

Partial tandem duplication (PTD) of the KMT2A (MLL) gene is detected in approximately 5-10% of cases of acute myeloid leukemia (AML)1–5 and within cases showing normal karyotype, confers a worse prognosis.2,6,7 In these patients, the duplications are variable in size and most commonly involve exons 2 or 3, spanning through exon 6 or exons 8–11.1 Unlike many other translocations involving the KMT2A gene, MLL-PTD cannot be detected using conventional cytogenetics.8 Other methods, such as the cytogenomic array, can be used to identify patients with this genetic alteration who may be potential candidates for therapy (e.g., demethylating agents, histone deacetylase inhibitors).3,9
As in AML, MLL-PTD can also be seen in a subset of patients with myelodysplastic syndrome (MDS) and its acquisition has been observed during leukemic transformation;4,10 however, the clinical significance of MLL-PTD at diagnosis, particularly with regard to survival, has not been well characterized in these patients. In the study herein, we describe unique pathologic and clinical characteristics in a series of MDS patients with MLL-PTD, and analyze the impact of MLL-PTD on therapy response and clinical outcomes.
We reviewed the records of consecutive patients with a diagnosis of MDS who had cytogenomic microarray studies performed between March 2014 and June 2017 (n=83). Cases with MLL-PTD were identified and compared to a control cohort of patients (n=38) without MLL-PTD who were derived from the same original cohort (Online Supplementary Tables S1–S2). Cases of AML with MLL-PTD were also reviewed. All patients had been evaluated and treated at the same institution during the same time period. Overall survival (OS) was calculated from the diagnosis date to date of death, censoring for patients alive at the time of study completion. Additional methods are described in Online Supplementary Methods.
A total of 14 cases with MLL-PTD were identified by the cytogenomic microarray (Table 1A,B). Excluding patients with therapy-related myeloid neoplasms, MLL-PTD was detected in 6–7% of patients with MDS that were screened by the array during the same time period. These patients were predominantly male, ranging in age from 39.9–68.7 years of age at diagnosis (mean 59.3 years, median 66.9 years).
Table 1A.
Pathologic characteristics of MLL-PTD cases.
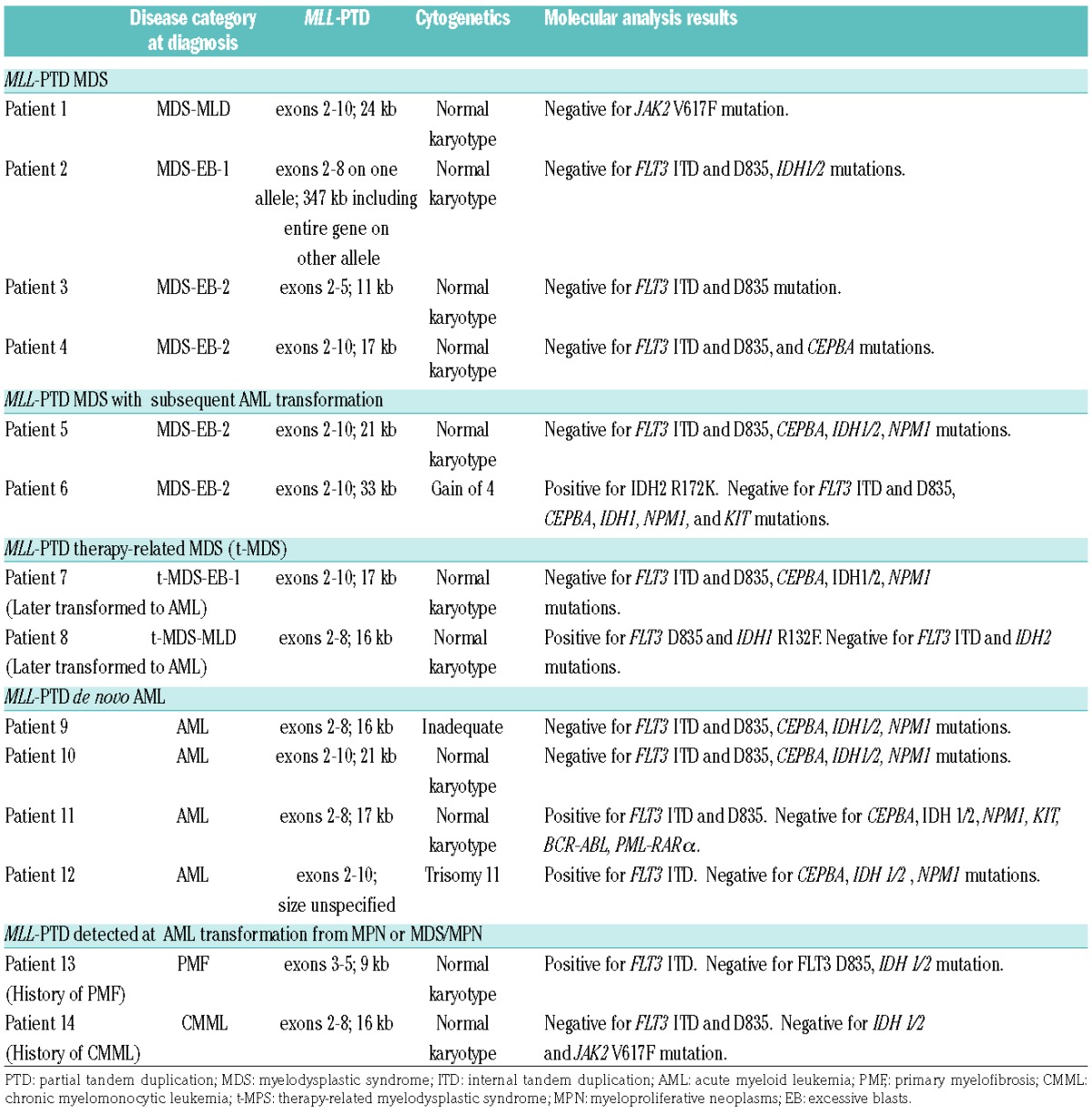
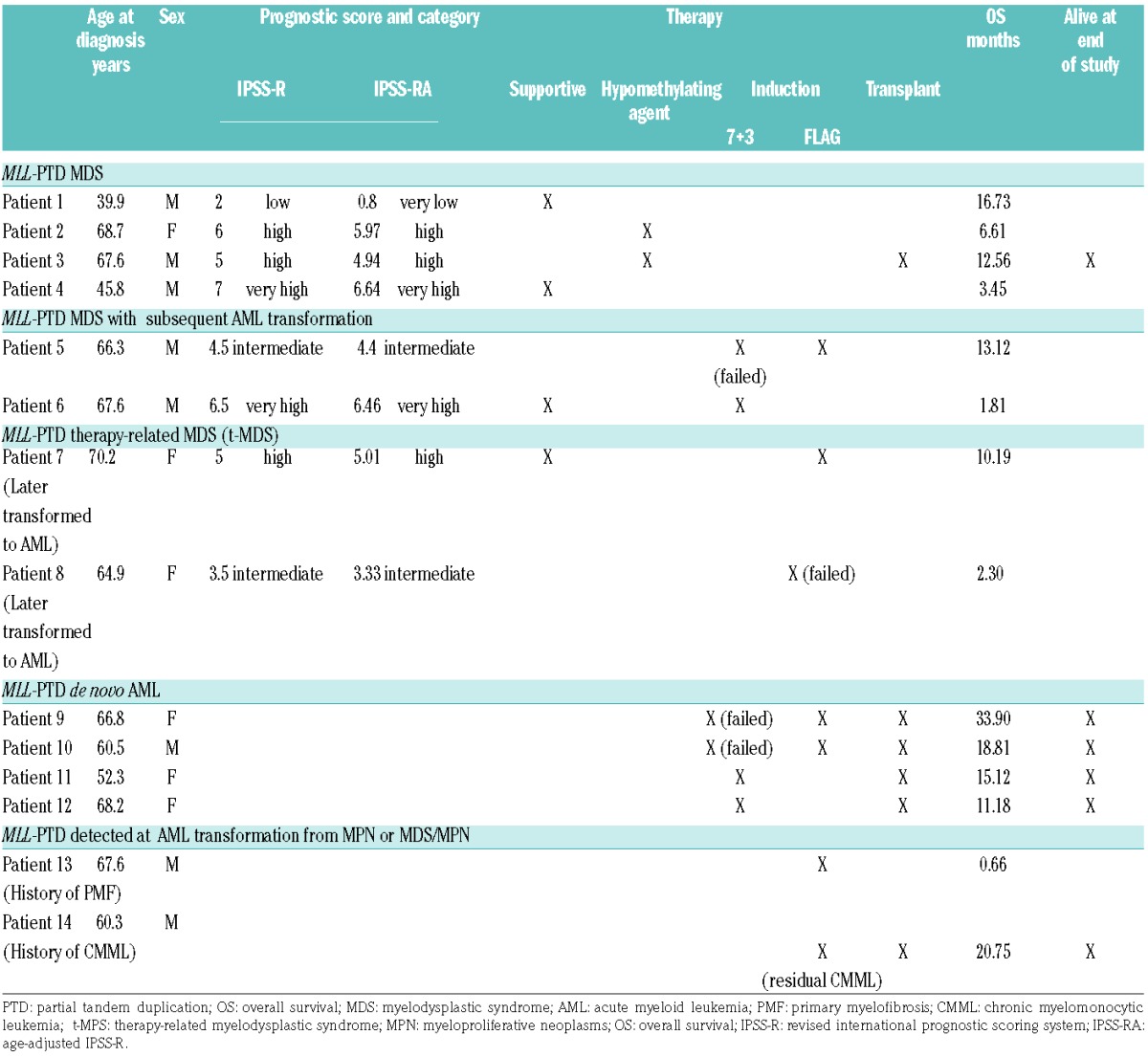
Table 1B.
Clinical characteristics of MLL-PTD cases.
The majority of MLL-PTD MDS cases were classified as MDS with excess blasts (MDS-EB; by WHO 2016 criteria),11 showed normal karyotype by conventional cytogenetics, and had variable revised international prognostic scoring system (IPSS-R) scores ranging from low to very high (Table 1A,B). Cases of MLL-PTD presented with lower absolute neutrophil count (P=0.012) and platelet count (P=0.046) compared to non-MLL-PTD MDS cases with high to very high age-adjusted IPSS-R (IPSS-RA), but comparable hemoglobin and bone marrow blast percentage (Figure 1A). Flow cytometric immunophenotyping did not reveal specific immunophenotypic aberrations distinguishing MLL-PTD cases from non-MLL-PTD cases (data not shown).
Figure 1.

MLL-PTD MDS presents with lower absolute neutrophil and platelet counts and displays worse prognosis than high risk non-MLL-PTD MDS. (A) Hemoglobin, absolute neutrophil count, platelet count, and bone marrow blast percentage were compared between MLL-PTD MDS and non-MLL-PTD MDS with high to very high IPSS-RA score [mean value and standard error of the mean are depicted; *(P=0.012) **(P=0.046)]. (B) Survival analysis comparing MLL-PTD MDS to non-MLL-PTD MDS based on IPSS-RA prognostic group (P<0.0001). (C) Survival analysis comparing MLL-PTD MDS to high or very high risk non-MLL-PTD MDS based on IPSS-RA score (P=0.002). (D) Survival analysis comparing MLL-PTD MDS to non-MLL-PTD MDS with poor to very poor cytogenetic and good cytogenetic groups (P=0.027). (E) Survival analysis comparing MLL-PTD MDS, transformed MLL-PTD MDS, and de novo MLL-PTD AML cases (P=0.043). PTD: partial tandem duplication; MDS: myelodysplastic syndrome; IPSS-RA: age-adjusted IPSS-R.
MLL-PTD MDS patients showed worse OS compared to MDS patients without MLL-PTD, even when compared to those with high to very high IPSS-RA (Figure 1B, P<0.0001; Figure 1C, P=0.002). The median OS for MLL-PTD MDS was 9.85 months from the time of diagnosis compared to 31.5 months for the high to very high IPSS-RA group. OS was also worse when comparing cases of MDS with poor to very poor cytogenetic features, which had a median OS of 18.2 months (Figure 1D, P=0.027). MLL-PTD MDS patients, with and without acute leukemic transformation, also showed worse OS compared to patients with MLL-PTD who presented with de novo AML (Figure 1E, P=0.043). All four patients with de novo AML were alive at the conclusion of this study.
Patients with MLL-PTD MDS who transformed to AML as well as de novo MLL-PTD AML received either 7+312 or FLAG13 induction therapy (Table 1B).
Three of the five MLL-PTD patients who received 7+3 induction therapy showed residual disease in day 14 marrows, whereas two de novo AML cases, both of which also had a FLT3-ITD mutation, achieved CR and were subsequently transplanted. All cases that failed initial 7+3 induction therapy showed complete response to FLAG re-induction therapy. One patient with therapy-related myelodysplastic syndrome (t-MDS) failed initial FLAG induction therapy. Two additional MDS patients received initial FLAG induction therapy but passed away due to infection before disease response evaluation could be completed.
All MLL-PTD patients who proceeded to transplant were still alive at the conclusion of this study, except for one patient with t-MDS who relapsed with AML (Table 1B). All patients who did not receive a transplant passed away, with the longest survival time being 16.7 months. The single MLL-PTD MDS patient who received a transplant remains alive at the time of writing.
The study herein identifies a subset of MDS defined by the presence of MLL-PTD that is associated with more advanced disease with excess blasts and worse outcome, compared to MLL-PTD PTD MDS, even those with high risk IPSS scores and complex karyotype. Though the sample size is small, the effect size is pronounced and statistically significant in all comparisons. The overall prevalence of MLL-PTD in MDS (6–7% of cases) is similar to that observed in AML and slightly higher than reported in a previous study of mutational analysis in MDS.10 It is of note that the control cohort of high and very high risk IPSS-RA in our study shows a better median OS of 31.5 months than has been reported in the larger multinational IPSS-R study, which showed a median survival of 19 months for high risk and 10.8 months for very high risk groups.14 This finding could potentially be reflective of differences in therapy and clinical practice or of the smaller numbers of patients in this single institution review. It is also significant because despite improved survival of high and very high risk MDS patients being treated at the same institution during the same time interval, patients with MLL-PTD still fared very poorly. Furthermore, their median survival of 9.85 months is even lower than very high risk MDS patients as predicted by the IPSS-R study.14 This finding suggests that the finding of MLL-PTD in an MDS patient is indeed a very poor prognostic factor and taken a step further, these patients may potentially benefit from expeditious transplantation if feasible. Supporting this is the observation that all patients with MLL-PTD who received a transplant (other than those with therapy-related myeloid neoplasms) were still alive at the conclusion of this study.
For those patients who developed MLL-PTD AML, in cases where 7+3 induction chemotherapy was not successful, complete response was achievable in most cases treated with FLAG re-induction therapy. The presence of additional FLT3-ITD mutation may possibly predict a better response to 7+3 induction therapy; however, the sample population is too small to draw a definitive conclusion. Co-occurrence of FLT3-ITD and MLL-PTD has been reported previously in AML, though the significance of this finding on outcome or response is currently unknown.15 The potential use of demethylating agents and histone deacetylase inhibitors, as has been proposed for cases of MLL-PTD AML,3,9 is one area of further exploration. Of note, de novo MLL-PTD AML patients, which exclude patients with underlying MDS, showed the best outcomes of patients in this study, possibly due to early transplantation, and may have better prognosis than has been previously reported.
In summary, our findings suggest that MLL-PTD MDS presents as high grade disease with excess blasts and typically normal karyotype. Testing for MLL-PTD might be considered in all patients with MDS, and its discovery could potentially warrant consideration for early transplantation. Because the size of our sample is limited, additional studies on a larger cohort of patients to further define the features of this subgroup and delineate appropriate therapy are needed.
Supplementary Material
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Schnittger S, Kinkelin U, Schoch C, et al. Screening for MLL tandem duplication in 387 unselected patients with AML identify a prognostically unfavorable subset of AML. Leukemia. 2000;14(5):796–804. [DOI] [PubMed] [Google Scholar]
- 2.Döhner K, Tobis K, Ulrich R, et al. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol. 2002;20(15):3254–3261. [DOI] [PubMed] [Google Scholar]
- 3.Whitman SP, Liu S, Vukosavljevic T, et al. The MLL partial tandem duplication: evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood. 2005;106(1):345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steudel C, Wermke M, Schaich M, et al. Comparative analysis of MLL partial tandem duplication and FLT3 internal tandem duplication mutations in 956 adult patients with acute myeloid leukemia. Genes Chromosomes Cancer. 2003;37(3):237–251. [DOI] [PubMed] [Google Scholar]
- 5.Strout MP, Marcucci G, Bloomfield CD, Caligiuri MA. The partial tandem duplication of ALL1 (MLL) is consistently generated by Alumediated homologous recombination in acute myeloid leukemia. Proc Natl Acad Sci U S A. 1998;95(5):2390–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shiah HS, Kuo YY, Tang JL, et al. Clinical and biological implications of partial tandem duplication of the MLL gene in acute myeloid leukemia without chromosomal abnormalities at 11q23. Leukemia. 2002;16(2):196–202. [DOI] [PubMed] [Google Scholar]
- 7.Caligiuri MA, Strout MP, Lawrence D, et al. Rearrangement of ALL1 (MLL) in acute myeloid leukemia with normal cytogenetics. Cancer Res. 1998;58(1):55–59. [PubMed] [Google Scholar]
- 8.Schichman SA, Caligiuri MA, Gu Y, et al. ALL-1 partial duplication in acute leukemia. Proc Natl Acad Sci USA. 1994;91(13):6236–6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kühn MWM, Hadler MJ, Daigle SR, et al. MLL partial tandem duplication leukemia cells are sensitive to small molecule DOT1L inhibition. Haematologica. 2015;100(5):e190–e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dicker F, Haferlach C, Sundermann J, et al. Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia. 2010;24(8):1528–1532. [DOI] [PubMed] [Google Scholar]
- 11.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009;361(13):1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson G, Taylor P, Smith GM, et al. A multicentre, open, non-comparative phase II study of a combination of fludarabine phosphate, cytarabine and granulocyte colony-stimulating factor in relapsed and refractory acute myeloid leukaemia and de novo refractory anaemia with excess of blasts in transformation. Br J Haematol. 2001;112(1):127–137. [DOI] [PubMed] [Google Scholar]
- 14.Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kihara R, Nagata Y, Kiyoi H, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014;28(8):1586–1595. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.