2D). We observed the highest increase in enrichment between rounds 5/6, 7/8 and 9/10. All of these large enrichment steps can be correlated to the experimental conditions applied in the corresponding rounds. In round 6, we switched from EDTA to CFX elution, in round 8 we applied the pre-elution step twice and drastically reduced the amount of eluted RNA and in round 10 we reduced the amount of immobilized CFX to one-tenth. Interestingly and counterintuitively, in round 9 we found nearly the same sequence and structure distribution compared to round 8 and the change in distance between 8 and 9 is almost zero. Although we could elute the highest amount of RNA from the column, the removal of the stringency (no pre-elution) led to an amplification-only round, where no further selection took place. These results suggest that selection pressure should be kept constant or increased over the experiment, but not omitted. Otherwise other factors can influence pool sequence distribution, such as RT-PCR or in vitro transcription, which may introduce bias. In sum, NGS is able to determine structural diversity and by doing so building a foundation for selection the best-suited rounds for in vivo screening. With respect to diversity, we have chosen round 6 and round 10 as the libraries to start screening. In round 6, we observed a prominent structural enrichment for the first time, whereas we consider round 10 the most enriched library.
NGS guided in vivo screening and riboswitch engineering
Aptamer sequences from round 6 and 10 were cloned into the 5′ UTR of a constitutively driven gfp+ gene. The pools were integrated into S. cerevisiae by homologous recombination and analyzed for CFX-dependent changes in fluorescence (21). We screened 6000 colonies in total for both rounds and discarded candidates with a fluorescence signal considered too low. Here, inserted aptamers were either already too structured or the insertion into the vector, which by default lacks a start codon, failed. Based on empirical knowledge, we also discarded candidates with very high fluorescence indicating the absence of a structured RNA. After this initial elimination procedure, 17% of the total that initially showed a GFP fluorescence in the desired range remained. For 599 and 435 clones for round 6 and round 10, respectively, fluorescence was measured in the absence and presence of CFX and the regulatory activity was calculated (Figure 3A). In round 6, no candidates showed any changes in fluorescence. In round 10, two candidates were identified with 1.7- and 1.5-fold decrease in GFP expression, respectively.
Figure 3.

In vivo screening for CFX riboswitches and refinement of the aptamer domain. (A) The boxplot summarizes the in vivo screening in S. cerevisiae for round 6 (R6) and round 10 (R10) and the screened doped pools with different degrees of randomization, starting with 30.0% (30.0) down to 3.0% (3.0). For each investigated clone, the regulatory activity was calculated as ratio of GFP fluorescence with and without 1 mM CFX (x-fold). The two regulatory active sequences 2B and 10A are highlighted. Based on 10A, a mutant was derived with a mutated AUG that was upstream of the original start codon (ΔAUG) and used for synthesizing four doped libraries (30.0–3.0%). Clones that showed better switching properties than ΔAUG (dotted line) were sorted into the gain-of-function group. The numbers written above the boxplot indicate the number of clones used for the particular box. (B) Heatmap based on sequencing the gain- and loss-of-function group. For each group, mutation rate was normalized to 1.0 and plotted as a heatmap as per-nucleotide function. Highlighted point mutations in the gain-of-function group are considered as directed mutations. All single data can be found in Supplementary Table S8. (C) Fluorescence measurements of the originally found candidate 10A, the mutated one ΔAUG and the CFX riboswitch (CFX-RS). Shown are the fluorescence values without (black bars) and with 1 mM CFX (white bars). Above each construct, the regulatory activity is written with standard deviation (SD) in brackets. (D) Investigation of the impact of each single mutation. As in C, the fluorescence values without and with 1 mM CFX are plotted and the regulatory activity and SD in brackets is shown on the right, respectively.
We decided to continue with candidate 10A and partially randomized each position of the 103-nt long sequence to different degrees to identify aptamer mutants with an improved phenotype. Before randomization, we deleted an upstream start codon within 10A (ΔAUG) to prevent premature translation initiation. We started with 30.0% and 9.0% randomization and analyzed around 2000 clones. With 30.0% randomization, we completely lost any regulation, whereas 13 clones with improved phenotype could be identified within the pool with 9.0% randomization (Figure 3A). The detailed sequence analysis of these clones revealed a maximum of only up to four nucleotide exchanges. Based on this, we repeated the analysis with two new doped libraries with 4.5% and 3.0% randomization, respectively. We screened about 1000 clones, out of which about 100 clones fell into the gain-of-function (GOF) group. Sequencing 100 clones from both GOF and also from the loss-of-function (LOF) group revealed that two regions are nearly invariant (nts 26–40 and 63–103), whereas two regions can acquire mutations (nts 1–25 and 41–62). Furthermore, we identified seven mutation hot spots (Figure 3B). The hot spots were defined not only based on the overall mutation rate, but also on the fact that these point mutations were directed into one specific base. By combining all directed GOF mutations (G1U, A11C, A25C, U47C, C51U, A56C, U61G), we considerably improved regulation. GFP measurements revealed an increased in vivo activity of 7.5-fold (Figure 3C). For the resulting construct, we chose the term CFX riboswitch. Investigating the impact of each single mutation, U61G can be highlighted as one of the mutations with the highest contribution to the enhanced switching property (Figure 3D). However, U61G alone is not fully responsible for the enhanced phenotype.
Taken together, initial screening, subsequent partial randomization and the combination of beneficial mutations led to the new synthetic CFX riboswitch. With 7.5-fold regulatory activity, the dynamic window is comparable to other synthetic riboswitches, e.g. the tetracycline or neomycin riboswitch (22,33) and other RNA-based devices that control gene expression in eukaryotes (48,49).
Secondary structure analysis of the CFX riboswitch by structural probing and mutational analysis revealed a pseudoknot structure
Next, we endeavoured to gain insight into the secondary structure of the CFX riboswitch. The RNA was in vitro transcribed, radiolabeled and subjected to an in-line probing analysis (41). The cleavage pattern is shown in Figure 4A. Interestingly, the riboswitch consists to a large extent of non-cleaved nucleotides, implicating a high degree of structured regions. RNA folding prediction with programs based on the Zuker algorithm (50), however, did not result in any structure that fit to the observed probing data. On the other hand, the assumption of a pseudoknot fold resolved all mismatches to the in-line probing pattern and resulted in a secondary structure prediction illustrated in Figure 4B. To prove the assumed pseudoknot, we mutated it and analyzed respective rescue mutations (M2/M2R) (Figure 4B). In addition, we introduced a mutation and its respective rescue into the closing stem P1 (M1/M1R) (Figure 4B). For both regions, disrupted base paring completely diminished regulation. Functionality could be restored by introduction of compensatory mutations. Only two regions showed significant flexibility, the first 25 nucleotides (nt 1–25) and the L2 region (nucleotides 44–59, blue in Figure 4B). Interestingly, gain-of-function mutants that improved regulation were exclusively found in these two regions (position 1, 11 and 25, U61G removes a mismatch within P2, and U47C, C51U and A56C located within the L2 region). There is no indication that nts 1–25 were involved in the aptamer structure, the-gain of-function may be attributed to context dependencies. The L2 region, however, seems to be important for regulation since three gain-of-function mutants were located in this region. In contrast, no gain-of-function mutant was identified in the central part formed by the pseudoknot, P3 and P1. This region harboured the only two positions with significant CFX-dependent changes in the in-line probing pattern indicating a role of U37 and G72, respectively, for ligand binding. We determined a dissociation constant for the CFX riboswitch of 60 nM by fluorescence titration spectroscopy and isothermal calorimetry (ITC) (Figure 4C and D). The analysis of the binding affinity of the mutants U37A and G72C, respectively, resulted in a dramatically reduced binding affinity. Simultaneously, both mutations lead to a complete loss of regulation in vivo (Figure 4E). We speculate that this region (the pseudoknot, P3 and the upper base pair of P1) may constitute the CFX binding pocket. Interestingly, the binding constant of the CFX riboswitch is similar to the initial candidates 10A and 10AΔAUG, although considerably enhanced in in vivo activity. It indicates that the improvement of regulation targeted the switching potential of the riboswitch rather than its ligand binding.
Figure 4.

Structure determination of the CFX riboswitch. (A) In-line probing experiment for CFX riboswitch. Shown is the cleavage pattern in the absence (–) and presence (+) of 10 μM CFX under alkaline conditions. As references and for nucleotide position assignment, non-reacted RNA (NR), hydroxyl reaction (OH) and nuclease T1 digestion (T1) were loaded onto the gel. G nucleotides and nucleotides that showed a change upon ligand addition are highlighted. Colour coding for identified stem and loop regions follows the coding for the proposed secondary structure in B. (B) Proposed secondary structure of the CFX riboswitch including three stems (P1, P2, P3), a loop-region (L2) and a pseudoknot fold (PS). Nucleotides with changes in the probing pattern are encircled. Mutations introduced to study structure–function relationships are indicated. (C) Fluorescence titration experiment data for the indicated RNAs. (D) Verification the binding constant of CFX-RS with ITC. Left panel: power required to maintain the temperature of the RNA solution recorded over the time until saturation was reached (baseline-corrected). Right panel: integrated heats of interaction plotted against the molar ratio of ligand over RNA and fitted to a single binding site model (MicroCal PEAQ-ITC Analysis Software 1.1.0). (E) In vivo data for point-mutated nucleotides U37 to A and G72 to C.
In sum, the CFX riboswitch presented here once more exemplifies the essential requirement for tight ligand binding, which is in line with previous work (3,22,33). Nucleotides involved in ligand binding were clustered around the pseudoknot fold, which supports the idea of the formation of a binding pocket that allows an initial binding of CFX to the aptamer. During a second binding step, the binding pocket is then closed through the P2/L2 region. We have demonstrated a similar two-step binding model for the tetracycline aptamer (51).
Ligand binding and recognition
We determined the binding affinities of seven different fluoroquinolones to further characterize the structure-function relationship of the CFX riboswitch. We analyzed ligand binding by fluorescence titration spectroscopy and determined their switching potential (Figure 5). The titration experiments revealed two side groups to be important for CFX binding: the carboxyl group on C3 and the fluorine group on C6. Here, decarboxy CFX (dCFX), pipedimic acid (PA) and 6-hydroxy-6-defluoro CFX (hCFX) showed a reduction in binding affinity of about 6- to 14-fold, respectively. Less relevant for binding is the cyclopropyl residue on N1, showing a 3-fold higher dissociation constant for norfloxacin (NFX) compared to CFX. Danofloxacin (DFX) and enrofloxacin (EFX), which have modifications on the piperazinyl residue, showed no significant change in affinity to the CFX riboswitch. This supports the assumption that immobilization of CFX for in vitro selection was most probably achieved by coupling the secondary amine to the activated epoxy group of the solid support (indicated by an arrow in Figure 1A on CFX).
Figure 5.

Specificity of molecular recognition of the CFX riboswitch and their impact on riboswitching. Left: Chemical structure of ciprofloxacin and seven selected fluoroquinolones that show binding to the CFX riboswitch. Chemical changes relative to CFX are shaded in light grey. Middle: Plot of log10KD values of the different fluoroquinolones and their activity in vivo (right), respectively (SD in brackets). Data shown in this graphical overview are summarized in Supplementary Table S9.
The in vivo activity is roughly related to the binding affinity of each ligand to the riboswitch, with the exception of EFX caused by its toxicity (reduction up to one tenth in yeast growth [data not shown]) and hCFX. hCFX may be directly converted into an intermediate of the CFX degradation pathway which gives an explanation for the missing in vivo activity (52,53).
Scalable and programmable control of cellular survival
The CFX riboswitch is capable to regulate GFP expression up to 7.5-fold in a nearly binary fashion due to the low OFF state. Taking this into account, we aimed to prove the potential of the CFX riboswitch to control survival by regulating genes necessary for cellular growth. For this purpose, we exchanged gfp+ with either the kanR or the URA3 gene. KanR codes for the aminoglycoside 3′-phosphotransferase that allows growth in the presence of the toxic compound geneticin (G418) (54). The URA3 gene encodes the orotidine 5′-phosphate decarboxylase that allows growth on synthetic drop-out media without uracil (55).
Both genes were cloned under the control the CFX riboswitch and the precursor 10AΔAUG. In addition, the respective positive control without the insertion of the riboswitch was analyzed (kanR+ or URA3+). As negative control, the start codon of the respective genes (kanR- or URA3-) was removed preventing gene expression and consequently cell growth.
Yeast strains with the respective plasmids were grown in the absence and in the presence of 1 mM CFX on appropriate plates and cell growth was analyzed by serial dilution growth assays. In the absence of CFX, no negative effect on cell growth could be detected upon riboswitch insertion controlling kanR (Figure 6A). This is interesting, since decreased expression level was observed in the GFP reporter gene assay compared to the control without riboswitch (Figure 3C). For controlling the URA3 gene, we detected a slight reduction of cellular growth upon introduction of the CFX riboswitch (Figure 6B). In the presence of 1 mM CFX, expression of both genes could be significantly reduced. For kanR, growth was reduced over three orders of magnitude and for URA3, hardly any growth could be detected after two days on plate.
Figure 6.

CFX riboswitch controls yeast growth. (A) Serial dilution growth assay were performed for both, 10AΔAUG (ΔAUG) and the engineered CFX riboswitch (CFX-RS). Additionally, two controls expressing the aminoglycoside 3′-phosphotransferase (kanR+) and a mutant without start codon ATG (kanR-) were spotted on SCD-ura plates (supplemented with G148) in the absence or presence of 1 mM CFX. Ten-fold serial dilutions were spotted from left to right (numbering above). Cells were grown for two days at 30°C. (B) Similar to the experiments with kanR, an analogous approach was performed by exchanging the kanR gene with URA3. Selection marker for plasmid maintenance were swapped.
In sum, the growth of yeast carrying essential genes under the control of the CFX riboswitch can be effectively and quantitatively controlled through addition of CFX. Furthermore, the effect of aptamer insertion on gene expression in a physiological context was absent or negligible, although expression levels in reporter gene assays responded to aptamer insertion.
The CFX riboswitch controls gene expression in HeLa cells
To prove CFX riboswitch functionality not only in yeast, but also in higher eukaryotes we exploited a dual luciferase system that expresses firefly and Renilla luciferase in HeLa cells. We cloned the CFX riboswitch in front of the start codon of the firefly luciferase in the pDLP vector system (56), analogous to the GFP variants in yeast. Previous work on tetracycline and neomycin riboswitches carried out in our lab indicated that the transfer of a yeast-optimized variant to higher eukaryotes can be challenging (3). One conceivable explanation could be the increased helicase activity of the ribosome in higher eukaryotes compared to yeast (57,58). Consequently, the underlying strategy was to stabilize the CFX riboswitch. Therefore, we exchanged loop L2 into a stable GAAA tetraloop (59). This stabilization of the riboswitch led to a nearly 2-fold regulation in HeLa cells (Figure 7). By application of the same partial randomization strategy outlined above, further improvement may be possible. Thus, we demonstrate for the first time to our knowledge that a riboswitch controlling translation initiation is portable between different species without major changes in sequence composition.
Figure 7.
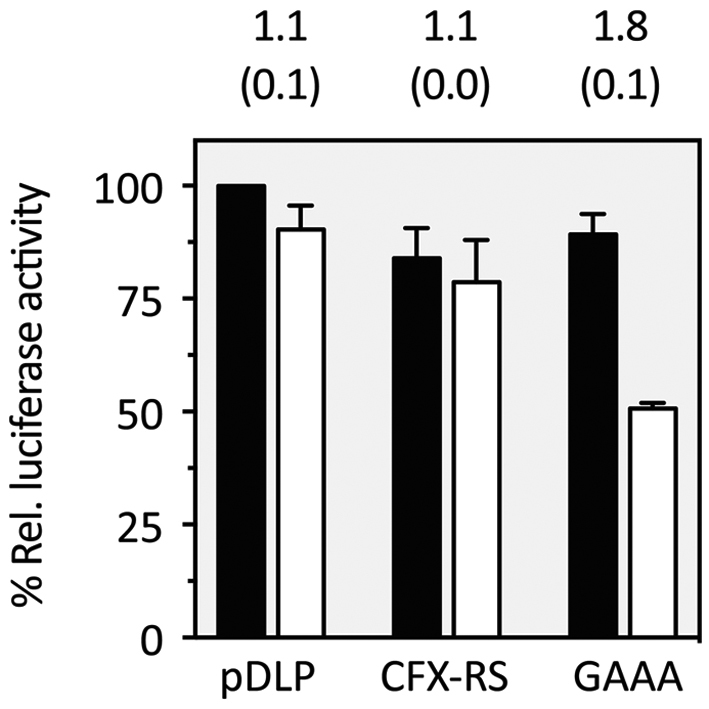
Application of the CFX riboswitch in a mammalian cell line. Dual luciferase assay of the CFX riboswitch (CFX-RS) and the stabilized GAAA mutant. The stabilized GAAA mutant, where the flexible L2 loop region was exchanged with a stable GAAA tetraloop, showed a significant reduction in firefly luciferase activity upon addition of 250 μM CFX. Black bars = w/o ligand, White bars = 250 μM CFX. pDLP is the vector without riboswitch. The experiments were repeated at least three times. SD are reported in brackets.
CONCLUSION
Riboswitches are associated with a range of advantages. These switches are often very simple as they consist of only one genetic element, RNA. As such, they are independent of transcription factors or other regulatory proteins. Leakiness, a phenomenon often described for transcriptional regulation due to position effects, does not affect riboswitches. Moreover, RNA-based sensor domains may in theory be selected for any desired ligand. The riboswitch field is presently on the cusp of the transition from proof-of-concept studies to the development of robust and applicable tools and switches (13).
The main obstacle limiting functional synthetic riboswitch development may currently be found in the fact that only a fraction of the selected aptamers are suitable for riboswitch design. Fit-for-purpose aptamers require not only excellent binding properties, but also the conformational flexibility essential for a switch. Only the first can be addressed by SELEX; for the latter, however, laborious screening is necessary.
Through the process of in vitro selection, subsequent NGS-guided in vivo screening and further optimization, we could identify and engineer a novel, CFX-responsive riboswitch with a dynamic range sufficient to allow for complete control of cellular behavior. Moreover, we created a riboswitch that is active both in lower and higher eukaryotes. Due to the digital behavior of the ON and OFF state, we predict a high performance in biological circuits.
In essence, our findings present the first evidence for the de novo design of a riboswitch in the last 10 years. We are convinced that our optimized protocol will allow the discovery of a multitude of new riboswitches. Further advancement of our methodology, e.g. optimized selection procedures that combine selection for binding performance and conformational switching ability or the application of microfluidic high-throughput screening, is under way. These developments open up novel and unforeseen perspectives, e.g. for control of gene expression. However, riboswitches could also find application as biosensors, e.g. for monitoring of cell metabolite concentrations and optimization of metabolic pathways or measurements of metabolic flux rates. Moreover, riboswitches would be effective as highly sensitive sensors for detection of pollutants. Thus, this novel RNA device has the potential to energize and inspire efforts for synthetic riboswitch development.
In essence, synthetic riboswitches have received relatively little attention so far, and they may have been slightly overlooked due to the fact that they have yet to reach a critical mass. They are not entirely uncontroversial, mainly due to the low number of functional switches available to date. However, we take the opposite view and propose that the novel CFX riboswitch and the associated tools developed in our study will be a turning point for the synthetic riboswitch field. In all likelihood, ongoing work in our group and other laboratories will lead to a breakthrough and widespread use of riboswitches in the near future.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
Deutsche Forschungsgemeinschaft [SFB902/A2]; ERASynBio ‘Intensify’, LOEWE CompuGene and H2020 ITN MetaRNA. Funding for open access charge: Deutsche Forschungsgemeinschaft.
Conflict of interest statement. None declared.
REFERENCES
- 1. Green A.A., Silver P.A., Collins J.J., Yin P.. Toehold switches: de-novo-designed regulators of gene expression. Cell. 2014; 159:925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chappell J., Takahashi M.K., Lucks J.B.. Creating small transcription activating RNAs. Nat. Chem. Biol. 2015; 11:214–220. [DOI] [PubMed] [Google Scholar]
- 3. Berens C., Groher F., Suess B.. RNA aptamers as genetic control devices: the potential of riboswitches as synthetic elements for regulating gene expression. Biotechnol. J. 2015; 10:246–257. [DOI] [PubMed] [Google Scholar]
- 4. Win M.N., Smolke C.D.. Higher-order cellular information processing with synthetic RNA devices. Science. 2008; 322:456–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Felletti M., Stifel J., Wurmthaler L.A., Geiger S., Hartig J.S.. Twister ribozymes as highly versatile expression platforms for artificial riboswitches. Nat. Commun. 2016; 7:12834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wittmann A., Suess B.. Selection of tetracycline inducible self-cleaving ribozymes as synthetic devices for gene regulation in yeast. Mol. Biosyst. 2011; 7:2419–2427. [DOI] [PubMed] [Google Scholar]
- 7. Gammage P.A., Gaude E., Van Haute L., Rebelo-Guiomar P., Jackson C.B., Rorbach J., Pekalski M.L., Robinson A.J., Charpentier M., Concordet J.-P. et al. . Near-complete elimination of mutant mtDNA by iterative or dynamic dose-controlled treatment with mtZFNs. Nucleic Acids Res. 2016; 44:7804–7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khalil A.S., Collins J.J.. Synthetic biology: applications come of age. Nat. Rev. Genet. 2010; 11:367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ellington A.D., Szostak J.W.. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990; 346:818–822. [DOI] [PubMed] [Google Scholar]
- 10. Tuerk C., Gold L.. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990; 249:505–510. [DOI] [PubMed] [Google Scholar]
- 11. McKeague M., McConnell E.M., Cruz-Toledo J., Bernard E.D., Pach A., Mastronardi E., Zhang X., Beking M., Francis T., Giamberardino A. et al. . Analysis of in vitro aptamer selection parameters. J. Mol. Evol. 2015; 81:150–161. [DOI] [PubMed] [Google Scholar]
- 12. McKeague M., Wong R.S., Smolke C.D.. Opportunities in the design and application of RNA for gene expression control. Nucleic Acids Res. 2016; 44:2987–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berens C., Suess B.. Riboswitch engineering—making the all-important second and third steps. Curr. Opin. Biotechnol. 2015; 31:10–15. [DOI] [PubMed] [Google Scholar]
- 14. Wachsmuth M., Findeiß S., Weissheimer N., Stadler P.F., Mörl M.. De novo design of a synthetic riboswitch that regulates transcription termination. Nucleic Acids Res. 2013; 41:2541–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lynch S.A., Desai S.K., Sajja H.K., Gallivan J.P.. A high-throughput screen for synthetic riboswitches reveals mechanistic insights into their function. Chem. Biol. 2007; 14:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weigand J.E., Suess B.. Tetracycline aptamer-controlled regulation of pre-mRNA splicing in yeast. Nucleic Acids Res. 2007; 35:4179–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beilstein K., Wittmann A., Grez M., Suess B.. Conditional control of mammalian gene expression by tetracycline-dependent hammerhead ribozymes. ACS Synth. Biol. 2015; 4:526–534. [DOI] [PubMed] [Google Scholar]
- 18. Breaker R.R. Riboswitches and the RNA World. 2012; CSHL Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lynch S.A., Gallivan J.P.. A flow cytometry-based screen for synthetic riboswitches. Nucleic Acids Res. 2009; 37:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Townshend B., Kennedy A.B., Xiang J.S., Smolke C.D.. High-throughput cellular RNA device engineering. Nat. Methods. 2015; 12:989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schneider C., Suess B.. Identification of RNA aptamers with riboswitching properties. Methods. 2016; 97:44–50. [DOI] [PubMed] [Google Scholar]
- 22. Weigand J.E., Sanchez M., Gunnesch E.-B., Zeiher S., Schroeder R., Suess B.. Screening for engineered neomycin riboswitches that control translation initiation. RNA. 2008; 14:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Levy S.E., Myers R.M.. Advancements in next-generation sequencing. Annu. Rev. Genom. Hum. Genet. 2016; 17:95–115. [DOI] [PubMed] [Google Scholar]
- 24. Ronald A.R., Low D.. Fluoroquinolone Antibiotics. 2003; Springer Science & Business Media. [Google Scholar]
- 25. Suto M.J., Domagala J.M., Roland G.E., Mailloux G.B., Cohen M.A.. Fluoroquinolones: relationships between structural variations, mammalian cell cytotoxicity, and antimicrobial activity. J. Med. Chem. 1992; 35:4745–4750. [DOI] [PubMed] [Google Scholar]
- 26. Azéma J., Guidetti B., Dewelle J., Le Calve B., Mijatovic T., Korolyov A., Vaysse J., Malet-Martino M., Martino R., Kiss R.. 7-((4-Substituted)piperazin-1-yl) derivatives of ciprofloxacin: synthesis and in vitro biological evaluation as potential antitumor agents. Bioorg. Med. Chem. 2009; 17:5396–5407. [DOI] [PubMed] [Google Scholar]
- 27. Yang Q., Nakkula R.J., Walters J.D.. Accumulation of ciprofloxacin and minocycline by cultured human gingival fibroblasts. J. Dent. Res. 2002; 81:836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paige J.S., Wu K.Y., Jaffrey S.R.. RNA mimics of green fluorescent protein. Science. 2011; 333:642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hall B., Micheletti J.M., Satya P., Ogle K., Pollard J., Ellington A.D.. Design, synthesis, and amplification of DNA pools for in vitro selection. Curr. Protoc. Mol. Biol. 88. 2009; 24.2.1–24.2.27. [DOI] [PubMed] [Google Scholar]
- 30. Vogel M., Suess B.. Label-free determination of the dissociation constant of small molecule-aptamer interaction by isothermal titration calorimetry. Methods Mol. Biol. 2016; 1380:113–125. [DOI] [PubMed] [Google Scholar]
- 31. Groher F., Suess B.. In vitro selection of antibiotic-binding aptamers. Methods. 2016; 106:42–50. [DOI] [PubMed] [Google Scholar]
- 32. Kötter P., Weigand J.E., Meyer B., Entian K.-D., Suess B.. A fast and efficient translational control system for conditional expression of yeast genes. Nucleic Acids Res. 2009; 37:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Suess B., Hanson S., Berens C., Fink B., Schroeder R., Hillen W.. Conditional gene expression by controlling translation with tetracycline-binding aptamers. Nucleic Acids Res. 2003; 31:1853–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Suess B., Weigand J.E.. Aptamers as artificial gene regulation elements. Methods Mol. Biol. 2009; 535:201–208. [DOI] [PubMed] [Google Scholar]
- 35. Levenshtein V.I. Binary codes capable of correcting deletions, insertions, and reversals. Sov. Phys. Dokl. 1966; 10:707. [Google Scholar]
- 36. Lorenz R., Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Andronescu M., Condon A., Hoos H.H., Mathews D.H., Murphy K.P.. Computational approaches for RNA energy parameter estimation. RNA. 2010; 16:2304–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Buß O., Jager S., Dold S.-M., Zimmermann S., Hamacher K., Schmitz K., Rudat J.. Statistical evaluation of HTS assays for enzymatic hydrolysis of β-keto esters. PLoS ONE. 2016; 11:e0146104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rio D.C. RNA: A Laboratory Manual. 2011; CSH Press. [Google Scholar]
- 40. Seetharaman S., Zivarts M., Sudarsan N., Breaker R.R.. Immobilized RNA switches for the analysis of complex chemical and biological mixtures. Nat. Biotechnol. 2001; 19:336–341. [DOI] [PubMed] [Google Scholar]
- 41. Regulski E.E., Breaker R.R.. In-line probing analysis of riboswitches. Methods Mol. Biol. 2008; 419:53–67. [DOI] [PubMed] [Google Scholar]
- 42. Pascault J.-P., Williams R.J.J.. Epoxy Polymers. 2009; John Wiley & Sons. [Google Scholar]
- 43. Davis J.H., Szostak J.W.. Isolation of high-affinity GTP aptamers from partially structured RNA libraries. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:11616–11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nutiu R., Li Y.. In vitro selection of structure-switching signaling aptamers. Angew. Chem. Int. Ed. Engl. 2005; 44:1061–1065. [DOI] [PubMed] [Google Scholar]
- 45. Uhlenbeck O.C. Tetraloops and RNA folding. Nature. 1990; 346:613–614. [DOI] [PubMed] [Google Scholar]
- 46. Hunsicker A., Steber M., Mayer G., Meitert J., Klotzsche M., Blind M., Hillen W., Berens C., Suess B.. An RNA aptamer that induces transcription. Chem. Biol. 2009; 16:173–180. [DOI] [PubMed] [Google Scholar]
- 47. Nguyen Quang N., Perret G., Ducongé F.. Applications of high-throughput sequencing for in vitro selection and characterization of aptamers. Pharmaceuticals (Basel). 2016; 9:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Culler S.J., Hoff K.G., Smolke C.D.. Reprogramming cellular behavior with RNA controllers responsive to endogenous proteins. Science. 2010; 330:1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wieland M., Fussenegger M.. Ligand-dependent regulatory RNA parts for synthetic biology in eukaryotes. Curr. Opin. Biotechnol. 2010; 21:760–765. [DOI] [PubMed] [Google Scholar]
- 50. Zuker M., Stiegler P.. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981; 9:133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Förster U., Weigand J.E., Trojanowski P., Suess B., Wachtveitl J.. Conformational dynamics of the tetracycline-binding aptamer. Nucleic Acids Res. 2012; 40:1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Singh A., Ward O.P.. Biodegradation and Bioremediation XVII. 2014; Springer Science & Business Media. [Google Scholar]
- 53. Liao X., Li B., Zou R., Dai Y., Xie S., Yuan B.. Biodegradation of antibiotic ciprofloxacin: pathways, influential factors, and bacterial community structure. Environ. Sci. Pollut. Res. 2016; 23:7911–7918. [DOI] [PubMed] [Google Scholar]
- 54. Webster T.D., Dickson R.C.. Direct selection of Saccharomyces cerevisiae resistant to the antibiotic G418 following transformation with a DNA vector carrying the kanamycin-resistance gene of Tn903. Gene. 1983; 26:243–252. [DOI] [PubMed] [Google Scholar]
- 55. Lacroute F. Regulation of pyrimidine biosynthesis in Saccharomyces cerevisiae. J. Bacteriol. 1968; 95:824–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kemmerer K., Weigand J.E.. Hypoxia reduces MAX expression in endothelial cells by unproductive splicing. FEBS Lett. 2014; 588:4784–4790. [DOI] [PubMed] [Google Scholar]
- 57. McCarthy J.E. Posttranscriptional control of gene expression in yeast. Microbiol. Mol. Biol. Rev. 1998; 62:1492–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Babendure J.R., Babendure J.L., Ding J.-H., Tsien R.Y.. Control of mammalian translation by mRNA structure near caps. RNA. 2006; 12:851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bottaro S., Lindorff-Larsen K.. Mapping the universe of RNA tetraloop folds. Biophys. J. 2017; 113:257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.