

Abstract
We have previously described the safety and efficacy of pegylated interferon-α2a therapy in a cohort of 62 patients with myeloproliferative neoplasm-associated myelofibrosis followed in centers affiliated to the French Intergroup of Myeloproliferative neoplasms. In this study, we report their long-term outcomes and correlations with mutational patterns of driver and non-driver mutations analyzed by targeted next generation sequencing. The median age at diagnosis was 66 years old, the median follow-up since starting pegylated interferon was 58 months. At the time of analysis, 30 (48.4%) patients were alive including 16 still being treated with pegylated interferon. The median survival of patients with intermediate and high-risk prognostic Lille and dynamic International Prognostic Scoring System scores treated with pegylated interferon was increased in comparison to that of historical cohorts. In addition, overall survival was significantly correlated with the duration of pegylated interferon therapy (70 versus 30 months after 2 years of treatment, P<10−12). JAK2V617F allele burden was decreased by more than 50% in 58.8% of patients and two patients even achieved complete molecular response. Next-generation sequencing analyses performed in 49 patients showed that 28 (57.1%) of them carried non-driver mutations. The presence of at least one additional mutation was associated with a reduction of both overall and leukemia-free survival. These findings in a large series of patients with myelofibrosis suggest that pegylated interferon therapy may provide a survival benefit for patients with intermediate- or high-risk Lille and dynamic International Prognostic Scoring System scores. It also reduced the JAK2V617F allele burden in most patients. These results further support the use of pegylated interferon in selected patients with myelofibrosis.
Introduction
Primary myelofibrosis is a Philadelphia chromosome-negative myeloproliferative neoplasm characterized by splenomegaly, constitutional symptoms and cytopenia and/or proliferative features in peripheral blood. Secondary myelofibrosis may develop from either polycythemia vera or essential thrombocythemia.1,2 The main causes of death of patients with myelofibrosis include disease progression leading to cachexia or infection, and acceleration and transformation of their disease into acute myeloid leukemia.3 The JAK2V617F mutation can be found in about half of myelofibrosis patients, while 20–30% carry a calreticulin (CALR) mutation and 5–10% a mutation in the thrombopoietin receptor gene MPL. These three driver mutations influence the clinical presentation and outcome: for example, CALR-mutated myelofibrosis patients are predominantly male, have higher platelet and lower leukocyte and red cell counts, and longer survival than those with the JAK2V617F mutation.4,5
In addition to these three driver mutations, other mutations are frequently found in myelofibrosis patients, mainly in genes involved in epigenetic regulation or the splicing machinery. Some of these mutations have been associated with poorer survival and Vannucchi et al. have defined five “high molecular risk” genes: ASXL1, EZH2, SRSF2, and IDH1/2. Mutations in any of these genes dramatically decreased overall and event-free survival of the affected patients and the presence of more than one additional mutation conferred an even worse outcome.6,7
Several strategies have been used to alleviate the proliferative aspects of myelofibrosis (e.g., hydroxyurea, pipobroman, 6-mercaptopurine) or the cytopenic ones (e.g., thalidomide and its derivatives, androgens, recombinant erythropoietin), as well as to manage splenomegaly (e.g., hydroxyurea, radiotherapy, splenectomy). The efficacy of these approaches is generally modest, especially with regards to cytopenia and does not clearly modify disease evolution.2
Ruxolitinib, a non-specific JAK1/JAK2 inhibitor approved for the treatment of symptomatic myelofibrosis patients, was recently shown to be very effective in reducing the inflammatory component of these diseases with significant improvement of pruritus, fever, weight loss and splenomegaly. Although still debated, results of the COMFORT I and II studies also suggest that ruxolitinib may increase overall survival of high-risk myelofibrosis patients compared to that of patients treated in the placebo or “best available therapy” arms.8–12 To date, however, allogeneic stem cell transplantation remains the only curative option for these patients.13–16
We have previously reported on the feasibility and hematologic results of myelofibrosis treatment with pegylated interferon-α2a in a prospective observational study conducted by the French Intergroup of Myeloproliferative neoplasms (FIM).17,18 Herein, we report the long-term outcomes of this large cohort of patients, focusing on survival and incidence of acute leukemia. In addition, next-generation sequencing data enabled us to assess the impact of interferon treatment on the prognosis associated with mutational patterns, and with the presence of non-driver mutations.
Methods
Patients’ recruitment
Between December 2006 and April 2011 we prospectively recruited 62 patients treated with pegylated interferon-α2a in 17 centers affiliated to the FIM group. The inclusion criteria and methodology have been described elsewhere.18 This study was approved by the local Institutional Review Board and registered in ClinicalTrials.gov (NCT02910258). All participants gave written informed consent. The patients treated in the CHRU of Brest were also registered in the OBENE observatory (NCT02897297).
Pegylated interferon-α2a was initiated by physicians in accordance with local and national guidelines. During the period of this study, ruxolitinib was only available through clinical trials (approval for use in myelofibrosis in France was obtained in August 2012).
Molecular analyses
Samples from all the patients were characterized for the three driver mutations. Genomic DNA was extracted from blood neutrophils or total leukocytes using the Flexigene DNA kit (Qiagen, Germany) according to the manufacturer’s recommendation.
JAK2V617F was quantified by real-time quantitative polymerase chain reaction analysis according to previously described methods.19 MPLW515K/L mutations were screened for using the MPLW515L/K MutaScreen Kit (Qiagen) according to manufacturer’s instructions. Quantitative polymerase chain reactions were performed on ABI7500 instruments (Applied Biosystems). CALR exon 9 mutations were screened for by fragment analysis according to published methods.4 Polymerase chain reaction products were analyzed on an ABI3130 instrument (Applied Biosystems).
Next-generation sequencing
Targeted next-generation sequencing was performed in 49 samples collected at the time of starting pegylated interferon-α2a treatment (34 JAK2V617F-positive, 12 CALR-positive, 3 triple-negative). The next-generation sequencing panel included 26 genes (ASXL1, BCOR, CBL, CSF3R, DNMT3A, ETNK1, ETV6, EZH2, IDH1, IDH2, JAK2, KRAS, MPL, NRAS, PDGFRA, RUNX1, SETBP1, SF3B1, SH2B3, SRSF2, STAG2, TET2, TP53, U2AF1, ULK1, and ZRSR2) and the sequencing was performed using AmpliseqTM (Thermo Fisher Scientific, Foster City, CA, USA) custom design. Library preparation and sequencing using PGMTM (Thermo Fisher Scientific) were performed according to the manufacturer’s instructions.
Mutations were detected using the Variant Caller v4.2 plugin from Torrent Suite Software and IonReporter v5.2 (Life Technologies). For mutation calling, arbitrary filters were fixed with variant allele frequencies >2% and depth >50X. False positive variants were dropped after BAM analysis on Alamut® (Interactive Biosoftware). Only exonic non-synonymous mutations were analyzed.
Statistical analyses
The Student t-test, chi-squared test and Kaplan-Meier curves were applied using the R-project (3.1.2 version, BiostaGV website, hosted by the Institute for Statistics and Mathematics of Wirtschaftuniversität Wien, Austria). Results were considered statistically significant if the P value was less than 0.05, and each value was expressed plus or minus the standard deviation. Overall survival was defined as the period between diagnosis of myelofibrosis or, when indicated, initiation of pegylated interferon-α2a treatment and last visit or death. Leukemia-free survival was defined as survival without transformation to acute leukemia.
Univariate analyses were performed based on either an exact Fisher test or Wilcoxon rank sum test. Variables that were found to be associated with the outcome at the 10% level were then introduced into a multivariate logistic model.
Results
Patients’ characteristics
Sixty-two patients with primary myelofibrosis (n=29, 46.8%) or secondary myelofibrosis (n=33, 53.2%) were included in this study. The median age of these patients at the time their myelofibrosis was diagnosed was 66 years old (range, 33–81) and the mean interval between the diagnosis of myelofibrosis and the beginning of pegylated interferon-α2a treatment was 19.1 months. Forty-two patients (68%) had been previously treated and 40 of them (95%) had received hydroxyurea. The patients’ characteristics are summarized in Table 1.
Table 1.
Characteristics of the patients and their disease.
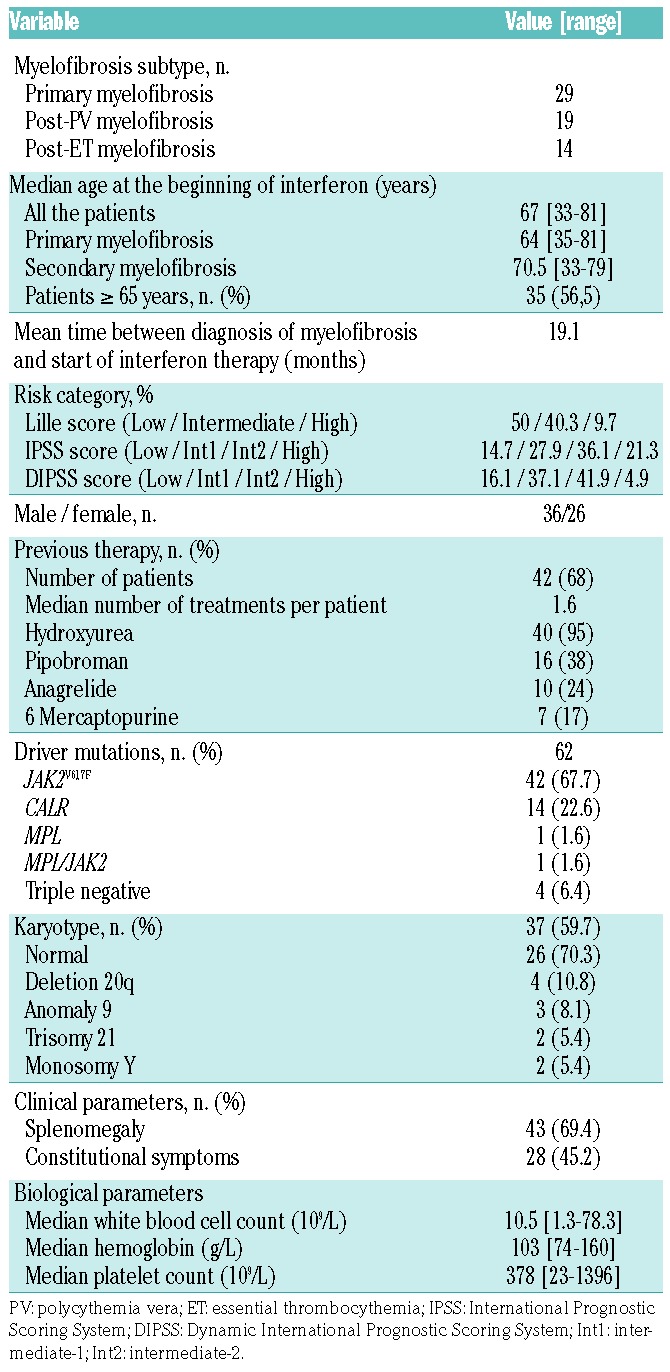
At the time of starting treatment with pegylated interferon-α2a, the median age of the patients with primary myelofibrosis was 64 years, whereas that of the patients with secondary myelofibrosis was 70.5 years old. The majority of the patients were over 65 years old (35/62, 56.5%). The male/female sex ratio was 1.38. Splenomegaly was present in 43 patients (69.4%), constitutional symptoms were documented in 28 (45.2%). More than two-thirds of the patients (44/62, 71%) were in a proliferative phase (leukocytosis and/or thrombocytosis). However, 36 patients were anemic (58.1%) and 13 were transfusion-dependent (36%). Most of the patients were classified in the “Intermediate-2” risk category according to International Prognostic Scoring System (IPSS) or Dynamic IPSS (DIPSS) scores.
The mutational status for driver mutations was identified for all patients: 42 (68%) had JAK2V617F, 14 had CALR exon 9 mutation (9 with type 1, 3 with type 2, and 2 with other mutations), one had MPLW515, four were triple-negative (3NEG) and one had coexisting JAK2 and MPL mutations. Karyotype was available for 37 (59.7%) patients, and was normal in 70.3% (Table 1).
Survival and leukemic transformation
The median follow-up after starting treatment with pegylated interferon-α2a was 58 months (range, 9–107), whereas the median follow-up after having been diagnosed with myelofibrosis was 69.6 months (range, 10–178). The median duration of pegylated interferon-α2a treatment was 39 months (range, 6–107).
At the time of the analysis, 30 patients (48.4%) were still alive. The median overall survival of the cohort was 7.4 years from the diagnosis of myelofibrosis whereas the median leukemia-free survival had not been reached (Figure 1A,B). The 5-year actuarial survival rate for the whole cohort was 69.4% from diagnosis and 54.8% from the first prescription of pegylated interferon-α2a. The duration of pegylated interferon-α2a therapy had a significant impact on overall survival: the median overall survival was 30 months in patients who received less than 2 years of treatment compared to 70 months for patients who received the drug for more than 2 years (P<0.0001).
Figure 1.

Survival of the whole study cohort. (A) Overall and (B) leukemia-free survival of the whole cohort and survivals according to the prognostic (C) Lille and (D) DIPSS scores.
As expected, the Lille and DIPSS scores differentiated patients treated with pegylated interferon-α2a in terms of overall survival (Figure 1C,D). However, the median survival observed in this cohort was clearly longer than that reported in the reference cohorts used to establish the prognostic scores, especially in higher risk categories: according to the Lille score (8.9 versus 7.75 years for low risk, 5.42 versus 2.17 years for intermediate rsik and 4.46 versus 1.08 years for high-risk) and to the DIPSS score (6.9 versus 4 years for intermediate-2 and 4.58 versus 1.5 years for high-risk patients).20,21 For patients with IPSS intermediate-2 or high-risk, the 5-year actuarial survival rate was 60% from diagnosis and 48.6% from the first prescription of interferon. We did not observe any differences between patients with primary and secondary myelofibrosis with regards to median overall survival (7.4 versus 7 years, P=0.82) or leukemia-free survival (not reached for both, P=0.95). The type of driver mutation had a statistically significant impact on survival: the median overall survival was 13.5 years for CALR-mutated patients compared to 7 years for JAK2-mutated patients (P<0.0001).
Causes of death were documented in 29/32 patients (90.6%): eight had a secondary malignancy including transformation to AML in seven and secondary cancer in one, seven died of complications of myelofibrosis/cytopenia, five transplanted patients had fatal graft-versus-host disease (GvHD), five had cardiovascular events and four died of infections.
Overall, the disease evolved to acute myeloid leukemia in eight patients (13%), only one of whom was alive at the time of the analysis. Transformation to AML occurred during pegylated interferon-α2a treatment in three patients at a median time of 1.2 years after initiation of pegylated interferon-α2a therapy (1.3 years since the diagnosis of myelofibrosis). In five patients, the transformation to acute myeloid leukemia occurred after discontinuation of pegylated interferon-α2a, at a median of 4.2 years after initiation of interferon and 6.8 years since the diagnosis of myelofibrosis (the median duration of interferon treatment in these 5 patients was 2.1 years).
Discontinuation of pegylated interferon-α2a
At the time of analysis, 16 patients (25.8% of the entire cohort, 53.3% of the living patients) were still being treated with pegylated interferon-α2a. Forty-five patients (72.6%) had discontinued interferon treatment: 25 (55.6%) due to resistance and 20 (44.4%) due to intolerance. Resistance was defined by myelofibrosis progression (n=19), transformation to acute myeloid leukemia (n=3) or failure of the disease to improve (n=3). Intolerance included occurrence of new cytopenia (n=8), psychiatric complications (n=6), fatigue (n=2), cutaneous porphyria (n=1), type 2 diabetes mellitus (n=1) or other (n=2). The median duration of pegylated interferon-α2a treatment was 20 months in patients with resistance compared to 12 months in those with intolerance. Patients developing intolerance to pegylated interferon-α2a had longer median overall survival and leukemia-free survival than patients with resistance (P=10−5 and P=0.048, respectively) (Figure 2A,B).
Figure 2.

Survival according to treatment status. Kaplan-Meier estimated (A) overall and (B) leukemia-free survival differentiating patients who were still being treated with pegylated-interferon from patients who had stopped interferon because of intolerance or resistance.
Of the 45 patients who stopped interferon treatment, 15 (33.3%) were given ruxolitinib, seven (15.6%) underwent allogeneic stem cell transplantation (ASCT) and 23 patients (51.1%) were treated with different drugs or received no further medicine (Figure 3). The median survival after cessation of pegylated interferon-α2a was 17 months (range, 3–62). The median survival of patients who received ruxolitinib was 22 months compared to 14 months for those who did not (P=0.12) or 10 months for patients who underwent ASCT (P=0.003).
Figure 3.

Patients’ treatment. ASCT: allogeneic stem cell transplantation; disc: discontinuation (of Peg-Ifn); dur: duration; FU: follow-up; m: months; n: number; Peg-Ifn: pegylated-interferon.
Nineteen patients (30.6%) received an erythropoietin-stimulating agent during interferon therapy; we did not observe that these patients, compared to those not given such agents, had increased resistance to pegylated interferon-α2a, greater occurrence of acute myeloid leukemia or a difference in overall or leukemia-free survival.
Evolution of the allele burden of driver mutations
The median mutant allele burdens at the time of starting pegylated interferon-α2a treatment, studied in 31 JAK2-mutated and eight CALR-mutated patients were 66.8% (range, 8.9–98.3) and 41.2% (range, 32–46.1), respectively.
The JAK2V617F allele burden was quantified serially in 27/31 patients. In this group, the median allele burden prior to pegylated interferon-α2a treatment was 57.3%; the burden remained stable during the first year and then decreased to 47.1% at 24 months and 29% at 36 months. We observed a decrease of mutant allele burden with a more than 10% reduction in 17/27 patients (63%), more than 20% in 15/27 patients (55.6%), and more than 50% in 10/27 patients (37%). Four patients (15%) achieved a reduction of more than 95%, including two patients who had complete molecular responses (below the detection threshold of 0.1% in our assay). Among the other patients, 7/27 (26%) had a stable allele burden (±10%) and three patients had a more than 10% increase in allele burden (Figure 4). We did not observe any difference of outcome (death or acute myeloid leukemia evolution) between patients whose JAK2V617F allele burden did or did not decrease.
Figure 4.

Variations of the JAK2V617F allele burden during the follow-up. Relative variation of the JAK2V617F allele burden for each of the 27 patients for whom sequential testing was done.
Sequential quantification of mutant CALR allele burden was available in only four patients. The median value remained essentially stable, altering from 42.4% to 46.8%. Only one patient experienced a reduction of mutant CALR allele burden, which decreased by 33%.
Impact of non-driver mutations
Of the 49 patients analyzed with targeted next-generation sequencing, 28 (57.1%) carried at least one additional mutation different from the driver mutation. Overall 44 mutations were identified (1.6 per patient) in 16 different genes (Figure 5). Of these mutations, 47% affected epigenetic regulators, 21% signaling and 16% splicing or other categories (Figure 5). The most frequent mutations involved ASXL1 and TET2 genes (7 cases each). The number of patients harboring non-driver mutations was similar between JAK2-mutated (21/34, 61.8%) and CALR-mutated (6/12, 50%) patients (P=0.51). Additional mutations were found in 68% (23/34) of patients who discontinued pegylated interferon-α2a treatment (9/15, 60% for intolerance and 14/19, 74% for resistance) compared to only 33% (5/15) of patients who remained on pegylated interferon-α2a treatment (P=0.02).
Figure 5.

Non-driver mutations identified by next-generation sequencing among 49 tested patients. The black color indicates high molecular risk (HMR) mutations. (A) Number of patients with each mutation; (B) number of additional mutations identified per patient. The percentages correspond to the proportion of HMR mutations among additional mutations.
Patients with at least one non-driver mutation had shorter overall survival than those with only driver mutations (6.1 years versus not reached, P=0.06) (Figure 6A). The same was true for leukemia-free survival (not reached in both groups, P=0.026) (Figure 6B). In detail, leukemia-free survival was significantly different between patients carrying no (median not reached), one (median not reached) or several additional mutations (median 6.7 years) (P=0.026). A similar trend was observed for overall survival (not reached, 7 years and 6 years, respectively), but the difference did not reach statistical significance.
Figure 6.

Survival according to non-driver mutation status. (A) Overall survival and (B) leukemia-free survival according to the presence of at least one mutation. (C) Overall survival and (B) leukemia-free survival according to the presence of one of the high molecular risk mutations. High molecular risk is defined by the presence of one of the five following mutations: ASXL1, SRSF2, EZH2 or IDH1/2.
Nine patients (18% of the tested patients, 32% of those with non-driver mutations) carried at least one of the mutations belonging to the high molecular risk (HMR) group: six patients had one mutation and three had two or more mutations. Carrying a mutation in these HMR genes was associated with reduced overall and leukemia-free survival but, surprisingly, HMR mutations did not have a stronger impact than any other additional mutations (Figure 6C,D).
HMR mutations were found in five (24%) JAK2-mutated patients, three (50%) CALR-mutated patients, and one triple-negative patient (P=0.32). Mutations in ASXL1 were identified in three (14%) JAK2–mutated and three (50%) CALR-mutated patients (P=0.1). The presence of ASXL1 mutations had no impact on either the leukemia-free survival or the overall survival of CALR-positive patients, and it was not relevant whether the mutation pattern was CALR-positive/ASXL1-negative or CALR-negative/ASXL1-positive.
Discussion
This study reports long-term outcomes of the largest cohort of interferon-treated myelofibrosis patients to our knowledge. The first finding is an unexpectedly long median overall survival of 89 months after myelofibrosis diagnosis in this population of patients, of whom the majority had intermediate or high-risk disease according to the DIPSS (84%) and Lille (50%) scoring systems. For these categories, the observed overall survival in this study was clearly longer than that expected according to the DIPSS (6.9 versus 4 years for intermediate-2 risk patients and 4.58 versus 1.5 years for high-risk patients) and the Lille (5.42 versus 2.17 years for intermediate-risk and 4.46 versus 1.08 years for high-risk) score categories.20,21 The 5-year actuarial survival rate was 69.4% from diagnosis for the whole cohort, and 60% for patients with intermediate-2 or high risk according to the IPSS. These findings suggest a positive impact of interferon therapy on both overall and leukemia-free survivals in addition to the high rate of clinical and hematologic responses that we previously reported.17,18 By comparison, two prospective trials (COMFORT-I and II) have reported the results of the use of ruxolitinib in myelofibrosis patients with IPSS intermediate-2 or high-risk score.8,9 In a recent actualization after 5 years of the COMFORT-II study,22 Harrison et al. observed an overall survival of 59.4% (median not reached). However, the study was not originally designed to assess survival, and these results have been debated.12,23,24
At the time of the analysis, 16 patients (25.8%) were still being treated with pegylated interferon-α2a whereas 45 (72.6%) had stopped treatment, 25 (55.6%) due to resistance and 20 (44.4%) due to intolerance. The overall survival of patients who continued their therapy was longer than that of patients who stopped (P<10−6). The patients who were identified as resistant to pegylated interferon-α2a had the worst survival, suggesting that resistance to interferon is a marker of aggressive disease. However, these results should be interpreted in the light of subsequent treatment that clearly affected survival. For example, patients who received ruxolitinib after discontinuing pegylated interferon-α2a had a median survival of 22 months compared to 14 months for patients treated with other therapies.
All seven patients who underwent ASCT died within a median of 10 months, mainly from GvHD (5/7 patients). Although numbers are small, this is in stark contrast with previous studies of ASCT in myelofibrosis patients reporting 5-year overall survival rates between 41 and 55%.25,26 There is no available study of the impact of interferon on the outcome of ASCT in Philadelphia chromosome-negative myeloproliferative neoplasms. However, in chronic myeloid leukemia, Pigneux and colleagues showed that interferon therapy increased the incidence of GvHD (65 versus 38%, P=0.01) and decreased disease-free and overall survival rates at 5 years (33 versus 41%, P=0.005% and 41 versus 55%, P=0.002, respectively).27 Collectively, our results suggest that interferon therapy should not be initiated in patients with myelofibrosis who have a high probability of undergoing ASCT within a few months.
Interferon has been shown to decrease the mutant allele burden of JAK2 or CALR driver mutations in both polycythemia vera and essential thrombocythemia. Accordingly, we observed a greater than 10% reduction of the JAK2V617F allele burden in 63% of the patients and a greater than 95% reduction in four (15%). This molecular response in myelofibrosis patients is, however, less than the 89.6% JAK2V617F allele burden reduction and 24% complete molecular responses reported by Kiladjian et al. in patients with polycythemia vera.28,29 Very good molecular responses were also recently reported by Masarova et al. after long-term follow-up of pegylated interferon-α2a therapy in patients with polycythemia vera or essential thrombocythemia.30 Among 63 JAK2V617F-positive patients, they observed a molecular response rate of 63% (including 16% complete molecular responses), with a reduction of the median mutant allele burden from 41 to 12%. The reduction of the JAK2V617F allele burden did not have any impact on overall survival or leukemia-free survival in our study. Silver et al. also recently reported such an absence of correlation between molecular response and clinical outcomes in a series of 30 myelofibrosis patients treated with interferon.31 Such a level of response seems unique to interferon since in the COMFORT-II study, ruxolitinib achieved a greater than 20% reduction of JAK2V617F burden in only 30% of the patients (compared to 55.6% in our series), and none reached a complete molecular response.22 We were also able to evaluate the CALR molecular response in a few patients and found that only one patient had a significant decrease in CALR mutant allele burden. This is in contrast with the results obtained by Verger et al. in patients with essential thrombocythemia in whom a reduction of the median mutant CALR allele burden from 41 to 26% was observed in a cohort of 31 patients.32
Besides the classical driver mutations, we identified at least one additional mutation in 28/49 patients with a mean of 1.6 additional mutations per patient. The presence of at least one mutation significantly reduced leukemia-free survival and the presence of more than one mutation was associated with a decrease of both overall survival and leukemia-free survival. Such a negative impact of additional mutations on survival is in line with previous findings in myelofibrosis patients.7 Vannucchi et al. have reported that mutations in ASXL1, EZH2, SRSF2 or IDH1/2 carried a stronger adverse prognostic impact, defining a high molecular risk profile.6 In our study, the presence of these HMR mutations affected outcome, but not more than other non-HMR mutations did. ASXL1 mutations alone did not significantly affect survival, but this may be due to the limited number of patients carrying this mutation in our series. However, our data could indicate that the higher risk associated with mutations affecting ASXL1, EZH2, SRSF2 or IDH1/2 could be in part reduced by interferon therapy. Another possible explanation for the discrepancy between our results regarding HMR mutations and those published by Vannucchi and colleagues is that our cohort of patients included a higher proportion with secondary myelofibrosis. Indeed, Rotunno et al. reported that only SRSF2 mutations affected survival in secondary myelofibrosis.33 Lastly, additional mutations were more frequently found in patients intolerant of or resistant to interferon, a finding in agreement with the results published by Silver et al. indicating that additional mutations are more frequent in patients who could not remain on pegylated interferon therapy.31 They also reported that a higher number of mutations, including HMR mutations, is associated with poorer response to interferon.
The limitations of our study include the absence of evaluation of symptoms and measurements of cytokine levels. Such studies were not possible due to the lack of a validated specific tool in French for symptom assessment in myelofibrosis at the time the study was initiated, and to the absence of stored plasma or serum given the observational nature of the study. Other important information would have been gained from sequential evaluation of bone marrow biopsies since it has been shown that interferon therapy may reduce fibrosis in selected cases.34 Although all patients had a biopsy for the diagnosis of their disease, investigators did not perform new biopsies after interferon therapy, so the impact of the treatment on this aspect of the disease could not be studied in this cohort of patients.
In conclusion, while we have previously reported the clinical and hematologic efficacy of pegylated interferon-α2a treatment in myelofibrosis patients, this long-term analysis suggests that interferon therapy may also improve overall survival and leukemia-free survival. In contrast, interferon therapy before ASCT could increase the risk of GvHD and should probably be avoided in this context. Intolerance of or resistance to interferon identifies a group of patients with a dismal outcome, as does the presence of additional mutations. These results indicate that even in the ruxolitinib era, the place of pegylated interferon-α2a should be discussed in patients with myelofibrosis, the optimal target population possibly being high-risk myelofibrosis patients without the prospect of ASCT and with proliferative disease.
Supplementary Material
Acknowledgments
The authors would like to thank all the clinical research team members who participated in data collection. We also thank the “Ligue contre le cancer” for their continuous support of research into myeloproliferative neoplasms at Brest Hospital. This study is part of the “CTIM3” project supported by a grant from the French Cancer Institute (INCa), TRANSLA13-140 and of the FIMBANK project (INCa BCB 2013).
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/3/438
References
- 1.Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): consensus on terminology by the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT). Leuk Res. 2007;31(6):737–740. [DOI] [PubMed] [Google Scholar]
- 2.Tefferi A. Primary myelofibrosis: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91(12):1262–1271. [DOI] [PubMed] [Google Scholar]
- 3.Cervantes F, Tassies D, Salgado C, Rovira M, Pereira A, Rozman C. Acute transformation in nonleukemic chronic myeloproliferative disorders: actuarial probability and main characteristics in a series of 218 patients. Acta Haematol. 1991;85(3):124–127. [DOI] [PubMed] [Google Scholar]
- 4.Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. [DOI] [PubMed] [Google Scholar]
- 5.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861–1869. [DOI] [PubMed] [Google Scholar]
- 7.Guglielmelli P, Lasho TL, Rotunno G, et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. 2014;28(9):1804–1810. [DOI] [PubMed] [Google Scholar]
- 8.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. [DOI] [PubMed] [Google Scholar]
- 9.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vannucchi AM, Kantarjian HM, Kiladjian JJ, et al. ; COMFORT Investigators. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica. 2015;100(9):1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Passamonti F, Vannucchi AM, Cervantes F, et al. Ruxolitinib and survival improvement in patients with myelofibrosis. Leukemia. 2015;29(3):739–740. [DOI] [PubMed] [Google Scholar]
- 12.Cervantes F, Pereira A. Does ruxolitinib prolong the survival of patients with myelofibrosis? Blood. 2016;129(7):832–837. [DOI] [PubMed] [Google Scholar]
- 13.Alchalby H, Kröger N. Allogeneic stem cell transplant vs. Janus kinase inhibition in the treatment of primary myelofibrosis or myelofibrosis after essential thrombocythemia or polycythemia vera. Clin Lymphoma Myeloma Leuk. 2014;14(Suppl):S36–41. [DOI] [PubMed] [Google Scholar]
- 14.Kröger NM, Deeg JH, Olavarria E, et al. Indication and management of allogeneic stem cell transplantation in primary myelofibrosis: a consensus process by an EBMT/ELN International Working Group. Leukemia. 2015;29(11):2126–2133. [DOI] [PubMed] [Google Scholar]
- 15.Kröger N, Giorgino T, Scott BL, et al. Impact of allogeneic stem cell transplantation on survival of patients less than 65 years of age with primary myelofibrosis. Blood. 2015;125(21):3347–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robin M, Porcher R, Wolschke C, et al. Outcome after transplantation according to reduced-intensity conditioning regimen in patients undergoing transplantation for myelofibrosis. Biol Blood Marrow Transplant. 2016;22(7):1206–1211. [DOI] [PubMed] [Google Scholar]
- 17.Ianotto JC, Kiladjian JJ, Demory JL, et al. PEG-IFN-alpha-2a therapy in patients with myelofibrosis: a study of the French Groupe d’Etudes des Myelofibroses (GEM) and France Intergroupe des syndromes Myéloprolifératifs (FIM). Br J Haematol. 2009;146(2):223–225. [DOI] [PubMed] [Google Scholar]
- 18.Ianotto JC, Boyer-Perrard F, Gyan E, et al. Efficacy and safety of pegylated-interferon α-2a in myelofibrosis: a study by the FIM and GEM French cooperative groups. Br J Haematol. 2013;162(6):783–791. [DOI] [PubMed] [Google Scholar]
- 19.Lippert E, Boissinot M, Kralovics R, et al. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood. 2006;108(6):1865–1867. [DOI] [PubMed] [Google Scholar]
- 20.Dupriez B, Morel P, Demory JL, et al. Prognostic factors in agnostic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood. 1996;88(3):1013–1018. [PubMed] [Google Scholar]
- 21.Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703–1708. [DOI] [PubMed] [Google Scholar]
- 22.Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30(8):1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barosi G, Zhang MJ, Gale PR. Does ruxolitinib improve survival of persons with MPN-associated myelofibrosis? Should-it? Leukemia. 2014;28(11):2267–2270. [DOI] [PubMed] [Google Scholar]
- 24.Martí-Carvajal AJ, Anand V, Solà I. Janus kinase-1 and Janus kinase-2 inhibitors for treating myelofibrosis. Cochrane Database Syst Rev. 2015;(4):CD010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scott BL, Gooley TA, Sorror ML, et al. The Dynamic International Prognostic Scoring System for myelofibrosis predicts outcomes after hematopoietic cell transplantation. Blood. 2012;119(11):2657–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta V, Malone AK, Hari PN, et al. Reduced-intensity hematopoietic cell transplantation for patients with primary myelofibrosis: a cohort analysis from the center for international Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2014;20(1):89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pigneux A, Tanguy ML, Michallet M, et al. ; Société Française de Greffe de Moelle. Prior treatment with alpha interferon does not adversely affect the outcome of allogeneic transplantation for chronic myeloid leukaemia. Br J Haematol. 2002;116(1):193–201. [DOI] [PubMed] [Google Scholar]
- 28.Kiladjian JJ, Cassinat B, Turlure P, et al. High molecular response rate of polycythemia vera patients treated with pegylated interferon alpha-2a. Blood. 2006;108(6):2037–2040. [DOI] [PubMed] [Google Scholar]
- 29.Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. 2008;112(8):3065–3072. [DOI] [PubMed] [Google Scholar]
- 30.Masarova L, Patel KP, Newberry KJ, et al. Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: a post-hoc, median 83 months follow-up of an open-label, phase 2 trial. Lancet Haematol. 2017;4(4):e165–e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silver RT, Barel AC, Lascu E, et al. The effect of initial molecular profile on response to recombinant interferon-α (rIFNα) treatment in early myelofibrosis. Cancer. 2017;123(14):2680–2687. [DOI] [PubMed] [Google Scholar]
- 32.Verger E, Cassinat B, Chauveau A, et al. Clinical and molecular response to interferon-α therapy in essential thrombocythemia patients with CALR mutations. Blood. 2015;126(24):2585–2591. [DOI] [PubMed] [Google Scholar]
- 33.Rotunno G, Pacilli A, Artusi V, et al. Epidemiology and clinical relevance of mutations in post polycythemia vera and post essential thrombocythemia myelofibrosis: a study on 359 patients of the AGIMM group. Am J Hematol. 2016;91(7):681–686. [DOI] [PubMed] [Google Scholar]
- 34.Pizzi M, Silver RT, Barel A, Orazi A. Recombinant interferon-α in myelofibrosis reduces bone marrow fibrosis, improves its morphology and is associated with clinical response. Mod Pathol. 2015;28(10):1315–1323. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.