

Ribosomal RNA sequence variants are conserved, exhibit tissue-specific expression, and are found in actively translating ribosomes.
Abstract
The ribosome, the integration point for protein synthesis in the cell, is conventionally considered a homogeneous molecular assembly that only passively contributes to gene expression. Yet, epigenetic features of the ribosomal DNA (rDNA) operon and changes in the ribosome’s molecular composition have been associated with disease phenotypes, suggesting that the ribosome itself may possess inherent regulatory capacity. Analyzing whole-genome sequencing data from the 1000 Genomes Project and the Mouse Genomes Project, we find that rDNA copy number varies widely across individuals, and we identify pervasive intra- and interindividual nucleotide variation in the 5S, 5.8S, 18S, and 28S ribosomal RNA (rRNA) genes of both human and mouse. Conserved rRNA sequence heterogeneities map to functional centers of the assembled ribosome, variant rRNA alleles exhibit tissue-specific expression, and ribosomes bearing variant rRNA alleles are present in the actively translating ribosome pool. These findings provide a critical framework for exploring the possibility that the expression of genomically encoded variant rRNA alleles gives rise to physically and functionally heterogeneous ribosomes that contribute to mammalian physiology and human disease.
INTRODUCTION
Ribosomopathies are diseases arising from aberrations in the assembly, composition, or function of the ribosome, the two-subunit RNA-protein complex responsible for cellular protein synthesis (1). Varied and tissue-specific disease phenotypes associated with the perturbation of ribosomal proteins as well as posttranscriptional and posttranslational modifications of ribosomal components (1, 2) have renewed interest in the hypothesis that functionally distinct ribosome subpopulations may exist in the cell (3–5), often referred to as specialized ribosomes (6). While physical heterogeneities in the ribosome arising from differences in the sequences and stoichiometries of ribosomal and ribosome-associated proteins have recently drawn significant attention (7–9), the existence and contribution of sequence variation in ribosomal RNA (rRNA) as a potential source of ribosome heterogeneity has received less consideration.
rRNA serves as the binding site for ribosomal proteins within the assembled ribosome and contributes to central aspects of the translation mechanism (10). rRNA also contributes to the binding of the myriad extra-ribosomal factors required to synthesize proteins from messenger RNA (mRNA) and ribosome-associated proteins, the “ribo-interactome,” implicated in gene expression (11). Sequence variation in rRNA may therefore alter the structure and function of the ribosome as well as its dynamic interplay with components of the cellular milieu.
Little is presently known about the extent of genetically encoded sequence variation in rRNA and whether such heterogeneities contribute to the ribosome’s potential capacity to exhibit specialized functions or ribosomopathies. Recent studies have, however, suggested a potentially critical role for rRNA sequence variation in gene expression. For example, nutritional stress during pregnancy affects the epigenetic status of a subset of ribosomal DNA (rDNA) operons delineated by a variant in the rDNA promoter sequence (12). Variant rRNA alleles have also been hypothesized to play a role in long-term memory formation (13). Furthermore, somatic genome rearrangements of rDNA are observed in >50% of adult solid tumors (14), raising the possibility that rRNA sequence and copy number variation contribute to cancer (15).
Human and mouse genomes encode hundreds of copies of the rDNA operon, which are arranged in tandem arrays on five chromosomes (13, 14, 15, 21, and 22 in human; 12, 15, 17, 18, and 19 in mouse) (Fig. 1A) (16, 17). Each rDNA operon encodes a 47S pre-rRNA that is posttranscriptionally processed into the 18S rRNA of the small ribosomal subunit and the 5.8S and 28S rRNAs of the large ribosomal subunit. Human and mouse also have tens to hundreds of copies of 5S rRNA, a third rRNA component of the large subunit, on chromosomes 1 and 8, respectively (Fig. 1A) (18). In humans, sequence variations in the rDNA promoter and externally transcribed spacer (ETS) regions are associated with tissue-specific transcriptional activities (19, 20). In mouse, enhancer elements in the rDNA intergenic spacers (IGSs) determine heterochromatin formation and the silencing of distinct rDNA subtypes, contributing to cell cycle–dependent expression patterns and cellular differentiation (21, 22). Evidence of altered rDNA operon expression patterns in the life cycles of both bacteria and parasites (5) argues that these control mechanisms may be evolutionarily conserved.
Fig. 1. rDNA operons in the human genome.
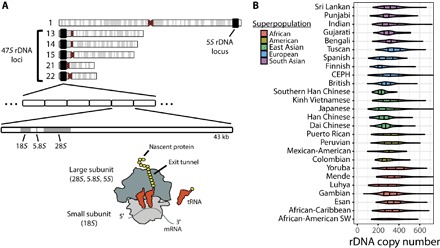
(A) Chromosomal locations of rDNA operons and organization of the rRNA genes within the 47S rDNA operon in the human genome. Together with the 5S rRNA, the 18S, 5.8S, and 28S rRNAs form the RNA core of the ribosome. tRNA, transfer RNA. (B) Per-individual rDNA copy number estimation in humans, grouped by population.
The notion that ribosomes are homogeneous and consequently passive players in gene expression (1, 8, 23) stems in part from early restriction digest and nuclease protection studies demonstrating relatively low levels of rDNA sequence variation (19, 24–26). Contemporary efforts to discover rDNA sequence variation with high-throughput sequencing technologies are challenged by the absence of rDNA sequences from mammalian reference genome assemblies (27) and the inherent difficulty of variant discovery and sequence assembly in highly repetitive regions (28). To begin to understand the role of rRNA sequence variation in animal physiology and gene regulation, we have developed rDNA copy number estimation and rRNA variant discovery strategies that we use to analyze all 2546 human individuals from the 1000 Genomes Project (29) and 32 common mouse laboratory strains from the Mouse Genomes Project (30). In human genomes, we find rDNA copy number to vary by nearly two orders of magnitude. We also observe rDNA operons to encode thousands of sequence variants in the rRNA components of the assembled ribosome. As expected for an inherited trait under adaptive selection, hundreds of these variants are population-stratified. Our analyses further reveal that variants are evolutionarily conserved between mouse and human and map to functional centers of the ribosome, including defined interaction sites with translation factors. rDNA variants are also shown to be present in actively translating ribosomes and to exhibit tissue-specific expression. These findings reveal that the expression of rRNAs of distinct sequences gives rise to an extensive landscape of inherited intraindividual ribosome heterogeneity in mammals, suggesting that rRNA alleles may template alterations in ribosome composition or affect ribosome function in a manner that influences gene expression and cellular physiology.
RESULTS
Identifying reads generated from rRNA genes
Modern analyses of genome variation based on high-throughput sequencing data, including such comprehensive studies as the 1000 Genomes Project (29) and the Mouse Genomes Project (30), systematically exclude rDNA from consideration because of its absence from reference genome assemblies. We therefore developed a bioinformatics strategy designed to (i) identify reads from rRNA genes in whole-genome sequencing (WGS) data; (ii) reliably estimate rDNA copy number; and (iii) detect sequence variation while controlling for errors, systematic bias, mapping artifacts inherent to short-read data analysis, and homology to regions of the genome outside of rDNA (fig. S1 and Materials and Methods).
Rationalizing that the full diversity of rRNA variants is best captured by actively translating ribosomes, we performed RNA sequencing (RNA-seq) on polysomes isolated from proliferating mouse and human cells to use as bait (Materials and Methods). Polysomes represent the cellular fraction of ribosomes actively engaged in translating single mRNAs (31–34) and are physically separated from the individual ribosomal subunits and precursors of ribosome biogenesis by sucrose gradient fractionation (34). Candidate WGS reads from rRNA regions were then extracted by a computational hybridization approach designed to identify those reads from mouse and human with homology to the RNA-seq reads from translating ribosomes or the corresponding “prototype” 5S, 5.8S, 18S, and 28S rRNA sequences (fig. S1, A and B, and Materials and Methods). Reads that mapped with fewer errors to the assembled reference genome were discarded to minimize false-positive calls arising from reads generated outside of rDNA operons but incorrectly mapped to the rDNA prototypes, such as those generated from regions with homology to rDNA outside of the known, unassembled clusters of rDNA (fig. S1, C and D, and Materials and Methods) (35).
rDNA copy number varies widely across individuals in human and mouse
Experimental estimates of rDNA copy number derived from polymerase chain reaction amplicons from 27 human tissue samples and computational analyses of 168 individuals suggest that humans have between 200 and 600 rDNA copies (17, 35). Because rDNA regions have been reported to have >10% meiotic rearrangement frequency (17) and to be hotspots for genome rearrangement (14), we sought to examine the full extent of human rDNA copy number diversity by assessing all 2546 human genomes from the 1000 Genomes Project. The approach that we used accounts for biases in read depth in short-read data, where regions exhibiting high GC content, such as rRNA, are systematically underrepresented (36).
To estimate rDNA copy number in a single individual, we developed a statistically rigorous method that accounts for sample-specific GC content biases that affect read coverage in short-read sequencing data (fig. S1, E and F, and Materials and Methods) (36). This approach was validated by (i) accurate estimation of 50,000 randomly sampled internal control regions of the genome of known copy number, (ii) the high correlation (r = 0.99) observed between rDNA copy number estimates obtained from 18S, 5.8S, and 28S rDNA genes, and (iii) verification that rDNA copy number estimates were not correlated with GC content (fig. S2, A and B, and Materials and Methods). Applying this method to the analysis of the 2546 sequenced human genomes, we estimate rDNA copy number in humans to vary over a range spanning two orders of magnitude, from 61 to 1590 copies per individual (mean, 315; SD, 104; median, 301). These findings are in strong agreement with early experimental estimates of 320 rDNA operons per individual (37) and with recent results obtained for subpopulations (17, 35).
Allele frequencies (AFs) that differ by population, termed population stratification, may indicate genetic loci under selection. Using the Vst statistic (38), an analog of the fixation index that quantifies multicopy number population stratification, we found that 19 of 26 populations represented were differentiated by rDNA copy number (Vst > 0.2, max Vst = 0.40) (Fig. 1B and table S1). Consistent with rDNA existing only on autosomes, rDNA copy number did not stratify by sex (fig. S2C). However, rDNA copy number was not stratified by continental populations (max Vst = 0.09) (fig. S2D), suggesting that rDNA copy number is dynamic, changing on a relatively rapid scale such that differentiation is only apparent between smaller, local populations.
In line with previous results (15, 18) and as expected from the relationship between rDNA copy number and genome size (39), we found mouse to have fewer rDNA copies than human on average, varying over 5.7-fold across 32 strains, from 74 to 419 rDNA copies per strain (mean, 222; SD, 74; median, 218) (fig. S2E). The wide range of rDNA copy numbers observed in both mouse and human, representing differences of approximately 66 million nucleotides (nt) of rDNA between individuals at the extremes, together with the observed population stratification, is consistent with high rates of meiotic rDNA recombination (14, 17).
rDNA sequence variation can be detected from WGS data
We next sought to determine the extent of rDNA sequence variation from WGS data. To minimize spurious variant calls arising from instrument error, the base qualities of the extracted reads were empirically recalibrated (fig. S1G and Materials and Methods) (40, 41). To prevent false-positive variant calls arising from ambiguities in read alignments, especially at read termini (42), reads were locally realigned around putative indels, and the base qualities of the 8 nt at each read end were reduced to quality score zero (Q0) to preclude read termini from analysis and thus the possibility of misalignment contributing to variant calls (fig. S1H). We then used LoFreq (43), a base quality–aware algorithm designed for conservative detection of rare sequence variants that implements a strand bias test with Bonferroni correction, to call statistically significant variants in rDNA (fig. S1I). As a positive control, this variant discovery strategy detected the known rDNA sequence variants using WGS data from Escherichia coli K-12 with high specificity and sensitivity, where the estimated AF was highly correlated with the true AF (r > 0.98, P < 1 × 10−6) (Materials and Methods).
Humans exhibit pervasive inter- and intraindividual sequence variation in the rRNA genes
Applying this variant discovery strategy to WGS data from the 1000 Genomes Project, we detected 1790 variant alleles at 1662 positions of the 7184 nt (23%) in human 5S, 5.8S, 18S, and 28S rDNA (Table 1). Measuring the intraindividual AF of each rRNA sequence variant, that is, the fraction of operons in an individual’s genome containing a specific rRNA sequence variant, we identified 497 variants that occur in at least one individual with intraindividual AF > 20% (Fig. 2 and fig. S3). Single individuals were found to encode 32 high-frequency variants (intraindividual AF > 20%) on average. Across individuals, 128 positions of the 5S, 18S, and 28S were found to be multiallelic. The vast majority of variants (1739 of 1790) were single-nucleotide polymorphisms (SNPs), which were overrepresented at adenine (A) (P = 2.4 × 10−8) and cytosine (C) (P = 1.5 × 10−5) positions and underrepresented at guanine (G) (P = 2.2 × 10−19) positions. In line with reliable SNP identification (41), the mean transition/transversion (Ti/Tv) ratio per individual was 1.96 (fig. S4A), which is substantially higher than the Ti/Tv ratio for SNPs observed in exomes, introns, and intergenic regions of similarly extreme GC content (44, 45). Accounting for low-complexity regions of the rRNA for which read depth was extremely low because of GC content bias, variant positions were overrepresented in 18S rRNA (P = 2.7 × 10−14) and underrepresented in 28S rRNA (P = 9.5 × 10−17) both per individual and across populations. Consistent with previous results (25, 26), the 51 distinct indel variants, ranging in length from 1- to 12-nt insertions or 1- to 9-nt deletions, largely consisted of GC-rich repeat sequences (fig. S4B and table S2). These findings reveal pervasive inter- and intraindividual rRNA sequence variation in the human population.
Table 1. rDNA variant allele counts and variant position counts for each rRNA component of the ribosome.
| SNP | Deletion | Insertion | Total | |||||
| Variant alleles | Positions | Variant alleles | Positions | Variant alleles | Positions | Variant alleles | Positions | |
| Rn18s | 626 | 579 | 2 | 2 | 2 | 2 | 630 | 583 |
| Rn28s | 1023 | 954 | 13 | 12 | 33 | 27 | 1069 | 993 |
| Rn5.8s | 50 | 48 | 0 | 0 | 0 | 0 | 50 | 48 |
| Rn5s | 40 | 37 | 1 | 1 | 0 | 0 | 41 | 38 |
| Total | 1739 | 1618 | 16 | 15 | 35 | 29 | 1790 | 1662 |
Fig. 2. High-frequency genomic rRNA variants detected in human.
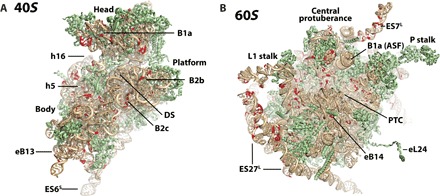
Subunit interface views of the (A) small-subunit (40S) and (B) large-subunit (60S) tertiary structures of the human ribosome showing positions (red) with variant alleles exhibiting intraindividual AF > 20% in any individual analyzed. Structural features within the 40S and 60S subunits are indicated. PTC, peptidyl transferase center.
Intraindividual rRNA variant AFs ranged over a broad spectrum, from a single copy to all rDNA copies in an individual’s genome. At one extreme, we observed 19 variants occurring in >50% of humans with a maximum intraindividual AF of <5% (fig. S4C). That is, these variants were found in more than half of individuals tested, but within any single individual, at most 5% of the individual’s rDNA operons contained the variant. For instance, the G928A 18S rRNA variant occurred with a maximum intraindividual AF of 3.7%. This residue occurs in helix 22 (h22) of 18S rRNA at a known contact point with the C subunit (Gly344) of initiation factor eIF3 (46). Similarly, the G1233A 18S rRNA variant, which occurs with a maximum intraindividual AF of 4.6%, is found in h30, a component of the peptidyl-tRNA binding domain (47). These findings suggest that low intraindividual AFs at these functional positions may be evolutionarily advantageous. At the other extreme, 62 rRNA variants were found to occur with a maximum intraindividual AF of >75% (fig. S4C), meaning that at least 75% of the rDNA operons within at least one individual contained the variant. Such high intraindividual AFs suggest that most of the expressed ribosomes in these respective individuals likely contain these variants. Further, 22 variants in the 18S and 28S rRNA were found to occur at a minimum intraindividual AF of >10% in more than 500 individuals (fig. S4D).
Posttranscriptionally modified rRNA positions exhibit intraindividual heterogeneity
Eukaryotic rRNA is posttranscriptionally modified in functional centers of the ribosome (48–50). Of the 1790 rDNA variants identified in the 5S, 5.8S, 18S, and 28S rRNA, 61 localized to 59 of the 256 (23%) positions known to be modified (table S3) (48–51). Variants at these positions were identified with high intraindividual AFs. For instance, the C462T variant in 18S rRNA, a posttranscriptionally modified position that is a known site of interaction with eukaryotic release factor 1 (eRF1) during translation termination and ribosome release (52), is found with intraindividual AF up to 61%. The A1183G variant in 18S rRNA, found with intraindividual AF as high as 27%, occurs at a position located in intersubunit bridge eB14 near the decoding center of the ribosome that is 2′-O-methylated by the small nucleolar RNA snR41 (50, 53). Twenty-nine positions reported as sites of pseudouridine modification exhibited SNP variants with intraindividual AF as high as 41%, suggesting that a substantial population of ribosomes in human cells lack this functionally important modification (50). These findings collectively suggest that rRNA modifications in these individuals may be absent or made on nucleotides of distinct identities, perhaps implying alternative ribosome biogenesis machinery.
Variants occur in intersubunit bridge elements
The formation, breakage, and dynamic remodeling of the intersubunit bridges that establish the binding interface linking the small and large ribosomal subunits influence nearly every aspect of the translation mechanism (33, 54, 55). Lending support to the notion that rRNA variants may affect ribosome function, rRNA variants with high intraindividual AFs (>20%) localized to intersubunit bridge elements or to the binding sites of ribosomal proteins that mediate them (B1a, B2b, B2c, B2e, B4, B7b, B7c, eB11, eB14, and eB8b). In contrast, no variants were observed in intersubunit bridge B2a, B5, or B5b (table S4). Notably, variants were found on both sides of intersubunit bridges B1a, B2c, B2e, and eB13. Intersubunit bridge B1a, often referred to as the A-site finger (ASF), extends from the 28S rRNA across the tRNA binding sites to the small-subunit head domain and is implicated in peptidyl-tRNA positioning within the mammalian aminoacyl (A) site and substrate translocation (56, 57). Bridge B2c contributes to the central intersubunit bridge structure, bridge B2, that links the site of peptide bond formation within the large subunit to the site responsible for aminoacyl-tRNA decoding in the small-subunit decoding center (54). Bridge eB13, located at the leading edge of the translating ribosome and established through ribosomal protein eL24, extends from the base of the large subunit to ribosomal protein eS6, a small-subunit protein implicated in mammalian target of rapamycin (mTOR)–dependent regulation of the translation mechanism (58, 59). Together, we speculate that variants spanning bridge elements may affect the process of translation initiation by altering intersubunit bridge formation or the dynamic remodeling of intersubunit bridge elements that accompany the mechanism of protein synthesis (33, 54, 55). Variants on both sides of an intersubunit bridge may also template a mechanism for coordinating the pairing of specific 40S and 60S subunits composed of distinct rRNA alleles in the cellular milieu.
rRNA variants stratify by ancestry
SNP and copy number variations outside of rDNA regions are known to stratify by ancestry, which can be indicative of selective pressures (29, 60). Although the genes encoding rRNA represent only ~0.1% of the human genome, they are a known hotspot for genomic rearrangements (14, 17). We therefore sought to probe the relationship between intraindividual rRNA variant AF and population. Strikingly, we found the intraindividual AF of 327 rRNA variants (18% of the total) to stratify by population (Vst > 0.2) (table S5). Population-stratified variants spanned all four rRNA genes. In total, 44 variants exhibited signatures of extreme population stratification (Vst > 0.5), such as the A2538G 28S variant (max Vst = 0.56) (Fig. 3A), of which 32 were from the 18S rRNA and 12 were from the 28S rRNA. Only four rRNA variants were stratified by continental groups (max Vst = 0.30).
Fig. 3. rRNA variants with population-stratified intraindividual AF.
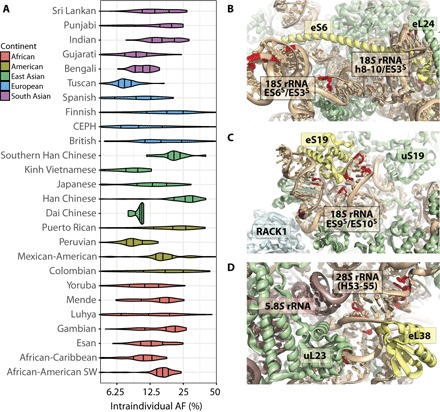
(A) Intraindividual AF of 28S rRNA sequence variant A2538G, by population (max Vst = 0.56). X axis has log2 scaling. (B to D) Tertiary structures of the ribosome showing strongly population-stratified variants (Vst > 0.5) with high intraindividual AF (>20%) in any individual analyzed that are located (B) in expansion segments (ES6S and ES3) in the surface near ribosomal protein eS6, (C) proximal to the binding site for ribosomal protein eS19, and (D) proximal to the binding site for ribosomal protein eL38. Ribosome tertiary structures [Protein Data Bank (PDB) ID 4V6X (67)] show rRNA (tan), ribosomal proteins (green), and key structural landmarks.
In total, 239 of all 325 possible population pairs (74%) stratified at least one rRNA variant. Any two populations stratified 30 to 239 variants. The number of stratified variants across populations was not unusually distributed by continental group (Kruskal-Wallis test, P = 0.11) (fig. S5). By contrast, population studies of variation outside of rDNA regions have found African populations to exhibit more variation compared to other populations because of higher genetic diversity among Africans and the derivation of the reference genome from European lineage samples (29, 60). Consistent with a >10% recombination rate per rDNA cluster per meiotic cycle (17), the observed disparity in stratification at the population and continental levels suggests that genetic variation in rDNA loci occurs at relatively high rates such that isolated populations quickly diverge with respect to rRNA sequence.
Notably, 24 of the most highly population-stratified variants occurring with intraindividual AF >20% in at least one individual cluster in three distinct regions of the ribosome. Seven population-stratified variants cluster within the rRNA region comprising the base of the small-subunit body domain that includes expansion segments (ESs) ES6S and ES3 as well as the surface surrounding ribosomal protein eS6, which is implicated in myriad translational dysfunctions in cancer (Fig. 3B) (61). Eight population-stratified variants cluster on the apical, solvent-exposed surface of the small-subunit head domain, immediately proximal to ribosomal protein eS19, which is causally linked to Diamond-Blackfan anemia (Fig. 3C) (62, 63). Finally, seven population-stratified variants cluster in immediate proximity of the ribosomal protein eL38 binding site, which is implicated in the translation of Hox genes and body pattern formation during development (Fig. 3D) (64). These observations support the notion that rRNA sequence variants may contribute to the regulation of gene expression in human physiology and disease.
rRNA variant AFs are correlated
The potential for rRNA variants to affect ribosome function may be greater if they occur on the same rRNA or in the same ribosome. Unambiguous determination of the occurrence of rRNA variants on the same operon is prohibited by the large number of highly homologous rDNA operons in the genome and the short insert size of the paired-end reads (28). We therefore assessed correlation between intraindividual rRNA variant AFs to provide a first approximation of whether variants may be genetically linked. To do so, we calculated all pairwise intraindividual AF correlations for variants occurring in >75% of 1887 unrelated individuals. By this method, we found 118 highly correlated (|r| > 0.75) variants in 18S rRNA that were separated by less than 500 nt, which formed multiple nexus of correlated variants within each 18S rRNA subdomain (fig. S6A). Another 16 variants were highly correlated (|r| > 0.75) and separated by more than 500 nt. For instance, two highly correlated variant clusters link a ~200-nt region near h21 in the small-subunit body domain to a ~150-nt region spanning h34 through h40 in the small-subunit head domain, which are distant in both primary and tertiary structure (fig. S6B). Correlations of this kind may exist if variants occurred within the same ancestral rDNA operon, which has since experienced extensive duplication and deletion rearrangements through meiotic recombination, or if direct or indirect functional linkages exist between these distal regions of the ribosome.
rRNA variants are evolutionarily conserved
Human and mouse reference rRNA sequences are highly homologous (percent identity: 5S = 100%, 5.8S = 99%, 18S = 99%, 28S = 85%). Applying our variant discovery strategy to the 32 strains sequenced by the Mouse Genomes Project, we detected 285 distinct variant alleles at 276 positions of the rRNA genes in mouse rDNA (table S6). Individual strains had between 39 and 191 rRNA variants (mean, 125; SD, 31; median, 131). Given the relatively small number of available mouse strains and their limited depth of coverage, these results likely represent a lower bound for rDNA heterogeneity.
Eighty of the 276 rRNA variant positions identified in mouse (29%) were also variant in human. Of these, 48 had the same variant alleles, the majority of which (45 of 48; ~94%) also had the same prototype allele (table S7). Many of these 45 variants occurred at positions of known functional relevance, including interaction sites between rRNA and extraribosomal factors involved in a wide range of translation-related processes, including mRNA quality control and degradation (Dom34, Hbs1) (65), ribosome-associated protein quality control (nuclear export mediator factor NEMF) (66), translation elongation (eEF2) (67), translation initiation (eIF5B, eIF1A/eIF1) (47, 68), ribosome biogenesis (snR87, HBII-10, U16, and U103) (69), translation termination (eRF1) (70), and selenoprotein synthesis (SBP2) (71). For instance, the C543T variant in h16 of 18S rRNA, which has an intraindividual AF as high as 22% in humans, is thought to directly contact the mRNA helicase DHX29 (46), an extraribosomal factor implicated in translation initiation (Fig. 4A). The G480A variant in h5 of 18S rRNA, which has an intraindividual AF as high as 65% in humans, resides at a position thought to contribute to guanosine triphosphate (GTP) hydrolysis during tRNA selection (72), and thus the rate and fidelity of protein synthesis (Fig. 4B). The G1764A variant of H38 of the 28S rRNA occurs in the ASF (intersubunit bridge B1a), which is implicated in peptidyl-tRNA positioning and substrate translocation (Fig. 4C) (56, 57). These findings suggest that rDNA sequence variants occurring in functionally important regions of the ribosome are conserved across species.
Fig. 4. Evolutionarily conserved rRNA variants in functionally important centers of the human ribosome.
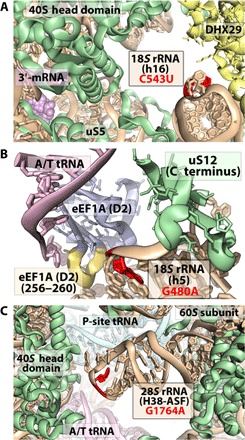
(A) 18S C543U variant (red) of helix h16 contacts DHX29 (yellow), a component of translation initiation. (B) 18S G480A variant (red) of helix h5 occurs near contact points with residues 256 to 260 within domain 2 (yellow) of eEF1A (blue). (C) 28S G1764A variant (red) of helix H38 in the central protuberance. The structural models shown are based on EMD-3056-3058 (46) and PDB IDs 5LZS (87) and 4V6X (67), respectively, where eIF3 and DHX29 from EMD-3056-3058 were modeled onto PDB ID 5LVS.
rRNA variants are differentially expressed between organs
To investigate whether the identified rDNA variants are expressed, we harvested total RNA from four major organs—brain, lung, liver, and ovary—representing all three germ layers from three 8-week-old BALB/cJ littermates and performed RNA-seq without rRNA depletion (Materials and Methods). Among the four tissue types examined, 70 distinct rRNA variants were reproducibly detected: 19 in the 18S, 1 in the 5.8S, and 50 in the 28S. Consistent with a direct relationship between rDNA variant copy number and expression level, 31 of these variants (44%) were also detected in the rDNA of the BALB/cJ mouse strain. For these variants, intraindividual genomic AF and rRNA expression level were highly correlated (r = 0.97, P = 1.8 × 10−19). Hence, consistent with their high intraindividual AF, sequence variants present in rRNA genes are actively expressed. Another 14 expressed rRNA variants were nonoverlapping with mouse rDNA variants and instead coincided with positions of known posttranscriptional modification (48–51). The remaining 25 variants were neither detected in BALB/cJ rDNA nor found to coincide with positions of known modification. This subset may reflect undetected positions of rDNA variation or posttranscriptionally modified positions in rRNA that have yet to be annotated.
Tissue-specific histone marks on rDNA (22, 73), the methylation of nonexpressed rDNA regions (12, 20), and tissue-specific expression of variants in the ETS region of rDNA (24) are well documented. This precedent suggests that rDNA gene variants, and thus ribosomes assembled from distinct rRNA sequences, may exhibit tissue-specific expression. Consistent with such a model, we observed that 26 of the 70 expressed rRNA variants (37%) were differentially expressed in at least one tissue (Fig. 5 and Table 2). Each pair of tissues was distinguished by 6 to 17 differentially expressed rRNA variants. These differentially expressed rRNA variants consisted of substitutions, deletions, and insertions that localized to regions of functional and structural importance, including intersubunit bridge B4 (fig. S7A) and the binding sites for eIF3 (ES7) (fig. S7B), deacylated tRNA (fig. S7C), ribosomal proteins eL33 and eL44, as well as ribosome biogenesis factors nucleolin and Arx1 (74–76). Of the 26 differentially expressed rRNA variants identified, 15 (58%) were detected in the BALB/cJ rDNA and another 4 coincided with known positions of rRNA modification: positions 1032 and 1248 of the 18S and positions 1136 and 4083 of the 28S (50, 51). Because reverse transcriptase error profiles are dependent on modification type (77) and multiple types of modification have been observed at 18S positions 1032 and 1248 and 28S position 1136 (49–51), these results suggest that modification profiles at these positions differ across tissues, either by overall modification levels or by the distribution of modification types at the same position.
Fig. 5. Tissue-specific expression of rRNA variants.
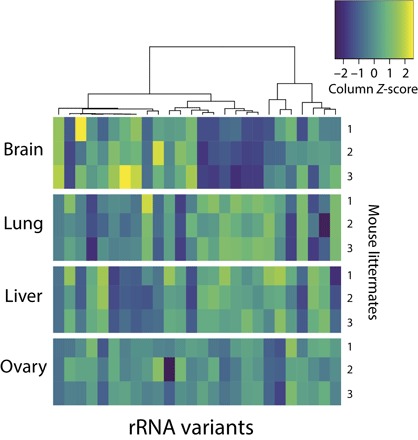
rRNA variant expression heat map and hierarchical clustering of the 26 variants detected to be differentially expressed among pairs of tissues. Each row represents a biological replicate. Rows are grouped by tissue source (three biological replicates, that is, rows, per tissue source). Each column represents an rRNA variant. Expression is normalized per rRNA variant (that is, by column) across all replicates and tissues (that is, 12 samples per each column). For example, the rRNA variant represented by the leftmost column has higher relative expression in brain, whereas the variant represented by the rightmost column has the lowest relative expression in liver.
Table 2. The top 10 differentially expressed rRNA variants between mouse organs with the largest absolute changes in estimated number of ribosomes.
| rRNA | Position | Prototype | Variant | Tissue A | Tissue B |
Expression level A |
Expression level B |
Expression difference |
Difference (millions of ribosomes) |
Region annotation |
Detected in rDNA |
| 28S | 2234 | G | GT | Lung | Ovary | 0.77 | 0.47 | 0.30 | 3.01 | ES15 (H45) | Yes |
| 28S | 755 | C | CGG | Brain | Lung | 0.11 | 0.31 | 0.20 | 1.99 | ES7B (H25) | No |
| 28S | 3359 | A | G | Brain | Ovary | 0.17 | 0.01 | 0.16 | 1.6 | ES27B (H63) | No |
| 28S | 3359 | A | AC | Brain | Lung | 0.11 | 0.26 | 0.15 | 1.51 | ES27B (H63) | Yes |
| 28S | 984 | C | T | Brain | Lung | 0.20 | 0.33 | 0.13 | 1.27 | ES7C/D (H25) |
Yes |
| 28S | 3410 | CGT | C | Lung | Ovary | 0.47 | 0.58 | 0.11 | 1.1 | ES27B (H63) | Yes |
| 28S | 4967 | A | G | Brain | Lung | 0.21 | 0.11 | 0.11 | 1.09 | ES41 (H99) | Yes |
| 28S | 3579 | C | T | Lung | Liver | 0.77 | 0.87 | 0.10 | 1.02 | ES27 (H63) | Yes |
| 28S | 652 | G | GC | Lung | Liver | 0.70 | 0.79 | 0.09 | 0.88 | ES27A (H25) | Yes |
| 28S | 2657 | G | C | Brain | Lung | 0.07 | 0.15 | 0.09 | 0.87 | H58 | Yes |
In absolute terms, these findings show that a substantial fraction of the total pool of expressed ribosomes differs between organs (Table 2). For instance, given the estimate of 10 million ribosomes per mammalian cell (78), there are more than 3 million more ribosomes (30% of the total) bearing a U insertion in 28S rRNA at position 2234 in lung compared to ovary. Variants with the largest estimated differences in the absolute number of ribosomes between tissues were found in eukaryote-specific ESs, whose functional relevance has yet to be thoroughly elucidated (Table 2). Half of the 10 variants showing the largest tissue-specific expression differences were found in ES27 (H63) located on the back side of the 60S subunit. This highly dynamic ES plays an important role in stabilizing the ribosome-associated complex and coordinating extraribosomal factors implicated in cotranslational protein folding near the nascent peptide exit tunnel (75, 79). The full repertoire of ES27 contributions to the translation mechanism remains to be determined. Intriguingly, variants in these regions were also found in human genomes at high intraindividual AF, suggesting that their tissue-specific expression is conserved.
rRNA variants are incorporated into actively translating ribosomes
To assess whether expressed rRNA variants assemble into functional ribosomes, we sequenced rRNA from polysomes isolated from mouse mammary epithelial cells, which could be readily grown in sufficient quantities for quantitative polysome analyses (Materials and Methods). Consistent with expectation (31), quantitative mass spectrometry confirmed that these polysome fractions were principally ribosome components and devoid of ribosome biogenesis-related proteins (table S8 and Materials and Methods). Within this subpopulation of the total cellular ribosome pool, we detected 62 rRNA variants, of which 30 were found in the rDNA of at least one of the mouse strains analyzed and another 19 coincided with known positions of rRNA modification. The expression levels of the rRNA variants detected in the actively translating ribosome pool were also found to be highly correlated with intraindividual genomic variant AF (r = 0.96, P = 1.2 × 10−17) (fig. S8 and Materials and Methods). Moreover, 42 of the 70 variants identified as expressed in tissues were detected in actively translating ribosomes, where expression levels were again highly correlated (r = 0.99, P = 2.0 × 10−36). Eight of the 15 differentially expressed rRNA variants observed in both mouse tissues and BALB/cJ rDNA (53%) were also present in the actively translating ribosome pool. The substantial overlap between the variants detected in polysomes with those found in tissues and in genomic DNA, together with the high correlation between genomically encoded and expressed AF, provides compelling evidence that variant rRNAs are present in ribosomes actively engaged in protein synthesis.
To further elucidate the correspondence between rRNA variant levels in expressed versus actively translating ribosomes, we sequenced total RNA isolated from the same mouse epithelial cells (Materials and Methods). We found that 38 of 40 rRNA variants detected in expressed ribosomes were also present in actively translating ribosomes from the same cells, again exhibiting highly correlated expression levels (r = 0.98, P = 3.0 × 10−28). The strong correlations in rRNA variant AF among genomic, expressed, and actively translating ribosomes provide further evidence that genomically encoded rRNA variants are present in actively translating ribosomes at similar levels as their frequency in the genome. These findings support the notion that genomically encoded rRNA variants are assembled into functional ribosomes and have the potential to affect the translation process and consequently gene expression. The observed distinctions in the rRNA sequence composition of actively translating ribosomes may affect translation by affecting dynamic aspects of the protein synthesis mechanism, subcellular localization of the ribosome, or the ribosome’s association with extraribosomal factors, the ribo-interactome (6, 11).
DISCUSSION
rDNA is currently “dark matter” that is missing from contemporary genome assemblies (27). Consequently, the contribution of rRNA heterogeneities to ribosomopathies or functional distinctions in the pool of assembled ribosomes has yet to be explored. The rigorous rDNA copy number estimation and rRNA sequence variant discovery strategies that we describe here reveal pervasive intra- and interindividual rRNA sequence variation in the human genome, with at least 1662 of the 7184 nt of rRNA (23%) exhibiting sequence variation in the global population, and variation in rDNA copy number between individuals that spans nearly two orders of magnitude. We further observe tissue-specific rRNA variant expression and extensive overlap between the discovered rRNA variants and positions of known functional importance in the assembled ribosome, including sites identified as being posttranscriptionally modified (49–51). These findings collectively suggest that tissue-specific expression of distinct rDNA operons may contribute to important, undocumented aspects of cellular physiology.
There are at least two possible mechanisms of differential rDNA operon expression: (i) rDNA operon–specific regulation or (ii) tissue-to-tissue variation in the copy number of subclasses of rDNA operons bearing specific variant alleles. Although differences in the total rDNA copy number between mouse tissues have been reported (15), it is unknown how the copy number of specific subclasses of rDNA operons varies by tissue. Notably, we observe larger variations in tissue-specific rRNA variant expression than expected from rDNA copy variation alone. In this context, we note that the chromatin structure and epigenetic modifications of the rDNA promoter contribute to the specificity of rDNA operon transcription (73, 80, 81). The expression of rDNA is also correlated with nucleotide variants within the IGS region, rDNA promoter, and the 5′ ETS region upstream of the rRNA genes (12, 20, 24). Hence, tissue-specific rRNA variant expression is likely also regulated by rDNA promoter, IGS, and ETS sequence heterogeneities. IGS regions contain variable-length tandem repeats, transposable Alu repeat elements that are highly abundant in the human genome, and simple sequence microsatellites (16) and exhibit substantial structural variation (17, 82). Although these properties complicate the detection of sequence and structural variation in these regions (28), the present findings reveal that knowledge of the full repertoire of complete rDNA operon sequences will be necessary to comprehend the true diversity of these complex elements and the mechanisms by which IGS and ETS regions contribute to the regulation of rDNA operon expression.
The extensive catalog of rRNA variants revealed by the present analysis solidifies and substantially extends reports of rRNA sequence heterogeneity in mammals (19, 25, 26). Despite the conservative nature of the analytic approach applied, we observe that nearly 25% of the functional rRNA sequences can vary. Approximately 7% of the detected variants are more than 20% penetrant in at least one sequenced individual. Given the observed correlation between genomic AF and expression, these data suggest an unanticipated level of rRNA sequence variation in the actively translating ribosome pool.
Our finding that positions of conserved sequence variation map to functional regions of the assembled ribosome, including the binding sites for ribosomal proteins implicated in ribosomopathies, suggests that specific alleles may be maintained for yet unknown reasons. Heterogeneities in rRNA sequence may affect ribosome function by altering the binding of core ribosomal proteins, transient associations of the ribosome with extraribosomal factors, or conformational changes in the ribosome that are important to the mechanism of protein synthesis. For instance, rRNA sequence variants in ribosomal protein binding domains may template the biogenesis of ribosomes with distinct core protein compositions, a feature that has been recently connected to the translation of distinct mRNAs (83). Because heterogeneities of this kind are sufficient to define subclasses of physically distinct ribosomes, we conclude that rDNA copy number and rRNA sequence variation may provide a means to template specialized functions that enable the ribosome to actively participate in determining the cellular proteome (2, 12).
Importantly, the present analysis likely represents a lower bound on the true diversity of rDNA sequences in the mouse and human genomes and thus sequence variation within the assembled ribosome. Read coverage is low in regions of extremely high GC content (36), which applies to rRNA in general and especially for regions of the 28S (>80% GC) for which we observed nearly zero read depth. These regions include those identified as interspecies variable domains that are expressed and present in actively translating ribosomes (19, 25, 26, 84). Such considerations argue for in-depth investigations into the nature and extent of sequence diversity among the multitude of full-length rDNA operons. They also warrant focused investigations into the mechanisms regulating rDNA operon expression, rRNA assembly into functional ribosomes as well as how, and to what extent, such variation contributes to the regulation of gene expression in normal physiology and disease.
MATERIALS AND METHODS
RNA isolation from mouse epithelial cell polysomes
Mouse mammary epithelial cells, NMuMG cells, were obtained from the American Type Culture Collection and were grown in T225 flasks (Corning) in high-glucose (4.5 g/liter) Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% fetal bovine serum (Atlanta Biologicals), 1% penicillin/streptomycin (Gibco), and insulin (10 μg/ml). Human embryonic kidney (HEK) 293T cells were grown in the same manner and medium with the exception of insulin. Both cell lines were routinely checked for mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza). To stabilize polysomes before harvest, cells were treated with 350 μM cycloheximide (Sigma-Aldrich) for 30 min at 37°C. Cells were then released from the surface using 0.05% trypsin-EDTA (Gibco), centrifuged at 750 rpm for 5 min at 4°C in an Allegra X-30R centrifuge (Beckman Coulter), washed in cold 1× phosphate-buffered saline (pH 7.4) (Gibco), pelleted at 10,000 rpm for 1 min at +4°C in a microcentrifuge, and flash-frozen on liquid nitrogen. Frozen cell pellets were thawed on ice and resuspended in lysis buffer (1 g of cells per milliliter of lysis buffer) containing 20 mM tris-HCl (pH 7.5), 5 mM MgCl2, 10 mM KCl, 1 mM dithiothreitol (DTT), 5 mM putrescine, 350 μM cycloheximide (Sigma-Aldrich), RNaseOUT (1:10,000) (Thermo Scientific), and 1× Halt Protease Inhibitor Cocktail (EDTA-free) (Thermo Scientific). Resuspended cells were incubated on ice for 5 min before the addition of NP-40 (0.5% final concentration), sodium deoxycholate (0.5% final concentration), and TURBO DNase (1:100, Invitrogen). Samples were then incubated on a rotator for 20 min at +4°C before the lysate was clarified in a microcentrifuge (14,000 rpm for 15 min at +4°C). The clarified lysate was then loaded onto precooled 10 to 50% sucrose gradients buffered in the same manner as the lysis buffer, but without ribonuclease inhibitors, protease inhibitors, or deoxyribonuclease. Sucrose gradients were spun at 30,000 rpm for 3 hours at +4°C in an SW 32 rotor in an Optima XPN-100 ultracentrifuge (Beckman Coulter). Gradients were then fractionated using a BR-188 density gradient fractionation system (Brandel). Fractions corresponding to polysomes were collected and pelleted at 35,700 rpm for 18 hours at +4°C in a Ti70 rotor in an Optima XPN-100 ultracentrifuge (Beckman Coulter). Polysome pellets were resuspended in buffer before subunits were separated by increasing KCl concentration to 0.5 M and treating with 5 mM puromycin for 30 min on ice and 15 min at 37°C. Separated subunits were laid atop a 37% sucrose cushion containing 20 mM tris-HCl (pH, 7.5), 50 mM KCl, 1.5 mM MgCl2, and 1 mM DTT and spun at 100,000 rpm for 6 hours at +4°C in a TLA-100.3 rotor in an Optima MAX-XP ultracentrifuge (Beckman Coulter). Pelleted subunits were resuspended in buffer, and RNA was extracted using QIAzol Lysis Reagent (Qiagen) and purified using RNeasy (Qiagen).
Mass spectrometry of mouse epithelial cell polysomes
NMuMG polysomes, isolated as described above in biological triplicate, were denatured, reduced, and alkylated followed by proteolytic digestion with LysC (Wako Chemicals) and trypsin (Promega). Approximately 120 fmol of each desalted (85) sample was analyzed by a nano–liquid chromatography–tandem mass spectrometry system (UltiMate 3000 coupled to Q Exactive Plus, Thermo Scientific). Peptides were separated using a 12 cm × 75 μm C18 column (3-μm particles, Nikkyo Technos Co. Ltd.) at a flow rate of 200 nl/min, with a 5 to 40% gradient over 130 min (buffer A, 0.1% formic acid; buffer B, 0.1% formic acid in acetonitrile). Data were quantified and searched against an E. coli database (July 2014) using MaxQuant (version 1.5.3.28) (86). Oxidation of methionine and protein N-terminal acetylation were allowed as variable modifications, cysteine carbamidomethyl was set as a fixed modification, and two missed cleavages were allowed. The “Match between runs” option was enabled, and false discovery rates (FDRs) for proteins and peptides were set to 1%. Protein abundances were expressed as LFQ (label-free quantitation) values.
Total RNA isolation from mouse tissues
All mouse work was performed in accordance with institutional, Institutional Animal Care and Use Committee, and American Association for Laboratory Animal Science guidelines, by the animal protocol 0709-666A. Brain, lung, liver, and ovary tissue was dissected from three 8-week-old BALB/cJ littermates and flash-frozen before storage at −80°C. Tissue (50 to 100 mg) was mechanically lysed using a Mixer Mill 400 (Retsch) in 1.5-ml microcentrifuge tubes containing 1.4-mm ceramic beads (Omni) and Lysis/Binding Buffer (Ambion). Samples were lysed six times for 1 min at 30 Hz with a short incubation on ice between each cycle. The lysate was clarified for 30 s at 10,000 rpm and +4°C in a microcentrifuge, and RNA was extracted from the clarified lysate using the mirVana miRNA Isolation Kit (Ambion). RNA was then treated with TURBO DNase (Invitrogen) and purified using the RNeasy Mini Kit (Qiagen).
RNA sequencing
Before sequencing, the quantity and concentration of all RNA samples were determined using a NanoVue spectrophotometer (GE Healthcare). High RNA integrity was verified using 2100 Bioanalyzer (Agilent). Illumina-compatible libraries were prepared using a modified TruSeq RNA Sample Preparation method (Illumina) that omitted the polyadenylated enrichment step. Sequencing was performed using 100-nt paired-end sequencing on a HiSeq 4000 (Illumina) in the Genomics Resources Core Facility at Weill Cornell Medicine.
References, prototypes, and databases
GenBank IDs of the rRNA prototypes used are NR_003278.3, NR_003279.1, NR_003280.2, and NR_030686.1 (mouse) and NR_003285.2, NR_003286.2, NR_003287.2, and NR_023379.1 (human). GenBank IDs of the rDNA prototypes used were U13369.1 and X12811.1 (human) and BK000964.3 and chromosome 8 region [123538334, 123539354] (mouse). This region of chromosome 8 was chosen as a representative 5S rRNA and operon for mouse, as used previously (18), because no such sequence could be found in GenBank. The whole-genome reference sequence assemblies used were GRCm38 for mouse and GRCh38 for human, downloaded from Ensembl. dbSNP release b147 was used for known variant locations in the reference genome assemblies. Read libraries from the 1000 Genomes Project and Mouse Genomes Project were downloaded from the National Center for Biotechnology Information Sequence Read Archive using the sra-toolkit.
Identification of putatively unannotated rRNA in the reference genome
The rRNA components of the rDNA prototypes were aligned to the reference genome using BLAST. For the 18S and 28S, all hits to the assembled chromosomes with sequence identity > 75% and length > 75% with respect to the respective prototype were considered to be unannotated rRNA within the reference genome. The same criteria were used for the 5.8S and 5S, except with a length threshold of 85% of the respective prototype.
Recalibration of base qualities
Reads were mapped to the whole-genome reference with BWA (Burrows-Wheeler Aligner) mem with the “-M” flag, sorted, and converted to BAM format with samtools. We then followed the preprocessing steps of the Genome Analysis Toolkit (GATK) Best Practices pipeline for variant discovery. Namely, the BAM file was further prepared with the tools CleanSam and AddOrReplaceReadGroups in Picard, and read duplicates were marked with biobambam. Reads were realigned using the tool RealignerTargetCreator from GATK, where both the dbSNP variant database and the individual-/strain-specific variant sites were used for the “-known” option. Base quality score recalibration tables were obtained using the BaseRecalibrator from GATK with a mismatch context size of 3 and using the dbSNP and individual-/strain-specific variant sites for the “-knownSites” option, as well as the reference regions declared to be unannotated rRNAs, as defined above. Paired-end read insert size distributions were computed with the CollectInsertSizeMetrics tool of Picard.
Computational hybridization
Only read libraries containing paired-end reads with each mate having a length of ≥70 were used. For each read library, reads with an exact, contiguous 30-nt match to any of the rRNA prototypes for that species or to the reads obtained from RNA-seq of polysomes for that species (above) were identified using MUMmer. For all reads satisfying this criterion, both the read and its mate were extracted for downstream analysis.
Calling rRNA variants
Candidate rRNA paired-end reads identified by computational hybridization were aligned to the 47S/45S and 5S operon rDNA prototypes for the appropriate species using bowtie2 with parameters “-q --phred33 -N 1 -L 10 -I C,3,0 --n-ceil 0 --dpad 30 --ignore-quals --end-to-end --mp 1,1 --rdg 4,1 --rfg 4,1 --score-min L,0,-0.12 -I a -X b --fr,” where a and b were set as 4 median absolute deviations below and above the insert size mean, respectively, as calculated from the whole-genome data (above). The options were set as such to ensure that the entire length of the read aligned to the prototype, up to a certain edit distance, and that the edit distance calculation was independent of base quality. Indels in the reads were normalized using LeftAlignIndels and IndelRealigner of GATK. Read base qualities were then recalibrated using the recalibration tables obtained as described above.
Reads identified via computational hybridization were also aligned to the reference genome using bowtie2 with parameters “-q --phred33 -I a -X b --fr --end-to-end,” where a and b were set as 4 median absolute deviations below and above the insert size mean, respectively, as calculated from the whole-genome data (above). A paired-end read was discarded if it did not map concordantly to the prototype rDNA or if there existed a concordant alignment in the reference genome on one of the fully assembled chromosomes that did not overlap with any of the unannotated rRNA regions that had a strictly smaller edit distance than the edit distance of the paired-end read on the rDNA prototype. Here, the edit distance was computed as the sum of the edit distances for each mate. Of the remaining reads, the base qualities for the 8 nt on each end of each mate were manually reduced to Q0. Finally, variants were called with LoFreq, restricted to the regions of the rDNA consisting of the transcribed rRNA.
The accuracy of variant detection was evaluated using publicly available Illumina whole-genome sequence libraries SRR3226042, SRR447638, SRR447643, and SRR447662 for Escherichia coli K-12 MG1655. rRNA variants and their true AFs were defined from multiple alignments of each rRNA component (16S, 5S, and 23S) of the annotated rDNA operons in the MG1655 genome, designating rDNA operon rrnB as the “prototype.” For each library, no false-positive variant calls were made, and all true SNP variants were detected with minor exceptions (below). The estimated AF and true AF were highly correlated (r > 0.98, P < 1 × 10−6) for all libraries tested. These results are consistent with the reported extremely high specificity of LoFreq (43).
Several “true” MG1655 variants were not detected by the pipeline. In positions 2547, 2548, and 2549 of the 23S, reported to be “GAT” in one of the operons, zero reads were found matching this sequence, indicating that it is an error in the reference genome for MG1655. Additionally, variants at positions 3 and 117 of the 5S were not detected because of edge effects; namely, being on the extremities of the 5S, variants at these positions are supported by nucleotides in the ends of reads, but read termini are excluded from supporting variants by the manual reduction of read termini base qualities to Q0. Thus, these false negatives are expected from the conservative design of the method.
Imputation of rRNA variant AFs
As expected from LoFreq’s limited sensitivity at low sequencing depth (43), our variant detection strategy had limited statistical power to detect all rDNA variants in each individual due to (i) the low sequencing depth of most individuals from the 1000 Genomes Project, (ii) our restriction to paired-end reads with each read ≥70 nt in length, and (iii) low relative read depth in GC-rich regions, which includes all rRNA. To prevent false negatives from biasing interindividual comparisons such as population stratification analyses, we imputed rRNA variant AFs for the purposes of interindividual comparisons. Variant AFs were imputed as follows. Suppose the variant discovery strategy detected a variant allele defined by variant nucleotide v at position x in individual A. For another individual B, let f be the fraction of reads for individual B overlapping position x that exhibit nucleotide v, and let n be the estimated rDNA copy number of individual B. We then impute the variant AF in individual B as f if f*n ≥ 1, and 0 otherwise.
Copy number estimation
Short-read sequencing data read depth distribution is biased according to the GC content of the fragment that generates the read(s) (36). For each library, the median fragment length was computed with Picard “CollectInsertSizeMetrics.” GC content–specific fragmentation rates λg as defined by Benjamini and Speed (36) were empirically computed for each possible GC content g = 0, …, L using all properly aligned reads on the autosomes, and excluding regions with structural variation as reported by the 1000 Genomes Project or Mouse Genomes Project for the given sample, or annotated as segmental duplications (http://humanparalogy.gs.washington.edu/ and http://mouseparalogy.gs.washington.edu/) or with homology to rRNA (above). Thus, given a specific position p in the haploid genome with GC content x in the region [p, p + L − 1], the expected number of fragments generated at p (that is, with 5′ end at p) is λx/2. We assume that the number of fragments generated at a position p of the haploid genome with GC content x in the region [p, p + L − 1] follows a Poisson distribution and that the process of fragment generation is conditionally independent given the reference genome
where G(p) gives the GC content of the region [p, p + L − 1].
The expected number of fragments generated from a region R = [p1, …, pn] (that is, the number of fragments with 5′ end in R) is then given by
With this model, we can estimate the copy number of any region of the genome. Namely, given a region R with observed fragment count X, the maximum likelihood estimate of the copy number C of R is given by
This method was validated in several ways. For each individual analyzed, 50,000 regions of width 4000 nt on autosome that were not overlapping segmental duplications or regions reported to have structural variation for that individual were randomly sampled. Hence, these regions are all putatively of copy number C = 2. Copy number predictions for each sample were computed as described above. For each individual, the Pearson correlation coefficient r for correlation between the predicted copy number and GC content of the region was computed using the 50,000 randomly sampled regions for that individual. These correlations were low for all individuals (fig. S2A). This reconfirms that the GC content–specific fragmentation rate method implemented here controlled for GC content–specific biases in read libraries. Second, for each individual, the 90% quantile q of the absolute error in copy number estimates of the 50,000 sampled regions was computed (fig. S2B), showing that for nearly every single individual analyzed, the copy number estimates of 90% of the randomly sampled control regions differed from the true copy number by less than 0.5. Third, rDNA copy number predictions based on 18S and 28S, separately, were highly correlated per individual (r = 0.99) and uncorrelated with library depth (r = −0.02, P = 0.33) despite the distinct GC contents of the 18S and 28S.
Differential expression of rRNA variants in mouse organs
RNA-seq reads were aligned against the rRNA prototypes with bowtie2 with parameters as described above (see the “Calling rRNA variants” section). To prevent false-positive variant calls due to alignment artifacts and poorly aligned read termini, reads were realigned with GATK, and the quality scores of read termini were manually reduced to Q0 to preclude their contribution to variant calling, again as described above (see the “Calling rRNA variants” section). LoFreq, which implements the strand bias test and accounts for quality scores and has been proven to have an extremely low false-positive rate, was then used to call sequence variants. Per-sample AFs for detected variants were then computed from read alignment pileups. Here, each sample represents RNA-seq reads obtained from a single organ of a single mouse. Thus, for each organ, there were three observations—one from each of the three mice analyzed. Differential expression of variant AFs between pairs of tissues was calculated using limma, which uses a moderated t statistic to account for common biological variation, with an FDR threshold of 0.05.
Supplementary Material
Acknowledgments
We would like to thank V. Borcherding and D. Duckworth for their expert technical support as members of the Weill Cornell Medicine Scientific Computing Unit. We also thank R. Altman, G. Gregorio, E. Rundlet, M. Holm, and all Blanchard laboratory members for helpful comments during the preparation of the manuscript. We would also like to acknowledge H. Molina and M. Tešić Mark for their assistance with mass spectrometry experiments. Funding: This work was supported by the Tri-Institutional Stem Cell Initiative funded by the Starr Foundation, the NIH (GM079238 to S.C.B.), the Swedish Research Council (C.T.V.), and the Jacques Cohenca Predoctoral Fellowship (C.M.K.). Author contributions: M.M.P. and S.C.B. conceived and planned the project. S.C.B., C.T.V., and M.M.P. contributed to the planning and analysis of all experiments. M.M.P. performed all computational and statistical analyses. D.L., C.T.V., and L.B. planned the experiments in mice. L.B. and C.M.K. performed the mouse tissue extraction experiments. C.M.K. and R.A.D. performed polysome purification experiments. M.M.P. and S.C.B. wrote the manuscript. All authors contributed to the revision of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/eaao0665/DC1
fig. S1. Bioinformatics strategy for rRNA copy number estimation and variant discovery.
fig. S2. rDNA copy number estimation in human and mouse.
fig. S3. Tertiary structure of rRNA variants.
fig. S4. Properties of detected rDNA variants detected in rRNA genes in human.
fig. S5. The number of variants stratified by each population.
fig. S6. rRNA variants with correlated AF in human.
fig. S7. rRNA variants that are differentially expressed between mouse tissues that localize to known functional centers of the ribosome.
fig. S8. Genomic AF and polysome expression.
table S1. Population stratification of rDNA copy number (Vst > 0.2).
table S2. Detected rRNA indel variants.
table S3. Detected rDNA variants in human that occur at positions of rRNA known to be modified in eukaryotes.
table S4. Human individuals with variants in intersubunit bridges.
table S5. rRNA variants whose AFs are stratified by populations.
table S6. Summary of rRNA variants detected in mouse strains from the Mouse Genomes Project.
table S7. Common rRNA variant positions in mouse and human.
table S8. Log intensity of proteins detected from quantitative mass spectrometry analysis.
REFERENCES AND NOTES
- 1.McCann K. L., Baserga S. J., Mysterious ribosomopathies. Science 341, 849–850 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xue S., Barna M., Specialized ribosomes: A new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 13, 355–369 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi Z., Barna M., Translating the genome in time and space: Specialized ribosomes, RNA regulons, and RNA-binding proteins. Annu. Rev. Cell Dev. Biol. 31, 31–54 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Mills E. W., Green R., Ribosomopathies: There’s strength in numbers. Science 358, eaan2755 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Sauert M., Temmel H., Moll I., Heterogeneity of the translational machinery: Variations on a common theme. Biochimie 114, 39–47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barna M., Specialized ribosomes: A New frontier in gene regulation, organismal biology, & evolution. FASEB J. 31, 387.1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briggs J. W., Dinman J. D., Subtractional heterogeneity: A crucial step toward defining specialized ribosomes. Mol. Cell 67, 3–4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Preiss T., All ribosomes are created equal. Really? Trends Biochem. Sci. 41, 121–123 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Slavov N., Semrau S., Airoldi E., Budnik B., van Oudenaarden A., Differential stoichiometry among core ribosomal proteins. Cell Rep. 13, 865–873 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noller H. F., The parable of the caveman and the Ferrari: Protein synthesis and the RNA world. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372, 20160187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simsek D., Tiu G. C., Flynn R. A., Byeon G. W., Leppek K., Xu A. F., Chang H. Y., Barna M., The mammalian ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell 169, 1051–1065.e18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holland M. L., Lowe R., Caton P. W., Gemma C., Carbajosa G., Danson A. F., Carpenter A. A. M., Loche E., Ozanne S. E., Rakyan V. K., Early-life nutrition modulates the epigenetic state of specific rDNA genetic variants in mice. Science 353, 495–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hernández A. I., Alarcon J. M., Allen K. D., New ribosomes for new memories? Commun. Integr. Biol. 8, e1017163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stults D. M., Killen M. W., Williamson E. P., Hourigan J. S., Vargas H. D., Arnold S. M., Moscow J. A., Pierce A. J., Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 69, 9096–9104 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Xu B., Li H., Perry J. M., Singh V. P., Unruh J., Yu Z., Zakari M., McDowell W., Li L., Gerton J. L., Ribosomal DNA copy number loss and sequence variation in cancer. PLOS Genet. 13, e1006771 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grozdanov P., Georgiev O., Karagyozov L., Complete sequence of the 45-kb mouse ribosomal DNA repeat: Analysis of the intergenic spacer. Genomics 82, 637–643 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Stults D. M., Killen M. W., Pierce H. H., Pierce A. J., Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 18, 13–18 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibbons J. G., Branco A. T., Godinho S. A., Yu S., Lemos B., Concerted copy number variation balances ribosomal DNA dosage in human and mouse genomes. Proc. Natl. Acad. Sci. U.S.A. 112, 2485–2490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuo B. A., Gonzalez I. L., Gillespie D. A., Sylvester J. E., Human ribosomal RNA variants from a single individual and their expression in different tissues. Nucleic Acids Res. 24, 4817–4824 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang S., Wang J., Tseng H., Basonuclin regulates a subset of ribosomal RNA genes in HaCaT cells. PLOS ONE 2, e902 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santoro R., Schmitz K.-M., Sandoval J., Grummt I., Intergenic transcripts originating from a subclass of ribosomal DNA repeats silence ribosomal RNA genes in trans. EMBO Rep. 11, 52–58 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zentner G. E., Balow S. A., Scacheri P. C., Genomic characterization of the mouse ribosomal DNA locus. G3 (Bethesda) 4, 243–254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holland M. L., Epigenetic regulation of the protein translation machinery. EBioMedicine 17, 3–4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tseng H., Chou W., Wang J., Zhang X., Zhang S., Schultz R. M., Mouse ribosomal RNA genes contain multiple differentially regulated variants. PLOS ONE 3, e1843 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leffers H., Andersen A. H., The sequence of 28S ribosomal RNA varies within and between human cell lines. Nucleic Acids Res. 21, 1449–1455 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez I. L., Sylvester J. E., Schmickel R. D., Human 28S ribosomal RNA sequence heterogeneity. Nucleic Acids Res. 16, 10213–10224 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.International Human Genome Sequencing Consortium , Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Treangen T. J., Salzberg S. L., Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 13, 36–46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.1000 Genomes Project Consortium , A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams D. J., Doran A. G., Lilue J., Keane T. M., The Mouse Genomes Project: A repository of inbred laboratory mouse strain genomes. Mamm. Genome 26, 403–412 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Reschke M., Clohessy J. G., Seitzer N., Goldstein D. P., Breitkopf S. B., Schmolze D. B., Ala U., Asara J. M., Beck A. H., Pandolfi P. P., Characterization and analysis of the composition and dynamics of the mammalian riboproteome. Cell Rep. 4, 1276–1287 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chassé H., Boulben S., Costache V., Cormier P., Morales J., Analysis of translation using polysome profiling. Nucleic Acids Res. 45, e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Behrmann E., Loerke J., Budkevich T. V., Yamamoto K., Schmidt A., Penczek P. A., Vos M. R., Bürger J., Mielke T., Scheerer P., Spahn C. M. T., Structural snapshots of actively translating human ribosomes. Cell 161, 845–857 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferguson A., Wang L., Altman R. B., Terry D. S., Juette M. F., Burnett B. J., Alejo J. L., Dass R. A., Parks M. M., Vincent C. T., Blanchard S. C., Functional dynamics within the human ribosome regulate the rate of active protein synthesis. Mol. Cell 60, 475–486 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibbons J. G., Branco A. T., Yu S., Lemos B., Ribosomal DNA copy number is coupled with gene expression variation and mitochondrial abundance in humans. Nat. Commun. 5, 4850 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Benjamini Y., Speed T. P., Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 40, e72 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmickel R. D., Quantitation of human ribosomal DNA: Hybridization of human DNA with ribosomal RNA for quantitation and fractionation. Pediatr. Res. 7, 5–12 (1973). [DOI] [PubMed] [Google Scholar]
- 38.Redon R., Ishikawa S., Fitch K. R., Feuk L., Perry G. H., Andrews T. D., Fiegler H., Shapero M. H., Carson A. R., Chen W., Cho E. K., Dallaire S., Freeman J. L., González J. R., Gratacòs M., Huang J., Kalaitzopoulos D., Komura D., MacDonald J. R., Marshall C. R., Mei R., Montgomery L., Nishimura K., Okamura K., Shen F., Somerville M. J., Tchinda J., Valsesia A., Woodwark C., Yang F., Zhang J., Zerjal T., Zhang J., Armengol L., Conrad D. F., Estivill X., Tyler-Smith C., Carter N. P., Aburatani H., Lee C., Jones K. W., Scherer S. W., Hurles M. E., Global variation in copy number in the human genome. Nature 444, 444–454 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prokopowich C. D., Gregory T. R., Crease T. J., The correlation between rDNA copy number and genome size in eukaryotes. Genome 46, 48–50 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Van der Auwera G. A., Carneiro M. O., Hartl C., Poplin R., del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J., Banks E., Garimella K. V., Altshuler D., Gabriel S., DePristo M. A., From FastQ data to high-confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 11, 11.10.1–11.10.33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DePristo M. A., Banks E., Poplin R., Garimella K. V., Maguire J. R., Hartl C., Philippakis A. A., del Angel G., Rivas M. A., Hanna M., McKenna A., Fennell T. J., Kernytsky A. M., Sivachenko A. Y., Cibulskis K., Gabriel S. B., Altshuler D., Daly M. J., A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng Z., Cheng Y., Tan B. C.-M., Kang L., Tian Z., Zhu Y., Zhang W., Liang Y., Hu X., Tan X., Guo J., Dong Z., Liang Y., Bao L., Wang J., Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 30, 253–260 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Wilm A., Aw P. P. K., Bertrand D., Yeo G. H. T., Ong S. H., Wong C. H., Khor C. C., Petric R., Hibberd M. L., Nagarajan N., LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J., Raskin L., Samuels D. C., Shyr Y., Guo Y., Genome measures used for quality control are dependent on gene function and ancestry. Bioinformatics 31, 318–323 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gudbjartsson D. F., Sulem P., Helgason H., Gylfason A., Gudjonsson S. A., Zink F., Oddson A., Magnusson G., Halldorsson B. V., Hjartarson E., Sigurdsson G. Th., Kong A., Helgason A., Masson G., Magnusson O. Th., Thorsteinsdottir U., Stefansson K., Sequence variants from whole genome sequencing a large group of Icelanders. Sci. Data 2, 150011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.des Georges A., Dhote V., Kuhn L., Hellen C. U. T., Pestova T. V., Frank J., Hashem Y., Structure of mammalian eIF3 in the context of the 43S preinitiation complex. Nature 525, 491–495 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lomakin I. B., Steitz T. A., The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature 500, 307–311 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Decatur W. A., Fournier M. J., rRNA modifications and ribosome function. Trends Biochem. Sci. 27, 344–351 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Ofengand J., Bakin A., Mapping to nucleotide resolution of pseudouridine residues in large subunit ribosomal RNAs from representative eukaryotes, prokaryotes, archaebacteria, mitochondria and chloroplasts. J. Mol. Biol. 266, 246–268 (1997). [DOI] [PubMed] [Google Scholar]
- 50.Sloan K. E., Warda A. S., Sharma S., Entian K.-D., Lafontaine D. L. J., Bohnsack M. T., Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 14, 1138–1152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maden B. E. H., The numerous modified nucleosides in eukaryotic ribosomal RNA. Prog. Nucleic Acid Res. Mol. Biol. 39, 241–303 (1990). [DOI] [PubMed] [Google Scholar]
- 52.Muhs M., Hilal T., Mielke T., Skabkin M. A., Sanbonmatsu K. Y., Pestova T. V., Spahn C. M. T., Cryo-EM of ribosomal 80S complexes with termination factors reveals the translocated cricket paralysis virus IRES. Mol. Cell 57, 422–432 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khatter H., Myasnikov A. G., Natchiar S. K., Klaholz B. P., Structure of the human 80S ribosome. Nature 520, 640–645 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Dunkle J. A., Wang L., Feldman M. B., Pulk A., Chen V. B., Kapral G. J., Noeske J., Richardson J. S., Blanchard S. C., Cate J. H. D., Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science 332, 981–984 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Melnikov S., Ben-Shem A., Garreau de Loubresse N., Jenner L., Yusupova G., Yusupov M., One core, two shells: Bacterial and eukaryotic ribosomes. Nat. Struct. Mol. Biol. 19, 560–567 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Wang L., Altman R. B., Blanchard S. C., Insights into the molecular determinants of EF-G catalyzed translocation. RNA 17, 2189–2200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Budkevich T., Giesebrecht J., Altman R. B., Munro J. B., Mielke T., Nierhaus K. H., Blanchard S. C., Spahn C. M. T., Structure and dynamics of the mammalian ribosomal pretranslocation complex. Mol. Cell 44, 214–224 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hsieh A. C., Liu Y., Edlind M. P., Ingolia N. T., Janes M. R., Sher A., Shi E. Y., Stumpf C. R., Christensen C., Bonham M. J., Wang S., Ren P., Martin M., Jessen K., Feldman M. E., Weissman J. S., Shokat K. M., Rommel C., Ruggero D., The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li J., Shin S., Sun Y., Yoon S.-O., Li C., Zhang E., Yu J., Zhang J., Blenis J., mTORC1-driven tumor cells are highly sensitive to therapeutic targeting by antagonists of oxidative stress. Cancer Res. 76, 4816–4827 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sudmant P. H., Mallick S., Nelson B. J., Hormozdiari F., Krumm N., Huddleston J., Coe B. P., Baker C., Nordenfelt S., Bamshad M., Jorde L. B., Posukh O. L., Sahakyan H., Watkins W. S., Yepiskoposyan L., Abdullah M. S., Bravi C. M., Capelli C., Hervig T., Wee J. T. S., Tyler-Smith C., van Driem G., Romero I. G., Jha A. R., Karachanak-Yankova S., Toncheva D., Comas D., Henn B., Kivisild T., Ruiz-Linares A., Sajantila A., Metspalu E., Parik J., Villems R., Starikovskaya E. B., Ayodo G., Beall C. M., Di Rienzo A., Hammer M. F., Khusainova R., Khusnutdinova E., Klitz W., Winkler C., Labuda D., Metspalu M., Tishkoff S. A., Dryomov S., Sukernik R., Patterson N., Reich D., Eichler E. E., Global diversity, population stratification, and selection of human copy-number variation. Science 349, aab3761 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma X. M., Blenis J., Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Horos R., IJspeert H., Pospisilova D., Sendtner R., Andrieu-Soler C., Taskesen E., Nieradka A., Cmejla R., Sendtner M., Touw I. P., von Lindern M., Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood 119, 262–272 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Flygare J., Aspesi A., Bailey J. C., Miyake K., Caffrey J. M., Karlsson S., Ellis S. R., Human RPS19, the gene mutated in Diamond-Blackfan anemia, encodes a ribosomal protein required for the maturation of 40S ribosomal subunits. Blood 109, 980–986 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kondrashov N., Pusic A., Stumpf C. R., Shimizu K., Hsieh A. C., Ishijima J., Shiroishi T., Barna M., Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 145, 383–397 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Becker T., Armache J.-P., Jarasch A., Anger A. M., Villa E., Sieber H., Motaal B. A., Mielke T., Berninghausen O., Beckmann R., Structure of the no-go mRNA decay complex Dom34–Hbs1 bound to a stalled 80S ribosome. Nat. Struct. Mol. Biol. 18, 715–720 (2011). [DOI] [PubMed] [Google Scholar]
- 66.Shao S., Brown A., Santhanam B., Hegde R. S., Structure and assembly pathway of the ribosome quality control complex. Mol. Cell 57, 433–444 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anger A. M., Armache J.-P., Berninghausen O., Habeck M., Subklewe M., Wilson D. N., Beckmann R., Structures of the human and Drosophila 80S ribosome. Nature 497, 80–85 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Yamamoto H., Qin Y., Achenbach J., Li C., Kijek J., Spahn C. M. T., Nierhaus K. H., EF-G and EF4: Translocation and back-translocation on the bacterial ribosome. Nat. Rev. Microbiol. 12, 89–100 (2014). [DOI] [PubMed] [Google Scholar]
- 69.van Nues R. W., Granneman S., Kudla G., Sloan K. E., Chicken M., Tollervey D., Watkins N. J., Box C/D snoRNP catalysed methylation is aided by additional pre-rRNA base-pairing. EMBO J. 30, 2420–2430 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.des Georges A., Hashem Y., Unbehaun A., Grassucci R. A., Taylor D., Hellen C. U. T., Pestova T. V., Frank J., Structure of the mammalian ribosomal pre-termination complex associated with eRF1.eRF3.GDPNP. Nucleic Acids Res. 42, 3409–3418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kossinova O., Malygin A., Krol A., Karpova G., The SBP2 protein central to selenoprotein synthesis contacts the human ribosome at expansion segment 7L of the 28S rRNA. RNA 20, 1046–1056 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Voorhees R. M., Schmeing T. M., Kelley A. C., Ramakrishnan V., The mechanism for activation of GTP hydrolysis on the ribosome. Science 330, 835–838 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zentner G. E., Saiakhova A., Manaenkov P., Adams M. D., Scacheri P. C., Integrative genomic analysis of human ribosomal DNA. Nucleic Acids Res. 39, 4949–4960 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Serin G., Joseph G., Faucher C., Ghisolfi L., Bouche G., Amalric F., Bouvet P., Localization of nucleolin binding sites on human and mouse pre-ribosomal RNA. Biochimie 78, 530–538 (1996). [DOI] [PubMed] [Google Scholar]
- 75.Bradatsch B., Leidig C., Granneman S., Gnädig M., Tollervey D., Böttcher B., Beckmann R., Hurt E., Structure of the pre-60S ribosomal subunit with nuclear export factor Arx1 bound at the exit tunnel. Nat. Struct. Mol. Biol. 19, 1234–1241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Petrov A. S., Bernier C. R., Hsiao C., Norris A. M., Kovacs N. A., Waterbury C. C., Stepanov V. G., Harvey S. C., Fox G. E., Wartell R. M., Hud N. V., Williams L. D., Evolution of the ribosome at atomic resolution. Proc. Natl. Acad. Sci. U.S.A. 111, 10251–10256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Helm M., Motorin Y., Detecting RNA modifications in the epitranscriptome: Predict and validate. Nat. Rev. Genet. 18, 275–291 (2017). [DOI] [PubMed] [Google Scholar]
- 78.R. Freitas, R. Merkle, Kinematic Self-Replicating Machines (Landes Bioscience, 2004). [Google Scholar]
- 79.Leidig C., Bange G., Kopp J., Amlacher S., Aravind A., Wickles S., Witte G., Hurt E., Beckmann R., Sinning I., Structural characterization of a eukaryotic chaperone–the ribosome-associated complex. Nat. Struct. Mol. Biol. 20, 23–28 (2013). [DOI] [PubMed] [Google Scholar]
- 80.Santoro R., Grummt I., Molecular mechanisms mediating methylation-dependent silencing of ribosomal gene transcription. Mol. Cell 8, 719–725 (2001). [DOI] [PubMed] [Google Scholar]
- 81.Santoro R., Li J., Grummt I., The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat. Genet. 32, 393–396 (2002). [DOI] [PubMed] [Google Scholar]
- 82.Caburet S., Conti C., Schurra C., Lebofsky R., Edelstein S. J., Bensimon A., Human ribosomal RNA gene arrays display a broad range of palindromic structures. Genome Res. 15, 1079–1085 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shi Z., Fujii K., Kovary K. M., Genuth N. R., Röst H. L., Teruel M. N., Barna M., Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol. Cell 67, 71–83.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maden B. E. H., Dent C. L., Farrell T. E., Garde J., McCallum F. S., Wakeman J. A., Clones of human ribosomal DNA containing the complete 18 S-rRNA and 28 S-rRNA genes. Characterization, a detailed map of the human ribosomal transcription unit and diversity among clones. Biochem. J. 246, 519–527 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rappsilber J., Mann M., Ishihama Y., Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007). [DOI] [PubMed] [Google Scholar]
- 86.Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M., Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011). [DOI] [PubMed] [Google Scholar]
- 87.Shao S., Murray J., Brown A., Taunton J., Ramakrishnan V., Hegde R. S., Decoding mammalian ribosome-mRNA states by translational GTPase complexes. Cell 167, 1229–1240.e15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/eaao0665/DC1
fig. S1. Bioinformatics strategy for rRNA copy number estimation and variant discovery.
fig. S2. rDNA copy number estimation in human and mouse.
fig. S3. Tertiary structure of rRNA variants.
fig. S4. Properties of detected rDNA variants detected in rRNA genes in human.
fig. S5. The number of variants stratified by each population.
fig. S6. rRNA variants with correlated AF in human.
fig. S7. rRNA variants that are differentially expressed between mouse tissues that localize to known functional centers of the ribosome.
fig. S8. Genomic AF and polysome expression.
table S1. Population stratification of rDNA copy number (Vst > 0.2).
table S2. Detected rRNA indel variants.
table S3. Detected rDNA variants in human that occur at positions of rRNA known to be modified in eukaryotes.
table S4. Human individuals with variants in intersubunit bridges.
table S5. rRNA variants whose AFs are stratified by populations.
table S6. Summary of rRNA variants detected in mouse strains from the Mouse Genomes Project.
table S7. Common rRNA variant positions in mouse and human.
table S8. Log intensity of proteins detected from quantitative mass spectrometry analysis.