

Abstract
Key points
An increase in the excitability of GABAergic cells has typically been assumed to decrease network activity, potentially producing overall anti‐epileptic effects. Recent data suggest that inhibitory networks may actually play a role in initiating epileptiform activity.
We show that activation of GABAergic interneurons can elicit synchronous long‐lasting network activity.
Specific interneuron subpopulations differentially contributed to GABA network synchrony, indicating cell type‐specific contributions of interneurons to cortical network activity.
Interneurons may critically contribute to the generation of aberrant network activity characteristic of epilepsy, warranting further investigation into the contribution of distinct cortical interneuron subpopulations to the propagation and rhythmicity of epileptiform activity.
Abstract
In the presence of the A‐type K+ channel blocker 4‐aminopyrdine, spontaneous synchronous network activity develops in the neocortex of mice of either sex. This aberrant synchrony persists in the presence of excitatory amino acid receptor antagonists (EAA blockers) and is considered to arise from synchronous firing of cortical interneurons (INs). Although much attention has been given to the mechanisms underlying this GABAergic synchrony, the contribution of specific IN subtypes to the generation of these long‐lasting discharges (LLDs) is incompletely understood. We employed genetically‐encoded channelrhodopsin and archaerhodopsin opsins to investigate the sufficiency and necessity, respectively, of activation of parvalbumin (PV), somatostatin (SST) and vasointestinal peptide (VIP)‐expressing INs for the generation of synchronous neocortical GABAergic discharges. We found light‐induced activation of PV or SST INs to be equally sufficient for the generation of LLDs, whereas activation of VIP INs was not. By contrast, light‐induced inhibition of PV INs strongly reduced LLD initiation, whereas suppression of SST or VIP IN activity only partially attenuated LLD magnitude. These results suggest neocortical INs perform cell type‐specific roles in the generation of aberrant GABAergic cortical network activity.
Keywords: neocortex, interneurone, synchronization
Key points
An increase in the excitability of GABAergic cells has typically been assumed to decrease network activity, potentially producing overall anti‐epileptic effects. Recent data suggest that inhibitory networks may actually play a role in initiating epileptiform activity.
We show that activation of GABAergic interneurons can elicit synchronous long‐lasting network activity.
Specific interneuron subpopulations differentially contributed to GABA network synchrony, indicating cell type‐specific contributions of interneurons to cortical network activity.
Interneurons may critically contribute to the generation of aberrant network activity characteristic of epilepsy, warranting further investigation into the contribution of distinct cortical interneuron subpopulations to the propagation and rhythmicity of epileptiform activity.
Introduction
Synchronous epileptiform activity, in the form of long‐lasting discharges (LLDs), occurs spontaneously and propagates through the neocortex in the presence of the A‐type K+ channel blocker 4‐aminopyridine (4‐AP) (Avoli & Perreault, 1987; Avoli et al. 1988). Optogenetic studies in the entorhinal cortex indicate that stimulation of somatostatin‐ (SST) or parvalbumin (PV) INs evokes epileptiform events similar to spontaneous 4‐AP induced LLDs (Shiri et al. 2015; Yekhlef et al. 2015). LLDs persist when excitatory glutamatergic neurotransmission is blocked with 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX) and d‐(–)‐2‐amino‐5‐phosphonopentanoic acid (d‐APV) (EAA blockers) and they are suppressed by the GABAA antagonist bicuculline (Aram et al. 1991; Michelson & Wong, 1991; Avoli et al. 1994). This suggests that the events arise from synchronous activity of inhibitory interneurons (INs) and represent propagating GABA‐mediated excitation (Perreault & Avoli, 1992; Lamsa & Kaila, 1997; Williams & Hablitz, 2015). Much of the work carried out aiming to identify and characterize the mechanism underlying this GABAergic network synchrony suggests an accumulation of extracellular K+ produces a shift in the Cl− equilibrium as a result of hypofunction of the K+‐Cl‐ cotransporter KCC2, causing activation of GABAA receptors to produce excitatory currents at more hyperpolarized membrane potentials than normal. (Morris et al. 1996; Louvel et al. 2001; Hamidi et al. 2015). In the neonatal hippocampus, spontaneous LLDs as a result of GABAergic network activity occur without pharmacological manipulations (Khazipov et al. 1997). In the present study, we used the 4‐AP plus EAA blockers model to investigate the role of specific IN classes in generating and regulating LLDs in the neocortex.
The cortical GABAergic IN population is composed of distinct subgroups that are often identified by the expression of specific cell markers (Hendry et al. 1989; Somogyi & Klausberger, 2005; Yuste, 2005). The most abundant subpopulations (i.e. those expressing PV or SST) each account for 30–50% of the cortical IN population (Lee et al. 2010; Miyoshi et al. 2010; Xu et al. 2010). These cell types have been shown to exhibit distinct physiological and morphological characteristics, as well as unique synaptic connectivity patterns, allowing them to differentially contribute to network activity (Kubota et al. 1994; Toledo‐Rodriguez et al. 2004, 2005; Miyoshi et al. 2007; Uematsu et al. 2008; Rudy et al. 2011). We have recently shown that 4‐AP alters the action potential (AP) and repetitive firing properties of Martinotti cells and fast‐spiking basket cells in neocortex, the most common SST‐ and PV‐expressing cells, respectively, making these cells prime candidates for involvement in LLD initiation (Williams & Hablitz, 2015). Vasointestinal peptide (VIP)‐expressing INs also comprise a substantial portion of the cortical IN population (Vucurovic et al. 2010; Rudy et al. 2011; Zeisel et al. 2015). A unique property of VIP cells is their ability to inhibit other INs, particularly SST‐expressing cells, thereby facilitating network disinhibition (von Engelhardt et al. 2007; Lee et al. 2013; Pfeffer et al. 2013; Pi et al. 2013). We therefore characterized LLD responses in these three IN classes and, using genetically encoded opsins driven by IN specific promoters, assessed the effect of their activation and silencing on LLDs. We found PV‐expressing cells to be both necessary and sufficient for the generation of LLDs, whereas SST and VIP were found to only partially contribute to LLD generation, with VIP cells showing the least involvement. These results demonstrate the cell type‐specific contribution of cortical INs to network activity and support a role for INs in the generation of aberrant neuronal synchrony.
Methods
Ethical approval
All of the experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals with protocols approved by the University of Alabama at Birmingham Institutional Care and Use Committee. Animals had ad libitum access to food and water. All available measures were taken to minimize pain or discomfort for research subjects.
Animals
Experiments were performed on mouse lines with IN subtype‐specific expression of genetically encoded opsins, achieved using the cre‐lox system. All mouse strains were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Homozygous SST‐IRES‐Cre (Ssttm2.1(cre)Zjh/J; stock no: 013044), PV‐Cre (B6;129P2‐Pvalbtm1(cre)Arbr/J; stock no: 008069) or Vip‐IRES‐Cre (Viptm1(cre)Zjh/J; stock no: 010908) mice were crossed with homozygous Ai32 (B6;129S‐Gt(ROSA)26Sortm32(CAG‐COP4*H134R/EYFP)HZE/J; stock no: 012569) or Ai35D (B6;129S‐Gt(ROSA)26Sortm35.1(CAG‐aop3/GFP)Hze/J; stock no: 012735) mice to produce animals with cell type‐specific expression of channelrhodopsin (ChR) or archaerhodopsin (Arch), respectively.
Slice preparation
Acute cortical slices containing the sensorimotor cortex were prepared from 6–10‐week‐old mice of either sex from each strain. Data from neurons from males and females were combined because no sex differences were observed. Animals were anaesthetized with isoflurane and decapitated. The brain was quickly removed and immediately placed in ice‐cold oxygenated (95% O2/5% CO2, pH 7.4) cutting solution consisting of (in mm): 135 N‐methyl‐d‐glucamine, 23 NaHCO3, 1.5 KH2PO4, 0.4 ascorbic acid, 1.5 KCl, 0.5 CaCl2, 3.5 MgCl2 and 10 d‐glucose (Tanaka et al. 2008). Coronal brain slices (300 μm thick) were made using a Microm HM 650 vibratome (Microm, Walldorf, Germany). Slices were stored in artificial CSF (aCSF) containing (in mm) 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 3.5 KCl, 2.0 CaCl2, 2.0 MgCl2 and 10 d‐glucose at 37°C for 45 min, and then kept at room temperature until recording for a minimum of 1 h.
Whole‐cell recording
Individual slices were transferred to a submerged recording chamber mounted on the stage of a Zeiss Axio Examiner D1 microscope (Carl Zeiss Inc., Thornwood, NY, USA), equipped with Dodt contrast optics, a 40× water immersion lens and infrared illumination to view neurons in the slices. The recording chamber was continuously perfused with oxygenated aCSF (3 mL min−1 at 30°C). Borosilicate patch electrodes controlled by a Sutter MP‐225 motorized micromanipulator (Sutter Instruments, Novato, CA, USA) that had an open tip resistance of 3–6 MΩ when filled with an intracellular solution containing (in mm): 125 K‐gluconate, 10 KCl, 10 Hepes, 10 creatine‐PO4, 2 Mg‐ATP, 0.2 Na‐GTP and 0.5 EGTA, which had an adjusted pH and osmolarity of 7.3 and 290 mosmol, respectively. Tight seals of 1 GΩ or greater were obtained under visual guidance before breaking into whole‐cell mode. Neurons in layer (L) 2/3 were located by their proximity to the pial surface. Using responses to depolarizing and hyperpolarizing pulses, cell types were readily discriminated based on AP half‐width, input resistance and repetitive firing properties.
Data acquisition and analysis
Whole‐cell recordings were obtained using an ELC‐03XS npi bridge balance amplifier (npi Electronic GmbH, Tamm, Germany). Signals were acquired using Clampex software with a Digidata 1322A interface (Molecular Devices LLC, Sunnyvale, CA, USA). Evoked responses were digitized at 10 kHz, filtered at 2 kHz, and analysed using Clampfit, version 9.0 (Molecular Devices). Spontaneous LLDs were identified using the event detection feature of Clampfit. The minimum duration for event detection was set at 250 ms and the minimum amplitude was set at 3–5 times the root mean square amplitude of baseline noise. The amplitude, duration and area under the curve (AUC) of spontaneous and evoked activity were assessed. Amplitude was measured as the maximum peak of the response relative to baseline, duration was defined as the period from the onset of stimulation until return to resting membrane potential, and the AUC was calculated over the response duration. Direct current injection was used to hold cells at their resting potential for comparison of evoked and spontaneous events. Cells were held at −60, −70, −80 and −90 mV for characterization of LLDs, with analysis being performed at −90 mV unless otherwise noted. For evoked responses that did not return to baseline before the onset of activity induced by light offset in Arch expressing animals, duration and area were calculated over the period before light offset.
Light stimulation
Light‐activation of opsins was achieved through full‐field illumination of the tissue via fluorescent light (Xcite 120Q; Excelitas Technologies, Waltham, MA, USA) passed through the microscope objective. Light was passed through a YFP filter (YFP‐2427B‐000, Semrock Inc., Rochester, NY, USA; excitation: 500/24‐25, emmission: 542/27‐25) for activation of ChR, or through a mCherry filter (mCherry‐C‐000; Semrock Inc.; excitation 562/40, emmission: 641/75) for activation of Arch. Unless otherwise noted, a 10 ms light pulse was used for activation of ChR. Arch was activated for 1100 ms, beginning 100 ms before electrical stimulation. Light exposure was regulated using a VS25 optical shutter (Vincent Associates, Rochester, NY, USA) controlled by a Uniblitz VCM‐D1 shutter driver (Vincent Associates). The timing of light pulses was governed by an isolated pulse stimulator (Model 2100; A‐M Systems, Sequim, WA, USA) as triggered by Clampex software. Electrically evoked synaptic responses were stimulated using a bipolar electrode consisting of Formvar insulated 25 μm nichrome wires positioned in L2/3, ∼100 μm from the recording electrode. A DS3 constant current stimulus isolator (Digitimer, Welwyn Garden City, UK) was used to generate current pulses of 30–150 μA with a duration of 100 μs. The intensity and frequency of stimulation were adjusted to match the amplitude and frequency of spontaneous epileptiform activity in each slice.
Drugs and drug application
After recording control responses in drug‐free aCSF, 4‐aminopyridine (100 μm) (4AP; Sigma, St Louis, MO, USA), CNQX (10 μm) (Abcam, Cambridge, MA, USA) and d‐APV (20 μm) (Abcam) were washed in for at least 10 min to allow spontaneous epileptiform activity to develop and stabilize (Aram et al. 1991; Avoli et al. 1994). In some experiments, bicuculline methiodide (10 μm) (Abcam), picrotoxin (100 μm) (Sigma) or SR95531‐hydrobromide (10 μm) (Tocris, Ellisville, MO, USA) was used to block GABAA mediated synaptic transmission. SCH50911 (2 μm) (Tocris) was used to block GABAB mediated transmission. All drugs were bath applied, with each neuron serving as its own control.
Experimental design and statistical analysis
Statistical and power analyses were performed using Prism, version 6 (GraphPad, La Jolla, CA, USA) and G*Power (Faul et al. 2007), respectively. Data are expressed as either dots representing each individual data point or as the mean ± SEM. Average traces shown are calculated from a minimum of three sweeps. Sample size (n) is the number of cells used for each experiment, with a minimum of three animals used per group. Comparisons were made between forms of LLD initiation and manipulation or between animal lines as described in the text. A statistical comparison of responses was performed using a one‐ or two‐tailed Student's t test or one‐way ANOVA with Tukey's multiple comparisons test. Paired t tests were used to compare different conditions within the same cell. For all tests, P < 0.05 was considered significant. Statistical power was calculated using compromise power analysis. All significant tests had a power (1 – β) of 0.85 or greater unless specifically stated otherwise.
Results
4‐AP + EAA blockers induces synchronized GABAergic network activity in PYR cells
In the present study, to isolate the contribution of IN subtypes to inhibitory network activity, the AMPA and NMDA receptor antagonists CNQX and APV (EAA blockers) were bath applied in combination with the A‐type K+ channel blocker, 4‐AP, to induce synchronous activity. Before examining the role of different IN populations in the generation of synchronized GABAergic network responses (termed LLDs), we characterized the activity induced by application of 4‐AP + EAA blockers in our mice. For all recordings, cells were initially patched in drug‐free aCSF. 4‐AP + EAA blockers were then bath applied following the recording of baseline intrinsic properties and responses to electrical stimulation. Spontaneous LLDs emerged in L 2/3 pyramidal cells (PYR) and INs several minutes following application of 4‐AP + EAA blockers and progressively developed over the next 20–30 min (Fig. 1 A). The amplitude and frequency of spontaneous activity stabilized and remained relatively consistent for the remaining duration of the experiment. As shown in the PYR specimen records in Fig. 1 B, following LLD stabilization, electrical stimulation was capable of evoking a response similar to spontaneous LLDs. Evoked responses were significantly smaller in duration and area (Amplitude – Spontaneous: 6.63 + 0.47 mV, Evoked: 5.78 + 0.56 mV; n = 58, P = 0.1324, paired t test; Duration – Spontaneous: 2513 + 142 ms, Evoked: 2135 + 166 ms; n = 58, P = 0.002, paired t test; Area – Spontaneous: 7856 + 726 mV*ms, Evoked: 4147 + 382 mV*ms; n = 58, P < 0.0001, paired t test). This suggests that LLDs represent network activity and not the intrinsic firing of the recorded cell. Spontaneous, electrically evoked and light evoked LLDs were blocked by bath application of GABA receptor antagonists (Fig. 1 C) in both PYRs (n = 5) and INs (n = 3) and had an experimental reversal potential near the chloride equilibrium in all cells (n = 52) (Fig. 1 D). This is consistent with previous data showing this activity to be GABA mediated (Aram et al. 1991; Michelson & Wong, 1991; Avoli et al. 1994).
Figure 1. Application of 4AP + EAA blockers induces GABA‐mediated synchronous network activity.
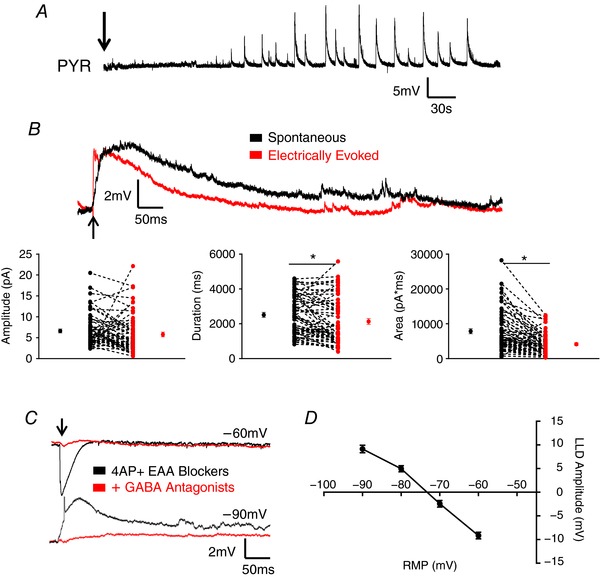
A, bath application of 4AP + EAA induces spontaneous synchronized network activity in PYRs. An arrow indicates the start of drug wash‐on. B, upper: representative traces showing spontaneous and electrically evoked LLDs in a PYR. Electrical stimulation evokes activity of similar amplitude to spontaneous LLDs but smaller in duration and AUC. An arrow indicates the time of stimulation. Lower: quantitative comparison of electrically evoked versus spontaneously occurring LLDs. Mean ± SEM are shown, as well as the results from individual cells. C, example traces of evoked LLDs in presence of 4AP + EAA blockers with and without GABA receptor antagonists. An arrow indicates the timing of stimulation. D, calculation of LLD reversal potential. * P < 0.05, paired t test. Each shape represents an individual cell. Error bars are the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
Cell type‐specific properties of spontaneous LLDs
We also aimed to characterize spontaneous LLDs on a cell type‐specific basis. Specimen records of spontaneous LLDs recorded from each cell type are shown in Fig. 2 A (left), with individual events shown on an expanded timescale in Fig. 2 A (right). Note that activity recorded from VIP INs was hyperpolarizing because the RMP of those cells was depolarized relative to the reversal potential for LLDs. LLDs produced a significantly greater number of APs in PV INs compared to all other cell types (PYR: 0.28 ± 0.10, n = 64; PV: 21.13 ± 5.03, n = 31; SST: 8.27 ± 3.95, n = 15; VIP: 0.11 ± 0.11, n = 9; P < 0.0001, one‐way ANOVA with Tukey's post hoc test) (Fig. 2 B). However, the frequency of LLDs was the same regardless of the cell type from which activity was recorded (PYR: 39.48 ± 2.28 s, n = 56; PV: 38.00 ± 3.15 s, n = 28; SST: 39.59 ± 4.93 s, n = 15; VIP: 25.89 ± 4.06 s, n = 9; P = 0.1673, one‐way ANOVA) (Fig. 2 C). Drug application caused a significant depolarization of all cell types, with a significantly larger depolarization being observed in PV cells compared to PYRs and SST INs (PYR: 1.47 ± 0.59 mV, n = 46; PV: 5.74 ± 0.76 mV, n = 36; SST: 1.98 ± 1.03 mV, n = 12; VIP: 2.16 ± 1.14 mV, n = 8; P = 0.0001, one‐way ANOVA with Tukey's post hoc test) (Fig. 2 D) and produced rhythmic spontaneous firing in PV cells (Fig. 2 A).
Figure 2. Cell type‐specific properties of spontaneous LLDs.
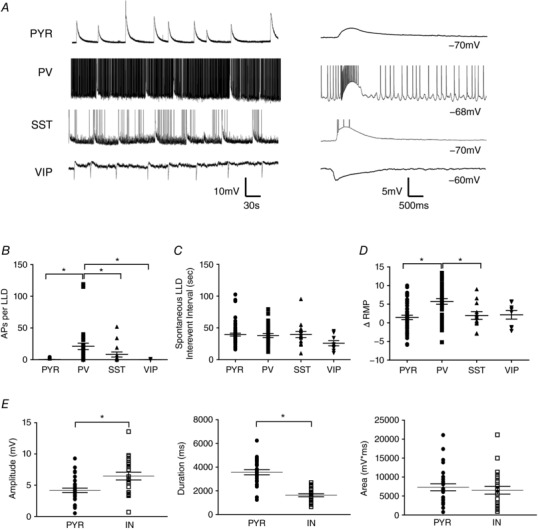
A, example traces of spontaneous LLDs recorded from a PYR cell and different IN subtypes. Continuously recorded events are shown on the left, with individual events shown on an expanded timescale to the right. LLDs recorded from VIP INs were hyperpolarizing because the RMP of those cells was depolarized relative to the LLD reversal potential. B, cell type comparison of the number of APs superimposed on spontaneous LLDs. C, spontaneous LLDs occurred with the same temporal profile in all cell types. D, application of 4AP + EAA blockers caused a depolarization of all recorded cell types. E, quantitative comparison of spontaneous LLD amplitude (left), duration (middle) and AUC (right) in PYRs and INs. * P < 0.05, Tukey's post hoc test. Each shape represents an individual cell. Error bars are the mean ± SEM.
Aside from the number of LLD‐induced APs, the magnitude of LLDs did not differ between IN subtypes; therefore, all INs were combined for further analysis and comparison with PYRs. As shown in Fig. 2 E, LLDs recorded from INs displayed a significantly larger amplitude compared to PYRs (PYRs: 4.18 ± 0.35 mV, n = 30; INs: 6.46 ± 0.62 mV, n = 23; P = 0.0014, two‐tailed t test) (Fig. 2 E, left), whereas the duration of LLDs in INs was shorter than those recorded from PYRs (PYRs: 3569 ± 214 ms, n = 30; INs: 1628 ± 123 ms, n = 23; P < 0.0001, two‐tailed t test) (Fig. 2 E, middle). As a result of these off‐setting differences, the total area (AUC) of LLDs did not significantly differ between INs and PYRs (PYRs: 7296 ± 912 mV*ms, n = 30; INs: 6534 ± 1016 mV*ms, n = 23; P = 0.5805, two‐tailed t test) (Fig. 2 E, right). Together, these results suggest LLDs are network generated events that manifest kinetics varying based on the intrinsic excitability of the cell from which they are recorded. As such, activity recorded from INs and PYRs was analysed separately to clearly identify the role of IN subtypes in LLD generation.
Activation and inhibition of SST INs
The experiments described above demonstrated the similarity between spontaneous and evoked LLDs. However, electrical stimulation excites numerous cellular elements and the exact contribution of specific cell types is unclear. Having characterized LLDs in these mice, we aimed to determine the sufficiency of SST cells to initiate synchronous network activity. Accordingly, mice expressing ChR restricted to SST‐positive cells were generated by crossing mice expressing a cre‐dependent ChR gene with SST:cre mice. The functional presence of ChR in SST cells was confirmed through whole‐cell, patch clamp recordings. SST INs were physiologically identified by their intrinsic membrane properties, namely, strongly accommodating firing patterns, and the presence of a voltage ‘sag’ upon membrane hyperpolarization, indicative of I h that is characteristically seen in SST‐expressing cells (Maccaferri & McBain, 1996; Zhu et al. 1999; Vervaeke et al. 2012; Williams & Hablitz, 2015) (Fig. 3 A, left). Additionally, opsin‐expressing cells co‐expressed YFP, allowing for visual identification of SST INs. In cortical slices from SST:ChR mice, whole field illumination of the tissue induced depolarization and AP firing in physiologically identified SST cells (Fig. 3 A, right). Light stimulation did not depolarize PYRs or PV INs in drug‐free aCSF at rest (Fig. 3 B, left). Control responses to a 10 ms light pulse at various membrane potentials for a PYR and PV IN are shown in Fig. 3 B (left, upper and lower, respectively). After application of 4AP + EAA blockers, enhanced responses were seen in both cell types (Fig. 3 B, right). Responses in all INs were associated with significant AP firing. As quantified in Fig. 3 C, the amplitude of light‐evoked responses was equivalent to spontaneous LLDs in both PYRs and INs (PYR Spon: 3.75 + 0.47 mV, PYR Light: 3.48 + 1.03 mV, n = 12; IN Spon: 6.36 + 1.23 mV, IN Light: 6.27 + 1.42 mV, n = 7; P = 0.0925, one‐way ANOVA), duration (PYR Spon: 3694 + 417 ms, PYR Light: 3345 + 391 ms, n = 12; IN Spon: 1383 + 201 ms, IN Light: 1258 + 173 ms, n = 7; P < 0.0001, one‐way ANOVA with Tukey's post hoc test) and AUC (PYR Spon: 6321 + 863 mV*ms, PYR Light: 5026 + 947 mV*ms, n = 12; IN Spon: 6161 + 1193 mV*ms, IN Light: 5507 + 1823 mV*ms, n = 7; P = 0.8103, one‐way ANOVA).
Figure 3. Activation of SST INs is sufficient to initiate LLDs.
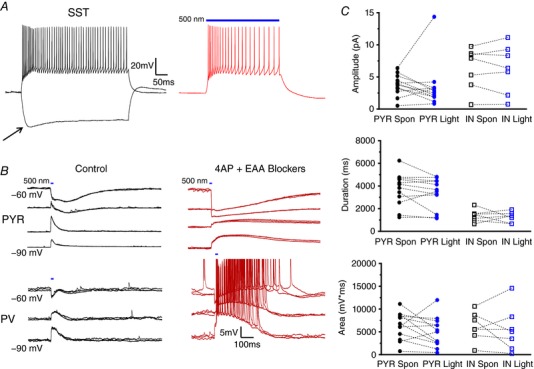
A, example traces of SST IN firing properties elicited by current injection (left) or light activation (right). Arrow indicates I h sag typical of SST cells. B, representative traces displaying activity evoked by a 10 ms light pulse in drug‐free aCSF (left) and after 4AP + EAA blockers wash‐on (right). Activity recorded in PYRs (top) and INs (bottom) was similar, although spontaneous APs were more frequently observed in INs. C, quantitative analysis comparing amplitude, duration and AUC of light‐evoked events with those occurring spontaneously. Bars indicate the timing and duration of light pulses. Each symbol represents an individual cell. Error bars are the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
The above results indicate that selective activation of SST:ChR INs is sufficient to evoke LLDs. We next aimed to determine the necessity of SST IN activation for LLD initiation using mice with genetically encoded expression of the proton pump Arch restricted to SST‐positive cells. Restriction of Arch expression to SST cells was achieved using the cre‐lox system, as with ChR. In whole‐cell recordings from physiologically identified SST cells, light activation of Arch produced a membrane hyperpolarization (Fig. 4 A, left) sufficient to block AP generation (Fig. 4 A, right). Light‐activation did not significantly affect evoked synaptic responses in PYRs or PV INs in drug‐free aCSF (Fig. 4 B, left). Specimen records of LLDs evoked by electrical stimulation in the presence or absence of whole‐field illumination are shown in Fig. 4 B (right). Responses were reduced during Arch activation. Events recorded with or without concurrent SST inactivation are quantified in Fig. 4 C, revealing that light‐mediated inhibition of SST INs caused a significant reduction of LLD amplitude in INs (Light Off: 10.98 + 1.08 mV, Light On: 7.63 + 0.90 mV; n = 18, P < 0.05, one‐way ANOVA with Tukey's post hoc test) but not PYRs (Light Off: 5.94 + 0.71 mV, Light On: 4.31 + 0.59 mV; n = 18, P > 0.05, one‐way ANOVA with Tukey's post hoc test). Conversely, LLD duration was significantly reduced in PYRs (Light Off: 1268 + 128 ms, Light On: 832 + 805 ms; n = 18, P < 0.05, one‐way ANOVA with Tukey's post hoc test) but not INs (Light Off: 861 + 65 ms, Light On: 6075 + 55 ms; n = 18, P > 0.05, one‐way ANOVA with Tukey's post hoc test). LLD AUC was reduced in all cell types recorded (PYR Light Off: 3186 + 392 mV*ms, PYR Light On: 1610 + 255 mV*ms; IN Light Off: 5095 + 532 mV*ms, IN Light On: 2668 + 332 mV*ms; P < 0.0001, one‐way ANOVA with Tukey's post hoc test).
Figure 4. Suppression of SST IN activity reduces evoked LLD magnitude.

A, example traces of membrane hyperpolarization induced by light activation of Arch (left) and superimposed traces showing SST cell responses to a depolarizing current injection with and without light activation (right). The SST IN was hyperpolarized by illumination and AP initiation blocked. B, superimposed traces of electrically evoked activity recorded from a PYR (top) and IN (bottom) with or without concurrent slice illumination in control aCSF (left) and after 4AP + EAA blockers wash‐on (right). Arrows indicate the time of stimulation. C, light inactivation of SST INs significantly reduced the amplitude, duration and AUC of evoked LLDs. * P < 0.05, Tukey's post hoc test. Bars indicate the timing and duration of light pulses. Each shape represents an individual cell. Error bars are the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
Role of PV INs in LLDs
We next assessed the contribution of PV INs to the generation of cortical GABAergic synchrony. As performed with SST INs, restricted expression of ChR to PV cells was employed to evaluate the sufficiency of PV INs to generate LLDs. As shown in Fig. 5 A, cell type‐specific expression of ChR in PV‐positive neurons was functionally confirmed by the induction of light‐induced membrane depolarizations and consequent AP firing in PV cells (Fig. 5 A, bottom) but not PYRs (Fig. 5 B, top trace) or SST INs. PV INs were differentiated from SST cells by their hallmark fast‐spiking firing properties and lower input resistance, and were visually identified by their expression of YFP. Figure 5 B shows specimen records of light‐induced responses in a PYR (top) and a PV IN (bottom) in the presence of 4AP + EAA blockers. Light‐activation of PV INs was sufficient to produce LLDs with amplitudes equivalent to spontaneous events in all cell types (Fig. 5 C) (PYR Spon: 5.69 + 0.92 mV, PYR Light: 3.28 + 0.56 mV, n = 8; IN Spon: 6.13 + 0.67 mV, IN Light: 4.82 + 1.06 mV, n = 6; P = 0.0831, one‐way ANOVA), durations (PYR Spon: 4032 + 300 ms, PYR Light: 3985 + 188 ms, n = 8; IN Spon: 2152 + 187 ms, IN Light: 1921 + 289 ms, n = 6; P < 0.0001, one‐way ANOVA with Tukey's post hoc test) and AUCs (PYR Spon: 13034 + 1921 mV*ms, PYR Light: 7414 + 1113 mV*ms, n = 8; IN Spon: 12268 + 2085 mV*ms, IN Light: 11221 + 3190 mV*ms, n = 6; P = 0.2076, one‐way ANOVA). Of note, and as shown in Fig. 5 B, kinetics of the light‐induced depolarization of opsin‐expressing cells could be readily discriminated from the slower onset LLD because a return towards baseline can be observed after light offset before the LLD peak amplitude.
Figure 5. PV IN activation is sufficient to initiate LLDs.
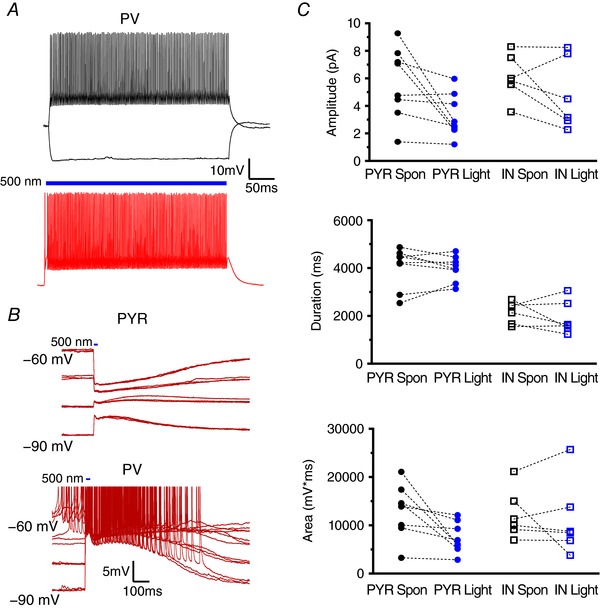
A, example traces of PV IN firing properties elicited by current injection (upper) or light activation (lower). B, representative traces displaying activity evoked, at arrow, by a 10 ms light pulse after 4AP + EAA blockers wash‐on. Activity recorded in PYRs (upper) and INs (lower) was comparable. C, quantitative comparison of the amplitude, duration and AUC of light‐evoked and spontaneously occurring LLDs. Bars indicate the timing and duration of light pulses. Each symbol represents an individual cell. Error bars are the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
Experiments were subsequently performed in slices from animals with Arch expression driven by the PV promoter to determine the necessity of PV INs for the generation of LLDs. Under whole‐cell recording conditions, slice illumination induced a membrane hyperpolarization in PV cells (Fig. 6 A, left) but not PYRs or SST INs (Fig. 6 B, left). Light activation was sufficient to block AP firing (Fig. 6 A, right). Specimen records of evoked synaptic potentials in a PYR at two membrane potentials under control conditions are shown in Fig. 6 B (top left). Responses with light on and off are shown superimposed. No significant difference was observed. Similar results were seen in recordings from SST INs (Fig. 6 B, bottom left). In the presence of 4AP + EAA blockers, comparison of electrically evoked events with or without simultaneous light‐induced PV IN inactivation revealed that LLDs are almost abolished when PV INs are silenced during event generation in all cell types (Fig. 6 C) (Amplitude – PYR Light Off: 6.84 + 0.55 mV, PYR Light On: 2.48 + 0.31 mV, n = 14; IN Light Off: 11.04 + 0.89 mV, IN Light On: 4.45 + 1.25 mV, n = 6; P < 0.0001, one‐way ANOVA with Tukey's post hoc test; Duration – PYR Light Off: 3536 + 197 ms, PYR Light On: 951 + 19 ms, n = 14; IN Light Off: 1846 + 379 ms, IN Light On: 710 + 109 ms, n = 6; P < 0.0001, one‐way ANOVA with Tukey's post hoc test; AUC – PYR Light Off: 8753 + 1108 mV*ms, PYR Light On: 940 + 148 mV*ms, n = 14; IN Light Off: 7463 + 1371 mV*ms, IN Light On: 1447 + 584 mV*ms, n = 6; P < 0.0001, one‐way ANOVA with Tukey's post hoc test).
Figure 6. Inactivation of PV INs prevents LLD initiation.
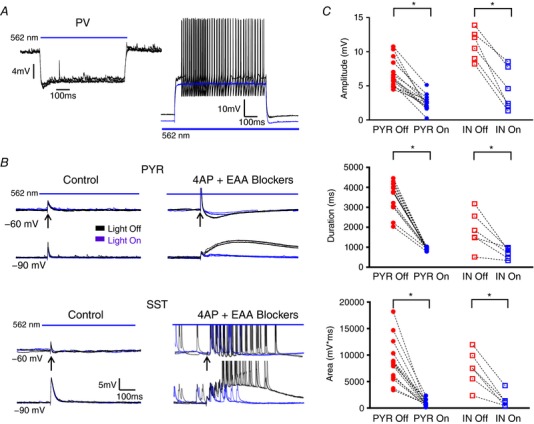
A, example traces of membrane hyperpolarization induced by light activation of Arch (left) and superimposed traces showing PV cell response to a depolarizing stimuli with and without concurrent tissue illumination (right). Arch activation hyperpolarized the PV IN and blocked AP generation. B, representative traces of electrically evoked activity recorded from a PYR (top) and IN (bottom) with and without concurrent slice illumination in control aCSF (left) and after 4AP + EAA blockers wash‐on (right). C, light inactivation of PV INs significantly reduced the amplitude, duration and AUC of evoked activity. * P < 0.05, Tukey's post hoc test. Bars indicate the timing and duration of light pulses. Arrows indicate time of electrical stimulation. Each shape represents an individual cell. Error bars are the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
Effects of altering activity in VIP INs
SST and PV cells provide robust inhibition of PYRs, whereas VIP‐expressing cells tend to facilitate disinhibition of PYRs via synaptic targeting of other INs, particularly SST‐positive cells (von Engelhardt et al. 2007; Lee et al. 2013; Pfeffer et al. 2013; Pi et al. 2013). As such, optogenetic manipulation of VIP‐positive cells was employed to determine the effect of this potential disinhibition on LLD generation. Cre‐dependent expression of ChR in VIP‐positive cells was confirmed via whole‐cell recordings from VIP INs (Fig. 7 A). As described above for PV and SST neurons, light activation of ChR in VIP INs produced a membrane depolarization with superimposed APs. Specimen records of LLDs in response to electrical stimulation or light activation of VIP:ChR are shown superimposed in Fig. 7 B. By contrast to SST and PV cells, activation of VIP INs alone was not sufficient to generate an LLD (Amplitude – PYR Spon: 3.49 + 0.28 mV, PYR Light: 0.77 + 0.15 mV, n = 10; IN Spon: 6.74 + 1.13 mV, IN Light: 0.93 + 0.21 mV, n = 10; P < 0.0001, one‐way ANOVA with Tukey's post hoc test; Duration – PYR Spon: 3049 + 283 ms, PYR Light: 450 + 68 ms, n = 10; IN Spon: 1486 + 170 ms, IN Light: 623 + 110 ms, n = 10; P < 0.0001, one‐way ANOVA with Tukey's post hoc test; AUC – PYR Spon: 3877 + 552 mV*ms, PYR Light: 153 + 41 mV*ms, n = 10; IN Spon: 3355 + 600 mV*ms, IN Light: 217 + 74 mV*ms, n = 10; P < 0.0001, one‐way ANOVA with Tukey's post hoc test) (Fig. 7 C).
Figure 7. VIP IN activation is not sufficient for the initiation of LLDs.
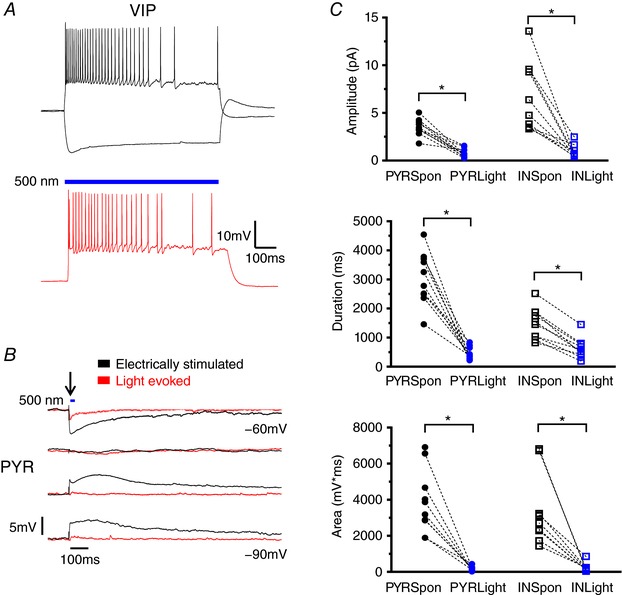
A, example traces of VIP IN firing properties elicited by current injection (top) or light activation (bottom). B, representative traces comparing electrically evoked activity with activity evoked, at arrow, by a 10 ms light pulse after 4AP + EAA blockers wash‐on in a PYR. Black arrow indicates the time of stimulation. C, the amplitude, duration and AUC of activity evoked by VIP IN activation were significantly smaller than spontaneous LLDs. Blue bars indicate the timing and duration of light pulses. * P < 0.05, Tukey's post hoc test. Each shape represents an individual cell. Error bars are the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 8 A illustrates the slice illumination‐induced membrane hyperpolarization in VIP cells expressing Arch. The light activated hyperpolarization was sufficient to block AP firing (Fig. 8 A, lower). As shown in Fig. 8 C, inhibition of VIP INs via light activation of Arch did not significantly attenuate LLD amplitude (PYR Light Off: 5.72 + 0.73 mV, PYR Light On: 4.39 + 0.76 mV, n = 11; IN Light Off: 7.87 + 1.89 mV, IN Light On: 5.90 + 1.44 mV, n = 6; P = 0.2034, one‐way ANOVA) or duration (PYR Light Off: 3257 + 306 ms, PYR Light On: 2519 + 284 ms, n = 11; IN Light Off: 1046 + 191 ms, IN Light On: 651 + 88 ms, n = 6; P < 0.0001, one‐way ANOVA with Tukey's post hoc test) in either PYRs or INs, but resulted in a significant reduction of the AUC in PYRs (PYR Light Off: 6869 + 912 mV*ms, PYR Light On: 3439 + 673 mV*ms, n = 11; IN Light Off: 2873 + 637 mV*ms, IN Light On: 1576 + 344 mV*ms, n = 6; P = 0.0003, one‐way ANOVA with Tukey's post hoc test).
Figure 8. Suppression of VIP IN activity minimally alters evoked LLD magnitude.
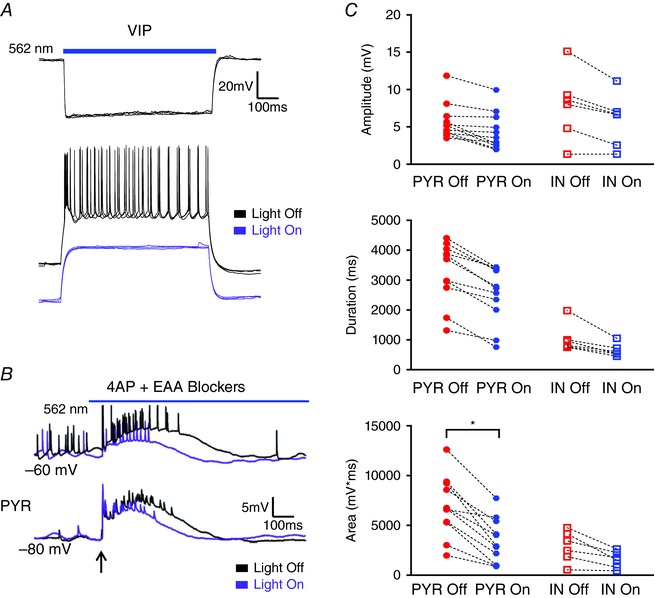
A, example traces of membrane hyperpolarization induced by light activation of Arch (top) and superimposed traces showing VIP cell response to a depolarizing current pulse with and without Arch activation (bottom). Light activation hyperpolarized the VIP IN and blocked AP generation. B, superimposed traces from a PYR cell comparing electrically evoked activity recorded with and without concurrent light illumination in the presence of 4AP + EAA blockers. Arrow indicates the time of stimulation. C, light inactivation of VIP INs significantly reduced only the AUC of evoked LLDs in PYRs. * P < 0.05, Tukey's post hoc test test. Blue bars indicate the timing and duration of light pulses. Each shape represents an individual cell. Error bars are the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
LLDs evoked at offset of Arch‐induced hyperpolarizations
Figure 9 A shows examples of evoked LLDs with and without Arch activation. In both SST:Arch (Fig. 9 A, upper) and PV:Arch (Fig. 9 A, lower) animals, light offset triggered a LLD in all cell types, presumably as a result of rebound excitation following release from light‐induced inhibition. These rebound LLDs were consistently activated with a latency less than 100 ms following light offset and had similar magnitude to spontaneous LLDs in both groups. However, the amplitude and AUC of responses to light offset were significantly larger in PV:Arch animals compared to SST:Arch animals (Amplitude – SST: 80 + 8%, n = 42, PV: 132 + 14%, n = 19; P = 0.0014, two‐tailed t test; Duration – SST: 149 + 18%, n = 42, PV: 123 + 28%, n = 19; P = 0.4167, two‐tailed t test; AUC – SST: 106 + 16%, n = 42, PV: 193 + 37%, n = 19; P = 0.0157, two‐tailed t test, 1 – β = 0.82) (Fig. 9 B). As shown in Fig. 9 C, rebound LLDs were also triggered at light offset without concurrent electrical stimulation, suggesting that rebound responses are light‐driven and not an artefact of prior tissue stimulation. Rebound LLDs in cells from VIP‐Arch animals (n = 16) were smaller than those in cells from PV:Arch and SST:Arch animals and even absent in some cases (quantified in Fig. 10 C), presumably as a result of the lack of an initial Arch effect on evoked network activity.
Figure 9. Light offset in Arch expressing INs is associated with triggering of LLDs.
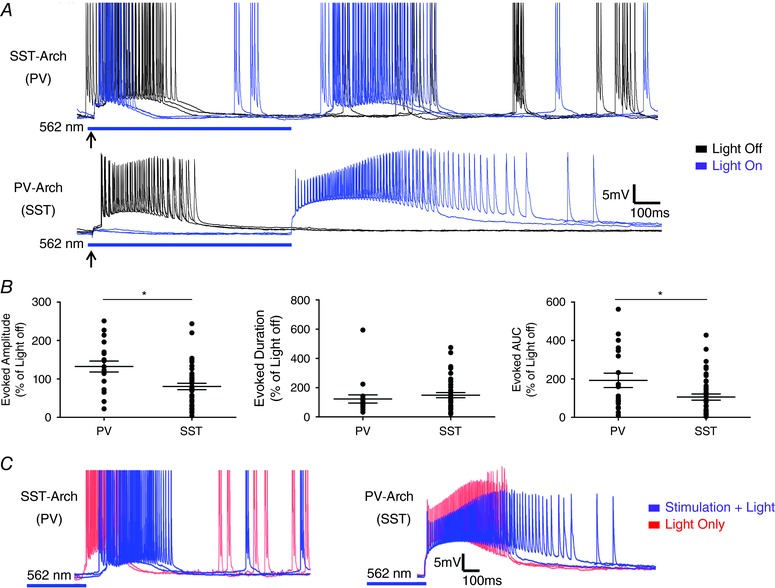
A, representative traces from SST‐Arch (top) and PV‐Arch (bottom) animals showing the generation of LLDs upon light offset. Upper traces show responses from a PV IN in a slice from a SST‐Arch animals, whereas lower traces show responses from a SST IN in a slice from a PV‐Arch animal. B, comparison of rebound LLDs induced by release of INs from light‐driven hyperpolarization. The amplitude and AUC of rebound LLDs evoked by PV IN synchronization were larger than those of SST IN induced activity. C, LLDs were evoked upon light offset in trials with and without electrical stimulation. * P < 0.05, two‐tailed t test. Each shape represents an individual cell. Error bars are the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 10. Distinct contribution of IN subtypes to LLD initiation.
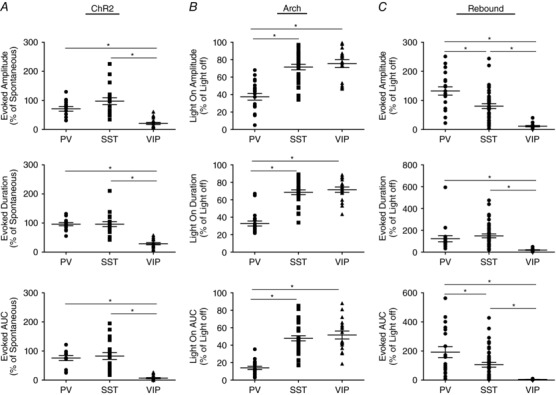
A, activation of VIP INs elicited activity of significantly smaller amplitude, duration and AUC compared to both SST and PV INs. B, suppression of VIP INs produced attenuation of the amplitude, duration and AUC of evoked activity comparable to SST INs but significantly smaller than PV INs. C, quantification of rebound activity evoked by Arch offset revealed that VIP IN synchronization produced activity of significantly smaller amplitude, duration and AUC than both SST and PV INs. * P < 0.05, Tukey's post hoc test. Each shape represents an individual cell. Error bars are the mean ± SEM.
Comparison of cell type‐specific effects
To directly compare the observed effects of SST, PV and VIP INs, the amplitude, duration and AUC of light‐driven responses were assessed relative to the properties of spontaneous LLDs recorded in each cell type (Fig. 10). As shown in Fig. 10 A, VIP INs were significantly less effective than both SST and PV cells in initiating LLDs (Amplitude – SST: 71 + 8%, n = 19; PV: 97 + 12%, n = 14; VIP: 21 + 4%, n = 20; P < 0.0001, one‐way ANOVA with Tukey's post hoc test; Duration – SST: 96 + 5%, n = 19; PV: 96 + 9%, n = 14; VIP: 29 + 4%, n = 20; P < 0.0001, one‐way ANOVA with Tukey's post hoc test; AUC – SST: 76 + 9%, n = 19; PV: 83 + 13%, n = 14; VIP: 7 + 2%, n = 20; P < 0.0001, one‐way ANOVA with Tukey's post hoc test). Silencing of SST‐expressing and VIP‐expressing cells produced a significantly smaller attenuation of LLDs compared to PV INs (Amplitude – SST: 72 + 3%, n = 36; PV: 37 + 4%, n = 20; VIP: 75 + 5%, n = 17; P < 0.0001, one‐way ANOVA with Tukey's post hoc test; Duration – SST: 69 + 3%, n = 36; PV: 33 + 3%, n = 20; VIP: 72 + 3%, n = 17; P < 0.0001, one‐way ANOVA with Tukey's post hoc test; AUC – SST: 48 + 3%, n = 36; PV: 14 + 2%, n = 20; VIP: 52 + 5%, n = 17; P < 0.0001, one‐way ANOVA with Tukey's post hoc test). VIP silencing produced an effect comparable to that of SST IN inactivation (Amplitude: P > 0.05, one‐way ANOVA with Tukey's post hoc test; Duration: P > 0.05, one‐way ANOVA with Tukey's post hoc test; AUC: P > 0.05, one‐way ANOVA with Tukey's post hoc test). In addition, as shown in Fig. 10 C, the amplitude of rebound activity triggered by light offset in VIP:Arch animals was significantly smaller than that of both SST:Arch and PV:Arch animals (VIP: 11 + 2%; n = 16, P < 0.0001, one‐way ANOVA with Tukey's post hoc test), duration (VIP: 19 + 3%; n = 16, P = 0.0003, one‐way ANOVA with Tukey's post hoc test) and AUC (VIP: 5 + 1%; n = 16, P < 0.0001, one‐way ANOVA with Tukey's post hoc test).
Discussion
We employed genetically encoded opsins to investigate the contribution of distinct IN classes to the generation of synchronous cortical GABAergic network activity. Using cell type‐specific activation of INs via ChR, we have shown that synchronized activation of either the cortical SST or PV IN population is sufficient to generate LLDs, whereas activation of VIP‐expressing INs is not. Cell type‐specific hyperpolarization of INs mediated by light activation of genetically encoded Arch showed that inactivation of SST or VIP INs resulted in ∼40% attenuation of LLDs, whereas inhibition of PV cells essentially blocked initiation of LLDs. Moreover, the synchronized depolarization of IN populations at Arch offset produced an LLD in SST:Arch and PV:Arch animals but not VIP:Arch animals. In conjunction with the data from VIP:ChR animals, this suggests that activation of VIP‐positive cells is insufficient to drive synchronous GABAergic network activity.
Within the cortex, INs have been shown to play a critical role in controlling PYR spike timing, generating cortical rhythms and synchronizing network activity (Pouille & Scanziani, 2001; Weher & Zadar, 2003; Haider & McCormick, 2009). A precise balance of excitation and inhibition (E:I balance) is required to permit information transfer at the same time as preventing unbridled excitation (McBain & Fisahn, 2001; Sun et al. 2006; Yizhar et al. 2011). As such, changes in neuronal excitability (as modelled in the present study by application of 4‐AP) that disrupt this E:I balance are assumed to underlie the development of epileptiform activity (Cossart et al. 2001; Noebels, 2003; Cobos et al. 2005; Trevelyan et al. 2006; Ascoli et al. 2008).
Aberrant cortical activity may arise from a shift in the E:I balance to favour either excitation or inhibition. A reduction in GABAergic IN output and subsequent inhibition of PYRs shifts the cortical E:I balance to favour excitation, resulting in an increase in PYR output (Bradford, 1995; Olsen & Avoli, 1997). This paradigm of unimpeded excitation is the most commonly considered source of epileptiform activity, generating much investigation into mechanisms underlying PYR excitability and disinhibition. Conversely, however, as seen in the 4‐AP + EAA blockers model used in the present study, an increase in IN excitability, which shifts the E:I balance to favour inhibition, can also produce aberrant cortical synchrony facilitated by the synaptic, and possibly ephaptic, coupling of IN networks. Pharmacological isolation of GABAergic networks was used in the present study to determine the contribution of distinct IN classes to cortical network activity and to clarify potential mechanisms underlying the generation of synchronous GABAergic activity.
Differences in the contribution of IN subclasses
Given that independent activation of SST or PV INs elicited similar LLDs, the finding that inactivation of PV cells has a greater effect on the generation of synchronous network activity is intriguing. One possible explanation for this discrepancy could be a relatively weaker silencing of SST INs expressing Arch. Light‐evoked hyperpolarizations were smaller in SST than PV INs, probably as a result of differences in input resistance. However, no correlation between the amplitude of light‐induced hyperpolarization and the degree of LLD attenuation was observed in either cell type. Moreover, VIP INs displayed considerably larger voltage deflections upon Arch activation compared to both PV and SST INs but were no more effective than SST INs at inhibiting the generation of LLDs. Together, these findings suggest thatg channel expression or conductance is probably not the defining factor. PV INs have been reported to be slightly more abundant than SST INs in the cortex (Lee et al. 2010; Miyoshi et al. 2010; Xu et al. 2010), making it possible that differences between cell types are partly a result of the number of cells activated. Cell abundance may partially contribute to the observed effects, although it would be expected that activation of VIP INs, which account for ∼25% of layer II/III INs (Vucurovic et al. 2010; Rudy et al. 2011; Zeisel et al. 2015), would produce a more robust response than that observed if simply the number of cells activated was a significant factor in determining the contribution of IN populations to LLD generation.
A third, and perhaps most convincing, explanation for the reported differences is the distinct synaptic connectivity characteristics of each cell type (Fig. 11). Fast‐spiking basket cells, which account for a vast majority of the PV‐positive cell population, densely innervate the soma and perisomatic regions of their postsynaptic target neurons (Kawaguchi & Kubota, 1993; Conde et al. 1994), allowing them to tightly regulate spike initiation (Pinto et al. 2000; Miller et al. 2001; Pouille & Scanziani, 2001; Lawrence & McBain, 2003; Gabernet et al. 2005; Cruikshank et al. 2007). By contrast, SST‐expressing cells generally form synapses on the dendrites of postsynaptic cells (Wang et al. 2004; Chiu et al. 2013; Hioki et al. 2013), which serves to regulate the somatopetal propagation of dendritic signals (Chen et al. 2015; Urban‐Ciecko et al. 2016). Moreover, a majority of PV IN efferents target other PV INs in addition to PYRs (Pfeffer et al. 2013; Jiang et al. 2015; Walker et al. 2016), whereas SST‐expressing cells predominantly innervate PYRs and dissimilar IN subtypes rather than other SST INs (Cottam et al. 2013; Pfeffer et al. 2013; Jiang et al. 2015; Walker et al. 2016). These factors establish PV INs as a prime candidate for driving cortical network activity.
Figure 11. Schematic representation of synaptic connectivity of cortical layer II/III interneurons.

Simplified diagram illustrating the synaptic connectivity patterns of PV, SST and VIP INs in layer II/III of the neocortex. Black lines indicate neuronal efferents with open circles representing synaptic terminals. The weight of black lines represents the relative prevalence of the indicated connection, with larger lines representing more prevalent connections. Dashed lines indicate output to INs of the same class. Note that PV efferents are somatic targeting, whereas SST and VIP INs typically synapse onto the dendrites of their postsynaptic targets. [Color figure can be viewed at wileyonlinelibrary.com]
In the 4AP + EAA blockers model, the Cl− equilibrium potential is assumed to be shifted, resulting in GABA‐activated currents being depolarizing (Aram et al. 1991; Morris et al. 1996; Lamsa & Kaila, 1997; Hamidi et al. 2015). Although the LLD reversal potential reported in the present study (Fig. 1 D) is near the normal GABA reversal, it should be noted those measurements were calculated from cells in which the intracellular ion concentrations were externally determined by the internal pipette solution used to dialyse the cells. Experiments using the perforated patch method, which does not disrupt intracellular ion concentrations, could be employed to more accurately assess LLD reversal. In an environment where GABA is depolarizing, the somatic targeting of PV cells would make them uniquely positioned to activate their postsynaptic targets, thereby recruiting those cells for the initiation of LLDs. Feed‐forward activation of PV cells by other PV Ins, as well as SST Ins, would further facilitate widespread network activity. With respect to the data reported in the present study, it is possible that the sufficiency of SST INs to generate LLDs when driven by light activation of ChR could be a result of feed‐forward activation of PV INs. In that case, inactivation of SST INs via Arch would not be expected to abolish LLDs because PV INs are still activated. The partial attenuation of LLDs by SST inhibition could then be attributed to a reduction in feed‐forward activation of SST INs by PV cells.
The failure of VIP IN activation to evoke an LLD further suggests that the diverse synaptic characteristics of IN populations underlie distinct contributions to network activity. Of the IN classes investigated in the present study, VIP INs are generally assumed to be the most excitable (Cauli et al. 2000; Lee et al. 2010; Miyoshi et al. 2010). This is supported by VIP INs displaying the highest input resistance in our recordings (SST: 177.5 ± 9.8 mΩ, n = 18; PV: 119.6 ± 7.4 mΩ, n = 42; VIP: 392.4 ± 19.9 mΩ, n = 17) and is also reflected in our observation that VIP INs displayed the largest voltage deflections upon activation of opsins (Fig. 8 A). Together with the aforementioned abundance of VIP INs, this suggests that the cell type‐specific contributions of INs are determined by a property other than their prevalence or excitability. With the exception of a small population of VIP‐positive (and PV‐negative) basket cells, VIP INs typically synapse onto the dendrites of their postsynaptic targets (Kawaguchi & Kubota, 1996; Kawaguchi & Kubota, 1997). VIP INs most often target SST‐expressing cells, with a much lower innervation of PV‐expressing cells being observed (von Engelhardt et al. 2007; Lee et al. 2013; Pfeffer et al. 2013; Pi et al. 2013). Accordingly, activation of VIP INs would be expected to produce less feed‐forward activation of PV cells compared to SST INs, accounting for the insufficiency of light‐driven VIP activation to generate LLDs. Marginal innervation of VIP INs by PV cells has been reported (Jiang et al. 2015; Walker et al. 2016); thus, the partial reduction in LLD magnitude upon VIP IN inhibition could be a result of reduced feed‐forward activation mediated by PV INs as proposed for SST cells. Future experiments in which the activity of multiple IN populations can be simultaneously modulated (e.g. activation of SST INs with concurrent PV IN silencing) are necessary to confirm this PV‐driven di‐synaptic paradigm of LLD generation.
INs in epilepsy
An increase in the excitability of GABAergic cells has historically been considered to decrease network activity, producing an anti‐epileptic effect. However, the data reported in the present study, along with other recent work characterizing the contribution of INs to seizure activity (Ellender et al. 2014; Sessolo et al. 2015; Yekhlef et al. 2015; Khoshkhoo et al. 2017; Shiri et al. 2017), suggest that the role of inhibitory networks is complex. INs are known to facilitate the generation of rhythmic activity within and between brain regions (McBain & Fisahn, 2001; Whittington & Traub, 2003; Traub et al. 2004; Bartos et al. 2007; Haider & McCormick, 2009). Synchronous GABAergic activity has been temporally linked to the onset of epileptiform activity (Ziburkus et al. 2006; Toyoda et al. 2015; Levesque et al. 2016), and also has been proposed as a potential mechanism underlying the generation of synchronous network discharges characteristic of epilepsy (de Curtis & Avoli, 2016). More work is necessary to fully characterize the role of GABA networks in epilepsy. Specifically, the interaction between GABA and glutamate‐driven activity must be dissected to identify the role of specific IN populations in the propagation and rhythmicity of synchronous network activity.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
ASB and JJH conceived and designed the experiments. ASB collected, assembled and analysed the data. ASB and JJH interpreted the data. ASB drafted the manuscript. ASB and JJH critically revised the article. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Acknowledgements
This research was supported by National Institutes of Health grants NS090041 and P30 NS047466. We thank K. Alison Margolies for technical assistance.
Biography
Andrew Bohannon received his PhD from the University of Alabama at Birmingham, studying the role of distinct interneuron subtypes in regulating cortical network activity in the laboratory of John Hablitz. He is currently employing his training in electrophysiology and molecular techniques as a postdoctoral scholar in the laboratory of Eiman Azim at the Salk Institute for Biological Studies. Andrew is investigating the meso‐ and macro‐circuitry contributing to the control of skilled forelimb movement, with the ultimate goal of identifying key intervention points for the treatment of movement disorders and design of brain–machine interfaces for the control of neuroprostheses.

Edited by: Jaideep Bains & Katalin Toth
References
- Aram JA, Michelson HB & Wong RKS (1991). Synchronized GABAergic IPSCs recorded in the neocortex after blockade of synaptic transmission mediated by excitatory amino acids. J Neurophysiol 65, 1034–1041. [DOI] [PubMed] [Google Scholar]
- Ascoli GA, Alonso‐Nanclares L, Anderson SA, Barrionuevo G, Benavides‐Piccione R, Burkhalter A, Buzsaki G, Cauli B, Defelipe J, Fairen A, Feldmeyer D, Fishell G, Fregnac Y, Freund TF, Gardner D, Gardner EP, Goldberg JH, Helmstaedter M, Hestrin S, Karube F, Kisvarday ZF, Lambolez B, Lewis DA, Marin O, Markram H, Munoz A, Packer A, Peterson CC, Rockland KS, Rossier J, Rudy B, Somogyi P, Staiger JF, Tamas G, Thomson AM, Toledo‐Rodriguez M, Wang Y, West DC & Yuste R (2008). Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nature Rev Neurosci 9, 557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M & Perreault P (1987). A GABAergic depolarizing potential in the hippocampus disclosed by the convulsant 4‐aminopyridine. Brain Res 400, 191–195. [DOI] [PubMed] [Google Scholar]
- Avoli M, Perreault P, Olivier A & Villemure J‐G (1988). 4‐Aminopyridine induces a long‐lasting depolarizing GABA‐ergic potential in human neocortical and hippocampal neurons maintained in vitro. Neurosci Letters 94, 327–332. [DOI] [PubMed] [Google Scholar]
- Avoli M, Mattia D, Siniscalchi P, Perreault P & Tomaiuolo F (1994). Pharmacology and electrophysiology of a synchronous GABA‐mediated potential in the human neocortex. Neuroscience 62, 655–666. [DOI] [PubMed] [Google Scholar]
- Bartos M, Vida I & Jonas P (2007). Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci 8, 45–56. [DOI] [PubMed] [Google Scholar]
- Bradford HF (1995). Glutamate, GABA and Epilepsy. Prog Neurobiol 47, 477–511. [DOI] [PubMed] [Google Scholar]
- Cauli B, Porter JT, Tsuzuki K, Lambolez B, Rossier J, Quenet B & Audinat E (2000). Classification of fusiform neocortical interneurons based on unsupervised clustering. Proc Natl Acad Sci USA 97, 6144–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SX, Kim AN, Peters AJ & Komiyama T (2015). Subtype‐specific plasticity of inhibitory circuits in motor cortex during motor learning. Nat Neurosci 18, 1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CQ, Lur G, Morse TM, Carnevale NT, Ellis‐Davies GC & Higley MJ (2013). Compartmentalization of GABAergic inhibition by dendritic spines. Science 10, 759–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC & Rubenstein JL (2005). Mice lacking Dlx1 show subtype‐specific loss of interneurons, reduced inhibition and epilepsy. Nature Neurosci 8, 1059–1068. [DOI] [PubMed] [Google Scholar]
- Conde F, Lund JS, Jacobowitz DM, Baimbridge KG & Lewis DA (1994). Local circuit neurons immunoreactive for calretinin, calbindin D‐28k or parvalbumin in monkey prefrontal cortex: Distribution and morphology. J Comp Neurol 341, 95–116. [DOI] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan‐Perez A, Defelipe J, Ben‐Ari Y, Esclapez M & Bernard C (2001). Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci 4, 52–62. [DOI] [PubMed] [Google Scholar]
- Cottam JCH, Smith SL & Hausser M (2013). Target‐specific effects of somatostatin‐expressing interneurons on neocortical visual processing. J Neurosci 33, 19567–19578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Lewis TJ & Connors BW (2007). Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat Neurosci 10, 462–468. [DOI] [PubMed] [Google Scholar]
- de Curtis M & Avoli M (2016). GABAergic networks jump‐start focal seizures. Epilepsia 57, 679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellender TJ, Raimondo JV, Irkle A, Lamsa KP & Akerman CJ (2014). Excitatory effects of parvalbumin‐expressing interneurons maintain hippocampal epileptiform activity via synchronous afterdischarges. J Neurosci 34, 15208–15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang AG & Buchner A (2007). G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behavior Res Methods 39, 175–191. [DOI] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DE, Carandini M & Scanziani M (2005). Somatosensory integration controlled by dynamic thalamocortical feed‐forward Inhibition. Neuron 48, 315–327. [DOI] [PubMed] [Google Scholar]
- Haider B & McCormick DA (2009). Rapid neocortical dynamics: Cellular and network mechanisms. Neuron 62, 171–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi S, D'Antuono M & Avoli M (2015). On the contribution of KCC2 and carbonic anhydrase to two types of in vitro interictal discharge. Pflügers Arch 467, 2325–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry SHC, Jones EG, Emson PYR, Lawson DEM, Heizmann CW & Streit P (1989). Two classes of cortical GABA neurons defined by differential calcium binding protein immunoreactivities. Exp Brain Res 76, 467–472. [DOI] [PubMed] [Google Scholar]
- Hioki H, Okamoto S, Konno M, Kameda H, Sohn J, Kuramoto E, Fujiyama F & Kaneko T (2013). Cell type‐specific inhibitory inputs to dendritic and somatic compartments of parvalbumin‐expressing neocortical interneuron. J Neurosci 33, 544–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Shen S, Cadwell CR, Berens P, Sinz F, Ecker AS, Patel S & Tolias AS (2015). Principles of connectivity among morphologically defined cell types in adult neocortex. Science 350, aac9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazipov R, Leinekugel X, Khalilov I, Gaiarsa J‐L Ben‐Ari Y (1997). Synchronization of GABAergic interneuronal network in CA3 subfield of neonatal rat hippocampal slices. J Physiol 498, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y & Kubota Y (1996). Physiological and morphological identification of somatostatin‐ or vasoactive intestinal polypeptide‐containing cells among GABAergic cell subtypes in rat frontal cortex. J Neurosci 16, 2701–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y & Kubota Y (1993). Correlation of physiological subgroupings of nonpyramidal cells with parvalbumin‐ and calbindin D28k‐immunoreactive neurons in layer V of rat frontal cortex. J Neurophysiol 70, 387–396. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y & Kubota Y (1997). GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex 7, 476–486. [DOI] [PubMed] [Google Scholar]
- Khoshkhoo S, Vogt D & Sohal V (2017). Dynamic, cell‐type‐specific roles for GABAergic interneurons in a mouse model of optogenetically inducible seizures. Neuron 93, 291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Hattori R & Yui Y (1994). Three distinct subpopulations of GABAergic neurons in rat frontal agranular cortex. Brain Res 649, 159–173. [DOI] [PubMed] [Google Scholar]
- Lamsa K & Kaila K (1997). Ionic mechanisms of spontaneous GABAergic events in rat hippocampal slices exposed to 4‐aminopryidine. J Neurophysiol 78, 2582–2591. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ & McBain CJ (2003). Interneuron Diversity series: Containing the detonation‐feedforward inhibition in the CA3 hippocampus. Trends Neurosci 26, 631–640. [DOI] [PubMed] [Google Scholar]
- Lee S, Hjerling‐Leffler J, Zagha E, Fishell G & Rudy B (2010). The largest group of superficial neocortical GABAergic interneurons expresses ionotropic serotonin receptors. J Neurosci 30, 16796–16808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kruglikov I, Huang ZJ, Fishell G & Rudy B (2013). A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat Neurosci 16, 1662–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque M, Herrington R, Hamidi S & Avoli M (2016). Interneurons spark seizure‐like activity in the entorhinal cortex. Neurobiol Dis 87, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvel J, Papatheodoropoulos C, Siniscalchi A, Kurcewicz I, Pumain R, Devaux B, Turak B, Esposito V, Villemure J‐G & Avoli M (2001). GABA‐mediated synchronizations in the human neocortex: Elevations in extracellular potassium and presynaptic mechanisms. Neuroscience 105, 803–813. [DOI] [PubMed] [Google Scholar]
- Maccaferri G & McBain CJ (1996). The hyperpolarization‐activated current (Ih) and its contribution to pacemaker activity in rat CA1 hippocampal stratum oriens‐alveus interneurones. J Physiol 497, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain CJ & Fisahn A (2001). Interneurons unbound. Nature Neurosci 2:11‐23. [DOI] [PubMed] [Google Scholar]
- Michelson HB & Wong RKS (1991). Excitatory synaptic responses mediated by GABAA receptors in the hippocampus. Science 253, 1420–1423. [DOI] [PubMed] [Google Scholar]
- Miller LM, Escabi MA & Schreiner CE (2001). Feature selectivity and interneuronal cooperation in the thalamocortical system. J Neurosci 21, 8136–8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, Butt SJB, Takebayashi H & Fishell G (2007). Physiologically distinct temporal cohorts of cortical interneurons arise from telencephalic Olig2‐expressing precursors. J Neurosci 27, 7786–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, Hjerling‐Leffler J, Karayannis T, Sousa VH, Butt SJB, Battiste J, Johnson JE, Machold RP & Fishell G (2010). Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J Neurosci 30, 1582–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris ME, Obrocea GV & Avoli M (1996). Extracellular K+ accumulations and synchronous GABA‐mediated potentials evoked by 4‐aminopyridine in the adult rat hippocampus. Exp Brain Res 109, 71–82. [DOI] [PubMed] [Google Scholar]
- Noebels JL (2003). The biology of epilepsy genes. Annu Rev Neurosci 26, 599–625. [DOI] [PubMed] [Google Scholar]
- Olsen RW & Avoli M (1997). GABA and epileptogenesis. Epilepsia 38, 399–407. [DOI] [PubMed] [Google Scholar]
- Perreault P & Avoli M (1992). 4‐Aminopyridine‐induced epileptiform activity and a GABA‐mediated long‐lasting depolarization in the rat hippocampus. J Neurosci 12,104‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer CK, Xue M, He M, Huang ZJ & Scanziani M (2013). Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat Neurosci 16, 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi H, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ & Kepecs A (2013). Cortical interneurons that specialize in disinhibitory control. Nature 503, 521–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto DJ, Brumberg JC & Simons DJ (2000). Circuit dynamics and coding strategies in rodent somatosensory cortex. J Neurophysiol 83, 1158–1166. [DOI] [PubMed] [Google Scholar]
- Pouille F & Scanziani M (2001). Enforcement of temporal fidelity in pyramidal cells by somatic feed‐forward inhibition. Science 293, 1159–1163. [DOI] [PubMed] [Google Scholar]
- Rudy B, Fishell G, Lee S & Hjerling‐Leffler J (2011). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Develop Neurobio 71, 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessolo M, Marcon I, Bovetti S, Losi G, Cammarota M, Ratto GM, Fellin T & Carmignoto G (2015). Parvalbumin‐positive inhibitory interneurons oppose propagation but favor generation of focal epileptiform activity. J Neurosci 35, 9544–9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiri Z, Manseau F, Levesque M, Williams S & Avoli M (2015). Interneuron activity leads to initiation of low‐voltage fast‐onset seizures. Ann Neurol 77, 541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiri Z, Levesque M, Etter G, Manseau F, Williams S & Avoli M (2017). Optogenetic low‐frequency stimulation of specific neuronal populations abates ictogenesis. J Neurosci 37, 2999–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P & Klausberger T (2005). Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol 562, 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QQ, Huguenard JR & Prince DA (2006). Barrel cortex microcircuits: Thalamocortical feedforward inhibition in spiny stellate cells is mediated by a small number of fast‐spiking interneurons. J Neurosci 26, 1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Tanaka Y, Furuta T, Yanagawa Y & Kaneko T (2008). The effects of cutting solutions on the viability of GABAergic interneurons in cerebral cortical slices of adult mice. J Neurosci Meth 171, 118–125. [DOI] [PubMed] [Google Scholar]
- Toledo‐Rodriguez M, Blumenfeld B, Wu C, Luo J, Attali B, Goodman P, Markram H (2004). Correlation maps allow neuronal electrical properties to be predicted from single‐cell gene expression profiles in rat neocortex. Cereb Cortex 14, 1310–1327. [DOI] [PubMed] [Google Scholar]
- Toledo‐Rodriguez M, Goodman P, Illic M, Wu C & Markram H (2005). Neuropeptide and calcium‐binding protein gene expression profiles predict neuronal anatomical type in the juvenile rat. J Physiol 567, 401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda I, Fujita S, Thamattoor AK & Buckmaster PS (2015). Unit activity of hippocampal interneurons before spontaneous seizures in an animal model of temporal lobe epilepsy. J Neurosci 35, 6600–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Bibbig A, LeBeau FEN, Buhl EH & Whittington MA (2004). Cellular mechanisms of neuronal population oscillations in the hippocampus in vitro. Annu Rev Neurosci 27, 247–278. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Watson BO & Yuste R (2006). Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci 26, 12447–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uematsu M, Hirai Y, Karube F, Ebihara S, Kato M, Abe K, Obata K, Yoshida S, Hirabayashi M, Yanagawa Y & Kawaguchi Y (2008). Quantitative chemical composition of cortical GABAergic neurons revealed in transgenic venus‐expressing rats. Cereb Cortex 18, 315–330. [DOI] [PubMed] [Google Scholar]
- Urban‐Ciecko J, Fanselow EE & Barth AL (2016). Neocortical somatostatin neurons reversibly silence excitatory transmission via GABAb receptors. Current Biology 25, 722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervaeke K, Lorincz A, Nusser Z & Silver RA (2012). Gap junctions compensate for sublinear dendritic integration in an inhibitory network. Science 335, 1624–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Engelhardt J, Eliava M, Meyer AH, Rozov A & Monyer H (2007). Functional characterization of intrinsic cholinergic interneurons in the cortex. J Neurosci 27, 5633–5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucurovic K, Gallopin T, Ferezou I, Rancillac A, Chameau P, van Hooft JA, Geoffroy H, Monyer H, Rossier J & Vitalis T (2010). Serotonin 3A receptor subtype as an early and protracted marker of cortical interneuron subpopulations. Cereb Cortex 20, 2333–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker F, Mock M, Feyerabend M, Guy J, Wagener RJ, Schubert D, Staiger JF & Witte M (2016). Parvalbumin‐ and vasoactive intestinal polypeptide‐expressing neocortical interneurons impose differential inhibition on Martinotti cells. Nature Commun 7, 13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Toledo‐Rodriguez M, Gupta A, Wu C, Silberberg G, Luo J & Markram H (2004). Anatomical, physiological and molecular properties of Martinotti cells in the somatosensory cortex of the juvenile rat. J Physiol 561, 65–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr M & Zador AM (2003). Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature 426, 442–446. [DOI] [PubMed] [Google Scholar]
- Whittington MA & Traub RD (2003). Interneuron diversity series: inhibitory interneurons and network oscillations in vitro. Trends Neurosci 26, 676–682. [DOI] [PubMed] [Google Scholar]
- Williams SB & Hablitz JJ (2015). Differential modulation of repetitive firing and synchronous network activity in neocortical interneurons by inhibition of A‐type K+ channels and Ih . Front Cell Neurosci 9, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Roby KD & Callaway EM (2010). Immunochemical characterization of inhibitory mouse cortical neurons: three chemically distinct classes of inhibitory cells. J Comp Neurol 518, 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekhlef L, Breschi GL, Lagostena L, Russo G & Taverna S (2015). Selective activation of parvalbumin‐ or somatostatin‐expressing interneurons triggers epileptic seizurelike activity in mouse medial entorhinal cortex. J Neurophysiol 113, 1616–1630. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno L, Prigge M, Schneider F, Davidson T, O'Shea D, Sohal V, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P & Deisseroth K (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R (2005). Origin and classification of neocortical interneurons. Neuron 48, 524–527. [DOI] [PubMed] [Google Scholar]
- Zeisel A, Munoz‐Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, Marques S, Munguba H, He L, Betsholtz C, Rolny C, Castelo‐Branco G, Hjerling‐Leffler J & Linnarsson S (2015). Cell types in the mouse cortex and hippocampus revealed by single‐cell RNA‐seq. Science 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Uhlrich DJ & Lytton WW (1999). Properties of a hyperpolarization‐activated cation current in interneurons in the rat lateral geniculate nucleus. Neuroscience 92, 445–457. [DOI] [PubMed] [Google Scholar]
- Ziburkus J, Cressman JR, Barreto E & Schiff SJ (2006). Interneuron and pyramidal cell interplay during in vitro seizure‐like events. J Neurophysiol 95, 3948–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]